
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering π–π interactions for enhanced photoluminescent properties: unique discrete dimeric packing of perylene diimides†
Yu Shao‡
 a,
Guang-Zhong Yin‡b,
Xiangkui Renc,
Xinlin Zhanga,
Jing Wangd,
Kai Guoe,
Xiaopeng Lif,
Chrys Wesdemiotise,
Wen-Bin Zhangb,
Shuguang Yanga,
Meifang Zhu*a and
Bin Sun*a
a,
Guang-Zhong Yin‡b,
Xiangkui Renc,
Xinlin Zhanga,
Jing Wangd,
Kai Guoe,
Xiaopeng Lif,
Chrys Wesdemiotise,
Wen-Bin Zhangb,
Shuguang Yanga,
Meifang Zhu*a and
Bin Sun*a
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai, 201620, P. R. China. E-mail: zmf@dhu.edu.cn; sunbin@dhu.edu.cn; Fax: +86 21 6779 2855; Tel: +86 21 6779 2849
bKey Laboratory of Polymer Chemistry & Physics of Ministry of Education, Center for Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China
cSchool of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China
dSouth China Advanced Institute of Soft Matter Science and Technology, South China University of Science and Technology, Guangzhou 510640, P. R. China
eDepartment of Chemistry, The University of Akron, Akron, OH 44325-3909, USA
fDepartment of Chemistry, University of South Florida, 4202 East Fowler Ave, Tampa, Florida 33620, USA
First published on 20th January 2017
Abstract
This article reports the enhanced photoluminescent properties observed in the crystals of a cube-plane-cube shape amphiphile composed of perylene diimide (PDI) tethered with polyhedral oligomeric silsesquioxanes (POSS) at the imide position via either rigid or flexible linkages (POSS-PDI-POSS). The fluorescence quantum yields (Φf) of the conjugates in crystal (∼0.48) are much higher than that of the more amorphous samples (∼0.17) and that of the reference compound without pendant POSS cage (∼0.12). The enhancement was understood in three ways. First, their self-assembly behaviors in gaseous phase were studied by tandem mass spectrometry coupled with traveling wave ion mobility separation; second, their self-assembly behaviors in solution were revealed by concentration- and solvent-dependent experiments using UV/vis absorption and fluorescence spectrometry; and third, the molecular packing in crystals was determined by wide angle X-ray diffraction and transmission electron microscope. The results demonstrate the conjugates' strong tendency toward dimer formation in solution and show a unique molecular packing of discrete dimeric motifs in the solid states. It suggests that the high Φf of these conjugates can be attributed to the discontinuous π–π stacking, which results in a weaker electron interaction between the dimers, and the relatively fixed position of the PDI, which prohibits the common structural relaxation of PDIs π-planes.
Introduction
Nature displays a variety of supramolecular structures within the length scale of 1 to 100 nm by putting relatively simple building blocks together using strategically arranged, delicately balanced non-covalent interactions.1,2 A good example is the light harvesting complex of purple bacteria in which hundreds of chlorophyll and carotene molecules are organized in highly symmetric cyclic structures to function as efficient antennas for maximum solar energy absorption.3 Engineering non-covalent interactions, particularly π–π interactions, has thus become one of the major research topics in the rational design of supramolecular structures for controlling the self-assembly processes in device performance optimization.3Perylene diimides (PDIs) have attracted extensive attention in the past decades as model dye molecules and organic semiconductors.4–12 They possess ideal fluorescence quantum yields (Φf) close to one hundred percent and excellent light absorption capability, and exhibit quite versatile functionalities via modifications at the N-position. Typically, these modifications involve linear or branched long chain alkyls with or without chiral centers to tune the π–π interactions while keeping the chromophore intact,13 alternatively, they can be modified at the bay area to become either p-type or n-type materials.7,8,10 In the assembly, PDIs usually form single dimensional seamless packed chromophores due to the directional π–π interaction. High π-orbital overlapping thus exists between neighboring molecules along the column,12,14 which leads to novel properties.15 However, the π–π interaction often decreases their solubility in common organic solvents and also causes a significant drop in fluorescence quantum yield (Φf) in the solid state.16–18 It is of fundamental interest to engineer the π–π interactions in these systems to achieve good solution processibility, high Φf values, good charge transport, and other optoelectronic properties. Key approaches include the ways to interrupt the continuous π–π interaction in the solid state, to prevent them from aggregating into a long stack, and to investigate the type of molecular packing that are helpful in achieving better photoluminescent properties.
One of the approaches is to introduce bulky side chain groups to trap the aggregation of PDI at various intermediate stages from isolated single molecules to dimers and oligomers. Lin and Würthner reported that dicyclohexyl-1,7-(2,6-diphenyl-phenol)perylene diimide possesses an isolated state in crystal due to the presence of bulky 2,6-terphenyl units that can shield the stacking of PDI, and prohibit any π–π interactions among the PDI units. This molecule shows almost identical UV/vis absorption profile in the solid state and in solution with an absolute Φf of ∼37%.19 Frischmann and Würthner further reported that N,N′-bis(4-bromo-2,6-diisopropylphenyl)perylene diimide can maintain monomeric at high concentration and exhibit unity quantum yields in different polarity solvents, CHCl3, THF, toluene and CH3CN. This compound can be efficient, multigram-scale synthesized by their novel synthetic route, suggesting that steric hindrance is a valuable approach to access soluble, non-aggregating, highly luminescent PDI derivatives.20 Mizoshita and Inagaki recently reported a PDI derivative with siloxane-based bulky and flexible substituents at the 2,5,8,11-positions. In the solid state film, the molecules can arrange into a crystalline G-form with monomer band green emission constructed by isolated molecules due to the bulky siloxane-based substituent's shielding effect. They can also arrange into an amorphous O-form with orange excimer-band emission due to intermolecular π–π interactions and loss of the periodic molecular arrangement. The film also exhibits conventional fluorescence mechanochromic behavior. Its fluorescence emission responds to mechanical and thermal stimuli, and could be recovered by treatment with solvent vapors or by adding small amount of silicone oil.21 Würthner et al. programs the formation of discrete self-assembled PDI dimers by protecting one side of the perylene plane with oligo(ethylene glycol) bridge at 1,7-position of the “bay area”.22,23 Recently, they designed a novel PDI that tends to form well-defined discrete π–π stacked dimers rather than oligomeric π stacks in good solvents. The formation of even larger aggregates composed of only π-dimeric units in poor solvents, but no additional contacts between PDI π faces is involved. These physical states are confirmed by combining 1H NMR spectra, UV/vis experiments, DOSY NMR and ROESY NMR studies, and single-crystal structure analysis.24 One intriguing question still remains: what are the structure–property relationships when the PDI packing becomes discrete in solution and the solid state?
Polyhedral oligomeric silsesquioxanes (POSS) are nano-sized cage compounds possessing a rigid, well-defined molecular shape decorated with versatile surface functional groups.25–29 The rigid conformation imparts molecular shape persistence, while the peripheral groups make POSS soluble in various organic solvents. POSS has been extensively used in polymer nano-composites to enhance their stability and mechanical properties by reactive blending or physical blending. More recently, it was proposed as a prototype “nano-elements” in the construction of giant molecular shape amphiphiles.30,31 Its conjugation with PDI via either flexible or rigid linkages leads to cube-plane-cube shape amphiphiles (POSS-PDI-POSS, compounds 1 and 2 in Scheme 1).32 The one with a rigid phenylene spacer (compound 2) leads to a shape-persistent molecule. Crystalline nano-belts could be assembled from solution and the structure has shown that the global π–π interactions were suppressed and the conjugates exist largely as dimers stabilized by local π–π interactions between two molecules. While it is known that the properties of the chromophores in the solid state are governed by the spatial arrangement of the chromophores,33–35 the minute and intricate relationship between molecular packing in the solid states and their photoluminescent properties remains to be clarified, especially for those with the same chromophore and for the different packing scheme of the exact the same molecules. This is important for the future engineering of solid state optical devices.
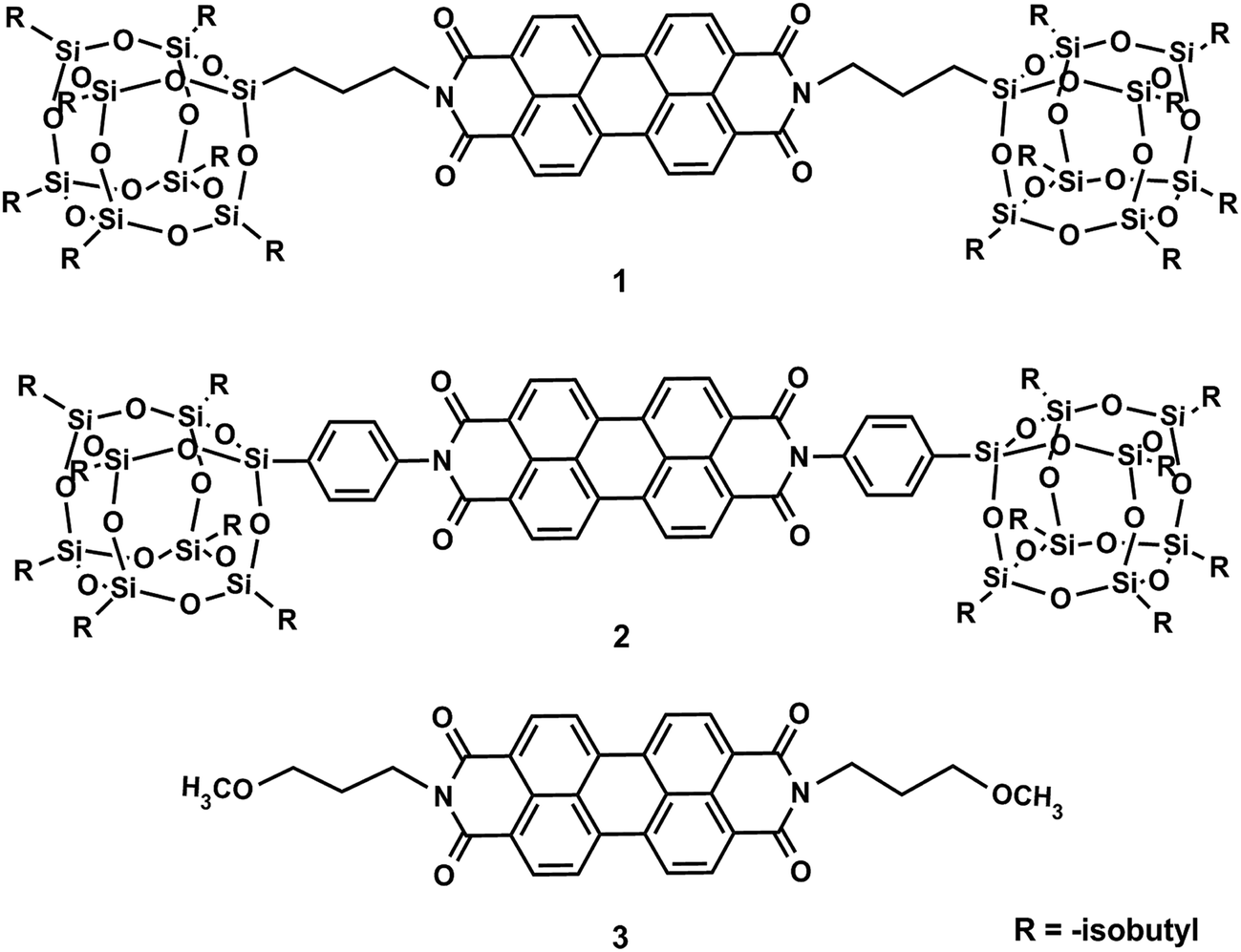 | ||
| Scheme 1 Chemical structures of the compounds. | ||
In this article, we report a systematic investigation on two POSS-PDI-POSS conjugates with rigid or flexible linkers (compounds 1 and 2, Scheme 1), compare their crystal structures and their self-assembly behaviors in gaseous phase, in solution, and in condensed state, and relate to the differences in their photoluminescent properties. In both cases, the presence of bulky POSS units has a striking effect on their self-assembly. The break of global continuous π–π stacking into discrete π–π dimers results in the high Φf of these conjugates in the solid states. As a reference, a PDI with short flexible linker (compound 3) was also designed and synthesized. Specially, we also compared the photoluminescent properties of the two compounds (1 and 2) in different packing scheme: the more ordered crystals and the more amorphous precipitated powders.
Results and discussion
Molecular design and synthesis
The molecular design of the conjugates was based on the following considerations: (1) linkage and (2) pendant. A POSS-tethered PDI connected with a rigid 1,4-phenylene linkage (compound 2) has been recently reported.32 The conjugate 2 shows a strong tendency of dimerization and a unique dimer packing scheme in crystals.32 Due to the rigid linkage, compound 2 is shape-persistent. It is thus of interest to study the effect of a more flexible linkage in between PDI and POSS on the final structure and material properties. The linkages are thus chosen to be a more flexible 1,3-propylene with similar length to the rigid one (compound 2) along the long axis of the molecule. To reveal the role of POSS cages, a control sample with the same linkage but no POSS units (compound 3) was also synthesized. Comparisons between these samples should illustrate unambiguously the effects of linkage and POSS.Compounds 1, 2 and 3 were synthesized using the conventional condensation methods as described previously.32 Compounds 1 and 2 are orange crystalline powders readily soluble in common organic solvents with solubility higher than 6 mg mL−1 in THF. This is much better than that of the PDI precursor, perylene dianhydride. In contrast, compound 3 lacks the POSS units and is only slightly soluble. All the compounds have been fully characterized to identify the chemical structure and purity. Compound 1 displays a weight loss plateau with the onset temperature 483 °C in thermo-gravimetric measurements, indicating its high thermal stability (Fig. S1†).
Analysis of the aggregation states in gaseous phase
To shed light on the structure of the aggregates, tandem mass spectrometry coupled with traveling wave ion mobility (TWIM) separation was utilized to determine the dominant aggregation species. Not only does it allow us to identify the dominant aggregated species, but also their relative amounts and the binding strength can be determined. The compound is first ionized from solution via electro-spray; collisionally activated disassociation (CAD) was then performed to destroy the specific aggregates. The resulting ionic species are then separated in ion mobility column according to their mass-to-charge ratios and sizes.Fig. 1a shows a full mass spectrum (MS) of compound 1. It is evident that monomer, dimer, trimer and tetramer with sodium ions can be identified. The strongest peak at m/z 2128 which could be a monomer with a single charge or a doubly-charged dimer. Fig. 1b illustrates the tandem mass spectrometry results for 1, obtained by breaking the dimeric species in the trap cell and subsequent ion mobility separation of the products. At relatively low collision energy, the undissociated doubly-charged dimeric species [2M·2Na+] is found to be the main species for ions at m/z 2128, while [M·Na+] is only slightly detected. No higher aggregates were detected. Upon increasing the collision energy from 6 to 45 eV, the signal of [2M·2Na+] is decreased and finally almost disappears at 42 eV; meanwhile, the signal of [M·Na+] fragment increases due to the dissociation of dimeric species. This result indicates that the dimers are not just a random cluster of two [M·Na+] but rather are held together by strong π–π interactions, and are not easily disassociated at the low collision CAD energy. It is remarkable that the π–π interaction in this case survives multiple collisions with center of mass energies as high as ∼0.78 eV. Therefore, similar to 2, compound 1 also favors the formation of dimers due to the strong π–π interaction and the steric hindrance between the adjacent bulky POSS units. It is further noted that the collision energy needed for complete dissociation of 2 (0.98 eV) is larger than that of 1 (0.78 eV), implying a stronger π–π interaction in 2. This may be due to the additional interaction from phenyl groups and/or because PDI planes can interact better with a rigid linker where the spatial confinement of the bulky POSS minimizes the interference with π–π interaction.
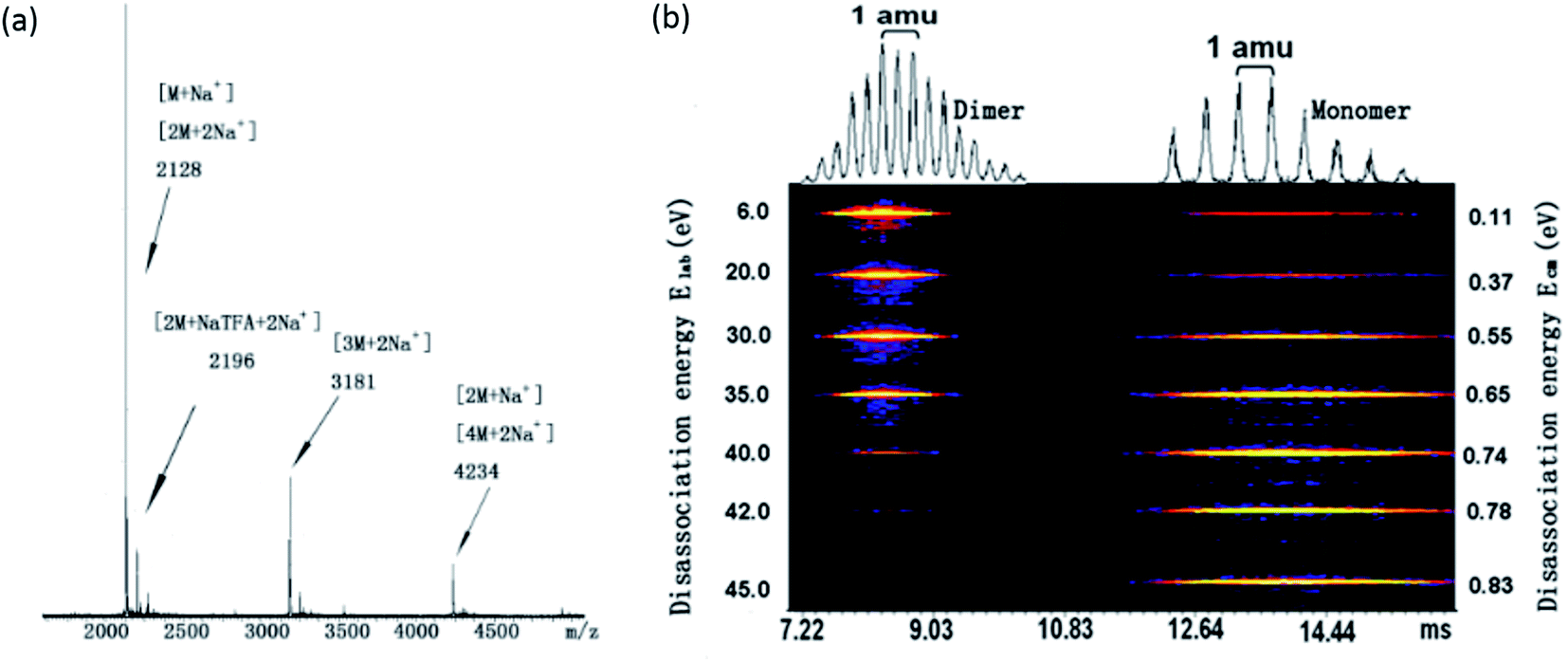 | ||
| Fig. 1 (a) Full mass spectrum of compound 1 with Na+ as the counter ion; (b) 2D ESI-TWIM-MS plot for m/z 2128, where brighter color indicates stronger intensity. Dimers disassociate into monomers with increasing CAD from 6 to 45 eV. | ||
The MS of compound 3 has been shown in Fig. S3† and its collision energy needed for complete disassociation of dimer has been compared with those of compound 1 and 2. Similar to 1 and 2, compound 3 also forms dimer and other higher oligomers. It shows that the disassociation energy for dimer of 3 (1.34 eV) is much stronger than that of compound 1 (0.78 eV) and 2 (0.98 eV), which is reasonable since the large sizes of the POSS and the steric hindrance may weaken the association between the π planes.
Self-assembly in solution
The photo physical properties are known to reflect the aggregation of π-conjugated compounds in solution. They are affected by many parameters such as concentration of the solute, solvent quality, etc. In good solvents like CHCl3, the compound always behaves as individual molecules across a broad concentration range; when methanol is used as a poor solvent, it is prone to form large aggregates and precipitate out of the solution even at low concentrations. The concentration-dependent UV-vis absorption and fluorescence emission spectra of compound 1 were finally obtained in a mixed solvent of dioxane/propanol with volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as shown in Fig. 2a and b. The choice of this mixed solvent is to induce aggregation at relatively low concentrations well before precipitation. At highly dilute concentrations (e.g., 5 × 10−5 mol L−1), the UV-vis spectra display typical monomeric absorption characteristics of PDI at 460 nm, 490 nm and 527 nm as shown in Fig. 2a, and they correspond to 0–2, 0–1, and 0–0 electronic transitions, respectively.36,37 Upon increasing the concentration, the molecules start to form aggregates, as evidenced by an apparent decrease in the absorption coefficient, increase in ε0–1/ε0–0 and the new absorption band at ∼572 nm. The emission spectra in Fig. 2b show good mirror symmetry with respect to the absorption spectra. Upon increasing the concentration to about 8 × 10−5 mol L−1, the emission peak at 540 nm decreases in its intensity and a hump at high wavelength appears (centered at around 624 nm). The aggregation kinetics was also investigated by the time-dependent spectra shown in Fig. 2c and d. It can be seen that the absorption peaks and emission peaks of monomer become lower while the peaks of aggregation gradually become stronger. The results indicate that the molecule tends to form aggregates upon increasing the concentration. It should also be noted that the emission peak of aggregation at 624 nm keeps essentially the same throughout the time course. Considering that compound 1 tends to form dimer as the motif for further packing, it is likely that the aggregates are also consisted of dimeric packing motifs with little electronic interaction between dimers.
:1 as shown in Fig. 2a and b. The choice of this mixed solvent is to induce aggregation at relatively low concentrations well before precipitation. At highly dilute concentrations (e.g., 5 × 10−5 mol L−1), the UV-vis spectra display typical monomeric absorption characteristics of PDI at 460 nm, 490 nm and 527 nm as shown in Fig. 2a, and they correspond to 0–2, 0–1, and 0–0 electronic transitions, respectively.36,37 Upon increasing the concentration, the molecules start to form aggregates, as evidenced by an apparent decrease in the absorption coefficient, increase in ε0–1/ε0–0 and the new absorption band at ∼572 nm. The emission spectra in Fig. 2b show good mirror symmetry with respect to the absorption spectra. Upon increasing the concentration to about 8 × 10−5 mol L−1, the emission peak at 540 nm decreases in its intensity and a hump at high wavelength appears (centered at around 624 nm). The aggregation kinetics was also investigated by the time-dependent spectra shown in Fig. 2c and d. It can be seen that the absorption peaks and emission peaks of monomer become lower while the peaks of aggregation gradually become stronger. The results indicate that the molecule tends to form aggregates upon increasing the concentration. It should also be noted that the emission peak of aggregation at 624 nm keeps essentially the same throughout the time course. Considering that compound 1 tends to form dimer as the motif for further packing, it is likely that the aggregates are also consisted of dimeric packing motifs with little electronic interaction between dimers.
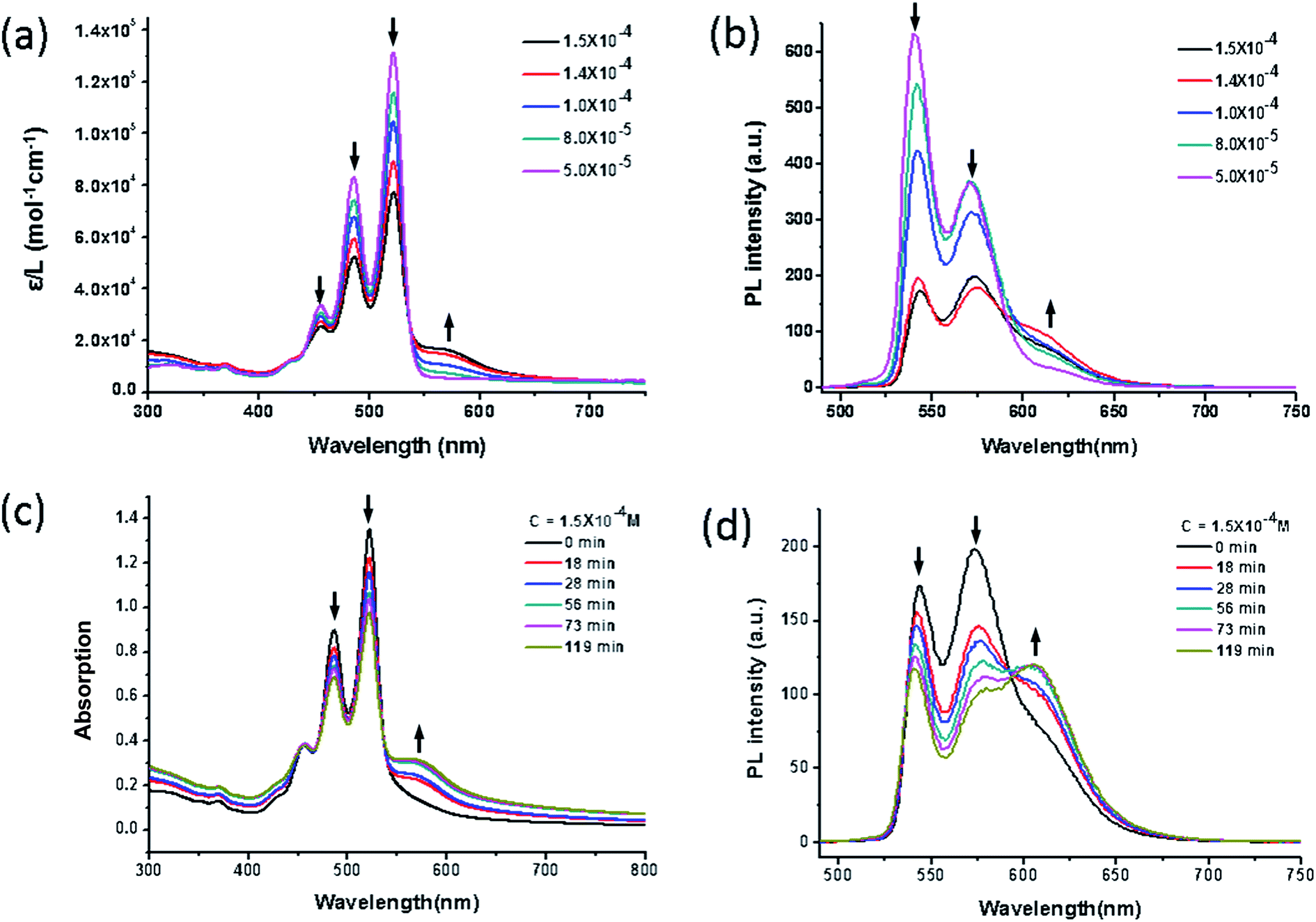 | ||
| Fig. 2 (a) Concentration-dependent UV-vis absorption spectra and (b) fluorescence emission spectra (λex = 497 nm) of compound 1 in a mixed solvent of 1,4-dioxane/propanol (v/v = 1/1) with different concentration (5 × 10−6 M to 1 × 10−4 M) at room temperature. (c) Time-dependent UV-vis absorption spectra and (d) emission spectra (λex = 497 nm) of 1 in a mixed solvent of 1,4-dioxane/propanol (v/v = 1/1) with a concentration of 1.5 × 10−4 M at room temperature. The label ε stands for extinction coefficient in the absorption spectra and PL stand for photoluminescence. Arrows indicate the direction of changes upon increasing the concentration or time. | ||
A qualitative comparison between the concentration-dependent UV-vis absorption and emission profile of 1, 2, and 3 suggests that the self-assemblies of 1 and 2 (ref. 32) are quite similar to each other and both of them are very different from 3 (Fig. S2†). The concentration-dependent UV-vis absorption and fluorescence spectra of compound 3 in CHCl3 shows an apparent decrease both in the absorption coefficient and photoluminescence intensity at concentrations larger than 2.1 × 10−4 M, indicating that the compound starts to form aggregates due to its limited solubility. The differences may lie in their different packing scheme in solid state: the former prefers a dimeric packing motif while the latter forms continuous stack of PDI planes.
Crystal formation and structure determination
Compound 1 could easily form ordered structures. Long ribbon-like species was found in the monolayer Langmuir–Blodgett film shown by AFM images,38 and single crystals have been reported for compound 1.39 We have also grown the single crystals of compound 1 as large as ∼1 centimeter in length using the slow-diffusion-induced crystallization method (see ESI†). The crystal formation was first confirmed by the observation under PLM (polarized light microscope) in Fig. S4a.† Corresponding 1D WAXD pattern was shown in Fig. S4b† further supports the formation of a crystalline lattice. However, this powder pattern was not sufficient to determine the structure due to the lack of information on the lattice symmetry. A large single crystal of 1 was then grown and used for single crystal X-ray diffraction experiments to determine the crystal unit lattice parameters and the molecular packing. The crystal structure was determined to be a triclinic unit cell with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, a = 1.14 nm, b = 2.09 nm, c = 2.31 nm and α = 89.9°, β = 81.9°, γ = 82.3°, which is essentially identical to a previous report.39
, Z = 2, a = 1.14 nm, b = 2.09 nm, c = 2.31 nm and α = 89.9°, β = 81.9°, γ = 82.3°, which is essentially identical to a previous report.39
Fig. S5† presents the molecular packing of the crystal lattice of compound 1 along three different axes. It is evident from these projections that the basic structural unit is the dimer. Each of the two molecules in the dimer has two POSS cages bent towards the same direction due to the steric hindrance between POSS in the complex and the relatively flexible linkages between POSS and PDI. The two POSS of each molecule are thus located in one side of the dimer. The dimers further pack into a double-layered structure in the crystal with two layers of POSS and one layer of PDI dimer (Fig. 3). As shown in this figure, the packing scheme of the dimer motifs shows little interdigitation, which is in sharp contrast to that of compound 2.32 The measured density of 1.28 g cm−3 matches well with the calculated value of 1.29 g cm−3 estimated from crystallographic data. We also carry out SAED experiments in TEM to investigate the single crystal grown from different methods (see ESI†). The results show that nano-belts and the bulk crystals shall have the same molecular packing scheme.
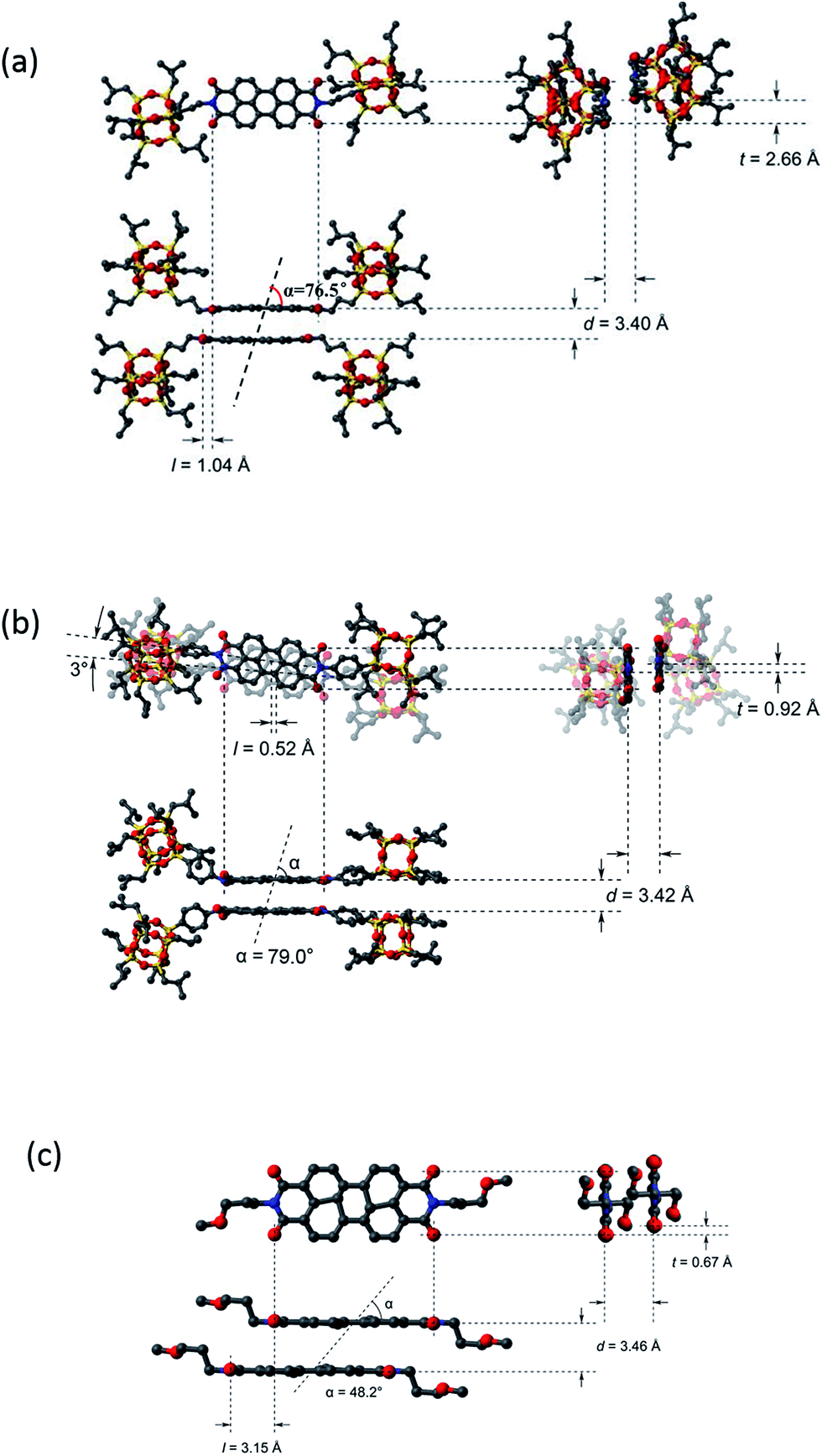 | ||
| Fig. 3 Dimer packing diagram of compound 1 (a), 2 (b), and 3 (c) in the unit cell of crystal with longitudinal offset, l, and transverse offset, t, and interplanar spacing, d. | ||
It should be noted that for compounds 1 and 2, dimers are the basic building blocks of the crystals due to the presence of the POSS molecular nanoparticle. The crosstalks or interactions between PDI molecules are somewhat “shielded” or “insulated” by POSS, resulting in a discontinuous π–π interaction in the crystals. On the contrary, compound 3 in which POSS groups are not attached, has been reported to have a continuous π–π stacking of PDIs along the c-axis within its crystal (Fig. S8†).40 What could result from such a distinct molecular packing scheme?
The rationale for the order of π–π interaction strength of 1 < 2 < 3 can be illustrated as follows. Compared to 1, compound 2 has additional interactions between the phenyl groups. In addition, PDI planes may interact better with a rigid linker since the spatial confinement of the bulky POSS minimizes their interference with π–π interaction. Compared to 3, the difference in disassociation is likely because of their difference in interplanar interactions of the perylene core: continuous packing of π-planes in 3 should result in a stronger interaction than 1. To illustrate the differences between the dimers of 1, 2, and 3 in condensed state, their structures are shown in Fig. 3 with packing parameters of the dimer, including longitudinal offset (l), transverse offset (t), interplanar spacing (d), and the angle between monomer–monomer centre-of-mass vector and the monomer planar vector (α) estimated based on the crystal parameters. Since the formation of the dimers of 2 involves an additional phenylene as well as certain rotation of the PDI planes which may result in difference in electronic interaction, a comparison between 1 and 3 would be more convincing. It was found that the bulky POSS substituent leads to a smaller d and l, but much bigger α and t in 1. Since smaller d, smaller l and bigger α generally imply stronger electronic interaction between dimers, it seems that the packing energy of perylene core may have a much more steeper dependence on the transverse offset t, which is consistent with literature reports.12,41 The packing parameters are listed in Table 1.32,34 There are two α values for compound 2 (each for a different PDI plane) due to the 3° rotational offsets between them.
| Component | d/nm | l/nm | t/nm | α/° |
|---|---|---|---|---|
| 1 | 0.340 | 0.104 | 0.266 | 76.5 |
| 2 | 0.342 | 0.052 | 0.092 | 79.0, 82.2 |
| 3 | 0.346 | 0.315 | 0.067 | 48.2 |
Photoluminescent properties in the solid state
Although PDIs are known to have high photo-stabilities and close-to-unity fluorescence quantum yields (Φf) in solution, their properties in solid are mostly compromised by fluorescence quenching which decreases the Φf. This is due to the continuous π–π stacking in the solid state aggregates, most of which are H-type, in the solid state.42 To improve the fluorescent Φf of PDI in the solid state, bulky side groups have been introduced to tune the π–π interaction. For example, the PDI derivative with symmetric 3,4,5-tris(1′-dodecynyl)-phenyl side groups but no bay substitutions shows a Φf of 47% for a 0.1 mmol L−1 aggregate solution in n-hexane but only 16% in thin films.15 The PDI derivative possessing symmetric long secondary alkyl chain (so-called “swallow tails”), named ST-PDI-ST, has been reported to have a Φf of 34% in the solid state, whereas PDI with asymmetric POSS and swallow tail, named POSS-PDI-ST, has a Φf as high as 70%.43 The chloro substituents at the bay area were found to be beneficial as demonstrated by a Φf of 40% in the solid state. With even larger bay-substituents, such as tert-butylphenoxy, the Φf from microcrystalline powders could be as high as 70%. Thus, it seems that by disrupting the π–π interaction, Φf could be dramatically improved. Yet there is no direct evidence relating crystal structure with the photoluminescent properties. Therefore, the photoluminescent properties of 1, 2, and 3 in condensed state were studied with respect to different packing states, namely, ordered crystals versus powders of less crystallinity.Fig. 4 shows the typical UV-vis absorption and fluorescence emission profile for compounds 1, 2, and 3 in both self-assembled crystalline powder and precipitated powder forms. The powder samples were obtained by direct rapid precipitation of samples into poor solvent CH3OH and may be more amorphous. The crystal form was obtained by slow-diffusion-induced crystallization method (see ESI†). Both forms were mechanically milled to powder so as to minimize the effect of reabsorption of fluorescence on Φf measurement due to the small Stokes shift.44 The results are summarized in Table 2. The effects of side-chain substitution on the absorption and fluorescence profile were first studied. In the crystalline solid state, compound 1 shows broadened absorption profiles with a maximum around 455 nm and a small peak around 524 nm. These absorption profile is similar to those PDI derivatives bearing two tridodecylphenyl side chains.15 It also agrees well with the calculated spectrum from the theoretical excitonic molecular model.15,34 So, it is proposed that these absorption are characteristic from discrete dimers and thus can be described by the excitonic molecular dimer model.34 Upon excitation (λ = 415 nm), an emission peak was observed at 624 nm. It should be noted this emission peak has almost the same position with those in 1,4-dioxane/propanol mixed solvent. Compared to compound 1, the crystal form of compound 2 shows similar absorption profile but the maximum of emission wavelength is red-shifted to 646 nm. This is due to the existence of additional phenyl groups in the π-conjugated system. The absorption profile of compound 3 is also broadened, but with a blue-shifted maximum at ∼410 nm, while the emission profile is much red-shifted to 642 nm in the crystal form. It suggests that the compound 3 forms larger aggregate with more extended π–π interaction beyond dimers. The crystal forms of compounds 1 and 2 are both quite fluorescent with a Φf of 48% and 35% (Table 2), respectively. By contrast, the crystal form of compound 3 has only modest fluorescence with a much lower Φf of only 12%. It suggests that in crystal states, the compounds 1 and 2 maintain the high fluorescence similar to that in solution whereas significant quenching occurs in the reference compound 3.
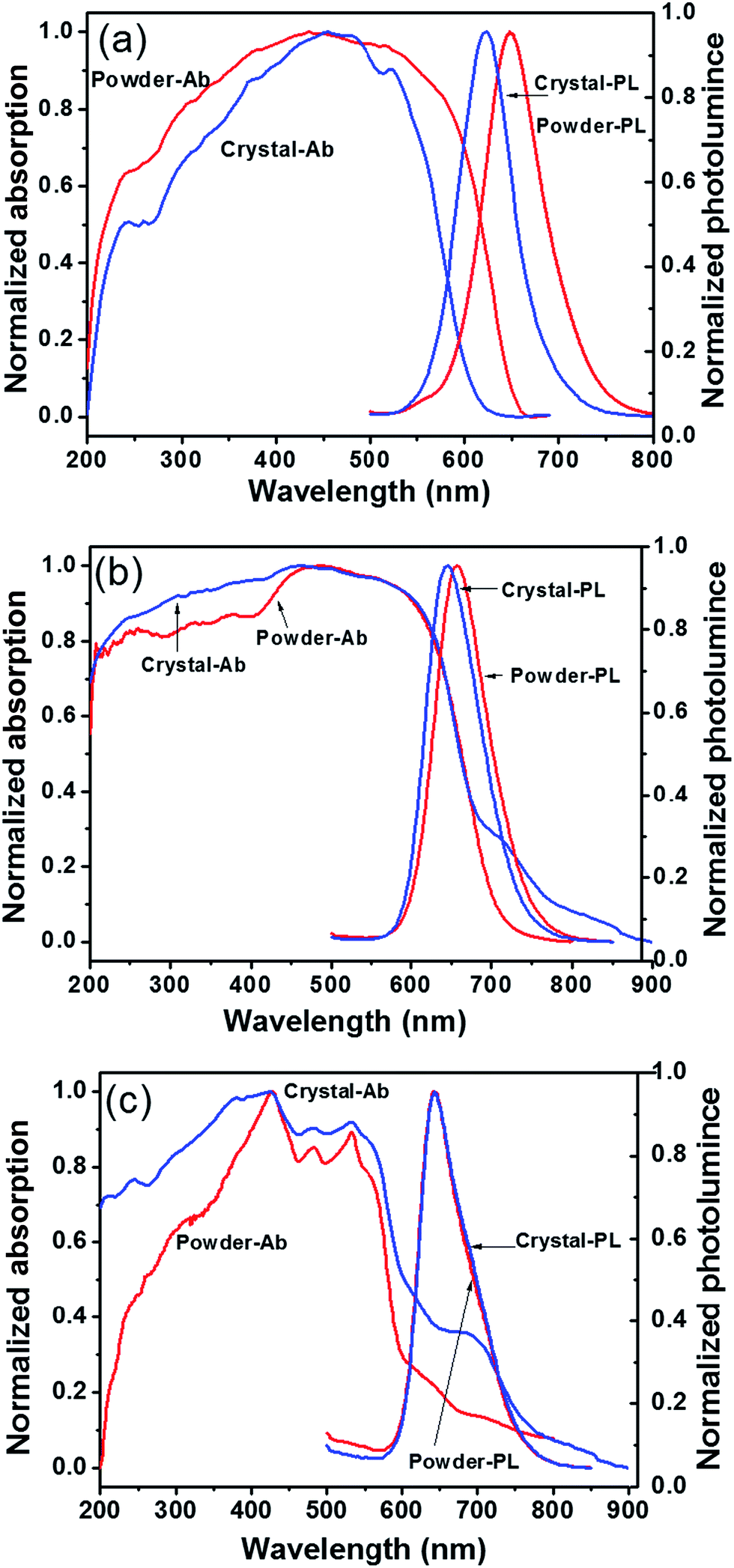 | ||
| Fig. 4 Absorption and fluorescence spectra of compounds 1 (a), 2 (b) and 3 (c) in powder and crystal state. | ||
| Component | λem,max (nm) | Φf (%) (λex/nm) | ||||
|---|---|---|---|---|---|---|
| Solution | Powder | Crystal | Solution | Powder | Crystal | |
| 1 | 538 | 649 | 624 | 100 (490) | 17 (410) | 48 (415) |
| 2 | 540 | 660 | 646 | 100 (490) | 14 (405) | 35 (410) |
| 3 | 538 | 642 | 642 | 100 (490) | 8 (420) | 12 (420) |
Even for the same molecule, different packing may lead to different properties. Precipitated powder forms are usually of less order and more amorphous than crystalline samples. This is evident from the WAXD results as shown in Fig. S9.† The precipitated powder samples show a reddish color, quite different from the orange color of the crystalline sample of 1. The color change implies slight difference in molecular packing. It is surprising that the precipitated powder sample has a Φf of 17%, a level that is comparable with that of compound 3 but much lower than 48% of crystal samples. The crystallinity of the samples was determined to be 59% for precipitated powders and 73% for crystals of compound 1. It is likely that in crystals, the POSS cages probably confine the PDI dimer in place, prohibiting further structural relaxation process commonly observed in other PDI derivatives and thus increasing the Φf. However, at lower crystallinity, molecules do not pack as regularly as they do in ideal crystals. Defects may exist, in which the PDIs may not be confined by the POSS and may have more interaction. Weak electron interactions between neighboring dimers and subsequent energy transfer and other relaxation process may occur, leading to red-shifted emission and fluorescence quench and a much lower Φf. Similar observations are also evident in compound 2, reflecting similar effects of molecular packing difference. By contrast, for compound 3, both amorphous powder sample and crystal sample show identical emission profile with comparable Φf. It may be explained by the long, continuous π–π stacking in both precipitated and crystalline samples.40 Their properties are thus not so sensitive to minor changes in packing. These results highlight the importance of structural control in tuning the optoelectronic properties of the materials.
Conclusions
In summary, a systematic study of the influence of tethered POSS on molecular packing in condensed state and related photoluminescent properties of PDI derivatives has been presented in this paper. The POSS was tethered to PDI via either rigid or flexible linkages. Crystal structure of compound 1 is determined, which exhibits a unique dimer packing scheme. This is also supported by the study of its self-assembly in gaseous phase, solution, and solid state. The discrete dimer packing scheme explains the enhanced fluorescence quantum yields (Φf) observed in the crystal form of compounds 1 and 2, but not in the control sample 3 or the amorphous powder form of 1 and 2. The attachment of bulky POSS groups effectively inhibits the continuous packing of the π–π interaction between the PDI planes, shields further PDI interactions, and prohibits structural relaxation process commonly observed in other PDI derivatives. This may be a general strategy to engineer π–π interactions for various applications. Consequently, the fluorescent crystal of compound 1 and 2 are ideal candidates for further investigations as discrete dimer models and self-assembled nano-emitters. We further emphasize that even for the same molecule, controlling condensed structure is critical in achieving high photoluminescent properties.Acknowledgements
This work was supported by the Program for Changjiang Scholars and Innovative Research Team in University (T2011079, IRT1221), Natural Science Foundation of Tianjin and Youth Science Foundation (14JCQNJC 02900), Science and Technology Commission of Shanghai Municipality (16JC1400700).Notes and references
- A. Klug, Angew. Chem., Int. Ed., 1983, 22, 565–582 CrossRef.
- X. Hu and K. Schulten, Phys. Today, 1997, 29, 381–389 Search PubMed.
- J. M. Lehn, Angew. Chem., 1990, 102, 1347–1362 CrossRef CAS.
- M. Treier, M. T. Nguyen, N. V. Richardson, C. Pignedoli, D. Passerone and R. Fasel, Nano Lett., 2009, 9, 126–131 CrossRef CAS PubMed.
- A. Graja, Low-dimensional organic conductors, World Scientific, Singapore, River Edge, NJ, 1992 Search PubMed.
- Y. S. Zhao, H. B. Fu, A. D. Peng, Y. Ma, D. B. Xiao and J. N. Yao, Adv. Mater., 2008, 20, 2859–2876 CrossRef CAS.
- Z. J. Chen, A. Lohr, C. R. Saha-Moller and F. Wurthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- A. J. Jiménez, M. J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Würthner, Chem. Sci., 2014, 5, 608–619 RSC.
- K. Sun, C. Y. Xiao, C. M. Liu, W. X. Fu, Z. H. Wang and Z. B. Li, Langmuir, 2014, 30, 11040–11045 CrossRef CAS PubMed.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hagele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS PubMed.
- D. Görl, X. Zhang and F. Würthner, Molecular Assemblies of Perylene Bisimide Dyes in Water, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- F. Wurthner, Chem. Commun., 2004, 35, 1564–1579 RSC.
- D. S. Correa, S. L. Oliveira, L. Misoguti, S. C. Zilio, R. F. Aroca, C. J. L. Constantino and C. R. Mendonca, J. Phys. Chem. A, 2006, 110, 6433–6438 CrossRef CAS PubMed.
- S. M. Lindner, N. Kaufmann and M. Thelakkat, Org. Electron., 2007, 8, 69–75 CrossRef CAS.
- Z. Chen, V. Stepanenko, V. Dhem, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Wurthner, Chem.–Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- R. Gomez, D. Veldman, R. Blanco, C. Seoane, J. L. Segura and R. A. J. Janssen, Macromolecules, 2007, 40, 2760–2772 CrossRef CAS.
- M. Adachi, Y. Murata and S. Nakamura, J. Phys. Chem., 1995, 99, 14240–14246 CrossRef CAS.
- M. J. Lin, Á. J. Jiménez, C. Burschka and F. Würthner, Chem. Commun., 2012, 48, 12050–12052 RSC.
- P. D. Frischmann and F. Würthner, Org. Lett., 2013, 15, 4674–4677 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Adv. Mater., 2012, 24, 3350–3355 CrossRef CAS PubMed.
- M. M. Safont-Sempere, P. Osswald, K. Radacki and F. Würthner, Chem.–Eur. J., 2010, 16, 7380–7384 CrossRef CAS PubMed.
- M. M. Safont-Sempere, P. Osswald, M. Stolte, M. Grüne, M. Renz, M. Kaupp, K. Radacki, H. Braunschweig and F. Würthner, J. Am. Chem. Soc., 2011, 133, 9580–9591 CrossRef CAS PubMed.
- C. Shao, M. Grüne, M. Stolte and F. Würthner, Chem.–Eur. J., 2012, 18, 13665–13677 CrossRef CAS PubMed.
- J. Wu and P. T. Mather, Polym. Rev., 2009, 49, 25–63 CrossRef CAS.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- R. Y. Kannan, H. J. Salacinski, P. E. Butler and A. M. Seifalian, Acc. Chem. Res., 2005, 38, 879–884 CrossRef CAS PubMed.
- H. Ghanbari, G. Cousins and A. M. Seifalian, Macromol. Rapid Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed.
- C. H. Ni, G. Wu, C. P. Zhu and B. L. Yao, J. Phys. Chem. C, 2010, 114, 13471–13476 CAS.
- X. Yu, S. Zhong, X. Li, Y. Tu, S. Yang, R. M. Van Horn, C. Ni, D. J. Pochan, R. P. Quirk, C. Wesdemiotis, W. B. Zhang and S. Z. D. Cheng, J. Am. Chem. Soc., 2010, 132, 16741–16744 CrossRef CAS PubMed.
- W. B. Zhang, Y. Li, X. Li, X. Dong, X. Yu, C. L. Wang, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Macromolecules, 2011, 44, 2589–2596 CrossRef CAS.
- X. K. Ren, B. Sun, C. C. Tsai, Y. F. Tu, S. W. Leng, K. X. Li, Z. Kang, M. V. H. Ryan, X. P. Li, M. F. Zhu, C. Wesdemiotis, W. B. Zhang and S. Z. D. Cheng, J. Phys. Chem. B, 2010, 114, 4802–4810 CrossRef CAS.
- J. Cornil, D. Beljonne, J.-P. Calber and J.-L. Bredas, Adv. Mater., 2001, 13, 1053–1067 CrossRef CAS.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- Y. Zhang, L. Zhang, H. Liu, Di Suna and X. Li, CrystEngComm, 2015, 17, 1453–1463 RSC.
- R. Gvishi, R. Reisfeld and Z. Burshtein, Chem. Phys. Lett., 1993, 213, 338–344 CrossRef CAS.
- D. Liu, S. De Feyeter, M. Cotlet, A. Stefan, U. M. Wiesler and A. Herrmann, Macromolecules, 2003, 36, 5918–5925 CrossRef CAS.
- X. D. Zhuang, Y. Chen, B. Zhang, Y. Li, B. Yu and W. Qiao, New J. Chem., 2010, 34, 1120–1124 RSC.
- D. Clarke, S. Mathew, J. Matisons, G. Simon and B. W. Skelton, Dyes Pigm., 2011, 92, 659–667 CrossRef.
- E. Hadicke and F. Graser, Acta Crystallogr., 1986, C42, 189–195 Search PubMed.
- P. M. Kazmaier and R. Hoffmann, J. Am. Chem. Soc., 1994, 116, 9684–9691 CrossRef CAS.
- F. Wurthner, T. E. Kaiser and C. R. Saha-Moller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- E. Lucenti, C. Botta, E. Cariati, S. Righetto, M. Scarpellini, E. Tordin and R. Ugo, Dyes Pigm., 2013, 96, 748–755 CrossRef CAS.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, detailed characterization of compound 1, the concentration-dependent UV-vis absorption spectra and fluorescence emission spectra of compound 3, and the molecular packing of compound 3 in the crystal. SAED experiments in TEM studying the nano-belts and the bulk crystals. See DOI: 10.1039/c6ra28147h |
| ‡ These authors contribute equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |