
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric vector-mediated delivery of an miR-21 inhibitor for prostate cancer treatment
Chuyi Chen†
a,
Xinghua Huang† a,
Yong Wang†b,
Liteng Linc,
Lei Liua,
Guanyi Lia,
Shangchao Wua,
Chaozhang Xua,
Jianhua Zhou*a and
Xintao Shuai*b
a,
Yong Wang†b,
Liteng Linc,
Lei Liua,
Guanyi Lia,
Shangchao Wua,
Chaozhang Xua,
Jianhua Zhou*a and
Xintao Shuai*b
aDepartment of Urology, Longgang District People's Hospital of Shenzhen, Shenzhen 518000, China. E-mail: gdjh.zhou@163.com; Tel: +86-755-28932833-6119
bPCFM Lab of Ministry of Education, School of Materials Science and Engineering, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: shuaixt@mail.sysu.edu.cn; Fax: +86-20-84112245; Tel: +86-20-84110365
cDivision of Vascular Interventional Radiology, The Third Affiliated Hospital, Sun Yat-Sen University, Guangzhou 510630, China
First published on 13th February 2017
Abstract
Prostate cancer is one of the most common male malignancies, and MiR-21 plays an important role in the pathogenesis of this cancer. The treatment of microRNAs has proven to be a viable strategy for tumor therapy. However, the delivery of genes remains a major challenge because of the lack of efficient carriers. In this study, a diblock copolymer PEG–PAsp(DETA) of biocompatible polyethylene glycol (PEG) and biodegradable poly(L-aspartic acid) grafted with diethylenetriamine (PAsp(DETA)) was introduced as a delivery vector for an miR-21 inhibitor (i.e. antisense oligonucleotides for miR-21). Using in vitro and in vivo animal experiments, we studied the transfection efficiency and mechanism of action of the PEG–PAsp(DETA)/miR-21 inhibitor towards prostate cancer PC-3 cells. The biodegradable polymer mPEG–PAsp(DETA) was successfully used as a gene carrier to effectively transport the miR-21 inhibitor into PC-3 cells, which resulted in miR-21 silencing, upregulation of PDCD4 gene expression, and induced apoptosis in PC-3 prostate cancer. Meanwhile, the cytotoxicity of biodegradable carriers is very low. This study demonstrates the potential of our novel nucleic acid nanomedicine for the effective treatment of prostate cancer.
Introduction
Prostate cancer (PCa), the second most common cancer among male malignancies in the world,1 takes hundreds of thousands of lives annually.2 Owing to the general androgen-sensitivity at the initial diagnosis of PCa, anti androgen therapy is widely used for most patients in the clinic. However, the eventual development of androgen-independent prostate cancers (AIPC) in patients will lead to metastasis and death.3 Besides, traditional therapies such as surgery, chemotherapy and radiotherapy are also far from ideal to cure the disease. Thus, novel therapeutic strategies are needed for the treatment of prostate cancer.MicroRNAs (miRNAs), a series of noncoding, single-stranded RNAs that play important roles in regulating gene expression of multiple kinds of human tumors, have drawn great attention in recent years.4,5 MiR-21, a miRNA dysregulated in a number of cancers including glioma, breast cancer, colorectal cancer and etc.,6 has been recently reported to be over-expressed in prostate cancer as well.7 E.g., Cordelier et al. detailedly reported a potential strategy for the therapy of pancreatic cancer by targeting miR-21 using lentiviral vectors.8 In addition, recent reports have shown that it promotes cell transformation by targeting the programmed cell death 4 (PDCD4) gene as well as enhances apoptosis resistance and invasion in prostate cancer cells by targeting myristoylated alanine rich protein kinase substrate (MARCKS).9,10 Therefore, miR-21 inhibition strategy will probably be an effective prostate cancer therapy.
Programmed cell death protein 4 (PDCD4), a tumor suppressor protein whose expression is increased during apoptosis,11 has been verified to be implicated in the development of lung, breast, colon, brain and liver cancers.12–16 PDCD4 inhibits the translation initiation factors of eIF4A and eIF4G, the main helicase required for cap-dependent translation, which leads to inhibition of pro-oncogenic factors.17 Furthermore, PDCD4 inhibits the translation of several specific mRNA targets such as p53 (ref. 18) and Bcl-xL.19 It has been tested that the over expression of PDCD4 inhibits tumor progression in mouse models.20 Due to the important role as PDCD4 plays in tumors, the regulation of PDCD4 as a therapeutic target by interfering the expression of related miR-21 for PCa therapy has drawn much interest in recent years.
Owing to the intractable problems for the direct application of nucleic acids in cancer therapy, including the inability to cross cell membrane and nuclease degradation, effective and safe RNA delivery vector are crucial for gene therapy of clinical diseases.21,22 Up to now, amine-contained cationic polymers, e.g. poly(ethylenimine) (PEI), poly(L-lysine), chitosan, and polyamidoamine (PAMAM) dendrimers, have shown great potential in nucleic acid delivery.23 In this work, a diblock copolymer PEG–PAsp(DETA) consisting of biocompatible polyethylene glycol (PEG) and biodegradable poly(L-aspartic acid) grafted with diethylenetriamine (PAsp(DETA)) was introduced as delivery vector of miR-21 inhibitor. DETA exerts a great efficiency of complexing nucleic acids own to its high density of amino groups.24
Materials and methods
Materials
α-Methoxy-ε-hydroxy-poly(ethylene glycol) (mPEG-OH, Mn = 2 kDa), dimethylformamide (DMF), dimethyl sulfoxide (DMSO) and diethylenetriamine (DETA) were purchased from Sigma-Aldrich and used as received. Dichloromethane (CH2Cl2) was dried over CaH2 and then distilled under ambient pressure. Methanol was of analytical grade and purchased from Guangzhou Chemical Reagent Factory, China. Dialysis bag (MWCO: 3.5 kDa) was purchased from Shanghai Green Bird Technology Development Co., Ltd., China. mPEG–NH2 and β-benzyl-L-aspartate (BLA–NCA) was synthesized as previously reported.25 Human prostate cancer cell lines PC-3 were obtained from the type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cell culture reagents were purchased from GIBCO (Carlsbad, CA, USA). mPEG–PAsp(DETA) was synthesized by Materials Science and Engineering of SUN YAT-SEN University. Cell Counting Kit (CCK-8/WST-8) was purchased from KeyGen (Guangzhou, China). The miR-21 inhibitor (i.e. antisense oligonucleotides for miR-21), miRNA inhibitor negative control (i.e. Nematode RNA that had almost no homology with human miRNA) and Cy3-labeled miRNA inhibitor were purchased from RiboBio (Guangzhou, China). The Taqman(r) microRNA RT Kit, TAQMAN UNIVERSAL MMIX II (NO UNG) and lipo2000 was purchased from Life Technologies (Carlsbad, USA). Antibodies against the following proteins were used: PDCD4 (Cell Signaling Technology, Danvers, MA, USA), GAPDH, Tubulin (Beyotime, Shanghai, China).Cell culture
Human PC-3 prostate cancer cells were cultured in 1640 medium containing 10% fetal bovine serum in a humidified atmosphere (37 °C, 5% CO2, 95% air). When the cell confluence of 80–90% was reached, they were trypsinized and subcultured.Synthesis of mPEG–PAsp(DETA)
