DOI:
10.1039/C6RA28229F
(Paper)
RSC Adv., 2017,
7, 8178-8185
Targeting selenium nanoparticles combined with baicalin to treat HBV-infected liver cancer†
Received
15th December 2016
, Accepted 5th January 2017
First published on 23rd January 2017
Abstract
As a traditional Chinese medicine, baicalin exhibits high antitumor activity towards hepatitis B virus (HBV)-infected liver cancer, but low toxicity towards normal tissues. However, the properties of baicalin, including its low hydrophilicity and poor biocompatibility, have limited its clinical application. Herein, in this study, we have designed and synthesized multifunctional selenium nanoparticles (SeNPs) with baicalin (B) and folic acid (FA) surface-modifications for the targeted treatment of HBV infected liver cancer. B–SeNPs–FA showed appropriate particle size distribution, high stability, and higher cellular uptake by tumor cells than normal cells. The nanoparticles primarily targeted lysosomes in HepG2215 cells through utilization of the main pathways of caveolae-mediated endocytosis and energy-dependent endocytosis. B–SeNPs–FA induced HepG2215 cell apoptosis by down-regulating the generation of reactive oxygen species (ROS) and the expression of the HBxAg protein. In addition, B–SeNPs–FA exhibited an excellent ability to inhibit cancer cell migration and invasion. Taken together, these results suggest that baicalin-loaded selenium nanoparticles with a folic acid targeting moiety could be a promising strategy for the design and synthesis of cancer-targeted nanomaterials to treat HBV-infected liver cancer.
Introduction
Compared with HepG2 cells, HepG2215 cells injected into nude mice particularly exhibit cellular mosaicism and clonal selection.1–4 Further, the de novo genetic alteration in the HepG2215 cells, which maintains their ability to replicate the hepatitis B virus (HBV), includes a new HBV integration site, several chromosome rearrangements and loss of heterozygosity (LOH) of one p53 allele.5–7 Chronic infection induced by the HBV is generally recognized as a risk factor in hepatocarcinogenesis.8–10 HBV belongs to the hepatotropic virus family, the gene structures of which have an open reading frame including S, C, P and X proteins.11,12 In particular, the X protein gene is about 462 bp long, and encodes a protein that contains 154 amino acids.13 The non-structural X protein of hepatitis B virus (HBxAg) is a principal regulator of virulence proteins, and plays a considerable role in the interaction point of infection, replication, pathogenesis and carcinogenesis induced by HBV.14 Now, HBxAg is recognized as a vital cofactor leading to chronic infection of liver cancer, and is regarded as one of the main proteins in liver disease that can activate various tumor genes directly or indirectly.15 Moreover, as a pleiotropic protein, HBxAg plays a considerable role in virus replication, cell cycle regulation, cell growth and cell apoptosis. So, down-regulating the expression levels of the HBxAg protein could be an important strategy in treating HBV-infected liver cancer and reducing the risk factor of hepatocarcinogenesis.
HCC (hepatocellular carcinoma) is the fifth most commonly diagnosed type of cancer, and HBV infection is one of the main causes of HCC.16 Chemotherapy is one of the effective methods in the treatment of HCC. Unfortunately, most anti-cancer drugs such as cisplatin and gemcitabine have serious side effects.17 Traditional Chinese medicines exhibit low toxicity and can have strong anti-cancer effects.18 Recent research has shown that some monomers extracted from Chinese medicine, such as desacetyluvaricin, have anti-HCC activity.19 Baicalin is a kind of flavonoid extracted from the Chinese medicine Scutellaria baicalensis. Research has shown that baicalin has therapeutic action for many diseases, such as virus infection, inflammation, cancer, etc.20–23 Considering that the secondary metabolite of baicalin exhibits low toxicity and protective effects on the liver, baicalin may have therapeutic prospects as an anti-HCC drug in the clinic.24 Because of the effects of baicalin in inhibiting tumor cells and its low toxicity to normal cells,25 it has been widely applied to treat viral hepatitis B in recent years in China.27 However, how to use baicalin at high concentrations is still a problem to researchers.26 Its low hydrophilicity and poor biocompatibility have limited its application in the field of biomedicine. For these reasons, finding drug carriers that can improve drug uptake and efficacy is important and urgent.
Nanomaterials, as antitumor drug carriers in cancer therapy, are playing a more and more important role in improving drug uptake and the efficacy of drugs in tumor cells.28 So far, a variety of nanosystems, such as selenium nanoparticles (SeNPs), mesoporous silica, polymers and liposomes, have been employed as drug carriers for cancer therapy in various ways.29–33 In particular, selenium (Se) is an essential trace element that has many important physiological functions, such as in resistance to disease, antioxidants and the enhancement of immunity in daily life.32,33 Selenium is also an important component of many kinds of enzymes in the human body. In addition, selenium participates in many biological functions, including protection of the liver, protecting against tumor formation, cardiovascular protection, regulation of hormone secretion, antagonism of heavy metals, free radical scavenging, and so on.32,34,35 A lack of Se will result in damage to the immune system and an increased risk of oncogenesis. SeNPs are regarded as novel nanosystems and have drawn increasing attention due to their higher effective antitumor ability, excellent antioxidant activity, low-toxicity to normal cells, and remarkable biocompatibility and degradability compared with other nanosystems.36–38
In order to effectively treat HBV-infected liver cancer, we designed and synthesized targeted selenium nanoparticles combined with baicalin in this study. The new nanosystem highly inhibited the expression of HBxAg protein, effectively treated HBV-infected liver cancer, and greatly reduced the risk factors of hepatocarcinogenesis (as described in Fig. 1B). Therefore, because of its excellent stability, prominent anti-cancer and anti-virus activity, and low toxicity to normal cells, this multifunctional nanosystem provides a promising strategy for treating HBV-infected liver cancer.
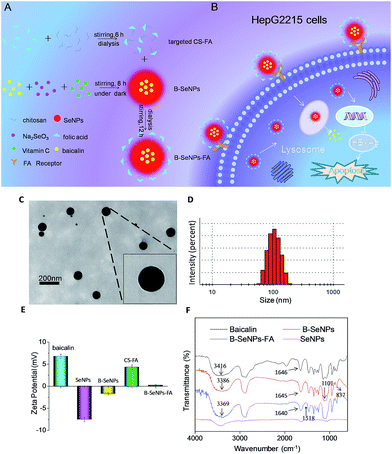 |
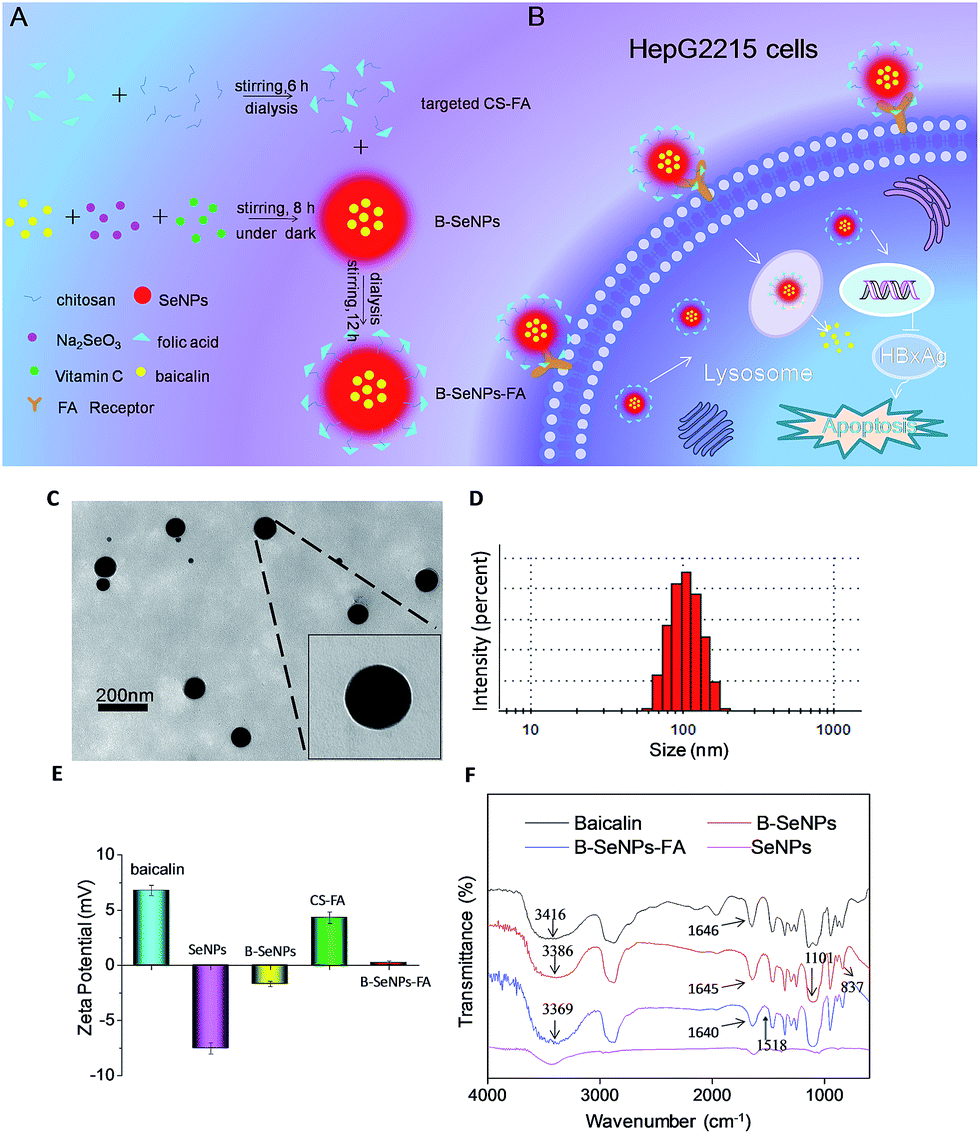
| | Fig. 1 (A) Synthesis process and structure of B–SeNPs–FA; (B) mechanisms of action of the targeted selenium nanoparticles combined with baicalin in the treatment of HBV-infected liver cancer; (C) TEM images of B–SeNPs–FA (the scale bar is 200 nm); (D) size distribution of B–SeNPs–FA; (E) zeta potential of baicalin, SeNPs, B–SeNPs, CS–FA and B–SeNPs–FA; (F) FT-IR spectra of baicalin, SeNPs, B–SeNPs and B–SeNPs–FA. | |
Experimental
Materials
Baicalin powder and polyethylene glycol (PEG) were purchased from Aladdin. Sodium selenite (Na2SeO3), chitosan (CS), folic acid (FA), dimethyl sulfoxide (DMSO), N-hydroxysuccinimide (NHS), N-(3-(dimethylamino)propyl)-N-ethylcarbodiimide (EDC), coumarin-6, thiazolyl blue tetrazolium bromide (MTT), propidium iodide (PI), dihydroethidium (DHE), and a bicinchoninic acid (BCA) kit were purchased from Sigma-Aldrich. Vitamin C was purchased from Guangzhou chemical reagent factory. Milli-Q water was applied in all experiments.
Preparation of B–SeNPs–FA
Preparation of targeted CS–FA. 5 mg of folic acid (FA) was dissolved in 1 mL of dimethyl sulfoxide (DMSO), and then N-hydroxysuccinimide (NHS) and N-(3-(dimethylamino)propyl)-N-ethylcarbodiimide (EDC) were also dissolved in the DMSO as an amidation catalyst to activate FA. After stirring the solution (containing FA, NHS and EDC) for 2 hours at ambient temperature and pressure, the activated FA solution was obtained. Then, chitosan (CS) solution was added to the activated FA solution, and then this mixed solution was stirred for 6 hours. Finally, the excess folic acid and other unreacted material were eliminated by dialysis for 24 hours. Thus, targeted CS–FA was obtained through the formation of an amido bond as previously described.36
Preparation of B–SeNPs–FA. First, 20 mg of baicalin (B) powder was dissolved in 10 mL of Milli-Q water containing 35% wt polyethylene glycol (PEG), and then the mixed solution was stirred in the dark until the baicalin powder was completely dissolved. Then, fresh Na2SeO3 solution (100 mM) and ascorbic acid (Vc) solution (100 mM) were prepared. The Na2SeO3 solution was added dropwise to the baicalin solution to a final concentration of 2 mM, and then the Vc solution was slowly added dropwise into the above mixed solution to a final concentration of 8 mM. After stirring for 8 hours with a magnetic stirrer, a B–SeNP solution was obtained. Finally, 500 μL of the targeted CS–FA was added into the B–SeNP solution, and then this solution was stirred for 12 hours to complete the reaction. After the unreacted material, including the excess baicalin and Na2SeO3 solution, has been removed by dialysis for 24 hours, B–SeNPs–FA was obtained.In this study, coumarin-6 was used as a fluorescent probe to detect the cellular uptake and intracellular trafficking of B–SeNPs–FA in vitro. The preparation method of 6-coumarin-B–SeNPs–FA was the same as the preparation of B–SeNPs–FA except that 5 μg mL−1 coumarin-6 was added into the Na2SeO3 and baicalin solution before addition of the Vc solution to form coumarin-6-loaded B–SeNPs–FA.
Characterization of B–SeNPs–FA
The B–SeNPs–FA was characterized as previously described by different kinds of spectroscopic and microscopic measurements, including transmission electron microscopy (TEM) (HitachiH-7650), Fourier transform infrared spectroscopy (FTIR) (Equinox 55 IR), UV-vis spectroscopy (Carry 5000) and Zetasizer particle size analysis (Malvern Instruments Limited).39
Cell culture and cell viability by MTT assay
The hepatocellular carcinoma cells (HepG2), HBV-transfected hepatocellular carcinoma cells (HepG2215) and normal hepatocyte cells (L02) were purchased from GuangZhou Jennio Biotech Co., Ltd and were cultured as described in our previous paper.41 To determine the relative cytotoxicity of B–SeNPs–FA to HepG2, HepG2215 and L02 cells in vitro, an MTT assay was performed as previously reported.42
Determination of B–SeNPs–FA cellular uptake and intracellular trafficking
The intracellular uptake of B–SeNPs–FA by HepG2 cells, HepG2215 cells and L02 cells was quantified by determining the selenium concentration using ICP-MS. The intracellular trafficking of coumarin-6-loaded B–SeNPs–FA in HepG2 and HepG2215 cells was monitored by incubating the cells with the lysosomal marker Lyso Tracker red and the nuclear marker DAPI. Finally, fluorescence images of the HepG2 and HepG2215 cells were taken by fluorescence microscopy (Nikon Eclipse 230 80i) as previously described.41
Cellular uptake pathways of B–SeNPs–FA
The cellular uptake pathway of B–SeNPs–FA through endocytosis was determined as previously reported.41
In vitro drug release of B–SeNPs–FA
The drug release of B–SeNPs–FA was measured as previously described.36,41
Flow cytometry analysis
As described in our previous study,42 flow cytometry analysis was utilized to examine the cell cycle distribution of HepG2 and HepG2215 cells that had been exposed to B–SeNPs–FA for 72 hours.
Determination of the reactive oxygen species (ROS) level
The variations in the intracellular ROS levels resulting from exposure to B–SeNPs–FA in HepG2 cells and HepG2215 cells were detected by using a fluorescence DHE assay as described previously.34
Western blot analysis
The influence of B–SeNPs–FA on the expression levels of various proteins in HepG2 cells, HepG2215 cells and L02 cells was analyzed by western blotting as previously reported.34
In vitro migration assay
HepG2 and HepG2215 cells (2 × 105 cells per mL) were seeded into 6-well plates. After incubation for 24 hours, these cells were starved with DMEM (containing 3% FBS) for 6 hours, scratched with pipette tips, and incubated in fresh medium with different concentrations of B–SeNPs–FA. Pictures of the migrated cells were taken using an Olympus inverted microscope after 24 hours.43
In vitro invasion assay
In order to analyze the effects of B–SeNPs–FA on the invasion of HepG2 and HepG2215 cells, Transwell Boyden chambers (8 μm pore, Corning, Lowel, MA) were utilized as previously described. Pictures of the migrated cells were taken using an Olympus inverted microscope.43
Statistical analysis
All experiments were repeated at least three times, and the data were expressed as the mean ± standard deviation (SD). The differences between the treatment groups were analyzed by one-way ANOVA with multiple comparisons using SPSS13.0 statistical software.
Results and discussion
Design and characterization of B–SeNPs–FA
B–SeNPs–FA was characterized by TEM, FTIR and Zetasizer particle size analysis. As shown in Fig. 1A, baicalin-loaded selenium nanoparticles with liver targeting ability were synthesized. We used TEM analysis to study the effect of baicalin on SeNP morphology. As shown in Fig. 1C and D, the B–SeNPs–FA particles were evenly dispersed in the solution and had a sphere-like morphology. The particle size of B–SeNPs–FA was around 100 nm. As Fig. 1E shows, the zeta potentials of baicalin and the SeNPs were about 6.79 mV and −7.53 mV, respectively. However, after the SeNPs were combined with baicalin, the zeta potential increased to −1.68 mV. This may be due to electrostatic interactions between baicalin and the SeNPs. In addition, the zeta potential of B–SeNPs–FA was 1.16 mV; this was higher than that of the B–SeNPs at −1.68 mV, which verified that the targeted CS–FA successfully modified the nanoparticles through electrostatic interactions. There were some similarities in the FTIR spectra of baicalin, the SeNPs, the B–SeNPs and B–SeNPs–FA, as shown in Fig. 1F. The strong absorption peaks of hydroxyl groups (–OH) were observed in the spectra for baicalin (in accordance with the structure of baicalin as shown in Fig. S1†), the B–SeNPs and B–SeNPs–FA, but the hydroxyl peaks were moved from 3416 cm−1 in baicalin to 3386 cm−1 in the B–SeNPs. These results indicated that a weak force was present between the hydroxyl groups and the selenium atoms. The peak due to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O– stretching vibration in baicalin at 1646 cm−1 moved slightly to 1645 cm−1 in the B–SeNPs, suggesting that a Se–O bond may be formed between baicalin and the SeNPs. In addition, in the FTIR spectrum of the B–SeNPs, the characteristic absorption peak at 1101 cm−1 was due to the C–O–C stretching vibration of the glycosidic bond, and the weak peak at 837 cm−1 corresponded to the H–C–C–C–C twisting vibration of the baicalin ring. These characteristic absorption peaks in baicalin appeared in the FTIR spectrum of the B–SeNPs, indicating that baicalin was successfully combined with the SeNPs and contributed to the high stability of the nanoparticles. Moreover, compared with the B–SeNPs, in the spectrum of B–SeNPs–FA, the peaks at 1518 cm−1 were assigned to the benzene rings from FA, indicating that the FA targeting moiety was successfully modified onto the nanoparticles. Taken together, these results confirmed that we had synthesized baicalin-loaded selenium nanoparticles with a folic acid targeting moiety. These could be a promising tool for the design and synthesis of cancer-targeted nanomaterials to treat HBV-infected liver cancer.
O– stretching vibration in baicalin at 1646 cm−1 moved slightly to 1645 cm−1 in the B–SeNPs, suggesting that a Se–O bond may be formed between baicalin and the SeNPs. In addition, in the FTIR spectrum of the B–SeNPs, the characteristic absorption peak at 1101 cm−1 was due to the C–O–C stretching vibration of the glycosidic bond, and the weak peak at 837 cm−1 corresponded to the H–C–C–C–C twisting vibration of the baicalin ring. These characteristic absorption peaks in baicalin appeared in the FTIR spectrum of the B–SeNPs, indicating that baicalin was successfully combined with the SeNPs and contributed to the high stability of the nanoparticles. Moreover, compared with the B–SeNPs, in the spectrum of B–SeNPs–FA, the peaks at 1518 cm−1 were assigned to the benzene rings from FA, indicating that the FA targeting moiety was successfully modified onto the nanoparticles. Taken together, these results confirmed that we had synthesized baicalin-loaded selenium nanoparticles with a folic acid targeting moiety. These could be a promising tool for the design and synthesis of cancer-targeted nanomaterials to treat HBV-infected liver cancer.
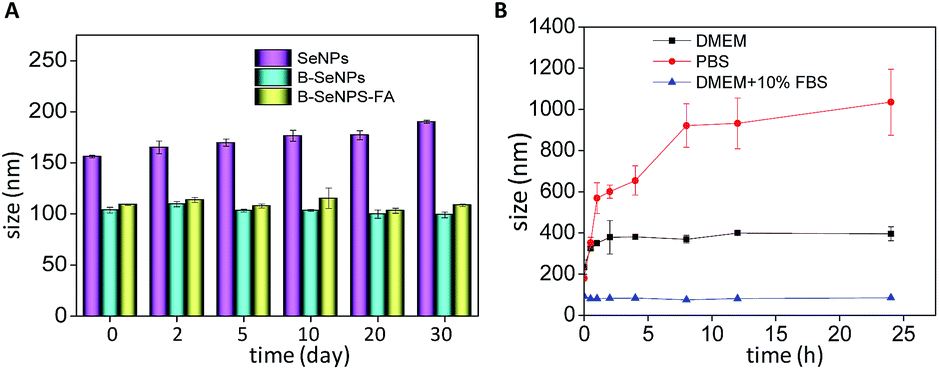
The stability of the nanoparticles is an important measure of the application prospects of a nanomaterial.41 Zetasizer particle size analysis was performed to detect the influence of baicalin on the stability of the nanoparticles. As shown in Fig. 2A, the particle size of B–SeNPs–FA and the B–SeNPs remained about 100 nm within 40 days, while the particle size of the SeNPs gradually increased from 156.1 nm to 190.2 nm. Preserved for the same amount time, B–SeNPs–FA and the B–SeNPs were more stable than the SeNPs, which suggested that baicalin could modify this nanosystem and improve its stability. In order to further determine the stability of B–SeNPs–FA, PBS, DMEM and DMEM containing 10% FBS were used to simulate the environment in the body. As shown in Fig. 2B, compared with in PBS and DMEM, B–SeNPs–FA was most stable, being stable for more than 24 h, when dispersed in DMEM containing 10% FBS, with the particle size remaining at about 100 nm. This was due to the stabilization effect of serum on the nanoparticles. These results for B–SeNPs–FA provided the basis for subsequent biological experiments.
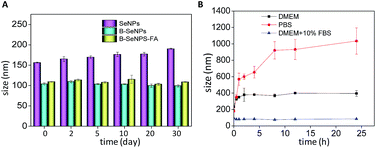 |
| | Fig. 2 (A) Stability of SeNPs, B–SeNPs and B–SeNPs–FA in aqueous solutions for 30 days; (B) changes in the particle size of B–SeNPs–FA following incubation in DMEM, PBS and DMEM containing 10% fetal bovine serum. | |
Selective cellular uptake of B–SeNPs–FA
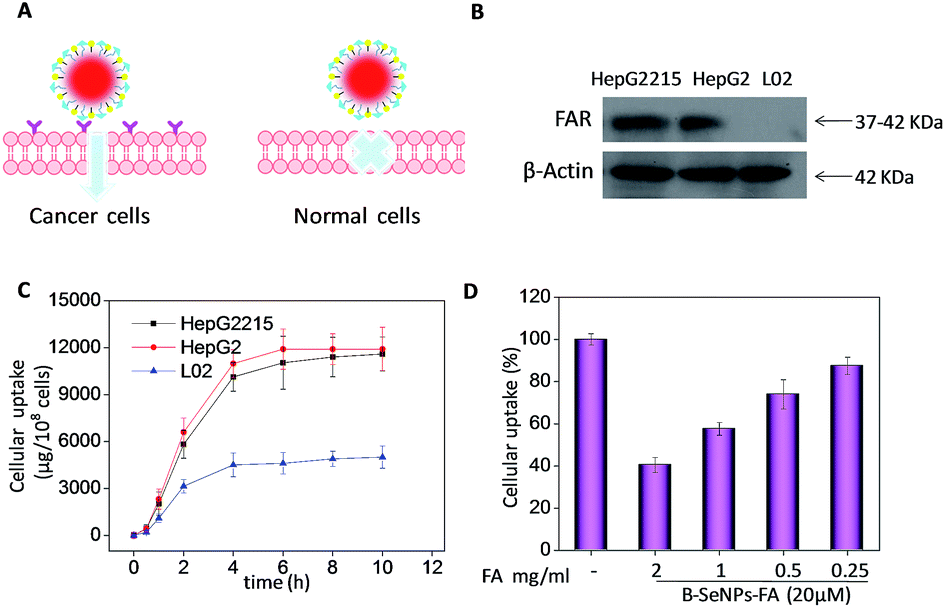
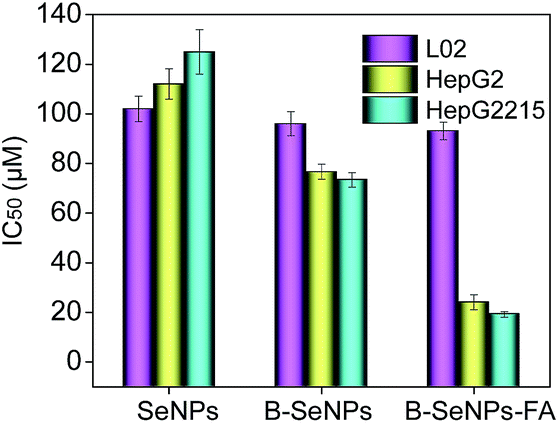
The efficacy of cellular uptake is a vital measurement of the bio-activity of a nanomaterial. To detect the cellular uptake in cancer cells and normal cells, internalization of B–SeNPs–FA by HepG2215, HepG2 and L02 cells was analyzed. As shown in Fig. 3C, the cellular uptake of B–SeNPs–FA by HepG2 cells and HepG2215 cells was much higher than that by L02 cells. To detect the contribution of FA to the selective cellular uptake of B–SeNPs–FA by the HepG2215, HepG2 and L02 cells, western blot analysis was carried out to investigate the expression level of FARs (folic acid receptors) on the cell membrane. As illustrated in Fig. 3B, the expression levels of FARs in the HepG2 and HepG2215 cells were much higher than that in the L02 cells. In order to further determine the role of FA-guided delivery, the HepG2215 cells were pretreated with different concentrations of FA for 2 h to block the FARs on the cell membranes, and then the cells were incubated with B–SeNPs–FA for 6 hours. As shown in Fig. 3D, the internalization of B–SeNPs–FA was obviously inhibited by the various concentrations of FA in a dose-dependent manner, which suggested that the selective cellular uptake of B–SeNPs–FA in HepG2215 cells was promoted by FA/FAR-mediated endocytosis. As expected, when exposed to B–SeNPs–FA for 36 hours, the IC50 values of the HepG2215, HepG2 and L02 cells were 19.13 μM, 23.89 μM and 93.14 μM, respectively, which were higher than those for the SeNPs (124.03 μM, 112.37 μM, 102.33 μM) and B–SeNPs (73.26 μM, 76.51 μM, 96.14 μM), as shown in Fig. 4. The results indicate that FA and baicalin significantly enhance selective cellular uptake by cancer cells (as described in Fig. 3A). B–SeNPs–FA had significantly decreased toxicity to L02 cells and dramatically enhanced antitumor effects on HepG2215 cells. Taken together, these results show that the combination of baicalin and FA surface decoration could improve the selective cellular uptake of nanoparticles by cancer cells rather than normal cells.
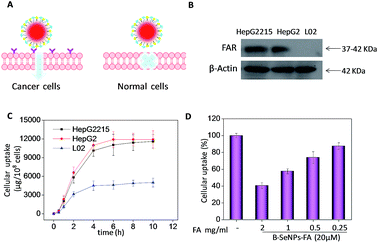 |
| | Fig. 3 Selective cellular uptake of B–SeNPs–FA. (A) A schematic illustration of selective cellular uptake; (B) the expression level of FARs in HepG2215 cells, HepG2 cells and L02 cells; (C) quantitative analysis of the cellular uptake efficiency of B–SeNPs–FA (30 mM) in HepG2215 cells, HepG2 cells and L02 cells; (D) the effects of FA on the cellular uptake of B–SeNPs–FA. The cells were treated with different concentrations of FA for 4 hours, and then incubated with 20 μM B–SeNPs–FA for 4 hours. | |
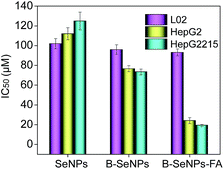 |
| | Fig. 4 Selective cytotoxicity of B–SeNPs–FA in L02 cells, HepG2 cells and HepG2215 cells for 36 hours. Each value represents the mean ± SD (n = 3). | |
Intracellular localization and uptake pathways of B–SeNPs–FA
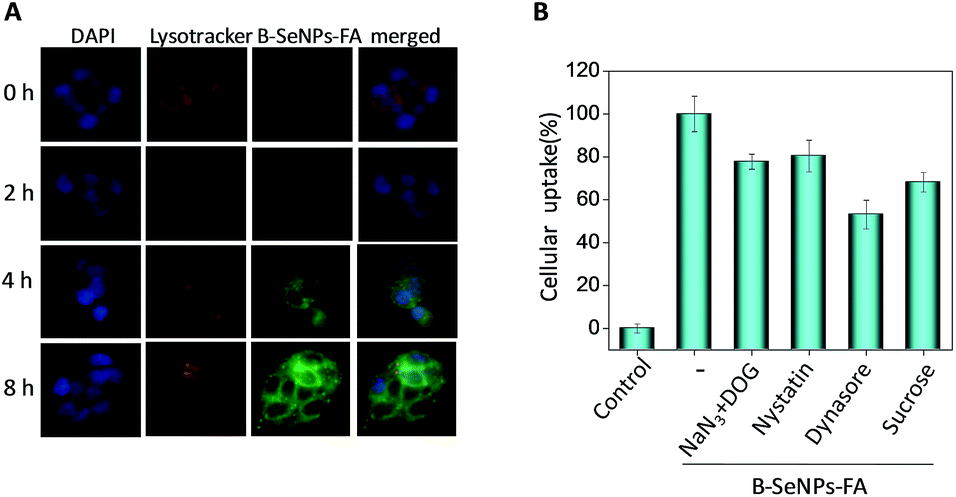
Endocytosis has been regarded as a crucial cellular uptake mechanism for nanosystems.40 According to our previous study, different from the traditional drug delivery nanosystems, selenium nanoparticles explode into smaller particles under the acidic environment of the lysosomes, which is helpful for the metabolism of selenium nanoparticles and the release of the loaded drugs.36,39,41 In this study, fluorescence microscopy analysis was used to trace the cellular localization of B–SeNPs–FA in HepG2215 cells. The coumarin-6-loaded B–SeNPs–FA was observed in HepG2 and HepG2215 cells that were stained with the lysosomal marker Lyso Tracker (red) and the nuclear marker DAPI (blue). As shown in Fig. 5A, with the increase of time, the green fluorescence intensity of B–SeNPs–FA gradually became strong, which suggested that more and more B–SeNPs–FA entered and accumulated in the HepG2215 cells. Also, there was lots of overlap of the green and red fluorescence in the HepG2215 cells, and this co-localization of B–SeNPs–FA and lysosomes in the cancer cells suggested that the primary target of B–SeNPs–FA is the lysosomes rather than the nucleus.
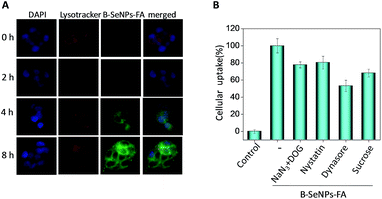 |
| | Fig. 5 (A) The cellular localization of coumarin-6-loaded B–SeNPs–FA in HepG2215 cells. The cells were exposed to coumarin-6-loaded B–SeNPs–FA (200 μM) for different time periods, and stained with Lyso Tracker (lysosome) and DAPI (nucleus); (B) intracellular uptake of B–SeNPs–FA in HepG2215 cells under different endocytosis-inhibited conditions. | |
The mechanism of cellular uptake was further analyzed using endocytosis inhibitor assays. As shown in Fig. 5B, nystatin significantly suppressed the endocytosis of B–SeNPs–FA to 80.43% of the control, which suggested that B–SeNPs–FA entered HepG2215 cells by caveolae-mediated endocytosis. In addition, the combination of sodium azide (NaN3) and 2-deoxy-D-glucose (DOG) treatment also inhibited the cellular internalization of B–SeNPs–FA to 77.77% of the control, indicating that B–SeNPs–FA is transported into HepG2215 cells via energy-dependent pathways. In addition, sucrose and dynasore, which inhibit the clathrin and dynamin-mediated lipid raft endocytosis mechanisms, reduced the amount of cellular uptake to 68.19% and 53.11% of the control, respectively. Taken together, caveolae-mediated endocytosis and energy-dependent endocytosis were the main pathways for the cellular uptake of B–SeNPs–FA in HepG2215 cells.
The pH responsive drug release of B–SeNPs–FA
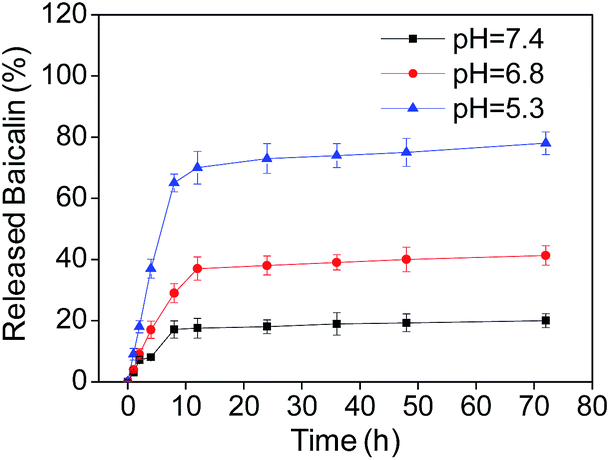
To understand the drug release ratio of B–SeNPs–FA, the nanoparticles were incubated in phosphate buffered solution (PBS) with pH 7.4, pH 6.8 and pH 5.3 to imitate different physiological environments. As shown in Fig. 6, the ratios of released baicalin in the different buffers all increased in a time-dependent manner. In addition, the ratio of released baicalin was 17.12% at pH 7.4, 29.01% at pH 6.8, and 65.45% at pH 5.3 within 8 h. The amount of baicalin release achieved was 20.30% at pH 7.4, 41.0% at pH 6.8, and 78.53% at pH 5.3 for the time of 72 h, and the amount of baicalin released at pH 5.3 was obviously higher than that released at pH 7.4 and pH 6.8 within the same time. These results demonstrate the pH-responsive drug release properties of the nanosystem.
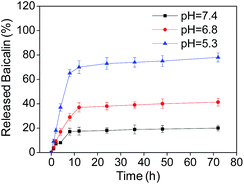 |
| | Fig. 6 The percentage of released baicalin at different pH values at different times. Each value represents the mean ± SD (n = 3). | |
B–SeNPs–FA inhibited HBxAg protein expression and induced cancer cell apoptosis
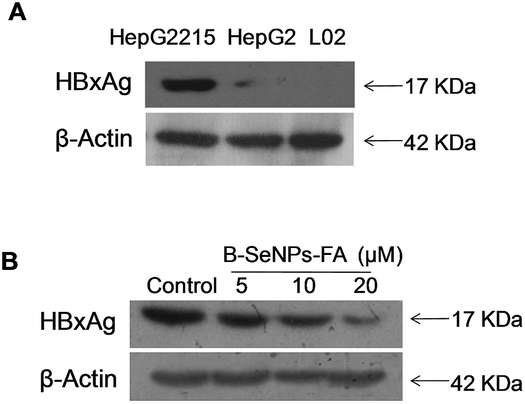
HBxAg, a major regulator of virulence proteins and a vital cofactor leading to chronic infection of liver cancer, plays a vital role in virus replication, cell cycle regulation, cell growth and cell apoptosis. Hence, the down-regulation of the expression of HBxAg would be a good way to treat HBV-infected liver cancer. First, we detected the expression level of HBxAg in the HepG2215 cells, HepG2 cells and L02 cells without B–SeNPs–FA. As shown in Fig. 7A, the expression level of the HBxAg protein in the HepG2215 cells was obviously higher than that in the HepG2 cells and L02 cells. We then further researched the influence of B–SeNPs–FA on the expression level of HBxAg in HepG2215 cells. We found that B–SeNPs–FA effectively inhibited the expression of HBxAg in HepG2215 cells in a dose-dependent manner (as shown in Fig. 7B). These results suggest that B–SeNPs–FA acts against HBV-transfected HepG2215 cells by inhibiting HBxAg protein expression.
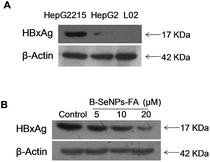 |
| | Fig. 7 (A) The western blot analysis of HBxAg in HepG2215, HepG2 and L02 cells; (B) the western blot analysis of HBxAg in HepG2215 cells induced with different concentrations of B–SeNPs–FA. β-Actin was used as a loading control. | |
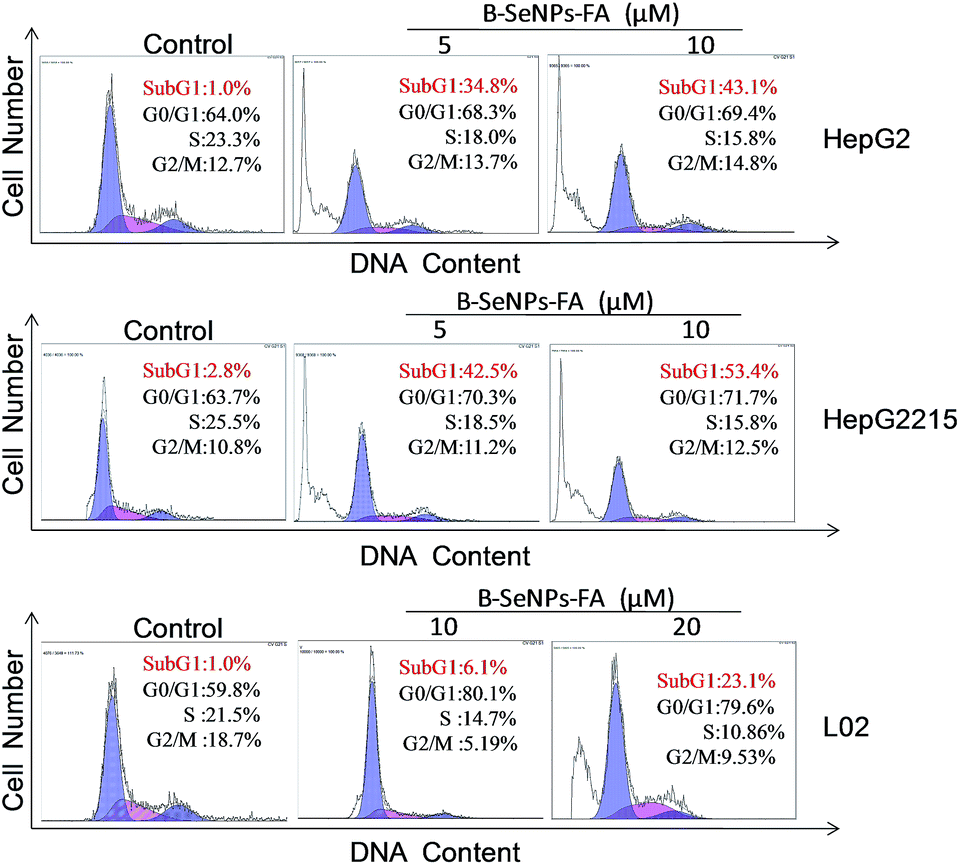
Apoptosis is a crucial mechanism contributing to the anti-cancer effects of SeNPs.39 In order to study the inhibition mechanism of B–SeNPs–FA on HepG2215 cells, flow cytometry analysis was used. As shown in Fig. 8, when HepG2215 cells and HepG2 cells were incubated with B–SeNPs–FA for 72 h, the number of cells distributed in the sub-G1 phase was significantly increased in a dose-dependent manner, which represented an increase in the level of cell apoptosis. After treatment with the same concentration of B–SeNPs–FA (10 μM), the percentage of apoptotic HepG2215 cells was 53.4%, which was significantly higher than the percentage of apoptotic HepG2 cells (43.1%), while no obvious apoptotic peak was detected in the L02 cells (normal liver cells). Additionally, there was no obvious change in the cell cycle distribution of these cells. These results suggested that B–SeNPs–FA had high toxicity to liver cancer cells (HepG2215 cells and HepG2 cells) via apoptosis induction. In particular, B–SeNPs–FA inhibited HBxAg protein expression, which accounted for HepG2215 cell apoptosis, and demonstrated the highly selective action of B–SeNPs–FA against cancer cells.
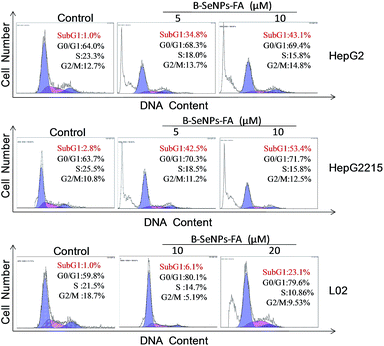 |
| | Fig. 8 Effects of B–SeNPs–FA on the cell cycle distribution of HepG2, HepG2215 and L02 cells after 72 hours. | |
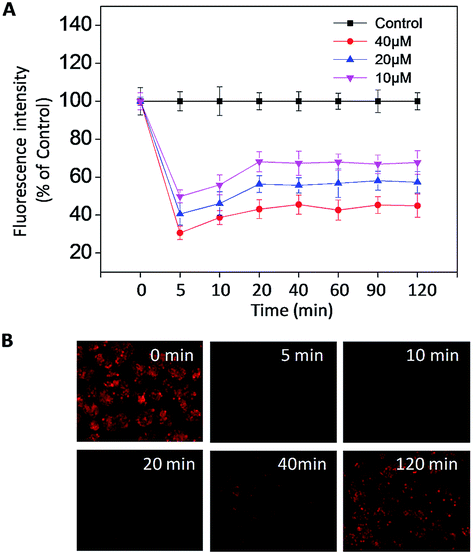
Imbalance of intracellular ROS induced by B–SeNPs–FA
Reactive oxygen species (ROS) are a series of intermediate products in the process of oxidation and reduction, including different forms of free radicals with high activities.44 ROS, mainly generated from oxidative stress, are considered to be an important factor in cancer development and recurrence.34 In recent years, many studies have found that changing the ROS level in cells could induce cell apoptosis and accelerate tumor cell death, and therefore could be used in treatment. In order to evaluate the intracellular ROS in the HepG2215 cells, a dihydroethidium (DHE) fluorescence assay was performed. As shown in Fig. 9A and B, when treated with different concentrations of B–SeNPs–FA, the levels of intracellular ROS all sharply declined within 5 min, and then slightly rose in the next 20 min before remaining nearly unchanged for 120 min. Moreover, we detected the dose-dependent down regulation of the ROS level by B–SeNPs–FA, and found that the level of ROS was decreased to 30.6% after 5 min when HepG2215 cells were exposed to B–SeNPs–FA (40 μM). Taken together, these results show that B–SeNPs–FA could lead to cell apoptosis by down-regulating the ROS level in HepG2215 cells.
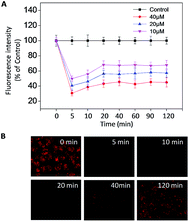 |
| | Fig. 9 (A) The intracellular ROS levels in HepG2215 cells at different time periods, determined from the DHE fluorescence intensity after the cells were treated with different concentrations of B–SeNPs–FA. All of the experiments were performed three times. (B) Fluorescence images of the ROS level change at 0, 5, 10, 20, 40, and 120 min in HepG2215 cells incubated with B–SeNPs–FA. | |
B–SeNPs–FA inhibited migration and invasion
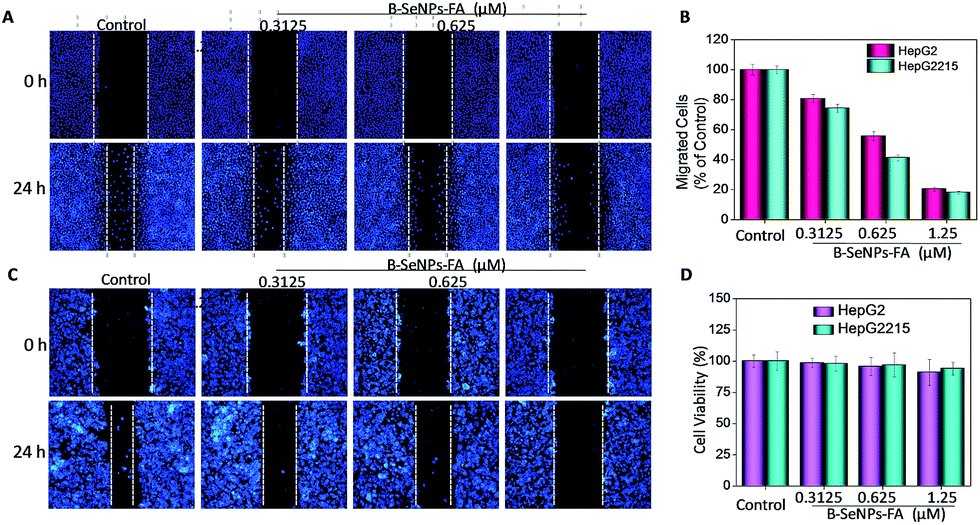
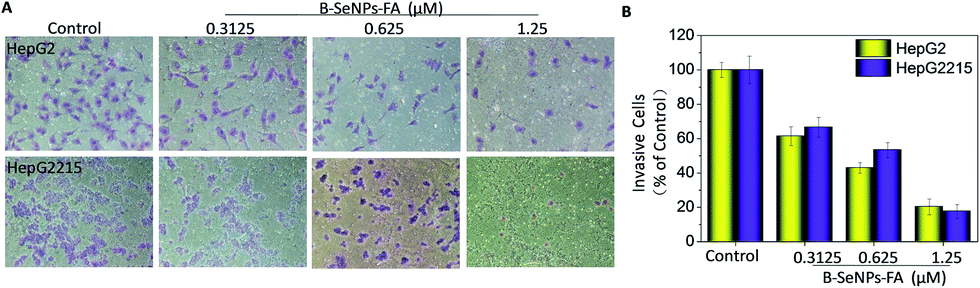
Inhibition of tumor cell migration and invasion, which involve a series of changes in biological cells including expression of adhesion molecules, formation of pseudopodia, alteration of the cytoskeleton, activation of associated signaling pathways, degradation of the basement membrane and extracellular substrates etc., is a key step in the treatment of cancer.41 Therefore, a scratch assay and a Transwell assay were performed to detect the inhibition effect of low toxicity concentrations of B–SeNPs–FA on the migration and invasion of HepG2 and HepG2215 cells (Fig. 10D). As shown in Fig. 10 and 11, compared with the control, the migration and invasion of HepG2215 and HepG2 cells were obviously inhibited by B–SeNPs–FA in a dose-dependent manner for 24 h (the quantitative data are given in Fig. 10B and 11B). These results indicate that B–SeNPs–FA exhibit excellent ability in inhibiting the migration and invasion of HepG2215 and HepG2 cells.
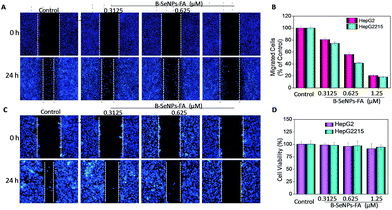 |
| | Fig. 10 (A) B–SeNPs–FA inhibited the migration of HepG2 cells; (B) the effect of B–SeNPs–FA on the relative number of migrating cells; (C) B–SeNPs–FA inhibited the migration of HepG2215 cells; (D) the low toxicity of B–SeNPs–FA to HepG2 and HepG2215 cells at the same concentration as used in the migration and invasion assay. | |
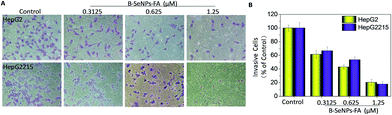 |
| | Fig. 11 (A) B–SeNPs–FA inhibited the invasion of HepG2 cells and HepG2215 cells; (B) the effect of B–SeNPs–FA on the relative number of invasive cells. | |
Conclusions
In summary, we designed and synthesized a multifunctional nanosystem to inhibit HBV-infected liver cancer. The selenium nanoparticles surface-modified with baicalin and FA targeting moieties showed appropriate particle size distribution, high aqueous solubility and good biocompatibility. The nanosystem exhibited excellent selectivity towards tumor cells by primarily targeting the lysosomes in HepG2215 cells, through utilizing the main pathways of caveolae-mediated endocytosis and energy-dependent endocytosis. In addition, B–SeNPs–FA could down-regulate the generation of ROS and inhibit the expression of HBxAg, which led to HepG2215 cell apoptosis. Moreover, B–SeNPs–FA exhibited excellent ability in inhibiting cancer cell migration and invasion. Taken together, baicalin-loaded selenium nanoparticles with an FA targeting moiety might be an innovative and feasible drug design for treating HBV-infected liver cancer. Our study also provides ideas for designing Chinese medicine monomers and effectively utilizing molecular targeting of traditional Chinese medicines.
Acknowledgements
This work was supported by the Natural Science Foundation of China (21401076, 21271002, 81273616, 81473557, 81403302), National High-level personnel of special support program, National High Technology Research and Development Program of China (SS2014AA020538), Science Foundation for Distinguished Young Scholars of Guangdong Province (S2013050014667), Foundation for High-level Talents in Higher Education of Guangdong, YangFan Innovative & Entrepreneurial Research Team Project (201312H05), Guangdong Special Support Program and Guangdong Frontier, Key Technological Innovation Special Funds (2014B050505012) and Fundamental Research Funds for the Central Universities.
References
- H. I. Yang, S. N. Lu, Y. F. Liaw, S. L. You, C. A. Sun, L. Y. Wang, C. K. Hsiao, P. J. Chen, D. S. Chen and C. J. Chen, N. Engl. J. Med., 2002, 347, 168–174 CrossRef CAS PubMed.
- K. W. Livezey, D. Negorev and D. Simon, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2000, 452, 163–178 CrossRef CAS PubMed.
- K. W. Livezey, D. Negorev and D. Simon, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2002, 505, 63–74 CrossRef CAS PubMed.
- Y. Q. Li and S. F. Wang, Biotechnol. Lett., 2006, 28, 837–841 CrossRef CAS PubMed.
- M. A. Sells, A. Z. Zelent, M. Shvartsman and G. Acs, J. Virol., 1988, 62, 2836–2844 CAS.
- B. Rehermann and M. Nascimbeni, Nat. Rev. Immunol., 2005, 5, 215–229 CrossRef CAS PubMed.
- L. EAftSot, J. Hepatol., 2012, 57, 167–185 CrossRef PubMed.
- E. E. Mast, H. S. Margolis, A. E. Fiore, E. W. Brink, S. T. Goldstein, S. A. Wang, L. A. Moyer, B. P. Bell and M. J. Alter, MMWR Recomm. Rep., 2005, 54, 1–31 Search PubMed.
- E. B. Keeffe, D. T. Dieterich, S. H. B. Han, I. M. Jacobson, P. Martin, E. R. Schiff, H. Tobias and T. L. Wright, Clin. Gastroenterol. Hepatol., 2006, 4, 936–962 CrossRef CAS PubMed.
- R. Puro and R. J. Schneider, J. Virol., 2007, 81, 7351–7362 CrossRef CAS PubMed.
- J. Pan, L.-X. Duan, B. S. Sun and M. A. Feitelson, J. Gen. Virol., 2001, 82, 171–182 CrossRef CAS PubMed.
- E. Szabó, C. Páska, P. K. Novák, Z. Schaff and A. Kiss, Pathol. Oncol. Res., 2004, 10, 5–11 CrossRef.
- A. S. Cheng, N. Wong, M. Y. Ada, K. Y. Chan, K. K. Chan, J. J. Sung and H. L. Chan, Cancer Lett., 2007, 253, 43–52 CrossRef CAS PubMed.
- J. Hoare, F. Henkler, J. J. Dowling, W. Errington, R. D. Goldin, D. Fish and M. J. McGarvey, J. Med. Virol., 2001, 64, 419–426 CrossRef CAS PubMed.
- X. Zhang, H. Zhang and L. Ye, J. Lab. Clin. Med., 2006, 147, 58–66 CrossRef CAS PubMed.
- M. Bouattour, O. Soubrane, A. de Gramont and S. Faivre, Trials, 2016, 17, 563 CrossRef PubMed.
- S. Deol, N. Weerasuriya and Y. S. Shon, J. Mater. Chem. B, 2015, 3, 6071–6080 RSC.
- M. Li-Weber, Cancer Treat. Rev., 2009, 35, 57–68 CrossRef CAS PubMed.
- J. Z. Yu, X. L. Wu, B. Yu, C. Q. Dai, K. L. Liu, G. Q. Qian, G. X. Zhou, S. H. Lu, D. H. Ju and X. Y. Chen, Pharmacogn. Mag., 2012, 8, 225–230 CrossRef CAS PubMed.
- Y. Chen, L. V. Minh, J. Liu, B. Angelov, M. Drechsler, V. M. Garamus, R. Willumeit-Römer and A. Zou, Colloids Surf., B, 2016, 140, 74–82 CrossRef CAS PubMed.
- D. E. Shieh, L. T. Liu and C. C. Lin, Anticancer Res., 1999, 20, 2861–2865 Search PubMed.
- C. Z. Wang, T. D. Calway, X. D. Wen, J. Smith, C. Yu, Y. Wang, S. R. Mehendale and C.-S. Yuan, Int. J. Oncol., 2013, 42, 1018–1026 CAS.
- W. C. Chen, T. H. Kuo, Y. S. Tzeng and Y. C. Tsai, Molecules, 2012, 17, 3844–3857 CrossRef CAS PubMed.
- S. Ikemoto, K. Sugimura, N. Yoshida, R. Yasumoto, S. Wada, K. Yamamoto and T. Kishimoto, Urology, 2000, 55, 951–955 CrossRef CAS PubMed.
- J. Feng, C. Guo, Y. Zhu, L. Pang, Z. Yang, Y. Zou and X. Zheng, Int. J. Clin. Exp. Med., 2014, 7, 4063 Search PubMed.
- F. L. Chan, H. L. Choi, Z. Y. Chen, P. S. F. Chan and Y. Huang, Cancer Lett., 2000, 160, 219–228 CrossRef CAS PubMed.
- R.-L. Huang, C.-C. Chen, H.-L. Huang, C.-G. Chang, C.-F. Chen, C. Chang and M.-T. Hsieh, Planta Med., 2000, 66, 694–698 CrossRef CAS PubMed.
- L. Z. He, Y. Y. Huang, H. L. Zhu, G. H. Pang, W. J. Zheng, Y. S. Wong and T. F. Chen, Adv. Funct. Mater., 2014, 24, 2754–2763 CrossRef CAS.
- Y. Y. Huang, Y. Luo, W. J. Zheng and T. F. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19217–19228 CAS.
- L. Z. He, T. F. Chen, Y. Y. You, H. Hu, W. J. Zheng, W. L. Kwong, T. T. Zou and C. M. Che, Angew. Chem., Int. Ed., 2014, 53, 12532–12536 CAS.
- E. M. Alexandrino, S. Ritz, F. Marsico, G. Baier, V. Mailänder, K. Landfester and F. R. Wurm, J. Mater. Chem. B, 2014, 2, 1298–1306 RSC.
- W. Liu, X. Li, Y.-S. Wong, W. Zheng, Y. Zhang, W. Cao and T. Chen, ACS Nano, 2012, 6, 6578–6591 CrossRef CAS PubMed.
- P. A. Tran and T. J. Webster, Int. J. Nanomed., 2011, 6, 1553–1558 CAS.
- W. T. Jiang, Y. T. Fu, F. Yang, Y. F. Yang, T. Liu, W. J. Zheng, L. L. Zeng and T. F. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 13738–13748 CAS.
- T. Chen, Y.-S. Wong, W. Zheng, Y. Bai and L. Huang, Colloids Surf., B, 2008, 67, 26–31 CrossRef CAS PubMed.
- T. Liu, L. L. Zeng, W. T. Jiang, Y. T. Fu, W. J. Zheng and T. F. Chen, Nanomedicine, 2015, 11, 947–958 CAS.
- H. Wu, X. Li, W. Liu, T. Chen, Y. Li, W. Zheng, C. W.-Y. Man, M.-K. Wong and K.-H. Wong, J. Mater. Chem., 2012, 22, 9602–9610 RSC.
- X. Gao, J. Zhang and L. Zhang, Adv. Mater., 2002, 14, 290 CrossRef CAS.
- Y. Huang, L. He, W. Liu, C. Fan, W. Zheng, Y.-S. Wong and T. Chen, Biomaterials, 2013, 34, 7106–7116 CrossRef CAS PubMed.
- B. Zhou, Y. Huang, F. Yang, W. Zheng and T. Chen, Chem.–Asian J., 2016, 11, 1008–1009 CrossRef CAS PubMed.
- L. L. Zeng, J. J. Chen, S. B. Ji, L. Chan, W. J. Zheng and T. F. Chen, J. Mater. Chem. B, 2015, 3, 4345–4354 RSC.
- Y. You, H. Hu, L. He and T. Chen, Chem.–Asian J., 2015, 10, 2744–2754 CrossRef CAS PubMed.
- H. Hu, Y. You, L. He and T. Chen, J. Mater. Chem. B, 2015, 3, 6338–6346 RSC.
- J. Su, H. Lai, J. Chen, L. Li, Y.-S. Wong, T. Chen and X. Li, PLoS One, 2013, 8, e63502 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28229f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a