
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structures and catalytic activity of cyclometalated rhenium complexes†
Ruichen Sun,
Tian Wang,
Shaowei Zhang,
Xiaodan Chu and
Bolin Zhu *
*
Tianjin Key Laboratory of Structure and Performance for Functional Molecules, Key Laboratory of Inorganic–Organic Hybrid Functional Material Chemistry, Ministry of Education, College of Chemistry, Tianjin Normal University, Tianjin 300387, PR China. E-mail: hxxyzbl@gmail.com; Fax: +86-22-23766532; Tel: +86-22-23766515
First published on 17th March 2017
Abstract
Thermal reactions of aryl-substituted phosphines or phosphinites with Re2(CO)10 in chlorobenzene resulted in the corresponding five-membered cyclometalated rhenium complexes (1–5) via an intramolecular activation of the C(sp2)–H or C(sp3)–H bond. The only exception occurred in the case of (1-naphthyl)diisopropylphosphinite, which gave a diphosphinite-substituted dinuclear rhenium complex 6. Competition reaction indicated that the aromatic C(sp2)–H bond is more likely to be activated than the C(sp3)–H bond under the same conditions. Photolysis of 1 or 2 in CHX3 led to the cleavage of the Re–C σ bond to yield corresponding phosphine-substituted tetracarbonyl rhenium halides 7–10. Complex 1 reacted with CF3COOH in CH2Cl2 to give addition product 11. Photolysis of cyclorhenated complexes 1–3 with a series of aryl halides in benzene resulted in the stoichiometric formation of biphenyl, together with corresponding phosphine-substituted tetracarbonyl rhenium halides (7, 9, 10, 12, and 13). When base was introduced into the above reaction, a catalytic system was established. Under optimized conditions, complex 1 provided moderate yield of biphenyl in a couple of hours at a [Re]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate ratio of 1:200. Molecular structures of complexes 1, 6, 9, and 11 were determined by X-ray diffraction.
:substrate ratio of 1:200. Molecular structures of complexes 1, 6, 9, and 11 were determined by X-ray diffraction.
Introduction
Cyclometalation has always been one of the most widely investigated reactions in the field of organometallic chemistry since it was first discovered in the early 1960s.1 The reason that cyclometalation has received such considerable attention is mainly due to the following three aspects: (i) it represents probably the mildest route for the intramolecular activation of C–H bonds, and forms a cyclometalated complex containing a new M–C σ bond. (ii) Cyclometalated complexes have wide applications in many fields, such as photosensitizers for solar cells2 and OLED materials.3 (iii) As catalysts for a wide range of reactions, including metathesis reactions,4 cross-coupling reactions,5 and transfer hydrogenation reactions.6 Of all transition metals, cyclometalated rhenium complexes have recently attracted more and more attention due to their luminescent properties, which may have potential as liquid crystals,7 and in particular, their indispensable role in many catalytic cycles, including the rhenium-catalyzed annulation reaction of aromatic imines with alkynes,8 alkenes,9 or aldehyde,10 and alkylation of 2-phenyl, or alkenyl-substituted pyridine.11 Generally, the most widely used precursors for cyclorhenation are Re(X)(CO)5 (X = H, Me, Cl, or Br) and Re(Cp)L3 (L = CO, PR3, or a mixture thereof). In this paper, we report the synthesis of a series of cyclometalated rhenium complexes starting from the precursor Re2(CO)10, as well as their catalytic activity in direct arylation of arenes with aryl halides.Results and discussion
Reaction of Re2(CO)10 with (1-naphthyl)diphenyl phosphine or diisopropyl(1-naphthyl) phosphine
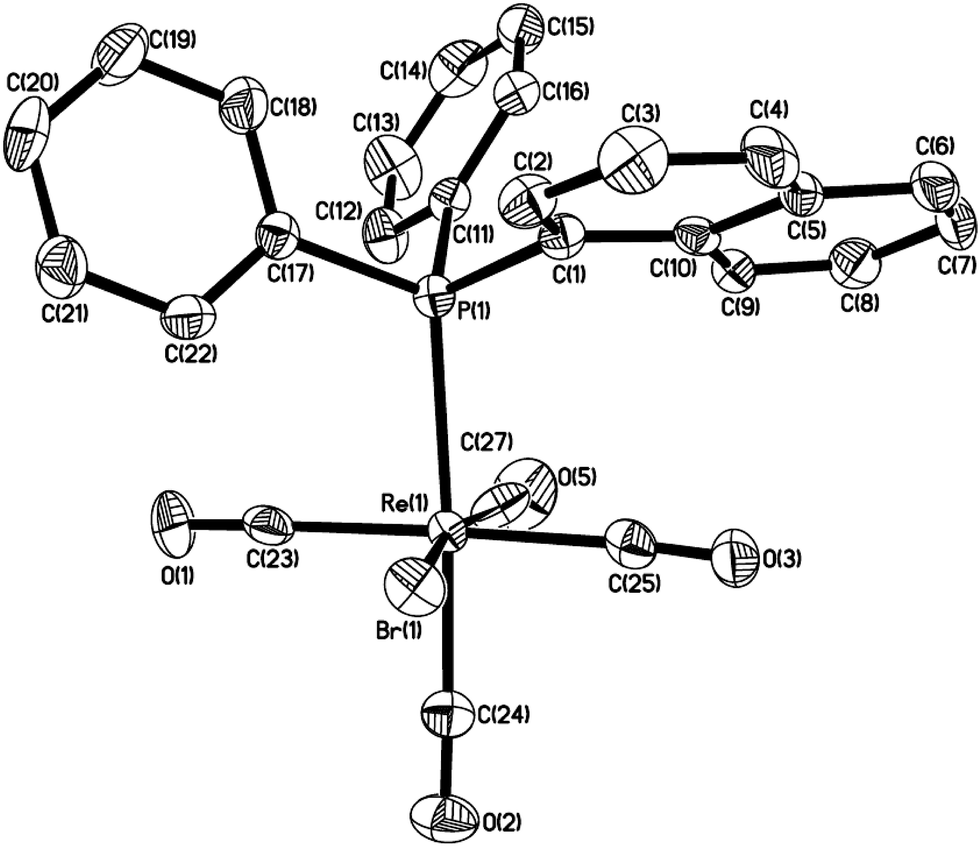
A solution of Re2(CO)10 and 2 equiv. of (1-naphthyl)diphenyl phosphine or diisopropyl(1-naphthyl) phosphine in chlorobenzene was refluxed overnight to result in corresponding five-membered cyclorhenated complex 1 or 2 in good yields (Scheme 1). Obviously, the formation of complexes 1 and 2 goes through an intramolecular activation of C(sp2)–H bond on the adjacent α position of naphthyl ring. The 1H NMR spectrum of 1 showed six peaks centered at δ 8.21 (1H), 7.95 (1H), 7.68 (1H), 7.60 (1H), 7.55–7.49 (1H, overlapping with peaks for protons on two phenyl rings), 7.35 (1H) corresponding to six protons of the metalated naphthyl group. More distinctly, the 1H NMR spectrum of 2 exhibited five well-separated multiplets at δ 8.16 (1H), 7.85 (1H), 7.58 (2H), 7.43 (1H), 7.28 (1H) for resonance of six protons on the metalated naphthyl group. The IR spectra of 1 and 2 (KBr film) indicated the expected strong ν(CO) absorptions in the range 2084–1925 cm−1 for terminal carbonyls. The structure of 1 was further confirmed by single-crystal diffraction analysis, which is shown in Fig. 1, with selected bond lengths and angles. It reveals an octahedral geometry with the rhenium(I) center coordinated by four carbonyl groups, one phosphorus atom P(1), and one metalated carbon atom C(3), in which P(1) and C(3) are mutually adjacent. Re–P bond length is 2.4413(9) Å, a litter longer than that (2.428(3) Å) in four-membered cyclorhenated analogue Re(CO)4(PPh2CH2CH2).12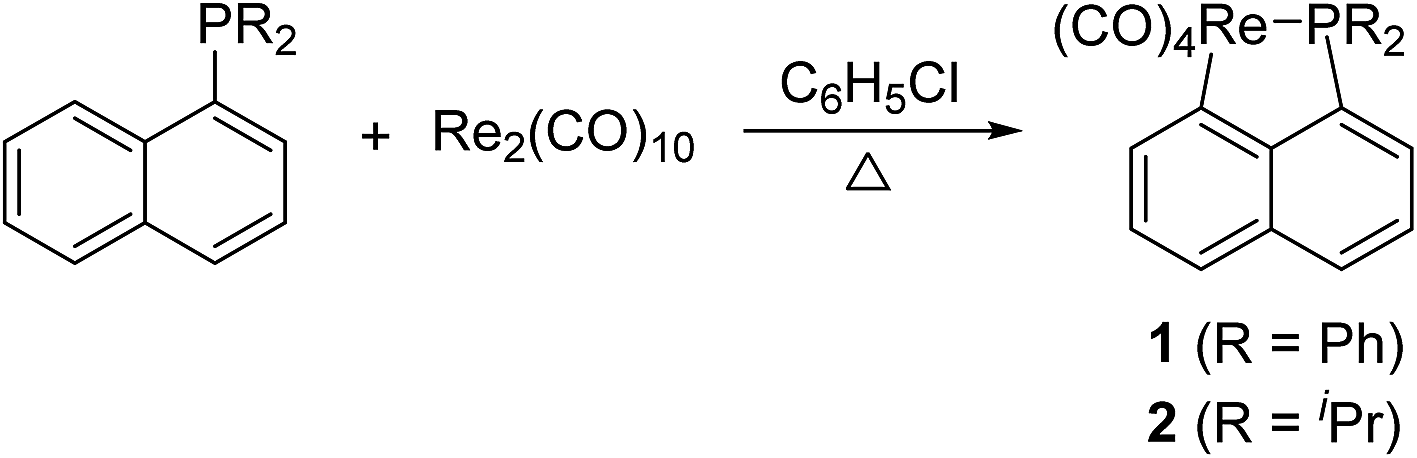 | ||
| Scheme 1 Reactions of 1-naphthyl-substituted phosphines with Re2(CO)10. | ||
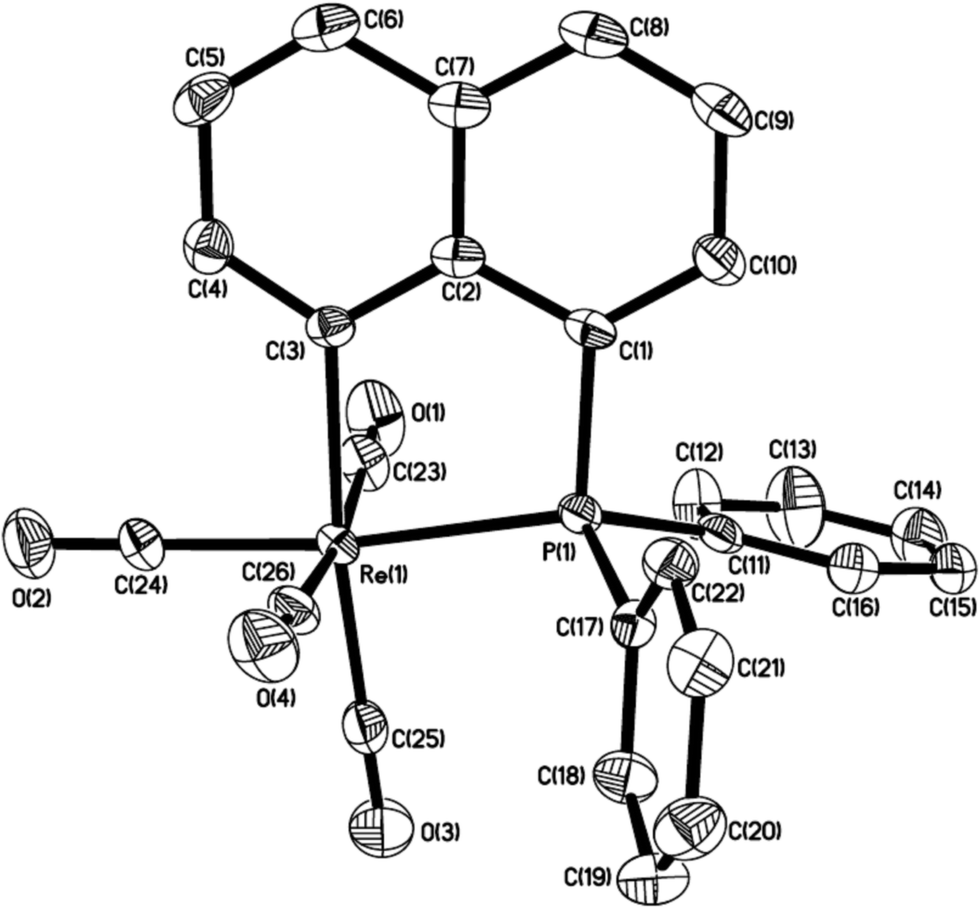 | ||
| Fig. 1 Thermal ellipsoid drawing of 1 showing the labeling scheme and 50% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Re(1)–P(1) 2.4413(9), P(1)–C(1) 1.812(3), Re(1)–C(23) 1.987(4), Re(1)–C(24) 1.960(4), Re(1)–C(25) 1.969(4), Re(1)–C(26) 2.003(4), ∠C(3)–Re(1)–C(25) 173.87(14), ∠C(24)–Re(1)–P(1) 170.15(12), ∠C(23)–Re(1)–C(26) 171.80(16), ∠C(3)–Re(1)–P(1) 79.05(9), ∠Re(1)–P(1)–C(1) 103.52(12). | ||
Reaction of Re2(CO)10 with diisopropyl(o-methylphenyl) phosphine
Having demonstrated that phosphine ligand could be employed as a donor group for metalation of a naphthyl C(sp2)–H bond in complexes 1 and 2, we decided to test whether it could also promote activation of a C(sp3)–H bond. The corresponding reaction of Re2(CO)10 with diisopropyl(o-methylphenyl) phosphine was carried out, which afforded a product 3 in 50% yield (Scheme 2). The 1H NMR spectrum of 3 showed three multiplets at δ 7.38 (1H), 7.24 (2H), 7.04 (1H) duo to four protons on the phenyl ring, one multiplet at δ 2.49 (2H) for methenyl signal, and two doublets of doublets at δ 1.20 (6H) and 1.05 (6H) for signals of four methyls on two isopropyl groups. However, there was no signal for the methyl on the phenyl ring; instead one multiple at δ 2.50, integrating to 2H, assigned to a metal-bound CH2. These resonances confirm the expected C(sp3)–H bond activation.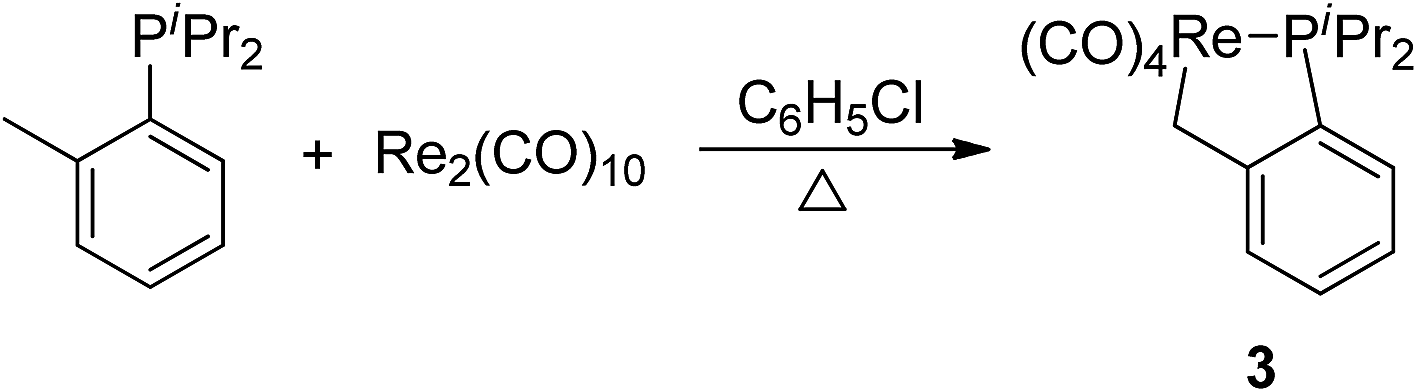 | ||
| Scheme 2 Reaction of diisopropyl(o-methylphenyl) phosphine with Re2(CO)10. | ||
Reactions of Re2(CO)10 with diisopropyl(2-methylnaphthyl) phosphine
Since both C(sp2)–H bond and C(sp3)–H bond could be activated in cyclometalation of Re2(CO)10 with phosphines, we were curious about the relative activity between them. Therefore, diisopropyl(2-methylnaphthyl) phosphine was chosen as the ligand, which provides both C(sp2)–H and C(sp3)–H bonds at the appropriate positions, to test the competition reaction. Therefore, reaction of Re2(CO)10 with diisopropyl(2-methylnaphthyl) phosphine under the above-mentioned conditions was performed, which gave a product 4 (Scheme 3). The 1H NMR spectrum of 4 showed four multiplets in the aromatic region, all integrating to 5H due to the naphthyl ring, and a doublet at δ 2.71, assigned to signal for the methyl on the naphthyl ring. These resonances indicated that the aromatic C(sp2)–H bond is selectively activated in the reaction to form the five-membered cyclometalated complex 4. The reason that the aromatic C(sp2)–H bond activation is favored over C(sp3)–H bond activation is probably owing to the higher thermal stability of aryl hydrogen activated product when compared to alkyl hydrogen activated product.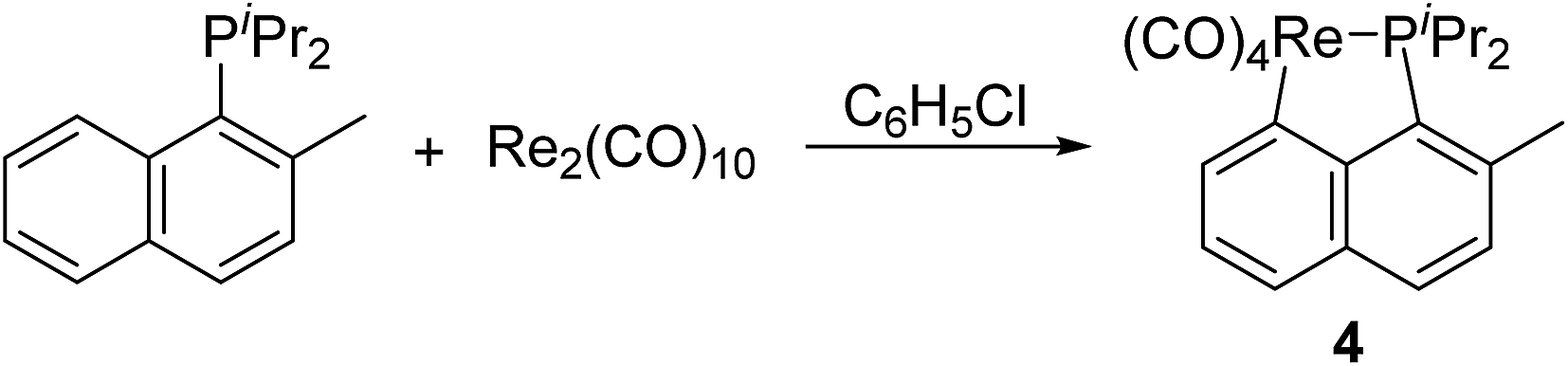 | ||
| Scheme 3 Reaction of diisopropyl(2-methylnaphthyl) phosphine with Re2(CO)10. | ||
Reactions of Re2(CO)10 with phosphinites
In order to explore applicable scope of this type of cyclometalation, ligand was extended to phosphinite. Therefore reaction of Re2(CO)10 with o-methylphenyl diisopropylphosphinite was examined, which produced the corresponding five-membered cyclometalated complex 5 (Scheme 4). The 1H NMR spectrum of 5 showed three multiplets (δ 7.55, 6.84, and 6.71) in the aromatic region, each integrating to 1H due to the phenyl ring, and a singlet at δ 2.25, assigned to signal for the methyl on the phenyl ring. These 1H NMR information confirms an ortho aromatic C(sp2)–H bond activation.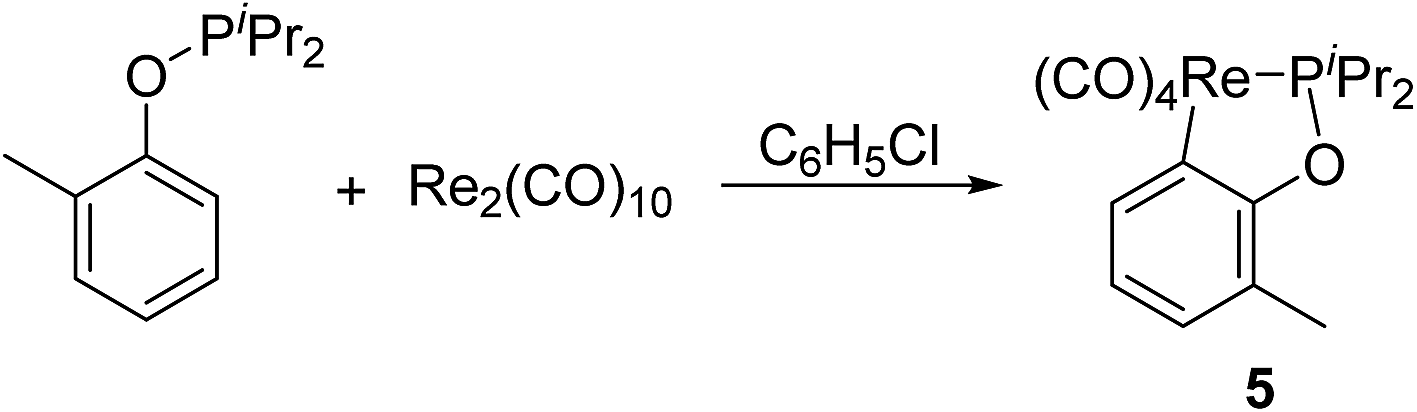 | ||
| Scheme 4 Reaction of o-methylphenyl diisopropylphosphinite with Re2(CO)10. | ||
However, when Re2(CO)10 reacted with another phosphinite, 1-naphthyl diisopropylphosphinite under the above-mentioned conditions, the reaction did not produce the similar five-membered cyclometalated complex, instead a dinuclear complex 6 was obtained (Scheme 5). The 1H NMR spectrum of 6 showed six multiplets in the aromatic region, all integrating to 7H corresponding to protons of the naphthyl ring. These resonances indicate the absence of aromatic C(sp2)–H bond activation. The structure of 6 was further confirmed by single-crystal diffraction analysis, which is shown in Fig. 2, with selected bond lengths and angles. It consists of a pair of square pyramidal Re(CO)4(P) units linked by a Re–Re bond. Each phosphinite ligand is located at the top of the pyramid. Two sets of carbonyl groups bounded to two rhenium atoms occur as the staggered conformation. Re–Re bond length is 3.0606(5) Å, a litter longer than that (3.04 Å) in Re2(CO)10.13
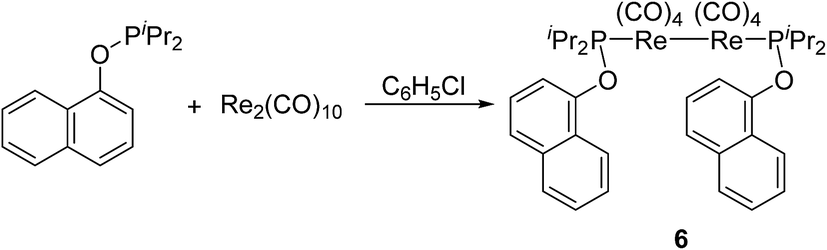 | ||
| Scheme 5 Reaction of 1-naphthyl diisopropylphosphinite with Re2(CO)10. | ||
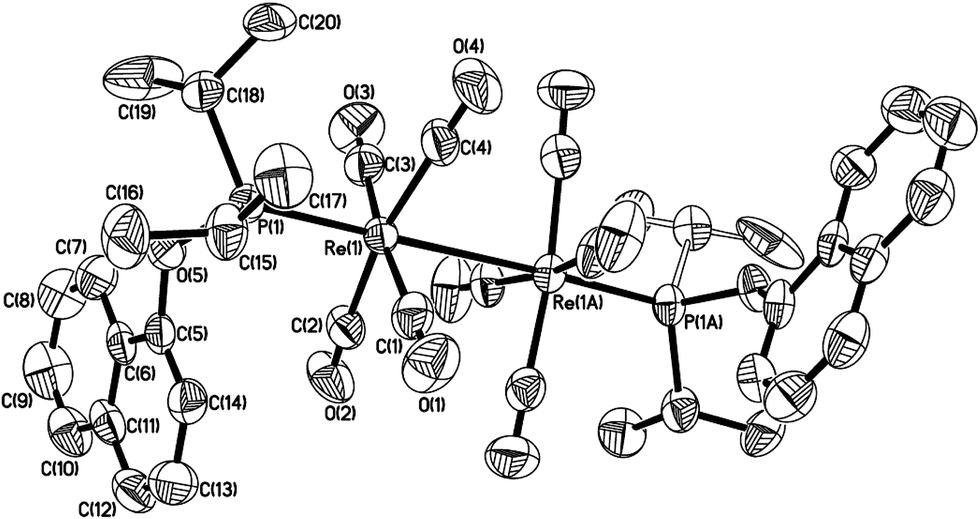 | ||
| Fig. 2 Thermal ellipsoid drawing of 6 showing the labeling scheme and 50% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Re(1)–Re(1A) 3.0606(5), Re(1)–P(1) 2.3419(15), Re(1)–C(1) 1.982(10), Re(1)–C(2) 1.977(7), Re(1)–C(3) 1.977(10), Re(1)–C(4) 1.958(8), ∠P(1)–Re(1)–Re(1A) 177.56(4), ∠C(1)–Re(1)–C(3) 173.0(3), ∠C(2)–Re(1)–C(4) 168.1(3). | ||
Cleavage of the cyclometalated Re–C bond of 1 or 2
Lu has reported the chemical behavior of the similar N-contained cyclorhenated complexes, including the electrophilic addition reactions of cyclorhenated complexes with HBr, RCO2H, or I2, which caused the opening of the metallacyclic ring to generate the corresponding rhenium products.14 For comparison of reactivity, we similarly treated the P-contained cyclorhenated complex 1 with excess I2 in CH2Cl2 at room temperature for 24 h, however, no reaction occurred. This result may indicate the higher stability of 1 when compared with N-contained analogues. Therefore we decided to try the decyclometalation of 1 or 2 under UV light conditions. A solution of complex 1 or 2 in CHCl3 or CHBr3 was irradiated by UV lamp to give corresponding phosphine-substituted tetracarbonyl rhenium halide 7–10 (Scheme 6). Obviously, the Re–C bond of the five-membered ring in 1 or 2 is cleavaged readily during the photolysis. Complexes 7–10 were fully characterized by spectroscopic analysis, as well as crystallographic analysis for 9. The molecular structure of 9 was shown in Fig. 3, with selected bond lengths and angles. Pseudooctahedral geometry is observed around the Re atom. The bromine and the (1-naphthyl)diphenyl phosphine groups are cis to each other. The Re(1)–P(1) bond length is 2.5016(12) Å, longer than that (2.4413(9) Å) in five-membered cyclorhenated structure of 1, which could be attributed to nonbonding interactions between the phosphine and Re(CO)4Br units.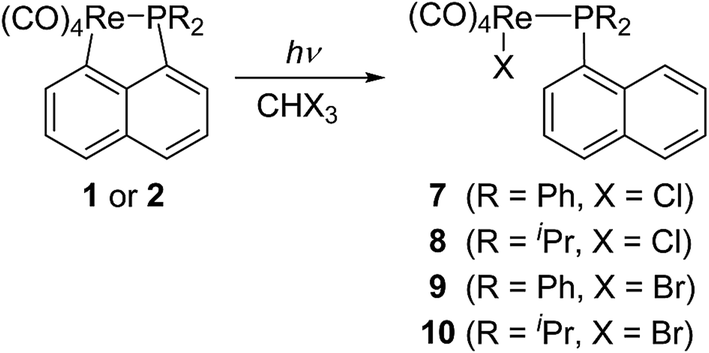 | ||
| Scheme 6 Photolysis of 1 or 2 in CHX3 (X = Cl, Br). | ||
 | ||
| Fig. 3 Thermal ellipsoid drawing of 9 showing the labeling scheme and 50% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Re(1)–P(1) 2.5016(12), Re(1)–Br(1) 2.620(2), Re(1)–C(23) 2.016(6), Re(1)–C(24) 1.968(5), Re(1)–C(25) 1.997(6), Re(1)–C(27) 1.952(8), ∠P(1)–Re(1)–C(24) 176.40(16), ∠C(23)–Re(1)–C(25) 178.0(2), ∠Br(1)–Re(1)–C(27) 169.1(6). | ||
Besides, for further comparison of reactivity, we also performed the photolysis of 1 with CF3COOH. After a solution of 1 with CF3COOH in CH2Cl2 was irradiated for 8 h, a white product 11 was obtained in moderate yield (Scheme 7). Complexes 11 was fully characterized by spectroscopic and crystallographic analysis. The molecular structure of 11 was shown in Fig. 4. Similarly to 9, the pseudooctahedral geometry is observed around the Re atom. The carboxylate (CF3CO2) and the (1-naphthyl)diphenyl phosphine groups are cis to each other. The Re(1)–P(1) bond length is 2.494(2) Å, close to (2.5016(12) Å) that in 9, but also longer than that (2.4413(9) Å) in five-membered ring structure of 1. The formation of 11 indicates that the reactivity of 1 is similar to that of N-contained analogues when reacted with CF3CO2H.14
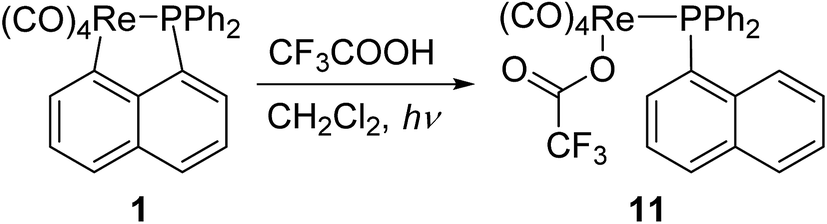 | ||
| Scheme 7 Photolysis of 1 with CF3COOH in CH2Cl2. | ||
 | ||
| Fig. 4 Thermal ellipsoid drawing of 11 showing the labeling scheme and 50% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Re(1)–P(1) 2.494(2), Re(1)–O(1) 2.145(5), Re(1)–C(23) 1.971(9), Re(1)–C(24) 2.007(8), Re(1)–C(25) 1.915(8), Re(1)–C(26) 2.003(8), ∠P(1)–Re(1)–C(23) 174.4(2), ∠C(24)–Re(1)–C(26) 176.4(3), ∠O(1)–Re(1)–C(25) 176.0(3). | ||
Photochemical reactions of 1–3 with aryl halides in benzene
We have reported the photochemical reactions of cyclopentadienylrheniumtricarbonyls [(η5-C5R5)Re(CO)3] (R = Me or H), or doubly-bridged biscyclopentadienyl dirhenium carbonyls with aryl halides in benzene, which generate biphenyl almost stoichiometrically, as well as the corresponding rhenium dicarbonyl products.15 In order to expand the reaction scope of this type, we used cyclometalated rhenium complexes 1–3 as starting materials and tried the similar photochemical reactions with aryl halides in benzene.Photolysis of 1–3 with 5 equiv. of bromobenzene in benzene under N2 for 1–4.5 h produced a yellow solution. After removal of the solvent under vacuum, the residue was carefully separated in air by column chromatography on silica gel, and afforded the corresponding phosphine-substituted tetracarbonyl rhenium bromides 9, 10, and 12 (Scheme 8). Besides, the reactions also stoichiometrically produced biphenyl (92–98%, entries 1–3 in Table 1). Further activation of C–I bond and the relatively inert C–Cl bond by 1 was also conducted by photolysis of 1 with iodobenzene and chlorobenzene in benzene, which similarly provided the corresponding phosphine-substituted tetracarbonyl rhenium iodide (13) and chloride (7), as well as the biphenyl (Scheme 8) (entries 4–5, Table 1). To expand the scope of the substrates, photolysis of several para-substituted aryl halides with 1 were examined, and the reactions gave similarly the rhenium halides 7 or 9, and the para-substituted biphenyl, no matter when the substituent is an electron-donating (MeO) or electron-withdrawing (CF3, CH3CO) group (entries 6–10, Table 1). Next, the source of the biphenyl was investigated. Photolysis of 1 with p-bromoanisole in benzene-d6 generated the 4-methoxybiphenyl-d5 (p-CH3O–C6H4–C6D5), completely no biphenyl or 4,4′-dimethoxybiphenyl were observed. It is revealed that the p-CH3O–C6H4 and C6D5 moieties in the resulting substituted biphenyl come from p-bromoanisole and solvent benzene-d6, respectively.
 | ||
| Scheme 8 Photochemical reactions of 1–3 with aryl halides in benzene. | ||
| Entry | Re | R′ | X | t [h] | Productb | Yieldc [%] | Re prod. |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: irradiation of 1–3 (0.10 mmol) and Ar–X (0.50 mmol) in 10 mL benzene solvent with a 500 W high-pressure Hg lamp (λmax = 365 nm) in the ice-water bath.b Biphenyl products were isolated by routine silica gel column chromatography, and identified by comparing their respective 1H NMR spectra with authentic samples.c Isolated yield (based on unrecovered 1–3). | |||||||
| 1 | 1 | H | Br | 1 | Biphenyl | >98 | 9 |
| 2 | 2 | H | Br | 1 | Biphenyl | 95 | 10 |
| 3 | 3 | H | Br | 1 | Biphenyl | 92 | 12 |
| 4 | 1 | H | Cl | 4 | Biphenyl | 85 | 7 |
| 5 | 1 | H | I | 1 | Biphenyl | 92 | 13 |
| 6 | 1 | p-MeO | Br | 4 | p-MeO–biphenyl | 98 | 9 |
| 7 | 1 | p-CF3 | Br | 1 | p-CF3–biphenyl | 97 | 9 |
| 8 | 1 | p-CH3CO | Br | 1 | p-CH3CO–biphenyl | 80 | 9 |
| 9 | 1 | p-MeO | Cl | 4.5 | p-MeO–biphenyl | 90 | 7 |
| 10 | 1 | p-CF3 | Cl | 1.5 | p-CF3–biphenyl | 87 | 7 |
Rhenium-catalyzed direct arylation of arenes with aryl halides
On the basis of the photochemical reactions described above, we considered if the products, the phosphine-substituted tetracarbonyl rhenium halides, could revert to the original cyclometalated rhenium complexes 1–3, then a catalytic cycle would be created. Generally, cyclometalation of the ligand-substituted rhenium halides could be realized in the presence of base. Lu has reported a cyclometalation reaction of ReBr(CO)4[C(NHR)(NHPh)] (R = iPr, or Pr) with Et2NH in CH2Cl2 to give Re(CO)4[η2-C(NHR)(NHPh)].14 For optimization of reaction conditions, the cross-coupling of 4-bromoanisole with benzene was selected as the model reaction. The reactions were carried out with 0.5 mol% of cyclometalated rhenium complex 1 in benzene and different bases were used. Only NaOH as the base afforded the biphenyl product in moderate yield (59%, entry 1 of Table 2), whereas other tested bases such as K2CO3, KOAc, tBuOK, and pyrrolidine gave rise to lower yields (26–50%, entries 2–5). Other two cyclometalated rhenium complexes 2 and 3 as catalysts were also screened under the optimized conditions, complex 2 afforded slightly higher yield (68%, entry 6) than 1, while complex 3 gave much lower yield (26%, entry 7). To expand the scope of the substrates, other aryl bromides, including bromobenzene, and 4-bromobenzotrifluoride were examined, and the reactions afforded the corresponding biphenyl in yields of 65% and 60% (entries 8 and 9). Besides, the substrates chlorobenzene and iodobenzene were also tested using the catalyst 1, which resulted in lower yields (26% and 35%, entries 10 and 11).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat | Base | R′ | X | t [h] | Productb | Yieldb,c [%] |
| a Reaction conditions: irradiation of 1–3 (0.01 mmol), Ar–X (2.0 mmol), and base (2.0 mmol) in 20 mL benzene solvent with a 500 W high-pressure Hg lamp (λmax = 365 nm) in the ice-water bath.b Biphenyl products were isolated by routine silica gel column chromatography, and identified by comparing their respective 1H NMR spectra with authentic samples.c Isolated yield (based on unrecovered 1–3). | |||||||
| 1 | 1 | NaOH | p-MeO | Br | 4 | p-MeO–biphenyl | 59 |
| 2 | 1 | K2CO3 | p-MeO | Br | 4 | p-MeO–biphenyl | 30 |
| 3 | 1 | KOAc | p-MeO | Br | 4 | p-MeO–biphenyl | 26 |
| 4 | 1 | tBuOK | p-MeO | Br | 4 | p-MeO–biphenyl | 50 |
| 5 | 1 | Pyrrolidine | p-MeO | Br | 6 | p-MeO–biphenyl | 29 |
| 6 | 2 | NaOH | p-MeO | Br | 5 | p-MeO–biphenyl | 68 |
| 7 | 3 | NaOH | p-MeO | Br | 5 | p-MeO–biphenyl | 26 |
| 8 | 1 | NaOH | H | Br | 4 | Biphenyl | 65 |
| 9 | 1 | NaOH | p-CF3 | Br | 3 | p-CF3–biphenyl | 60 |
| 10 | 1 | NaOH | H | Cl | 8 | Biphenyl | 26 |
| 11 | 1 | NaOH | H | I | 8 | Biphenyl | 35 |
Unfortunately, rhenium catalysts 1–3 did not show higher activity to direct arylation of arenes with aryl halide under our current conditions. When we performed the photolysis of phosphine-substituted tetracarbonyl rhenium bromide 9 in benzene in the presence of NaOH, the expected cyclometalated rhenium complex 1 was observed, at the same time, we also obtained some free ligand, (1-naphthyl)diphenyl phosphine. Obviously, complex 9 was decomposed partially under UV irradiation, which may explain the moderate catalytic performance of 1. Based on the results described above, a plausible catalytic mechanism is tentatively proposed: first, a CO is dissociated from cyclometalated rhodium complexes 1–3 under UV irradiation to give a 16e− unsaturated species; this is followed by oxidative addition of a C–X bond on aryl halides (Ph–X) to form the Re(III) intermediate. In the next step, a radical aromatic substitution that involves the aromatic radical with the solvent benzene might occur to give biphenyl and the corresponding cyclometalated rhenium hydride halide complexes, which undergo reductive elimination and recoordination of CO to afford the ring-opening complexes (7, 9, 10, 12, and 13). In the final step, cyclometalation of ring-opening complexes is achieved in the presence of base to generate the catalytic cycle. When we tried to synthesize the Re(III) intermediate to support the mechanism, unfortunately, we did not obtained the expected product either from photolysis of 1–3 in pure Ph–X or photolysis of 1–3 and Ph–X in cyclohexane.
Summary
A series of five-membered cyclometalated rhenium complexes (1–5) were synthesized by thermal treatment of aryl-substituted phosphines or phosphinites with Re2(CO)10 in chlorobenzene. The formation of cyclorhenated complexes went through an intramolecular activation of C(sp2)–H or C(sp3)–H bond. Competition reaction indicated that the aromatic C(sp2)–H bond is more likely to be activated than C(sp3)–H bond under the same conditions. The chemical behavior of the cyclorhenated complexes 1, 2 or 3 has been investigated, including photolysis of 1 or 2 in CHX3, reaction of 1 with CF3COOH, and photolysis of 1–3 with aryl halides in benzene, all of them caused the cleavage of Re–C σ bond and then the opening of metallacycle. Some reaction indicated the lower reactivity of the cyclometalated rhenium complexes in this work than that of N-contained cyclorhenated complexes. Besides, complexes 1–3 exhibited moderate catalytic activity on direct arylation of arenes with aryl halides under UV irradiation at a [Re]:substrate ratio of 1:200.
Experimental
General considerations
All reactions were carried out under nitrogen using standard Schlenk and vacuum line techniques; however the workup was carried out in air unless stated otherwise. All solvents were distilled from appropriate drying agents under nitrogen prior to use. 1H (400 MHz), 13C{1H} (100 MHz), and 31P{1H} (162 MHz) NMR spectra were recorded on a Bruker AV400 instrument at room temperature with CDCl3 as solvent. Chemical shifts were recorded in ppm, referenced to residual 1H and 13C signals of the non-deuterated CDCl3 (δ 7.26 and 77.16) as internal standards or to the 31P signal of PPh3 (δ −5.65) as an external standard. Elemental analyses were performed on a Perkin-Elmer 240C analyzer. IR spectra were recorded as KBr disks on a Nicolet 560 ESP FTIR spectrometer. Re2(CO)10 was purchased from Strem without further purification; the ligands diisopropyl(1-naphthyl) phosphine,16 (1-naphthyl)diphenyl phosphine,16 diisopropyl(o-methylphenyl) phosphine,17 diisopropyl(2-methylnaphthyl) phosphine,17 o-methylphenyl diisopropylphosphinite,18 and 1-naphthyl diisopropylphosphinite18 were prepared by literature methods.General procedure for the reactions of Re2(CO)10 with phosphines or phosphinites in chlorobenzene
A mixture of Re2(CO)10 (0.05 mmol) and phosphine or phosphinite (2.0 equiv.) in chlorobenzene (10 mL) was refluxed for 10 h and then concentrated under vacuum, the residue was chromatographed on a silica gel column with a mixture of petrol ether/CH2Cl2 as the eluent. The products were recrystallized from n-hexane/CH2Cl2 (1:1) at −10 °C to afford 1–6 as colorless crystals in yields of 45–85%.
1, (82% yield). 1H NMR: δ 8.21 (d, J = 6.0 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.60 (m, 1H), 7.55–7.49 (m, 5H), 7.44 (m, 6H), 7.35 (m, 1H). 13C{1H} NMR: δ 189.1, 189.0, 152.4, 149.7, 144.3, 138.2, 134.7, 134.3, 132.8, 132.6, 132.5, 131.6, 130.8, 129.0, 128.9, 127.9, 124.6, 123.9. 31P{1H} NMR: δ 26.4. IR (νCO): 2084 (m), 1998 (s), 1976 (s), 1955 (s), 1929 (s) cm−1. Anal. calcd for C26H16O4PRe: C, 51.23; H, 2.65. Found: C, 51.37; H, 2.42.
2, (85% yield). 1H NMR: δ 8.16 (m, 1H), 7.85 (m, 1H), 7.58 (m, 2H), 7.43 (m, 1H), 7.28 (m, 1H), 2.58 (m, 2H, CH), 1.33 (dd, 6H, CH3), 1.04 (dd, 6H, CH3). 13C{1H} NMR: δ 190.2, 189.8, 189.7, 189.4, 152.5, 149.7, 143.9, 138.8, 134.5, 132.2, 128.1, 127.5, 124.1, 123.8, 29.7, 29.5, 20.3, 20.1. 31P{1H} NMR: δ 49.2. IR (νCO): 2076 (w), 1970 (s), 1925 (s) cm−1. Anal. calcd for C20H20O4PRe: C, 44.36; H, 3.72. Found: C, 44.49; H, 3.48.
3, (56% yield). 1H NMR: δ 7.38 (m, 1H), 7.27–7.21 (m, 2H), 7.04 (m, 1H), 2.50 (m, 2H, CH2), 2.49 (m, 2H, CH), 1.20 (dd, 6H, CH3), 1.05 (dd, 6H, CH3). 13C{1H} NMR: δ 191.1, 190.3, 189.2, 162.8, 134.3, 131.4, 130.2, 129.5, 123.9, 77.4, 28.1, 27.8, 19.3, 18.9. 31P{1H} NMR: δ 48.8. IR (νCO): 2073 (s), 1986 (s), 1931 (s) cm−1. Anal. calcd for C17H20O4PRe: C, 40.39; H, 3.99. Found: C, 40.74; H, 3.75.
4, (63% yield). 1H NMR: δ 8.18 (m, 1H), 7.74 (m, 1H), 7.54 (m, 1H), 7.21 (m, 2H), 2.91 (m, 2H, CH), 2.71 (d, 3H, CH3), 1.49 (dd, 6H, CH3), 0.83 (dd, 6H, CH3). 31P{1H} NMR: δ 64.0. IR (νCO): 2074 (s), 1980 (s), 1927 (s) cm−1. Anal. calcd for C21H22O4PRe: C, 45.40; H, 3.99. Found: C, 45.27; H, 3.83.
5, (45% yield). 1H NMR: δ 7.55 (d, J = 7.2 Hz, 1H), 6.84 (d, J = 7.2 Hz, 1H), 6.71 (t, J = 7.2 Hz, 1H), 2.53 (m, 2H, CH), 2.25 (s, 3H, CH3), 1.30 (dd, 6H, CH3), 1.21 (dd, 6H, CH3). 31P{1H} NMR: δ 158.3. IR (νCO): 2080 (s), 1982 (s), 1955 (s) cm−1. Anal. calcd for C17H20O5PRe: C, 39.15; H, 3.87. Found: C, 39.43; H, 3.81.
6, (66% yield). 1H NMR: δ 8.20 (m, 2H), 7.80 (m, 2H), 7.58 (m, 2H), 7.48 (m, 4H), 7.34 (m, 2H), 7.15 (m, 2H), 2.85 (m, 4H, CH), 1.43–1.31 (m, 24H, CH3). 13C{1H} NMR: δ 200.2, 135.2, 128.0, 127.3, 126.6, 125.9, 125.4, 123.7, 122.2, 120.2, 115.8, 36.9, 32.8, 18.8, 18.5. 31P{1H} NMR: δ 159.0. IR (νCO): 1965 (s), 1933 (s), 1897 (s) cm−1. Anal. calcd for C40H42O10P2Re2: C, 43.01; H, 3.79. Found: C, 42.85; H, 3.62.
General procedure for photolysis of 1 or 2 in CHX3 (X = Cl, Br)
A solution of 1 or 2 (0.05 mmol) in CHX3 (5 mL) was irradiated by UV light for 8 h, then the solvent was removed under vacuum, the residue was chromatographed on a silica gel column with a mixture of petrol ether/CH2Cl2 as the eluent. The products were recrystallized from n-hexane/CH2Cl2 (1:1) at −10 °C to afford 7–10 as colorless crystals in yields of 54–81%.
7, (62% yield). 1H NMR: δ 8.01 (m, 1H), 7.96 (m, 2H), 7.78–7.73 (m, 4H), 7.54 (m, 1H), 7.50–7.39 (m, 8H), 7.36 (m, 1H). 31P{1H} NMR: δ 3.1. IR (νCO): 2105 (m), 1997 (s), 1943 (s), 1896 (m) cm−1. Anal. calcd for C26H17ClO4PRe: C, 48.34; H, 2.65. Found: C, 48.57; H, 2.49.
8, (54% yield). 1H NMR: δ 8.42 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 8.0 Hz, 2H), 7.73 (m, qH), 7.65 (m, 1H), 7.60–7.52 (m, 2H), 3.39 (m, 2H, CH), 1.58 (dd, 6H, CH3), 1.38 (dd, 6H, CH3). 31P{1H} NMR: δ 12.4. IR (νCO): 2105 (m), 2011 (s), 1922 (s) cm−1. Anal. calcd for C20H21ClO4PRe: C, 41.56; H, 3.66. Found: C, 41.35; H, 3.71.
9, (81% yield). 1H NMR: δ 8.00 (m, 1H), 7.95 (m, 2H), 7.76 (m, 4H), 7.53 (m, 1H), 7.48–7.38 (m, 8H), 7.35 (m, 1H). 31P{1H} NMR: δ −0.3. IR (νCO): 2105 (s), 2012 (s), 1947 (s), 1921 (s) cm−1. Anal. calcd for C26H17BrO4PRe: C, 45.23; H, 2.48. Found: C, 45.18; H, 2.36.
10, (78% yield). 1H NMR: δ 8.38 (m, 1H), 7.94 (m, 2H), 7.73 (m, 1H), 7.65 (m, 1H), 7.60–7.53 (m, 2H), 3.44 (m, 2H, CH), 1.58 (dd, 6H, CH3), 1.38 (dd, 6H, CH3). 31P{1H} NMR: δ 7.3. IR (νCO): 2106 (s), 2010 (s), 1919 (s) cm−1. Anal. calcd for C20H21BrO4PRe: C, 38.59; H, 3.40. Found: C, 38.43; H, 3.42.
Reaction of 1 with CF3COOH
A solution of 1 (0.05 mmol) in CH2Cl2 (5 mL) was treated with excess CF3COOH (0.3 mmol). The mixture was stirred for 24 h at room temperature. The solvent was removed under vacuum, and the residue was chromatographed on a silica gel column with a mixture of petrol ether/CH2Cl2 as the eluent. The product was recrystallized from n-hexane/CH2Cl2 (1:1) at −10 °C to afford 11 as colorless crystals in 48% yield.
11 1H NMR: δ 8.03 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.8 Hz, 1H), 7.56–7.41 (m, 12H), 7.38 (d, J = 8.8 Hz, 1H), 7.34 (m, 1H). 31P{1H} NMR: δ 9.8. IR (νCO): 2106 (s), 1998 (s), 1960 (s), 1691 (s) cm−1. Anal. calcd for C28H17F3O6PRe: C, 46.48; H, 2.37. Found: C, 46.63; H, 2.45.
General procedure for photolysis of 1–3 with aryl halides in C6H6
A solution of 1–3 (0.05 mmol) and aryl halides (0.25 mmol) in dry benzene (10 mL) in nitrogen flushed quartz tube was additionally degassed by bubbling nitrogen thru for 5 min. After irradiation for 1–4.5 h in the ice-water bath, the solvent was removed under the reduced pressure. The residue was chromatographed in air on silica gel using petroleum ether–CH2Cl2 mixture as eluent. The first band (colorless) afforded the corresponding biphenyl as white solids (80–98%). The second band (colorless) gave corresponding phosphine-substituted tetracarbonyl rhenium halides (7, 9, 10, 12, and 13).12, (32% yield). 1H NMR: δ 7.46 (m, 1H), 7.32 (m, 3H), 3.27 (m, 2H, CH), 2.60 (s, 3H, CH3), 1.58 (dd, 6H, CH3), 1.37 (dd, 6H, CH3). 31P{1H} NMR: δ 9.8. IR (νCO): 2102 (s), 2018 (s), 1980 (s), 1938 (s) cm−1. Anal. calcd for C17H21BrO4PRe: C, 34.82; H, 3.61. Found: C, 35.13; H, 3.55.
13, (53% yield). 1H NMR: δ 8.01–7.92 (m, 3H), 7.79–7.74 (m, 4H), 7.53 (m, 1H), 7.49–7.34 (m, 9H). 31P{1H} NMR: δ −6.2. IR (νCO): 2099 (s), 1992 (br), 1942 (s) cm−1. Anal. calcd for C26H17IO4PRe: C, 42.34; H, 2.32. Found: C, 42.47; H, 2.15.
General procedure for catalytic reactions of 1–3 with aryl halides in C6H6
A solution of 1–3 (0.01 mmol), aryl halides (2.0 mmol) and base (2.0 mmol) in dry benzene (20 mL) in nitrogen flushed quartz tube was additionally degassed by bubbling nitrogen thru for 5 min. After irradiation for 3–8 h in the ice-water bath, the solvent was removed under the reduced pressure. The residue was chromatographed in air on silica gel using petroleum ether as eluent. The corresponding biphenyl products were obtained as colorless solids in yields of 26–68%.Crystallography
Single crystals of complexes 1, 6, 9, and 11 (CCDC 1520723–1520726) suitable for X-ray diffraction were obtained by crystallization from n-hexane/CH2Cl2 (1:1). Data collection was performed on a Bruker SMART 1000, using graphite-monochromated Mo Kα radiation (ω–2θ scans, λ = 0.71073 Å). Semiempirical absorption corrections were applied for all complexes. The structures were solved by direct methods and refined by full-matrix least squares. All calculations were using the SHELXTL-97 program system. The crystal data and summary of X-ray data collection are presented in Table 3.
| 1 | 6 | 9 | 11 | |
|---|---|---|---|---|
| Formula | C26H16O4PRe | C20H21O5PRe | C26H17BrO4PRe | C28H17F3O6PRe |
| fw | 609.56 | 558.54 | 690.47 | 723.58 |
| T (K) | 173.00(10) | 296(2) | 173.00(10) | 154.5(9) |
| Cryst syst | Triclinic | Orthorhombic | Triclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Fdd2 | P |
P21/c |
| a (Å) | 11.9989(3) | 24.564(3) | 9.9390(5) | 24.3119(9) |
| b (Å) | 14.4984(3) | 42.547(5) | 11.0789(5) | 11.3315(3) |
| c (Å) | 14.8134(4) | 7.7934(8) | 11.2433(6) | 19.6695(6) |
| α (deg) | 64.239(2) | 90 | 94.119(4) | 90 |
| β (deg) | 84.8034(19) | 90 | 97.332(4) | 98.995(3) |
| γ (deg) | 74.8834(19) | 90 | 99.066(4) | 90 |
| V (Å3) | 2239.82(10) | 8145.1(15) | 1207.22(10) | 5352.1(3) |
| Z | 4 | 16 | 2 | 8 |
| Dcalc (g cm−3) | 1.808 | 1.822 | 1.900 | 1.796 |
| μ (mm−1) | 11.544 | 6.073 | 6.784 | 4.662 |
| F(000) | 1176.0 | 4336 | 660.0 | 2800.0 |
| Cryst size (mm) | 0.20 × 0.18 × 0.15 | 0.22 × 0.21 × 0.20 | 0.25 × 0.15 × 0.14 | 0.25 × 0.24 × 0.2 |
| Max. 2θ (deg) | 148.728 | 53.414 | 50.018 | 50.02 |
| No. of reflns collected | 16330 |
13167 |
7610 | 45685 |
| No. of indep reflns/Rint | 8839/0.0273 | 4177/0.0346 | 4240/0.0407 | 9416/0.0564 |
| No. of params | 577 | 254 | 320 | 703 |
| Goodness-of-fit on F2 | 1.061 | 1.001 | 0.958 | 1.087 |
| R1, wR2 [I > 2σ(I)] | 0.0294, 0.0764 | 0.0245, 0.0435 | 0.0329, 0.0571 | 0.0487, 0.1086 |
| R1, wR2 (all data) | 0.0313, 0.0780 | 0.0295, 0.0453 | 0.0394, 0.0608 | 0.0563, 0.1129 |
| Largest diff peak and hole (e Å−3) | 1.88, −0.99 | 0.578, −0.298 | 1.16, −1.42 | 1.92, −1.43 |
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21002069 and 21572160), the Natural Science Foundation of Tianjin (No. 14JCYBJC20300), and the Program for Innovative Research Team in University of Tianjin (TD12-5038).Notes and references
- J. P. Kleiman and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 1544 CrossRef CAS.
- P. G. Bomben, K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438 CrossRef CAS.
- H.-F. Chen, C. Wu, M.-C. Kuo, M. E. Thompson and K.-T. Wong, J. Mater. Chem., 2012, 22, 9556 RSC.
- (a) G. C. Volugioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef PubMed; (b) C. Samoilowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708 CrossRef PubMed.
- (a) D. A. Alonso and C. Nájera, Chem. Soc. Rev., 2010, 39, 2891 RSC; (b) A. Zapf and M. Beller, Chem. Commun., 2005, 431 RSC; (c) I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055 CrossRef CAS; (d) W. A. Herrmann, V. P. W. Böhm and C.-P. Reisenger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS; (e) I. Omae, Coord. Chem. Rev., 2004, 248, 995 CrossRef CAS.
- (a) H. Cao, L.-H. Cai, C.-X. Wang, X.-H. Zhu, Z.-M. Li and X.-F. Hou, J. Organomet. Chem., 2015, 775, 60 CrossRef CAS; (b) A. Grabulosa, A. Mannu, E. Alberico, S. Denurra, S. Gladiali and G. Muller, J. Mol. Catal. A: Chem., 2012, 363–364, 49 CrossRef CAS; (c) R. Cerón-Camacho, V. Gómez-Benítez, R. L. Lagadec, D. Morales-Morales and R. A. Toscano, J. Mol. Catal. A: Chem., 2006, 247, 124 CrossRef; (d) W. Baratta, P. Da Ros, A. Del Zotto, A. Sechi, E. Zangrando and P. Rigo, Angew. Chem., Int. Ed., 2004, 43, 3584 CrossRef CAS PubMed; (e) P. Dani, T. Karlen, R. A. Gossage, S. Gladiali and G. van Koten, Angew. Chem., Int. Ed., 2000, 39, 743 CrossRef CAS.
- (a) P. Spellane, R. J. Watts and A. Vogler, Inorg. Chem., 1993, 32, 5633 CrossRef CAS; (b) F. W. M. Vanhelmont, H. U. Güdel, M. Förtsch and H.-B. Bürgi, Inorg. Chem., 1997, 36, 5512 CrossRef CAS; (c) F. W. M. Vanhelmont, G. F. Strouse, H. U. Güdel, A. C. Stückl and H. W. Schmalle, J. Phys. Chem. A, 1997, 101, 2946 CrossRef CAS; (d) F. W. M. Vanhelmont, M. V. Rajasekharan, H. U. Güdel, S. C. Capelli, J. Hauser and H.-B. Bürgi, J. Chem. Soc., Dalton Trans., 1998, 2893 RSC; (e) X.-H. Liu, I. Manners and D. W. Bruce, J. Mater. Chem., 1998, 8, 1555 RSC.
- (a) Y. Kuninobu, Y. Nishina, A. Kawata, M. Shouho and K. Takai, Pure Appl. Chem., 2008, 80, 1149 CrossRef CAS; (b) Y. Kuninobu, H. Ueda, A. Kawata and K. Takai, J. Org. Chem., 2007, 72, 6749 CrossRef CAS PubMed; (c) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; (d) Y. Kuninobu, Y. Inoue and K. Takai, Chem. Lett., 2006, 35, 1376 CrossRef CAS; (e) Y. Kuninobu, A. Kawata and K. Takai, J. Am. Chem. Soc., 2005, 127, 13498 CrossRef CAS PubMed.
- (a) Y. Kuninobu, T. Nakahara, P. Yu and K. Takai, J. Organomet. Chem., 2011, 696, 348 CrossRef CAS; (b) Y. Kuninobu, Y. Nishina, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2008, 130, 14062 CrossRef CAS PubMed; (c) Y. Kuninobu, Y. Nishina, M. Shouho and K. Takai, Angew. Chem., Int. Ed., 2006, 45, 2766 CrossRef CAS PubMed.
- (a) Y. Kuninobu, T. Seiki, S. Kanamaro, Y. Nishina and K. Takai, Org. Lett., 2010, 12, 5287 CrossRef CAS PubMed; (b) Y. Kuninobu, Y. Nishina and K. Takai, Tetrahedron, 2007, 63, 8463 CrossRef CAS; (c) Y. Kuninobu, Y. Nishina, C. Nakagawa and K. Takai, J. Am. Chem. Soc., 2006, 128, 12376 CrossRef CAS PubMed.
- (a) Y. Kuninobu, Y. Fujii, T. Matsuki, Y. Nishina and K. Takai, Org. Lett., 2009, 11, 2711 CrossRef CAS PubMed; (b) Y. Kuninobu, Y. Nishina, K. Okaguchi, M. Shouho and K. Takai, Bull. Chem. Soc. Jpn., 2008, 81, 1393 CrossRef CAS.
- E. Lindner, G. Funk and S. Hoehne, Chem. Ber., 1981, 114, 2465 CrossRef CAS.
- N. I. Gapotchenko, N. V. Alekseev, N. E. Kolobova, K. N. Anisimov, I. A. Ronova and A. A. Johansson, J. Organomet. Chem., 1972, 35, 319 CrossRef CAS.
- K.-L. Lu, H.-H. Lee, C.-M. Wang and Y.-S. Wen, Organometallics, 1994, 13, 593 CrossRef CAS.
- (a) B. Zhu, X. Huang and X. Hao, Dalton Trans., 2014, 43, 16726 RSC; (b) B. Zhu, X. Huang and X. Hao, Eur. J. Inorg. Chem., 2014, 5932 CrossRef CAS.
- S. Zolezzi, S. A. Moya, G. Valdebenito, G. Abarca, J. Parada and P. Aguirre, Appl. Organomet. Chem., 2014, 28, 364 CrossRef CAS.
- H. Riihimäki, T. Kangas, P. Suomalainen, H. K. Reinius, S. Jääskeläinen, M. Haukka, A. O. I. Krause, T. A. Pakkanen and J. T. Pursiainen, J. Mol. Catal. A: Chem., 2003, 200, 81 CrossRef.
- R. B. Bedford and M. E. Limmert, J. Org. Chem., 2003, 68, 8669 CrossRef CAS PubMed.
Footnote |
| † CCDC 1520723–1520726. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28263f |
| This journal is © The Royal Society of Chemistry 2017 |