Poly(ethylene glycol)-block-poly(β-benzyl L-aspartate), i.e. mPEG–PBLA, was firstly synthesized by ring-opening polymerization of BLA–NCA with mPEG–NH2 as a macroinitiator.26 Briefly, 0.4 g of PEG–NH2 (0.2 mmol) was vacuum-dried at 70 °C for 4 h in a 100 mL flask, then dissolved in 50 mL of anhydrous CH2Cl2. Subsequently, 3.5 g BLA–NCA (14 mmol) dissolved in 5 mL of anhydrous DMF was added into the above solution under the protection of argon. The reaction was kept stirring for 72 h at 35 °C, then followed by precipitation into excessive cool diethyl ether. The precipitate was then filtered, washed with diethyl ether, and finally vacuum-dried until a constant weight was attained (mPEG–PBLA: Mn = 15.9 kDa, calculated from 1H NMR spectrum; yield: 96%). Afterwards, mPEG–PAsp(DETA) was synthesized by aminolysis of DETA with mPEG–PBLA as reported.24 In brief, 0.8 g of mPEG–PBLA (0.05 mmol) was dissolved in 10 mL of anhydrous DMSO at 35 °C, then 12.9 mL of DETA (120 mmol, about 50 eq.) was added into the solution and the reaction was stirred for 2 h. Subsequently, the mixture was dialyzed against methanol for 2 days and deionized water for 3 days and then freeze-dried to obtain mPEG–PAsp(DETA) (Mn = 18.2 kDa, calculated from 1H NMR spectrum; yield: 84%).Preparation of PEG–PAsp(DETA)/miR-21 inhibitor polyplex
mPEG–PAsp(DETA) and miR-21 inhibitor were dissolved in RNase-free water at concentration of 0.5 mg mL−1 and 100 μM, respectively. Then the polymer and RNA solution were mixed according to the designed N/P ratios (the molar ratio of polymer nitrogen to RNA phosphorus). After being vibrated vigorously for 30 seconds, the mixture was kept at room temperature for 30 minutes to form polyplex.Characterization of the polymer and polyplex
1H NMR spectra were carried out on a Varian Unity 300 MHz spectrometer using DMSO-d6 or D2O-d2 as solvent according to solubility of the polymer. FTIR spectral measurements were recorded using a Nicolet/Nexus 670 FTIR spectrometer with a resolution of 2 cm−1 and the powder samples were compressed into KBr pellets.The sizes and zeta potentials of polyplexes of mPEG–PAsp(DETA) and miR-21 inhibitor were determined using dynamic light scattering (DLS). Measurements were carried out at 25 °C on a 90 Plus/BI-MAS equipment (Brookhaven Instruments Corporation, USA). The data for particle size and zeta potential were collected on an autocorrelator with a detection angle of scattered light at 90°. For each sample, the data from five measurements were averaged to obtain the mean ± standard deviation (SD).
Transmission electron microscopy (TEM) was performed using a Hitachi model H-7650 TEM operated at 80 kV. The samples were prepared by drying a drop (5 μL, 0.5 mg mL−1) of the sample solution on a copper grid coated with amorphous carbon. For the negative staining of samples, a small drop of uranyl acetate solution (2 wt% in water) was added to the copper grid, which was then blotted with a filter paper after 1 min. The grid was finally dried overnight inside a desiccator before TEM observation.
Agarose gel electrophoresis
The experiment was conducted at pH 7.4 and the polyplexes were prepared in various N/P ratios (1, 2, 4, 6, and 8) as aforementioned. Electrophoresis was carried out on a 1% agarose gel with a current of 80 V for 15 min in a Tris–acetate–EDTA (TAE) buffer solution (40 mM Tris–HCl, 1% v/v acetic acid, and 1 mM EDTA). The migration of the complexes was visualized by staining with ethidium bromide (EB). The staining of single-stranded RNA is attributed to the binding of the EB to short intrastrand helical regions in the molecules.27 The images were acquired with a Gel Doc system.Cell viability assay
The PC-3 cell viabilities at various cell incubation conditions were measured using the CCK-8 assay kit. Approximately 5 × 103 cells were plated into each well of a 96-well plate with 100 μL of 1640 medium containing 10% FBS. The cells were incubated for 24 h at 37 °C and then divided into four groups as follows: blank control group, mPEG–PAsp(DETA)/miRNA inhibitor negative control group, PEG–PAsp(DETA)/miR-21 inhibitor group and lipo2000 group. Medium for every group was replaced with 100 μL fresh medium before the experiment began. According to the N/P ratios of 1.5, 3, 6, 15, 30, 60, 80, polyplexes containing different amounts of polymer were added into the medium, and then the culture plates were gently shaken and incubated for 48 h. Then, 10 μL of CCK-8 solution was added into each well to further incubate the cells for 2 h in the darkness. The Infinite F200 Mutimode Plate Reader (Tecan, Crailsheim, Germany) was used to detect the light absorption value of each well at wavelength of 450 nm. Experiments for all groups were performed in triplicate. The dose of the miR-21 inhibitor and miRNA inhibitor negative control was 100 nM in each well.In vitro transfection
Flow cytometry was used to evaluate the transfection efficiency of polyplexes in vitro. PC-3 cells were seeded onto a 6-well plate at a density of 5 × 104 cells per well and incubated for 24 h. After incubation with different polyplexes (N/P ratios: 6, 8, 12, 16) for 6 hours, the cells were washed three times with PBS added with 0.25% trypsin/EDTA. Finally, the cells were transferred into tubes for determining transfection efficiency with flow cytometry (Gallios, Beckmancoulter, USA). Normally cultured cells without transfection were measured for calibrating the background. The final dose of Cy3-labeled miRNA inhibitor was 100 nM per well.Laser confocal microscopy assay
PC-3 cells were seeded onto a Petri dish at a density of 3 × 103 cells per well and incubated for 24 h. After the polyplex (miRNA inhibitor labeled with Cy3) was added into the well and incubated for 6 h, the cells were washed with PBS for three times, and then the nuclei were stained with DAPI for 3 min. Finally, the cells were washed with PBS and visualized under the Zeiss LSM 710 confocal microscope (Carl Zeiss Co., Ltd., Gottingen, Germany). Free Cy3-miRNA inhibitor was introduced as control. The final dose of Cy3-labeled miRNA inhibitor was 100 nM per well.RT-PCR
PC-3 cells were seeded onto 6-well plates and normally incubated for 24 h. The cells were then divided into 4 groups as follows: blank control group, PEG–PAsp(DETA)/miRNA inhibitor negative control group (N/P = 8), PEG–PAsp(DETA)/miR-21 inhibitor group (N/P = 8) and lipo2000 group. Experiments for all groups were performed in triplicate. Total RNA was extracted using Tripure Isolation Reagent (Invitrogen, Carlsbad, CA, USA) after the cells were transfected for 24 h. Gene expression was determined by quantitative real time PCR (qPCR) as previously described using gene-specific primers as follows: miR-21 forward primer sequences: 5′-TAGCTTATCAGACTG-ATGTTGA-3′; PDCD4: 5′-TAAGTGACTCTCTCTTTTCCGGT-3′; (forward); 5′-TTTTTCCTTAGTCGCCTTTTTGC-3′ (reverse). For miRNA expression, total RNA was reverse-transcribed into first-strand cDNA which was then used as a template for the PCR reaction with a forward primer specific to the mature miRNA sequence as previously described. Gene expressions relative to U6 or β-actin expression were normalized for miRNA and mRNA, respectively. The programs were run at the thermal cycling conditions of 95 °C/10 min; 40 cycles of 95 °C/15 s, 60 °C/60 s.Western blotting assay
PC-3 cells seeded onto 6-well plates were divided into 4 groups: blank control group, PEG–PAsp(DETA)/miRNA inhibitor negative control group (N/P = 8), PEG–PAsp(DETA)/miR-21 inhibitor group (N/P = 8) and lipo2000 group. After transfection for 48 h, cells were harvested and than the total protein was collected to measure the concentration using a bicinchoninic acid protein assay kit (Shenggong Bio-Tech Co, Ltd, shanghai, China). Different protein samples were separated by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. After being blocked with 5% skim milk for 2 h at room temperature, the membranes were incubated overnight with primary antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution, CST, USA) at 4 °C and then with the secondary anti-rabbit Ig-G–horseradish peroxidase (1:2000 dilution, CST, USA) for 1 h at room temperature. The blots were then incubated with enhanced chemiluminescence for 2 min. The protein bands were imaged using a UV imaging system. In addition, the GAPDH was used as an endogenous control to normalize the data of protein expression.
:1000 dilution, CST, USA) at 4 °C and then with the secondary anti-rabbit Ig-G–horseradish peroxidase (1:2000 dilution, CST, USA) for 1 h at room temperature. The blots were then incubated with enhanced chemiluminescence for 2 min. The protein bands were imaged using a UV imaging system. In addition, the GAPDH was used as an endogenous control to normalize the data of protein expression.
In vivo efficacy studies
All in vivo experiments and animal care were approved by the National Center of Animal Science Experimental Teaching at the Institutional Animal Care and Use Committee of the Sun Yat-sen University and were in accordance with the “Guide for the Care and Use of Laboratory Animals”. Human prostate cancer xenografts in nude mice were used as the animal model in the in vivo studies. PC-3 cells were trypsinized, washed for three times with PBS and re-suspended. 2 × 107 cells in 100 μL PBS were then subcutaneously injected to the right back of the BALB/c nude mouse (male, 5 weeks, 18–20 g).To determine the potential of the PEG–PAsp(DETA)/miR-21 inhibitor polyplex to affect tumor growth in vivo, the mice bearing human PC-3 tumor were randomly divided into three groups (n = 5): (i) PBS group, (ii) control group, (iii) miR-21 inhibitor group. After the tumors had reached an average volume of 25 mm3, the tumors in three animal groups were directly injected with 50 μL of PBS alone (PBS group), polyplex containing 500 nM SCR in 50 μL of PBS as negative control (negative control group), polyplex containing 500 nM miR-21 inhibitor in 50 μL of PBS (miR-21 inhibitor group), respectively.
Tumors were treated intra-tumorally at every third day for 15 days. The tumor volume for each animal was measured with the caliper at each injection day. Volume = 0.5 × L × W2, in which “L” and “W” represented the length and the width of the tumors, respectively.
For the histological evaluation, the excised tumors were fixed with 4% paraformaldehyde and sectioned into 6 μm slices. Hematoxylin and eosin (H&E) staining, immunohistochemical staining, and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) assays were performed according to the manufacturer's protocols.
Statistical analysis
All experiments were repeated at least three times, and statistical analyses of the data were performed using SPSS18.0 software. The results were shown as the mean ± standard deviation (SD), and P < 0.05 was considered statistically significant.Results
Synthesis and characterization of polymer
The block polymer, mPEG–PAsp(DETA), of polyethylene glycol (PEG) and diethylenetriamine-grafted poly(L-aspartic acid) (PAsp(DETA)), was synthesized via two-step reactions (Scheme 1). The block copolymer and its prepolymer were characterized with 1H NMR and FTIR, as shown in Fig. 1A–C. The major resonance peaks of the copolymer in the 1H NMR spectrum fit well the expected chemical structure. For mPEG–PBLA, the peak at 2.50 was chemical shifts of protons from solvent DMSO, other speaks at a: 3.52, b: 4.61 and c: 2.56–2.86, d: 5.02, e: 7.27 and f: 8.15 ppm were attributed to protons of (–OCH2CH2–), (–NH–(CH2–)CH–CO–), (–NH(–CH2–)CH–CO–), (–CH2C6H5), (–CH2C6H5) and (–NH–CH–CO–), respectively, showing the success of polymerization. After ammonolyzed by DETA, the peaks of methylene protons in DETA (g: 2.40–2.83) appeared and meanwhile the peaks of benzyl protons in PBLA (d: 5.02, e: 7.27) disappeared, indicating the completeness of the ammonolysis reaction. The FTIR results showed that, after ammonolysis with DETA, stretching vibration absorption caused by carbonyl ester bond (1740 cm−1) and C–H of benzene (746 and 696 cm−1) disappeared, also indicating the block polymer was synthesized successfully.28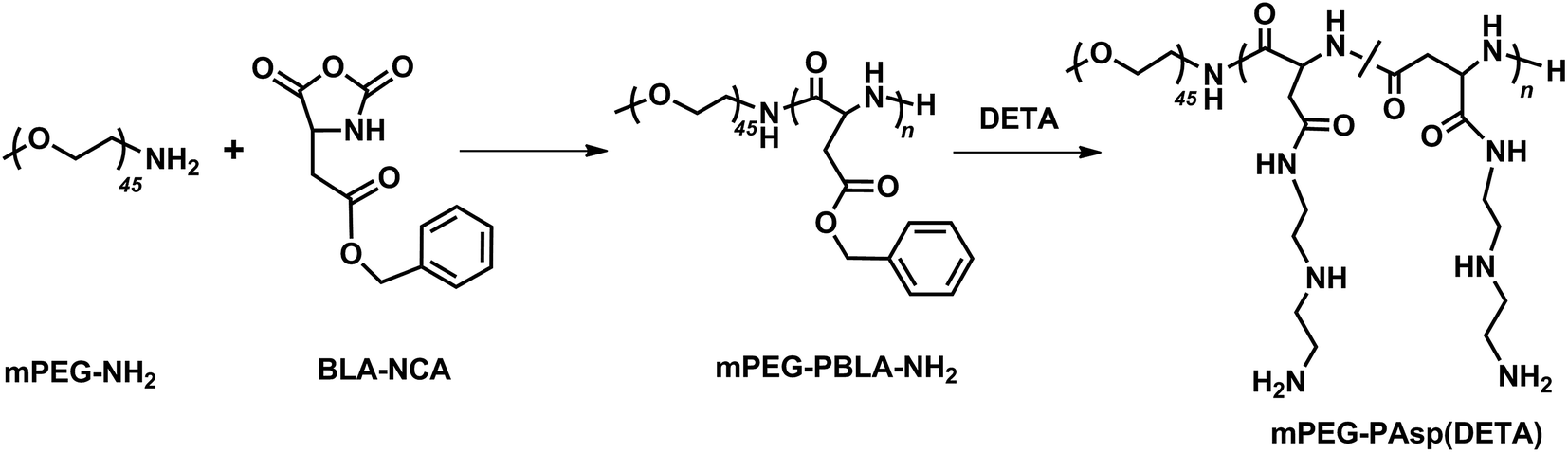 | ||
| Scheme 1 The synthesis of block polymer mPEG–PAsp(DETA). | ||
 | ||
| Fig. 1 (A) FTIR spectra of mPEG–PBLA (B) and mPEG–PAsp(DETA) (C). 1H NMR spectrum of mPEG–PBLA in DMSO-d6 (B) and mPEG–PAsp(DETA) in D2O-d2 (C). (D) Electrophoretic mobility of SCR (scrambled siRNA) in agarose gel after complexing with polymer mPEG–PAsp(DETA) at various N/P ratios in PBS 7.4. | ||
Agarose gel electrophoresis
MiR-21 inhibitor was bounded to PEG–PAsp(DETA) via electrostatic interaction. Complexation of the polymer with miRNA-21 inhibitor to form polyplexes was evaluated by electromobility shift assays on 1% agarose gels. As shown in Fig. 1D, the intensity of miRNA-21 inhibitor bands decreased with increasing N/P ratios from 0 to 8. At the N/P ratio of 4, the miRNA-21 inhibitor band disappeared completely, indicating that miRNA-21 inhibitor were fully complexed by PEG–PAsp(DETA).Characterization of polyplexes
The DLS detection showed that particle size of polyplexes was decreased to 79.6 ± 8.2 nm from 419.1 ± 20.6 nm when the N/P ratio was increased to 16 from 1 (Fig. 2A). In contrast, zeta potential increased along with the increase of N/P ratio because of the surplus of cationic amino.29 Aiming at realizing a well protection from enzymolysis meanwhile obtaining a high transfection efficiency, N/P ratio of 8 was chosen to be the complexing point for the subsequent tests since the suitable size and zeta potential.26,29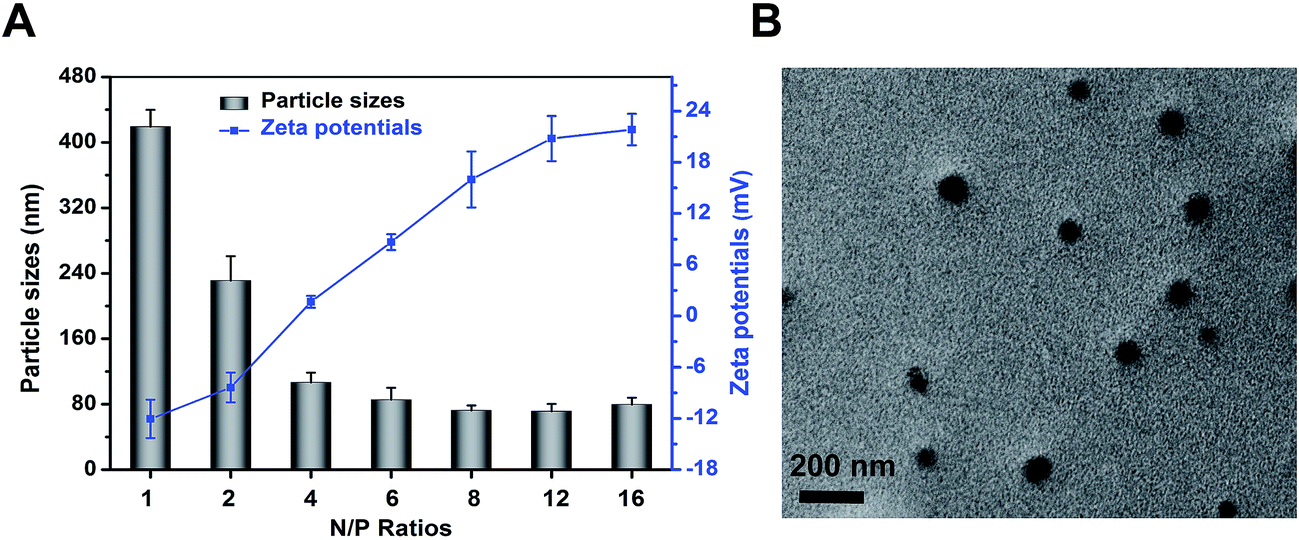 | ||
| Fig. 2 (A) Particle sizes and zeta potentials of mPEG–PAsp(DETA)/miR-21 inhibitor polyplexes formed at different N/P ratios. (B) TEM images of polyplex formed at N/P = 8 stained with uranyl acetate. | ||
Transmission electron microscopy analysis was conducted to reveal the morphology of polyplexes. As shown in Fig. 2B, polyplexes (N/P 8) were spherical and well dispersed. In addition, the mean diameter of the polyplexes was approximately 80 nm, according with that of DLS result.
Cytotoxicity of PEG–PAsp(DETA)
The CCK-8 assay was used to evaluate the cytotoxicity of polyplexes after 48 h co-incubation (Fig. 3). Generally, the cytotoxicity of PEG–PAsp(DETA) was also elevated. At the PEG–PAsp(DETA) concentration of 100 μg mL−1, the cell viability was 75.51 ± 6.85%. Above this concentration, the cell viability was remarkably decreased along with the increased polymer concentration. For the mPEG–PAsp(DETA)/miRNA inhibitor polyplexes, obvious cytotoxicity was shown only at very high N/P ratios above 15. Below this threshold value, the cells remained high viability above 80%. It is believed that complexation with RNA decreased the positive charge of the polymeric vector, which led to a decreased cationic cytotoxicity. Above N/P ratio of 15, large amount of excessive cationic PEG–PAsp(DETA) existed in the solution, which would inhibit cell growth again. For example, when the N/P ratio reached 80, the cell viability of PEG–PAsp(DETA)/miRNA inhibitor negative control group was decreased to 54.88 ± 2.93%. | ||
| Fig. 3 (A) Cytotoxicity of PEG–PAsp(DETA) in PC-3 cells determined by CKK-8 assay. PC-3 cells were treated with PEG–PAsp(DETA) at various concentrations from 0 to 1000 μg mL−1. (B) Cytotoxicity of PEG–PAsp(DETA)/miRNA inhibitor negative control polyplex in PC-3 cells at various N/P ratios from 1.5 to 80. The dose of miRNA inhibitor negative control was 100 nM in each well and incubation time was 48 h. | ||
In vitro transfection efficiency
The transfection efficiency of the polyplexes of mPEG–PAsp(DETA) and Cy3-labeled miRNA inhibitor in PC-3 cells was evaluated using flow cytometry. As shown in Fig. 4A, above N/P ratio of 6, the transfection efficiencies is more than 81.49 ± 1.57%.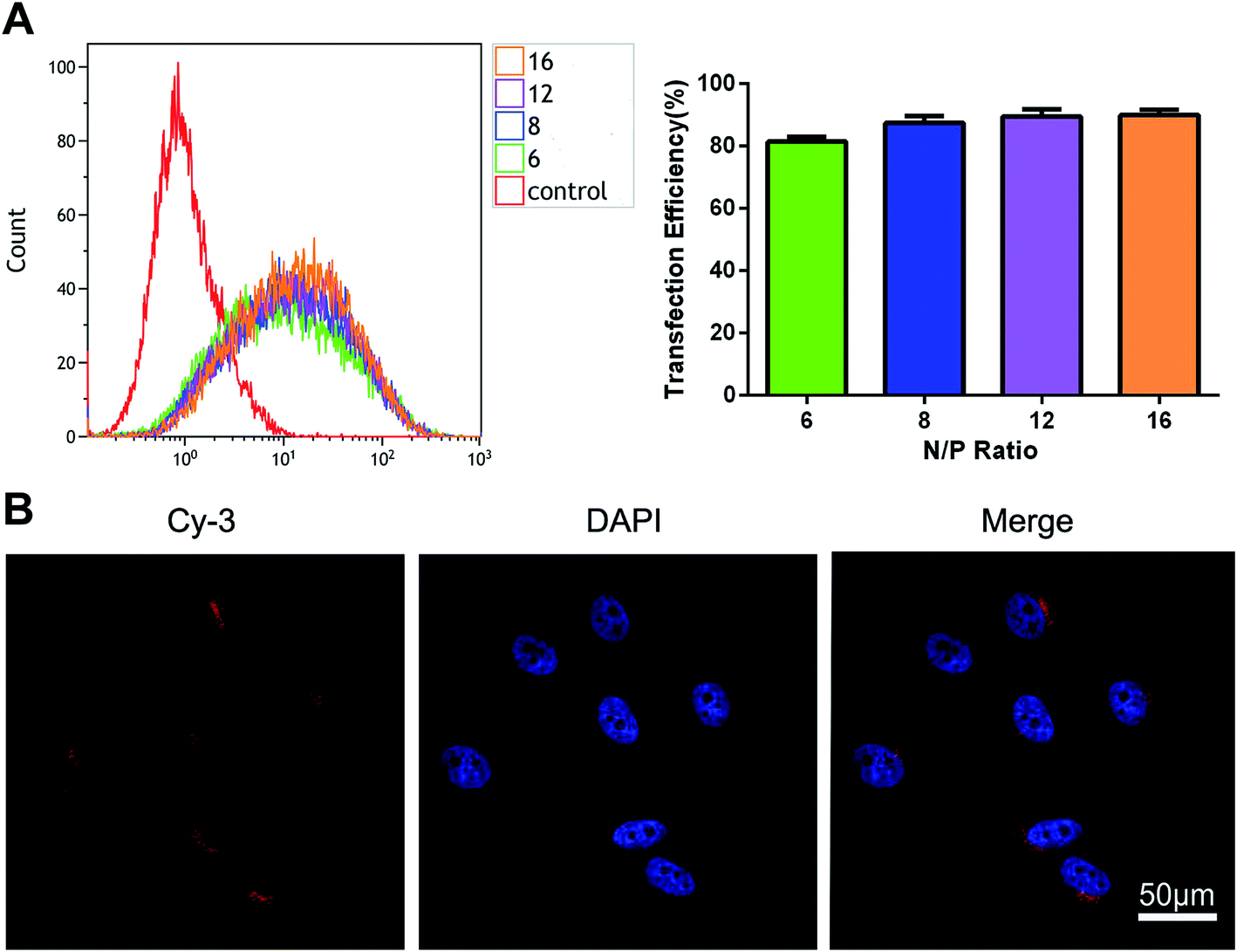 | ||
| Fig. 4 (A) The ratio of Cy3-positive cells detected by flow cytometry after treated with Cy3-labeled polyplexes formed at different N/P ratios (n = 3). Incubation time: 6 h. Dose: 100 nM miRNA inhibitor. (B) High magnification (360×) laser confocal microscopic images of PC-3 cells incubated with PEG–PAsp(DETA)/Cy3-labeled miRNA inhibitor polyplexes (N/P = 8). Dose of miRNA inhibitor: 100 nM. Blue: nuclei stained with DAPI. | ||
Cell uptake
Cell uptake of PEG–PAsp(DETA)/Cy3-labeled miRNA inhibitor polyplexes (N/P = 8) was evaluated using confocal laser scanning microscopy (CLSM). Furthermore, the nuclei were marked blue with DAPI for better visualization. As shown in Fig. 4B, red fluorescence was distributed in the cytoplasm and synapses of PC-3 prostate cancer cells.The cytotoxicity of PEG–PAsp(DETA)/miRNA-21 inhibitor determined by CCK-8 assay
CCK-8 assay was used to evaluate the cytotoxicity of mPEG–PAsp(DETA)/miRNA-21 inhibitor polyplex (N/P = 8) 48 h after cell transfection (Fig. 5A). Negative control group of PEG–PAsp(DETA)/miRNA inhibitor negative control polyplex was almost non-cytotoxic. The PC-3 cells incubated with it retained high viability of 93.24 ± 1.44%. In comparison, PEG–PAsp(DETA)/miRNA-21 inhibitor and lipo2000/miR-21 inhibitor showed cytotoxicities by revealing decreased viabilities down to 74.74 ± 3.60% and 71.04 ± 5.02%, respectively. This suppression of PC-3 cells growth is clearly due to the target gene regulation effect of miRNA-21 inhibitor, as will be further confirmed in the next section.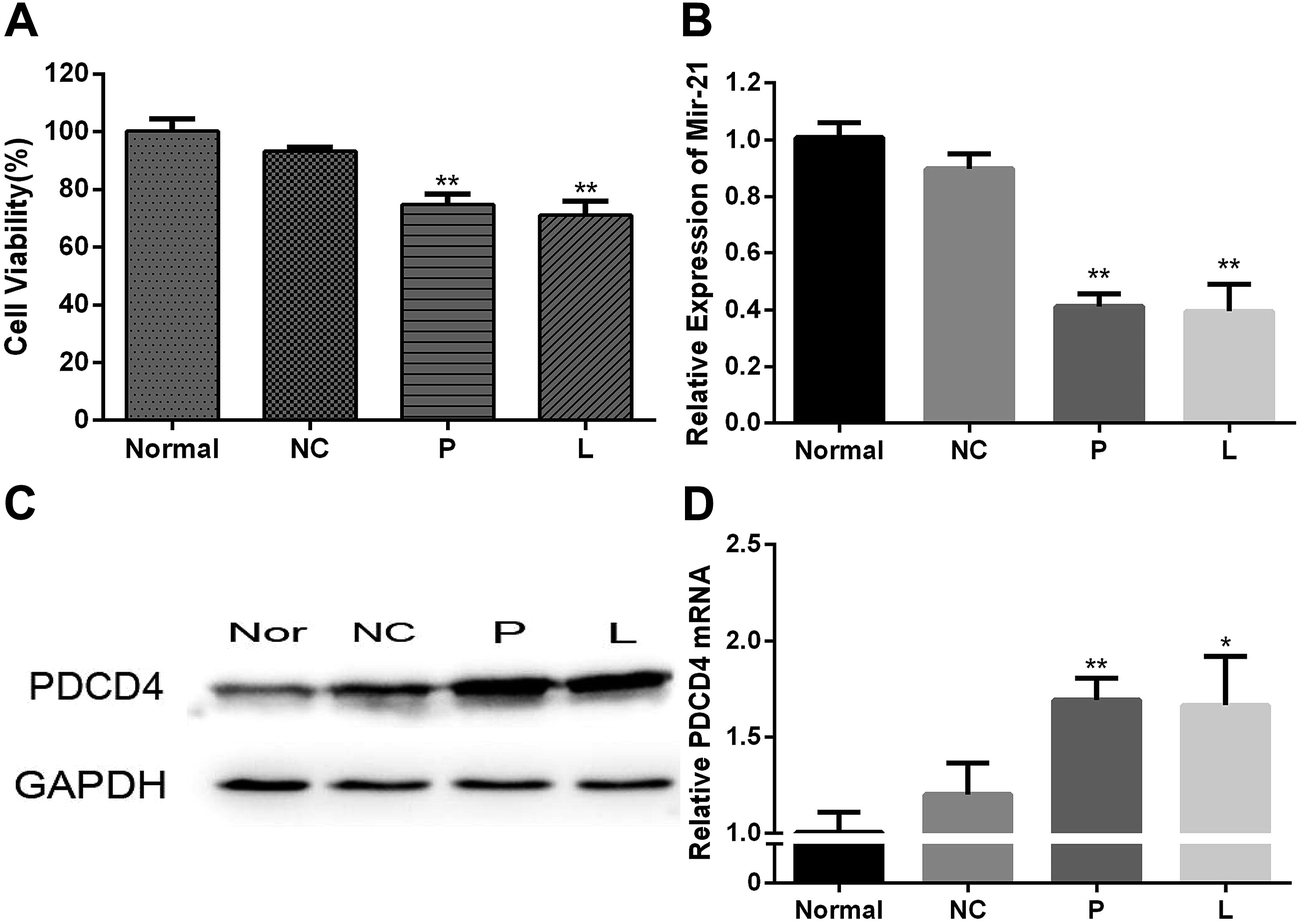 | ||
| Fig. 5 (A) Viabilities of PC-3 cells treated with various samples. (B) Analysis of miRNA-21 expression in PC-3 cells by RT-PCR. (C) Analysis of PDCD4 protein expression by western blot. (D) Analysis of PDCD4 gene expression in PC-3 cells by RT-PCR. Normal: blank group; NC: PEG–PAsp(DETA)/miRNA inhibitor negative control group; P: PEG–PAsp(DETA)/miRNA-21 inhibitor group; L: lipo2000/miR-21 inhibitor group. The polyplexes were formed at N/P = 8. Dose of RNA: 100 nM. | ||
miRNA-21 inhibitor enhances PDCD4
The PC-3 prostate cancer cells were transfected with PEG–PAsp(DETA)/miRNA-21 inhibitor polyplex, and then the miRNA-21 and target gene regulation was assessed by RT-PCR and western blot assays. As shown in Fig. 5B, the miR-21 expression in PC-3 cells of P and L groups are much lower than that in the normal and NC groups. The PC-3 cell transfection of miR-21 inhibitor in the P and L groups resulted in much higher PDCD4 expression at the mRNA level (Fig. 5D). Western blot analysis obtained consistent results at the protein level of PDCD4 expression. The PDCD4 protein levels in cells transfected with PEG–PAsp(DETA)/miRNA-21 inhibitor and lipo2000/miR-21 inhibitor were markedly increased, as compared to that in the normal and the NC groups (Fig. 5C).Anti-tumor effect of PEG–PAsp(DETA)/miRNA-21 inhibitor in mice bearing subcutaneous PC-3 tumor
The in vivo antitumor efficacy of mPEG–PAsp(DETA)/miRNA-21 inhibitor was examined with an PC-3 xenograft model in BALB/c nude mouse.30 As shown in Fig. 6A, tumor growth was completely arrested when the animals were treated with mPEG–PAsp(DETA)/miRNA-21 inhibitor. At the time point of day 30, the averaged tumor volume of mPEG–PAsp(DETA)/miRNA-21 inhibitor group was 97.76 ± 18.29 mm3, which is significantly smaller than that of PEG–PAsp(DETA)/miRNA inhibitor negative control group (233.00 ± 55.80 mm3, P < 0.0001) and PBS control group (256.83 ± 50.47 mm3, P < 0.0001).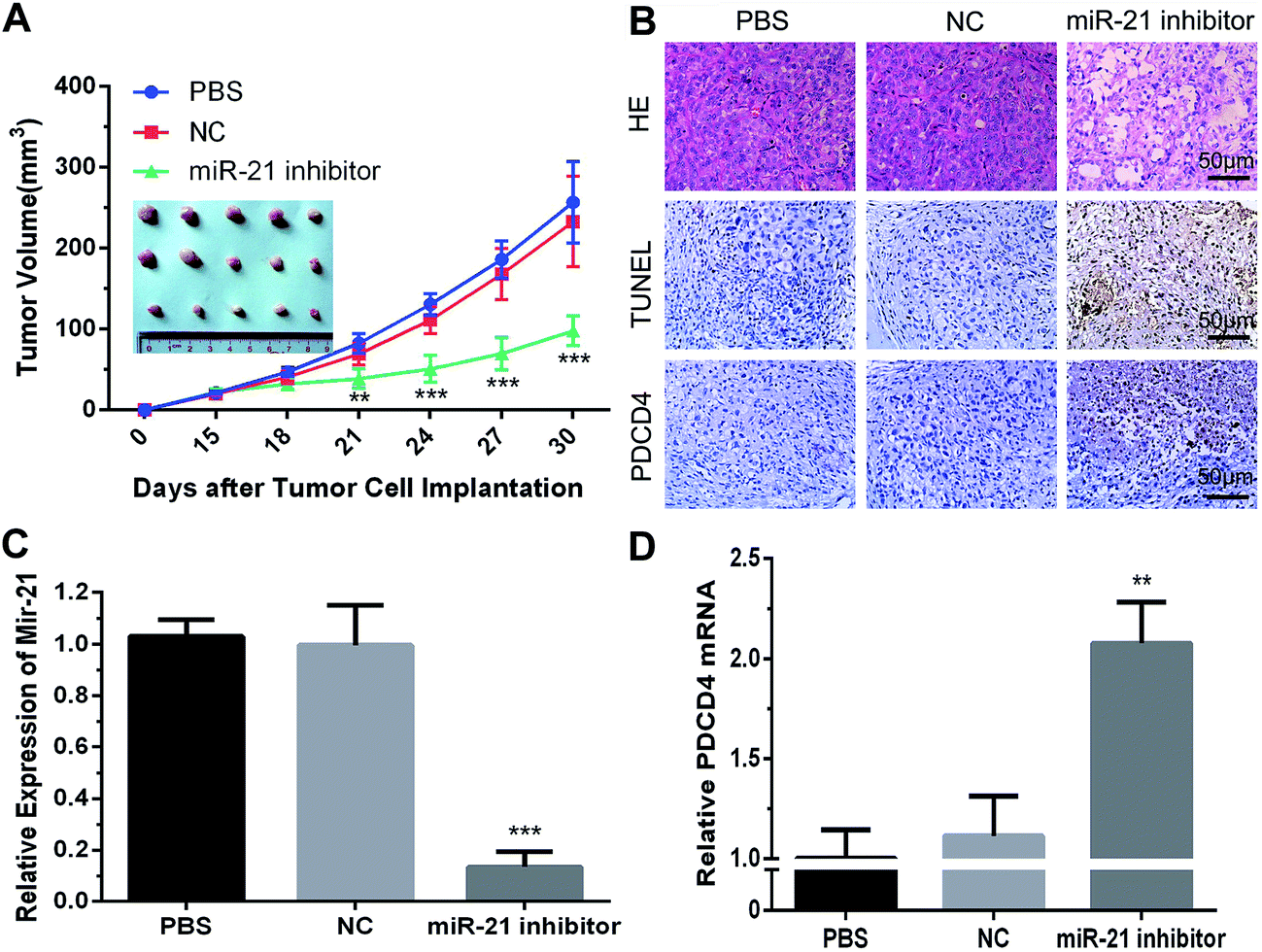 | ||
| Fig. 6 (A) Enhanced tumor growth inhibition of PC-3 xenografts by nanoparticles. After the tumors had reached an average volume of 25 mm3, the tumors in three animal groups were directly injected with 50 μL of PBS or polyplexes containing 500 nM SCR/miR-21 inhibitor dissolved in PBS, respectively. Data are mean ± SD (n = 5). (B) Representative images of histopathological analysis with H&E, TUNEL, and immunohistochemical staining for each dissected tumor tissue. Magnification: ×200 for H&E, TUNEL, PDCD4 respectively. Analysis of miRNA-21 (C) and PDCD4 (D) expressions in tumor xenograft by RT-PCR. Data are mean ± SD (n = 3). PBS: PBS injection group; NC: PEG–PAsp(DETA)/SCR polyplexes injection group; miR-21 inhibitor: PEG–PAsp(DETA)/miRNA-21 inhibitor polyplexes injection group. | ||
Real-time PCR results showed obviously decreased miR-21 levels in the mPEG–PAsp(DETA)/miRNA-21 inhibitor treated group compared to the negative control and PBS groups (P < 0.001). Meanwhile, the miR-21 inhibitor group exhibited much higher PDCD4 expression at the mRNA level (P < 0.01) (Fig. 6C and D).
Histological and immunohistochemical and TUNEL studies were performed to gain more direct evidence for the therapeutic activity of mPEG–PAsp(DETA)/miRNA-21 inhibitor polyplex. Tumors excised from mice treated by PEG–PAsp(DETA)/miRNA-21 inhibitor exhibited much fewer cancer cells than those excised from mice treated with the negative control or PBS control, as observed by H&E staining (Fig. 6B HE). TUNEL assay demonstrated that the mPEG–PAsp(DETA)/miRNA-21 inhibitor treatment resulted in the highest level of cancer cell apoptosis. In contrast, almost no apoptosis was observed in the PBS and NC groups (Fig. 6B TUNEL). The immunohistochemical assay detected apparently increased PDCD4 protein expression in the PEG–PAsp(DETA)/miRNA-21 inhibitor treated groups, as compared to other groups (Fig. 6B PDCD4).
Discussion
MiR-21 has been identified as an oncogenic miRNA31 over-expressed in various human cancers, including glioblastoma5 and breast,32 liver,33 and prostate cancers.34 Antisense studies of miR-21 in glioblastoma have shown that miR-21 controls cancer growth by inhibiting apoptosis of cells.5 Moreover, miR-21 played important roles in apoptosis and metastasis of prostate cancer.35 Recent studies have indicated that miR-21 can reduce programmed cell death 4 (PDCD4) expression by inducing the TGF-β pathway.36 Shi and Ye has confirmed that miR-21 can directly down-regulate the expression of PDCD4 by targeting its 3′UTR in prostate cancer cells.3 PDCD4 is expressed in normal tissues and its expression is suppressed in various tumors.12–14 Furthermore, PDCD4 is a novel suppressor of tumorigenesis, invasion and tumor progression.37–39 Admittedly, PDCD4 expression is related to the extent of malignancy in prostate cancer.20In this study, we attempted to deliver miR-21 inhibitor to PC-3 prostate cancer cells using a nonviral polymeric vector mPEG–PAsp(DETA). The introduced polymer, mPEG–PAsp(DETA), showed tremendous advantages compared with amphiphilic polymers/micelles for a long time application of RNAi in vivo. The introduction of hydrophobic groups may obstruct cationic polymer complex siRNA more or less, leading to a higher N/P ratio for transfection.40,41 On the contrary, mPEG–PAsp(DETA), with high density of amino groups and great flexibility of chains, exerts a considerable efficiency of complexing and transferring nucleic acids at low N/P ratios (Fig. 1D and 4A), which can reduce the polymeric cytotoxicity for the long term application in vivo. Moreover, the hydrophilic of mPEG–PAsp(DETA) is very easy to obtained.
Blocking the activity of miR-21 by miR-21 inhibitor led to a significant increase in PDCD4 protein expression in PC-3 cells. In constructing genetic nanomedicines, the N/P ratio of polymer against DNA or RNA is a key parameter affecting the polyplexes particle size, cytotoxicity, and transfection efficiency.29 High N/P ratio is favorable for cellular uptake through charge interaction-mediated interactions, but results in higher cytotoxicity as well. Thus, a balance between transfection efficiency and toxicity must be sought out. The agarose gel electrophoresis revealed that complete complexation of miR-21 inhibitor realized when the N/P ratios reached 4. Meanwhile, DLS data showed that the increase in N/P ratio resulted in a decrease in hydrodynamic diameter due to a better complexation of miRNA-21 inhibitor and polymer. Moreover, the CCK-8 assay indicated polyplexes formed below N/P ratio of 30 did not cause obvious cytotoxicity. Taking fully into account the transfection efficiency (Fig. 4A), polyplexes formed at N/P = 8 were chosen for subsequent biological experiments.
The cell uptake of polyplexes (N/P = 8) was then investigated with confocal laser scanning microscopy, which proved that the polymer based polyplexes were able to across the cell membrane and internalize into the PC-3 cells. Transfection of PC-3 cells with mPEG–PAsp(DETA)/miR-21 inhibitor polyplexes resulted in 74.74 ± 3.60% viability, which is close to that induced by and lipo2000/miR-21 inhibitor (71.04 ± 5.02%) and indicated the effective suppression of cell growth using the nano-sized miR-21 inhibitor. Furthermore, our results showed that polyplexes effectively silenced miR-21 expression and promoted the gene expression of PDCD4, acting effectively as that of the positive control group (lipo2000/miR-21 inhibitor).
We further verified the therapeutic effect of the mPEG–PAsp(DETA)/miR-21 inhibitor polyplexes in vivo, using a mouse model bearing subcutaneous human PC-3 xenografts.40,42 Our studies showed that the anti-tumor effects of mPEG–PAsp(DETA)/miR-21 inhibitor after intratumor injection in mice was obviously efficient than the negative control and PBS group. The tumor volume gradually increased for 15 days after injection in the two control groups. In contrast, mPEG–PAsp(DETA)/miR-21 inhibitor polyplexes significantly suppressed tumor growth. Moreover, the relative miR-21 level in tumor tissue at 15 day after injection of therapeutic group was markedly reduced in comparison with that of the negative control and PBS control groups.
Conclusions
In conclusion, we have demonstrated the enhanced inhibitory effect of miR-21 inhibitor on human prostate cancer cells using a polymeric vector-mediated strategy. It has been testified that the up-regulation of the PDCD4 gene by silencing miR-21 expression resulted in the enhanced inhibition of the proliferation of prostate cancer cells both in vitro and in vivo. These results suggested that the introduced miR-21 silencing strategy based on a polymeric vector-mediated delivery system was promising for prostate cancer treatment.Disclosure of conflict of interest
The authors declare that they have no conflicts of interest related to this work.Acknowledgements
This work was supported by National Basic Research Program of China (2015CB755500), the National Natural Science Foundation of China (51225305, U1401242, 51373203) and Natural Science Foundation of the Guangdong Province (2014A030312018), the Guangdong Innovative and Entrepreneurial Research Team Program (2013S086).Notes and references
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed.
- G. H. Shi, D. W. Ye, X. D. Yao, S. L. Zhang, B. Dai, H. L. Zhang, Y. J. Shen, Y. Zhu, Y. P. Zhu, W. J. Xiao and C. G. Ma, Acta Pharmacol. Sin., 2010, 31, 867–873 CrossRef CAS PubMed.
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef CAS PubMed.
- J. A. Chan, A. M. Krichevsky and K. S. Kosik, Cancer Res., 2005, 65, 6029–6033 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- S. Volinia, G. A. Calin, C. G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R. L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C. C. Harris and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2257–2261 CrossRef CAS PubMed.
- F. Sicard, M. Gayral, H. Lulka, L. Buscail and P. Cordelier, Mol. Ther., 2013, 21, 986–994 CrossRef CAS PubMed.
- I. A. Asangani, S. A. Rasheed, D. A. Nikolova, J. H. Leupold, N. H. Colburn, S. Post and H. Allgayer, Oncogene, 2008, 27, 2128–2136 CrossRef CAS PubMed.
- Y. Chen, W. Liu, T. Chao, Y. Zhang, X. Yan, Y. Gong, B. Qiang, J. Yuan, M. Sun and X. Peng, Cancer Lett., 2008, 272, 197–205 CrossRef CAS PubMed.
- K. Shibahara, M. Asano, Y. Ishida, T. Aoki, T. Koike and T. Honjo, Gene, 1995, 166, 297–301 CrossRef CAS PubMed.
- Y. Chen, T. Knosel, G. Kristiansen, A. Pietas, M. E. Garber, S. Matsuhashi, I. Ozaki and I. Petersen, J. Pathol., 2003, 200, 640–646 CrossRef CAS PubMed.
- O. Afonja, D. Juste, S. Das, S. Matsuhashi and H. H. Samuels, Oncogene, 2004, 23, 8135–8145 CrossRef CAS PubMed.
- H. Zhang, I. Ozaki, T. Mizuta, H. Hamajima, T. Yasutake, Y. Eguchi, H. Ideguchi, K. Yamamoto and S. Matsuhashi, Oncogene, 2006, 25, 6101–6112 CrossRef CAS PubMed.
- G. Mudduluru, F. Medved, R. Grobholz, C. Jost, A. Gruber, J. H. Leupold, S. Post, A. Jansen, N. H. Colburn and H. Allgayer, Cancer, 2007, 110, 1697–1707 CrossRef CAS PubMed.
- U. Liwak, L. E. Jordan, S. D. Von-Holt, P. Singh, J. E. Hanson, I. A. Lorimer, F. Roncaroli and M. Holcik, Oncotarget, 2013, 4, 1365–1372 CrossRef PubMed.
- H. Zakowicz, H. S. Yang, C. Stark, A. Wlodawer, N. Laronde-Leblanc and N. H. Colburn, RNA, 2005, 11, 261–274 CrossRef CAS PubMed.
- L. Wedeken, P. Singh and K. H. Klempnauer, J. Biol. Chem., 2011, 286, 42855–42862 CrossRef CAS PubMed.
- U. Liwak, N. Thakor, L. E. Jordan, R. Roy, S. M. Lewis, O. E. Pardo, M. Seckl and M. Holcik, Mol. Cell. Biol., 2012, 32, 1818–1829 CrossRef CAS PubMed.
- B. Dong, Z. Shi, J. Wang, J. Wu, Z. Yang and K. Fang, PLoS One, 2015, 10, e0134366 Search PubMed.
- L. Aagaard and J. J. Rossi, Adv. Drug Delivery Rev., 2007, 59, 75–86 CrossRef CAS PubMed.
- R. Niven, R. Pearlman, T. Wedeking, J. Mackeigan, P. Noker, L. Simpson-Herren and J. G. Smith, J. Pharm. Sci., 1998, 87, 1292–1299 CrossRef CAS PubMed.
- X. Qian, L. Long, Z. Shi, C. Liu, M. Qiu, J. Sheng, P. Pu, X. Yuan, Y. Ren and C. Kang, Biomaterials, 2014, 35, 2322–2335 CrossRef CAS PubMed.
- K. Miyata, M. Oba, M. Nakanishi, S. Fukushima, Y. Yamasaki, H. Koyama, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 16287–16294 CrossRef CAS PubMed.
- Y. Zhang, Z. Guan, B. Reader, T. Shawler, S. Mandrekar-Colucci, K. Huang, Z. Weil, A. Bratasz, J. Wells, N. D. Powell, J. F. Sheridan, C. C. Whitacre, A. G. Rabchevsky, M. S. Nash and P. G. Popovich, J. Neurosci., 2013, 33, 12970–12981 CrossRef CAS PubMed.
- Y. Wang, H. Xiao, J. Fang, X. Yu, Z. Su, D. Cheng and X. Shuai, Chem. Commun., 2016, 52, 1194–1197 RSC.
- Y. Zhao, L. Du and N. Zhang, Mol. Biotechnol., 2013, 54, 204–210 CrossRef CAS PubMed.
- J. Dai, S. Lin, D. Cheng, S. Zou and X. Shuai, Angew. Chem., Int. Ed., 2011, 50, 9404–9408 CrossRef CAS PubMed.
- Y. Yuan, F. Gong, Y. Cao, W. Chen, D. Cheng and X. Shuai, J. Biomed. Nanotechnol., 2015, 11, 668–679 CrossRef CAS PubMed.
- S. D. Selcuklu, M. T. Donoghue and C. Spillane, Biochem. Soc. Trans., 2009, 37, 918–925 CrossRef CAS PubMed.
- M. V. Iorio, M. Ferracin, C. G. Liu, A. Veronese, R. Spizzo, S. Sabbioni, E. Magri, M. Pedriali, M. Fabbri, M. Campiglio, S. Menard, J. P. Palazzo, A. Rosenberg, P. Musiani, S. Volinia, I. Nenci, G. A. Calin, P. Querzoli, M. Negrini and C. M. Croce, Cancer Res., 2005, 65, 7065–7070 CrossRef CAS PubMed.
- F. Meng, R. Henson, H. Wehbe-Janek, K. Ghoshal, S. T. Jacob and T. Patel, Gastroenterology, 2007, 133, 647–658 CrossRef CAS PubMed.
- J. Ribas, X. Ni, M. Haffner, E. A. Wentzel, A. H. Salmasi, W. H. Chowdhury, T. A. Kudrolli, S. Yegnasubramanian, J. Luo, R. Rodriguez, J. T. Mendell and S. E. Lupold, Cancer Res., 2009, 69, 7165–7169 CrossRef CAS PubMed.
- T. Li, D. Li, J. Sha, P. Sun and Y. Huang, Biochem. Biophys. Res. Commun., 2009, 383, 280–285 CrossRef CAS PubMed.
- Y. Yu, S. S. Kanwar, B. B. Patel, P. S. Oh, J. Nautiyal, F. H. Sarkar and A. P. Majumdar, Carcinogenesis, 2012, 33, 68–76 CrossRef CAS PubMed.
- A. Hilliard, B. Hilliard, S. J. Zheng, H. Sun, T. Miwa, W. Song, R. Goke and Y. H. Chen, J. Immunol., 2006, 177, 8095–8102 CrossRef CAS.
- A. P. Jansen, C. E. Camalier and N. H. Colburn, Cancer Res., 2005, 65, 6034–6041 CrossRef CAS PubMed.
- H. S. Yang, C. P. Matthews, T. Clair, Q. Wang, A. R. Baker, C. C. Li, T. H. Tan and N. H. Colburn, Mol. Cell. Biol., 2006, 26, 1297–1306 CrossRef CAS PubMed.
- Q. Q. Zhao, J. L. Chen, T. F. Lv, C. X. He, G. P. Tang, W. Q. Liang, Y. Tabata and J. Q. Gao, Biol. Pharm. Bull., 2009, 32, 706–710 CAS.
- C. Q. Mao, J. Z. Du, T. M. Sun, Y. D. Yao, P. Z. Zhang, E. W. Song and J. Wang, Biomaterials, 2011, 32, 3124–3133 CrossRef CAS PubMed.
- S. M. Sparks, C. L. Waite, A. M. Harmon, L. M. Nusblat, C. M. Roth and K. E. Uhrich, Macromol. Biosci., 2011, 11, 1192–1200 CrossRef CAS PubMed.
- J. G. Li, X. S. Yu, Y. Wang, Y. Y. Yuan, H. Xiao, D. Cheng and X. T. Shuai, Adv. Mater., 2014, 26, 8217–8224 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |