
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton-coupled electron transfer promotes the reduction of ferrylmyoglobin by uric acid under physiological conditions†
Andressa de Zawadzki ab,
Daniel R. Cardoso*a and
Leif H. Skibsted*b
ab,
Daniel R. Cardoso*a and
Leif H. Skibsted*b
aInstituto de Química de São Carlos, Universidade de São Paulo, Av. Trabalhador São-Carlense 400, CP 780, CEP 13560-970, São Carlos, SP, Brazil. E-mail: drcardoso@iqsc.usp.br
bDepartment of Food Science, University of Copenhagen, Rolighedsvej 30, 1958 Frederiksberg C, Denmark. E-mail: ls@food.ku.dk
First published on 23rd March 2017
Abstract
The hypervalent muscle pigment ferrylmyoglobin, MbFe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, is not reduced by urate monoanions at physiological conditions despite a strong driving force of around −30 kJ mol−1 while for low pH, uric acid was found to reduce protonated ferrylmyoglobin, MbFe(IV)O,H+, efficiently in a bimolecular reaction with k1 = 1.1 ± 0.1 × 103 L mol−1 s−1, ΔH‡ = 66.1 ± 0.1 kJ mol−1 and ΔS‡ = 35.2 ± 0.2 J mol−1 K−1. For intermediate pH, like for anaerobic muscles and for meat, proton-coupled electron transfer occurs in a transition state, {MbFe(IV)O⋯H+⋯urate}‡, which is concluded to be formed from uric acid and MbFe(IV)O rather than from urate and MbFe(IV)O,H+ with k3 = 9.7 ± 0.6 × 102 L mol−1 s−1, ΔH‡ = 59.2 ± 0.1 kJ mol−1 and ΔS‡ = 11.5 ± 0.3 J mol−1 K−1. The activation parameters as calculated from the temperature dependence of the pH-reduction profile in aqueous 0.067 mol L−1 NaCl (from 25 °C up to 40 °C), support a mechanism for reduction of hypervalent heme iron, where initial proton transfer to oxo-iron initiates the intermolecular electron transfer from urate to ferrylmyoglobin. The concentration of the powerful prooxidant ferrylmyoglobin increases strongly during digestion of red meat in the stomach. A concomitant increase in uric acid concentration may serve as an inherent protection against radical formation by ferrylmyoglobin.
O, is not reduced by urate monoanions at physiological conditions despite a strong driving force of around −30 kJ mol−1 while for low pH, uric acid was found to reduce protonated ferrylmyoglobin, MbFe(IV)O,H+, efficiently in a bimolecular reaction with k1 = 1.1 ± 0.1 × 103 L mol−1 s−1, ΔH‡ = 66.1 ± 0.1 kJ mol−1 and ΔS‡ = 35.2 ± 0.2 J mol−1 K−1. For intermediate pH, like for anaerobic muscles and for meat, proton-coupled electron transfer occurs in a transition state, {MbFe(IV)O⋯H+⋯urate}‡, which is concluded to be formed from uric acid and MbFe(IV)O rather than from urate and MbFe(IV)O,H+ with k3 = 9.7 ± 0.6 × 102 L mol−1 s−1, ΔH‡ = 59.2 ± 0.1 kJ mol−1 and ΔS‡ = 11.5 ± 0.3 J mol−1 K−1. The activation parameters as calculated from the temperature dependence of the pH-reduction profile in aqueous 0.067 mol L−1 NaCl (from 25 °C up to 40 °C), support a mechanism for reduction of hypervalent heme iron, where initial proton transfer to oxo-iron initiates the intermolecular electron transfer from urate to ferrylmyoglobin. The concentration of the powerful prooxidant ferrylmyoglobin increases strongly during digestion of red meat in the stomach. A concomitant increase in uric acid concentration may serve as an inherent protection against radical formation by ferrylmyoglobin.
1. Introduction
Uric acid is the final metabolite of purine bases in humans.1,2 Uric acid is an acid with a pKa1 value of 5.75 and a pKa2 of 9.8, and in plasma is mainly present as the urate monoanion.1,3–5 Urate/uric acid is considered to be the major antioxidant in human plasma, but the low aqueous solubility of both uric acid and sodium urate carries a risk of precipitation of crystals of uric acid or sodium urate in tissues and joints.1,2,6–8 Hyperuricemia may lead to gout and other diseases and is often related to dietary habits and lifestyle.8–11A high intake of red meat is frequently related to oxidative stress and radical damage in the gastrointestinal tract leading to pathological disorders.12–18 The oxidative damage seems to arise from the formation of the hypervalent meat pigments perferrylmyoglobin (˙MbFe(IV)O) and ferryl myoglobin (MbFe(IV)O).17,18 This activation of metmyoglobin (MbFe(III)) by H2O2 and/or organic hydroperoxides as present in foods initiates a pseudoperoxidase catalytic cycle of myoglobin.12,13,17–19 Activation of MbFe(III) thus yields the very short lived compound I (˙MbFe(IV)O) which subsequently decays to the long-lived compound II (MbFe(IV)O).20,21 Both ˙MbFe(IV)O and MbFe(IV)O are strong oxidants known to initiate lipid and protein oxidation in muscle tissues and in meat products during storage and digestion.12,13,19 Knowledge of the reaction kinetics and mechanisms by which toxic effects of hypervalent myoglobin species can be prevented or at least diminished by diet components such as natural antioxidants, are of relevance to improve human health through dietary recommendations.17
Uric acid is generated in the gastrointestinal tract through the breakdown of purine bases from the diet.2,3,7,22 Both serum and gastrointestinal levels of uric acid may thus be modulated by dietary factors and a high intake of red meat is traditionally considered a risk factor for hyperuricemia and diseases resulting from this condition.7–10,23,24 A higher level of uric acid formed in the digestive tract during food digestion could, however, also have positive health effects, since uric acid could be important as a reductant of hypervalent meat pigments.25 The capability of uric acid and urate to deactivate hypervalent myoglobin through the formation of iron(III) or iron(II) states in effect protecting the gastrointestinal tract and muscles against radical damage has, however, not been studied.
In this view, aiming to contribute to a better knowledge of the antioxidant role of uric acid and urate in biological system, the reactivity of uric acid toward the hypervalent meat pigment ferryl myoglobin under various pH and temperature conditions was investigated by fast kinetic absorption spectroscopy.
2. Materials and methods
2.1. Chemicals
Appropriate pH buffer solutions were prepared from K2HPO4/KH2PO4 (J. T. Baker, Phillipsburg, NJ, USA) and from CH3COONa/CH3COOH (Sigma-Aldrich, Steinheim, Germany) with deionized Milli-Q water (Merck Millipore Corporation, Darmstadt, Germany). Sodium chloride (Sigma-Aldrich) was used to keep the ionic strength constant at 0.067 mol L−1 for all pH investigated. Ethylenediaminetetraacetic acid disodium salt (EDTA), uric acid sodium salt, catalase from bovine liver and metmyoglobin, MbFe(III), (from horse heart; purity > 90%), were from Sigma-Aldrich (Steinheim, Germany). Catalase was used without purification. Myoglobin was chromatographically purified on a pD10 column (GE Healthcare Life Sciences, Chicago, USA) and eluted with Milli-Q water, using ε525 nm = 7700 M−1 cm−1 for spectrophotometric standardization of MbFe(III) solution.26 Hydrogen peroxide (30% v/v) was obtained from Sigma-Aldrich (Steinheim, Germany) and used without further treatment. Ferrylmyoglobin was prepared from metmyoglobin and hydrogen peroxide as previously described using catalase to remove the excess of hydrogen peroxide.27 Uric acid stock solutions were prepared in phosphate or acetate buffers and were diluted in the suitable buffer to get a final concentration of 2 × 10−3 mol L−1. The final pH and uric acid concentration were adjusted by dilution to the required value for the kinetic studies.2.2. Kinetic experiments
Optical absorption spectra were recorded at 25.0 ± 0.1 °C on a Varian Cary 100 Bio UV-visible Spectrophotometer (Agilent Technologies, Palo Alto, CA, USA) using quartz cuvettes of 1 cm × 1 cm (Hellma Analytics, Müllheim, Germany).The reduction of ferrylmyoglobin by uric acid was studied by stopped flow absorption spectroscopy under pseudo-first order conditions with an excess of uric acid. Kinetic studies were carried out in an Applied Photophysics SX20 sequential stopped-flow system coupled to a photodiode array detector (Applied Photophysics, Leatherhead, United Kingdom). Fresh solutions of ferrylmyoglobin and buffered uric acid were placed in two different syringes of the equipment and a regular mixing mode was used. The initial concentration was 15 × 10−6 mol L−1 MbFe(IV)O, and for uric acid concentrations in the range of 150–750 × 10−6 mol L−1 were used. The pH-jump technique was used as previously described in order to prevent protein denaturation during a long exposure of MbFe(IV)O to acidic conditions.28,29 Absorption spectra were recorded from 200 to 800 nm at different time intervals depending on the reaction rate. Kinetic traces were taken from single wavelength to calculate each observed rate constant by non-linear regression for each set of conditions.
In order to investigate the influence of temperature and pH on the rate constant for the reduction of ferrylmyoglobin by uric acid and urate, reactions were carried out at 25 °C, 30 °C, 35 °C and 40 °C using buffered uric acid solutions with varying pH at each temperature. All the solutions and the mixing chamber were thermostated using a circulating water bath system. For the pH range of 4.7–5.7, reactions were carried out in acetate buffer, while at higher pH values, phosphate buffer was preferred. Final pH of the mixture was measured at the end of the reactions at the respective temperatures.
2.3. Kinetic calculations
The OriginPro 2015 (OriginLab Corporation, Northampton, MA, USA) software package was used to process the experimental datasets. Pseudo-first order rate constants kobs were determined by single exponential parameter fitting. From the slope of the linear fit of kobs plotted against total uric acid concentration, values for second-order rate constants were obtained for the reactions at specific pH and temperature. From the pH-dependence of the second-order rate constants, k′ specific rate constants for the four possible reaction path identified were derived using non-linear regression analysis, see Results and discussion section.The activation parameters were determined from the specific rate constants at varying temperature through the use of the Eyring equation:30,31
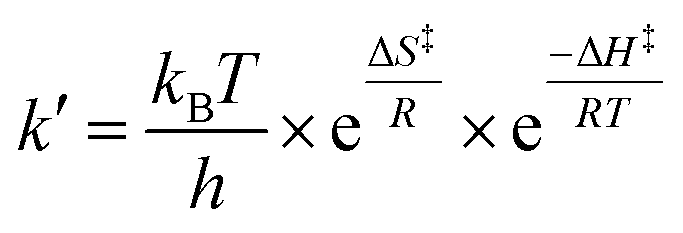
Plotting ln(k′/T) versus 1/T, where T is the temperature in Kelvin and fitting to an linear equation provided ΔH‡/R as the slope and ln(kB/h) + ΔS‡/R as intercept for each of the bimolecular reaction.
3. Results
Ferrylmyoglobin, MbFe(IV)O, the hypervalent muscle pigment, was not or only extremely slowly reduced by uric acid at physiological conditions with neutral pH, where uric acid with a pKa = 5.75 is deprotonated to form the urate anion, UA(NH)3−, despite a negative driving force of ΔG0 = −29 kJ mol−1 for the reaction
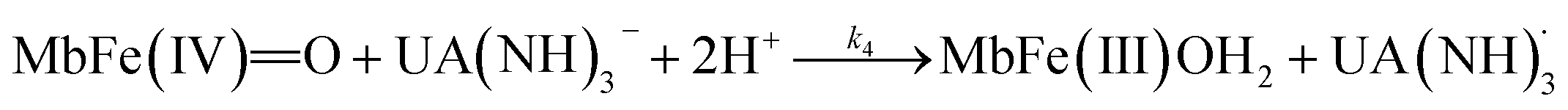 | (1) |
O/MbFe(III) couple and E0 = +0.59 V for the UA(NH)˙3/UA(NH)3− couple at pH = 7.3,32,33
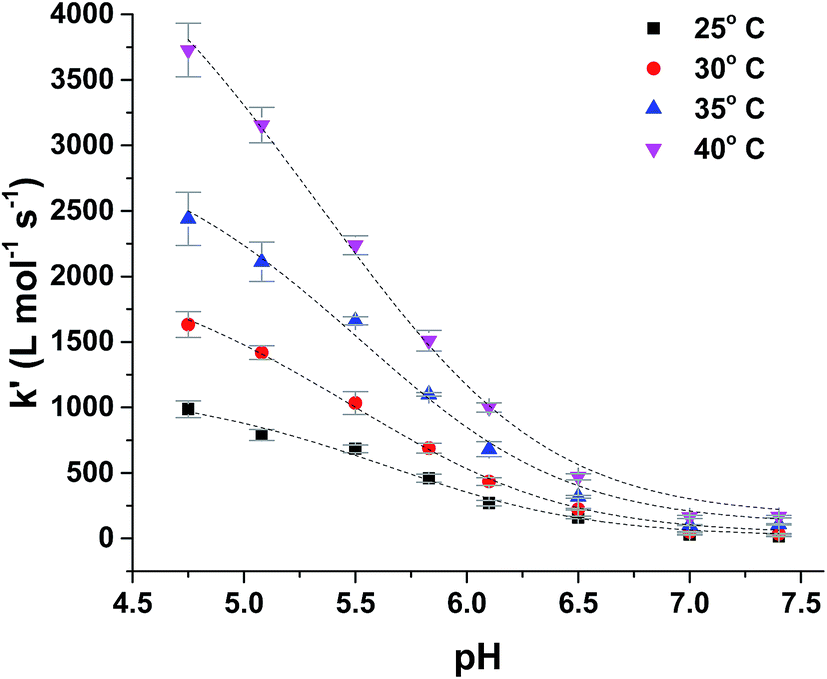
For more acidic conditions the reduction of MbFe(IV)O by uric acid occurred with increasing rate for decreasing pH. For all conditions investigated the initial reduction product metmyoglobin, MbFe(III)OH2, was not reduced further to ferrous myoglobin as was concluded from the UV-visible absorption spectra, see Fig. 1, in agreement with a potential for the MbFe(III)/MbFe(II) couple of E0 = +0.05 V.34,35
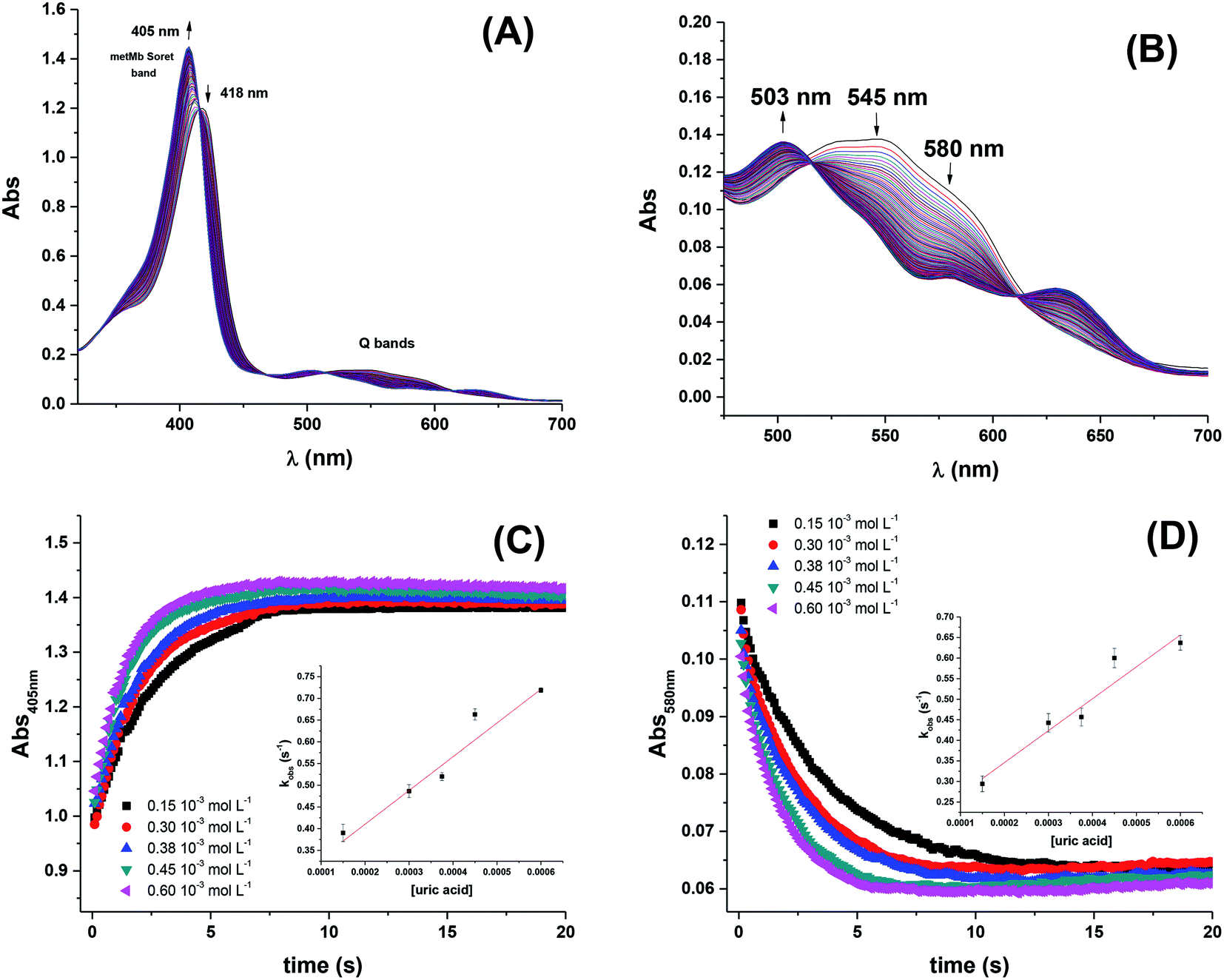 | ||
| Fig. 1 Spectral changes for (A) Soret band and (B) Q bands indicating reduction of MbFe(IV)O by uric acid. (C) Stopped-flow kinetic traces at 405 nm (Soret band) for the reduction of ferryl myoglobin by uric acid and dependence of kobs on uric acid concentration at 25 °C. Conditions: 5 × 103 mol L−1 acetate buffer pH 4.7 containing 1 × 103 mol L−1 of EDTA; [MbFe(IV)O]0 = 15 × 106 mol L−1. Second-order rate constants found by linear regression was 775 ± 109 L mol−1 s−1. (D) Stopped-flow kinetic traces at 580 nm (Q band) for conditions as for (C). Second-order rate constant found by linear regression was 791 ± 133 L mol−1 s−1. | ||
The reactivity of ferrylmyoglobin is known to be affected by pH.26,28,36 Protonation of ferrylmyoglobin (eqn (2)) and its intermediates has been shown to occur under acidic conditions to form highly reactive species that promptly oxidize relevant biologic reductants.19,26,28
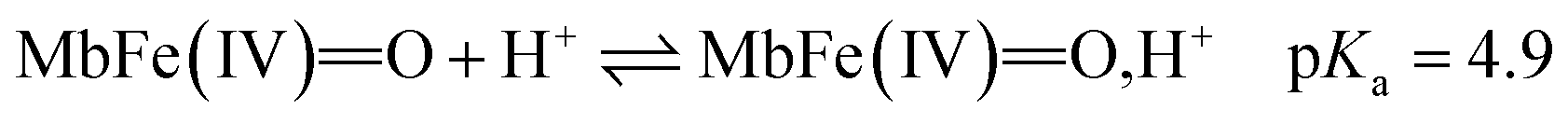 | (2) |
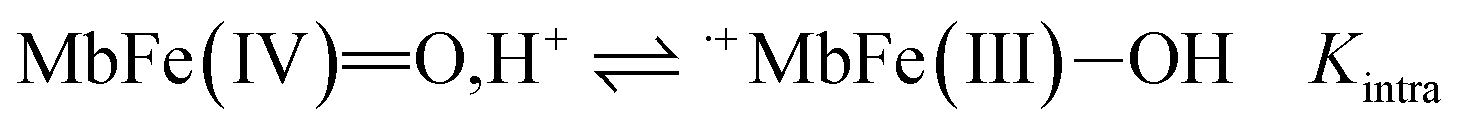 | (3) |
The increasing rate of reduction of MbFe(IV)O at decreasing pH has seen accounted for by protonation according to the equilibrium of eqn (2) as is also increasing the rate of the so-called autoreduction of MbFe(IV)O assigned to specific acid catalysis initiated through a intramolecular electron transfer to form a protein radical cation as seen in eqn (3). However, the increase in rate of reduction by urate at lower pH could also result from a protonization of urate to form uric acid as another reductant, and all four potential reactions of Scheme 1 need to be considered.
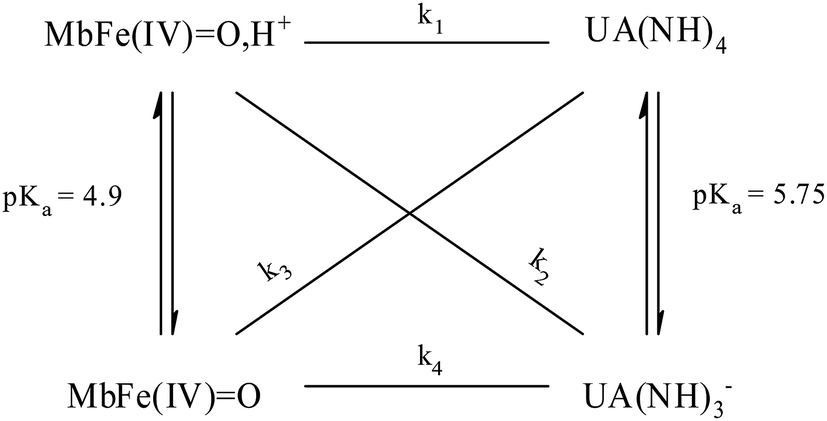 | ||
| Scheme 1 Possible reaction pathways for ferrylmyoglobin species reduction by uric acid/urate under different hydrogenionic conditions. | ||
Kinetic analysis for the reaction between the two acid/base couples using time-dependent spectral measurements for the reduction of ferrylmyoglobin to metmyoglobin is currently reported in literature based on the depletion in the absorbance at 580 nm.27–29,37 In order to get reliable measurements from this depletion it is necessary to use 30 × 10−6 mol L−1 as a minimum concentration of ferrylmyoglobin. However, the low aqueous solubility of uric acid especially under acidic conditions, requires the use of very low concentrations of ferrylmyoglobin to ensure pseudo first order conditions with an excess of uric acid.38 Using an initial concentration of ferrylmyoglobin below 15 × 10−6 mol L−1, the absorbance at 580 nm is almost close to the detection limit by standard visible spectroscopy. Then, it became necessary to use a more sensitive wavelength region to monitor the kinetics. The metmyoglobin Soret band has higher extinction coefficients (for example, ε405 nm = 1.8 × 104 L mol−1 cm−1) than the metmyoglobin Q bands and than the Q bands of ferrylmyoglobin. The reduction of ferrylmyoglobin was accordingly followed by the formation of MbFe(III)OH2 monitoring the spectral changes in the Soret region (390 nm to 430 nm). Fig. 1 shows the spectral changes for the reduction of ferrylmyoglobin at 25.0 ± 0.1 °C and pH 4.7 monitored both at 405 nm and 580 nm. The observed pseudo first-order rate constants kobs were found to depend linearly on the total uric acid concentration in agreement with a bimolecular rate-determining step, see insert in Fig. 1C and D. Consistent observations of isosbestic points, as may be seen in Fig. 1, for all the conditions investigated, indicate that no secondary reactions are occurring. The second-order rate constants, k′, determined for the reduction of ferrylmyoglobin using UV and visible wavelengths regions gave numerical values for two different spectral regions not differing significantly (ρ < 0.05). Then, for subsequent analysis, reactions were tracked by measuring the increase in the absorbance of metmyoglobin at 405 nm. The second-order rate constants were obtained at pH range of 4.7 to 7.5 at 25 °C (Fig. 2) and the reduction showed a pseudo first-order kinetics behavior in this pH region of physiological relevance for excess of urate/uric acid.
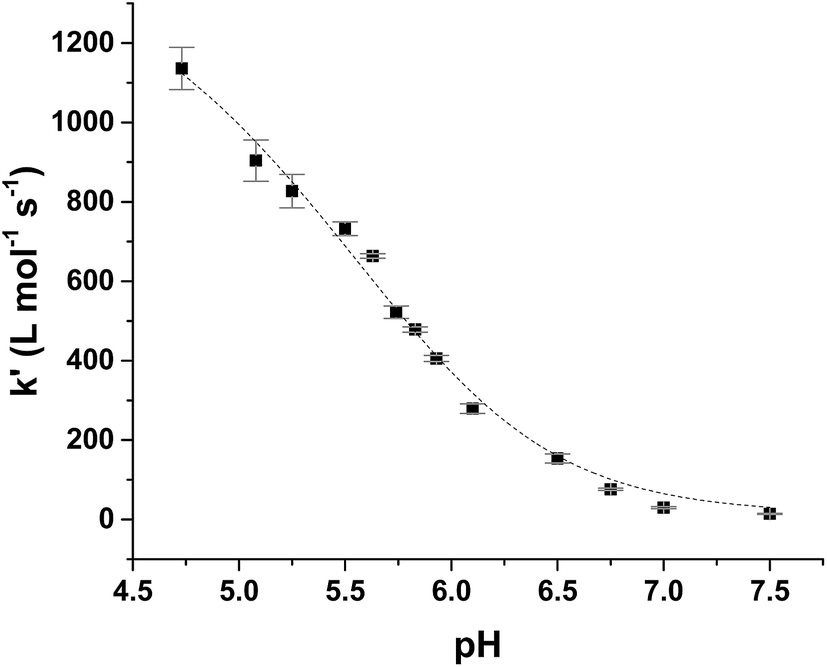 | ||
| Fig. 2 Dependence of second-order rate constant, k′, on pH for the reduction of ferrylmyoglobin by uric acid in aqueous 0.067 mol L−1 NaCl at 25.0 ± 0.1 °C with full line calculated by non-linear regression according to eqn (9). | ||
The four potential reactions according to the reactions of Scheme 1 are
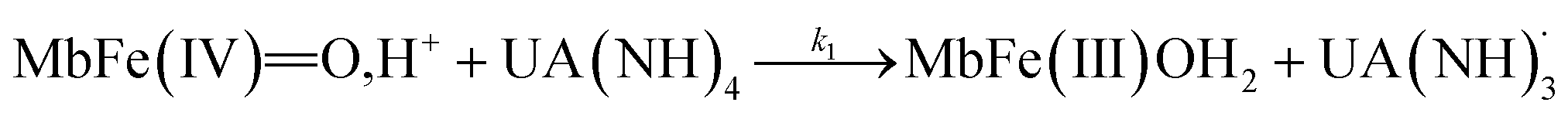 | (4) |
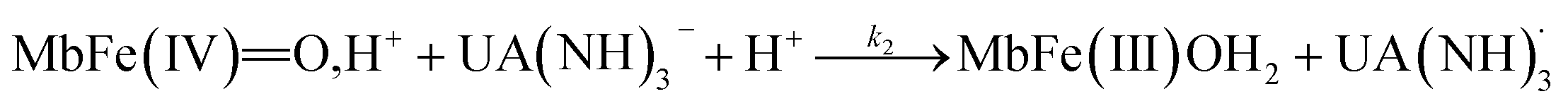 | (5) |
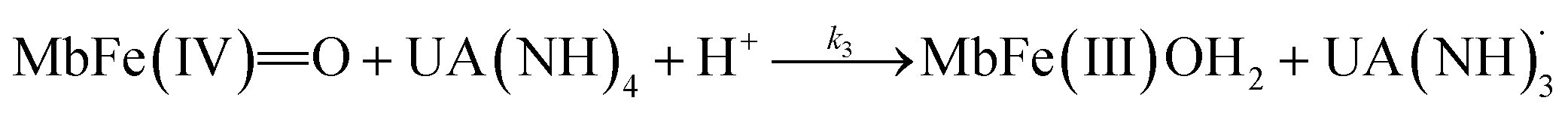 | (6) |
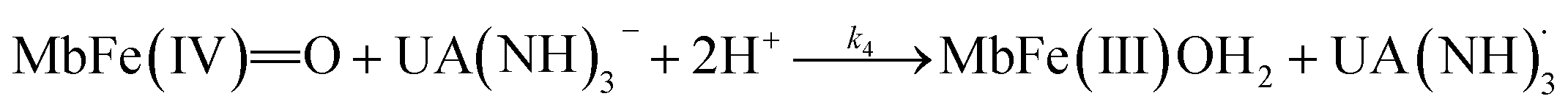 | (7) |
On the basis of the possible four reactions between the acid/base forms of the reactants, the second-order rate constant, k′, as dependent on pH was used to estimate the specific rate constants for these four reactions according to the following rate expression for conditions of excess of uric acid/urate:28,31
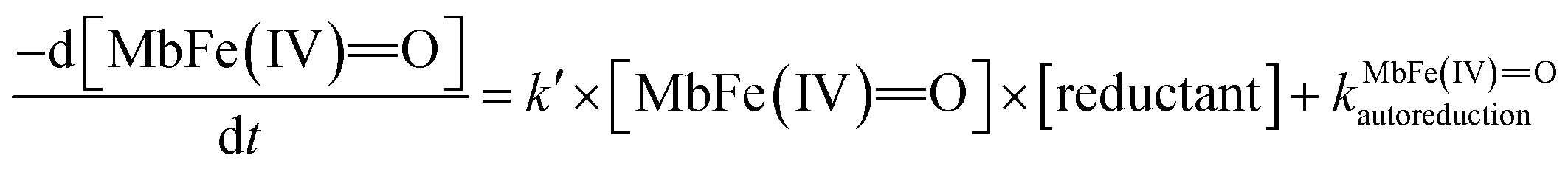 | (8) |
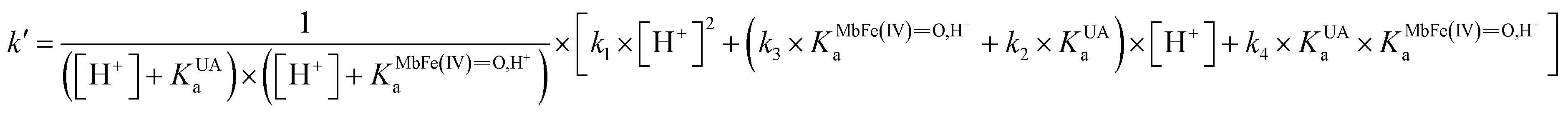 | (9) |
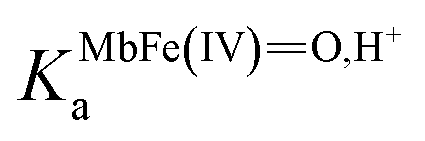 is the acid dissociation constant of MbFe(IV)O,H+ and KUAa is the acid dissociation constant of uric acid at 25 °C. The kMbFe(IV)Oautoreduction is known from previous studies and makes a negligible contribution to the overall rate in the actual pH-region.26
is the acid dissociation constant of MbFe(IV)O,H+ and KUAa is the acid dissociation constant of uric acid at 25 °C. The kMbFe(IV)Oautoreduction is known from previous studies and makes a negligible contribution to the overall rate in the actual pH-region.26
The rate constants for the two pathways, k1 and k4, were accordingly estimated by nonlinear fitting of k′ as function of pH at 25 °C together with the rate constant for the two reactions of eqn (5) and (6) according to eqn (9). The reactions of eqn (5) and (6) with the rate constants k2 and k3, respectively, notably have a common transition state {MbFe(IV)O⋯H+⋯urate}‡, and due to the proton ambiguity the experimental rate constant derived from eqn (9) can not be assigned to one of the two reactions alone based on reaction kinetics.26,28 The non-linear regression also included estimation of the pKa-value for uric acid and the pKa-value for ferrylmyoglobin from the experimental data, see Fig. 3 for the four temperatures. The estimated numerical values for the two pKa values were in agreement with the literature values.3,4,39,40 The distribution between acid/base forms of the two reactants as a function of pH in agreement with the determined pKa-values are shown in Fig. 4 for 25 °C together with the net charge on ferrylmyoglobin.
 | ||
| Fig. 3 Temperature dependence of pH-profile for second-order rate constant, k′, for reduction of ferryl myoglobin by uric acid in aqueous 0.067 mol L−1 NaCl at varying temperature. | ||
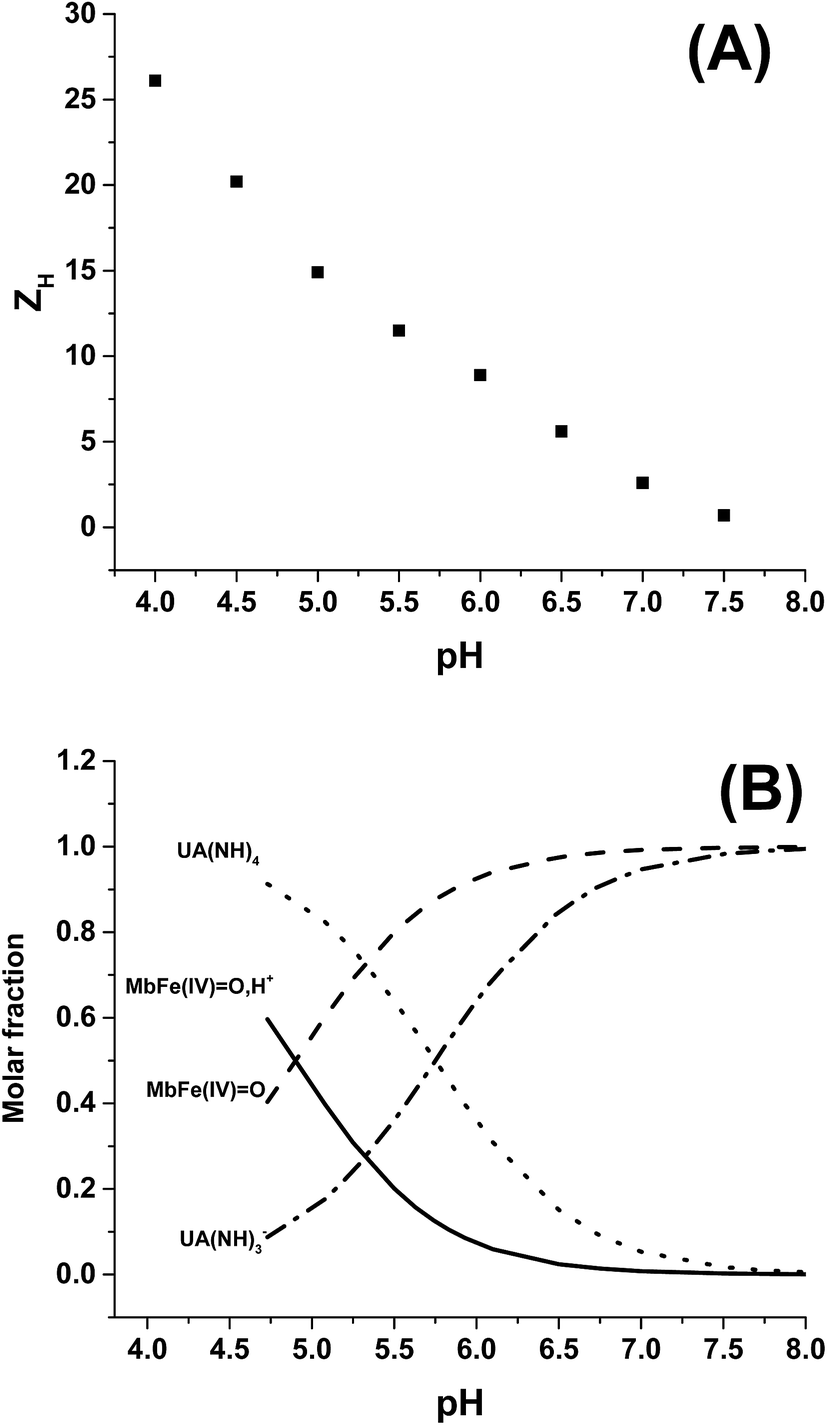 | ||
| Fig. 4 (A) Total protein charge ZH as function of pH for horse heart myoglobin calculated with Protein Calculator v3.4 (http://protcalc.sourceforge.net/cgi-bin/protcalc), sequences taken from pdb files (P68082.2). (B) Acid/base equilibrium distribution of uric acid and ferryl myoglobin as function of pH. | ||
Based on a number of spectroscopic techniques, the pKa-value of ferrylmyoglobin has been assigned to a value of pKa ≤ 2.7, which is significantly lower than the value obtained from analysis of reaction kinetics.28,36 The two methods for estimating the pKa-value may be related to two different chemical equilibria:
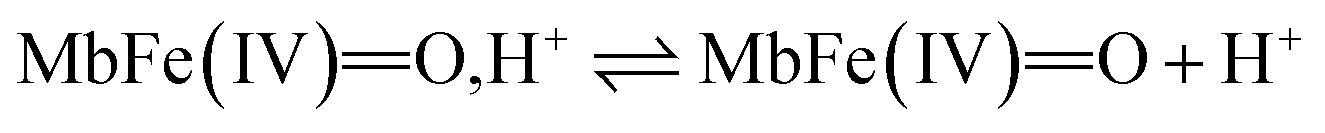 | (10) |
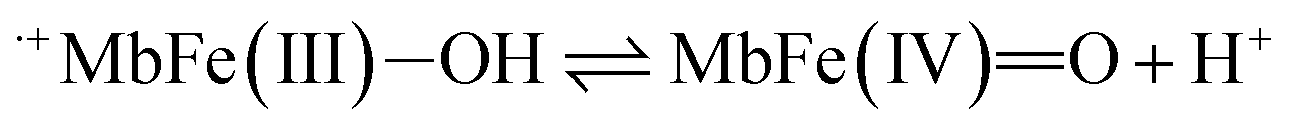 | (11) |
The rate constant for the reduction of eqn (7) was not significantly different from zero and the reaction of eqn (7) was accordingly concluded not to make any contribution to the reduction of ferrylmyoglobin. The reaction of eqn (4) on the other hand had the value of 1.12 ± 0.08 × 103 L mol−1 s−1 at 25 °C, which makes this reaction very fast compared to the autoreduction of ferrylmyoglobin which has a hydrogen ion dependent second-order rate constant of 7.9 ± 1.8 × 102 L mol−1 s−1 at 25 °C for pH 7 with 0.16 mol L−1 of ionic strength.26 As for the reaction dominating for intermediate pH, the calculated constant for the reaction on eqn (5) and the calculated rate constant for the reaction of eqn (6) can not be used to resolve the reaction into the individual reactions, since these two reactions has a common transition state.
The reduction of ferrylmyoglobin by uric acid was followed at four temperatures in the same pH-region, see Fig. 3, and for these four temperatures, the values for the specific rate constant and pKa-values presented in Table 1 were obtained by non-linear regression using eqn (9). The second-order rate constants for each of the three reactions were analyzed according to transition state theory as seen from the Eyring plot of Fig. 5, not considering the reaction of eqn (7). The experimental pKa-values showed little variation as demonstrated in the temperature-region investigated, see Table 1. The activation parameters calculated by linear regression from the three independent plots of Fig. 5 may be found in Table 1.
O by uric acid/urate in aqueous 0.067 M NaCl as calculated by non-linear regression at each temperature (see Fig. 3) together with pKa values of protonated ferryl myoglobin and uric acid as estimated from the temperature dependence of the pH-reduction profile
| k′ (L mol−1 s−1) | ΔH‡ (kJ mol−1) | ΔS‡ (J mol−1 K−1) | ||||
|---|---|---|---|---|---|---|
| 25° C | 30° C | 35° C | 40° C | |||
| k1 | 1.12 ± 0.08 × 103 | 2.10 ± 0.05 × 103 | 2.91 ± 0.12 × 103 | 4.80 ± 0.10 × 103 | 66.1 ± 0.1 | 35.2 ± 0.2 |
| k2 | 6.75 ± 0.39 × 103 | 9.10 ± 0.20 × 103 | 13.62 ± 0.04 × 103 | 19.73 ± 0.40 × 103 | 56.6 ± 0.1 | 17.5 ± 0.1 |
| k3 | 9.76 ± 0.56 × 102 | 1.56 ± 0.03 × 103 | 2.61 ± 0.16 × 103 | 3.35 ± 0.07 × 103 | 59.2 ± 0.1 | 11.5 ± 0.2 |
| pKa MbFe(IV)O,H+ |
4.90 ± 0.11 | 4.92 ± 0.06 | 4.94 ± 0.12 | 4.92 ± 0.07 | ||
| pKa UA(NH)4 | 5.74 ± 0.24 | 5.70 ± 0.14 | 5.66 ± 0.25 | 5.69 ± 0.16 | ||
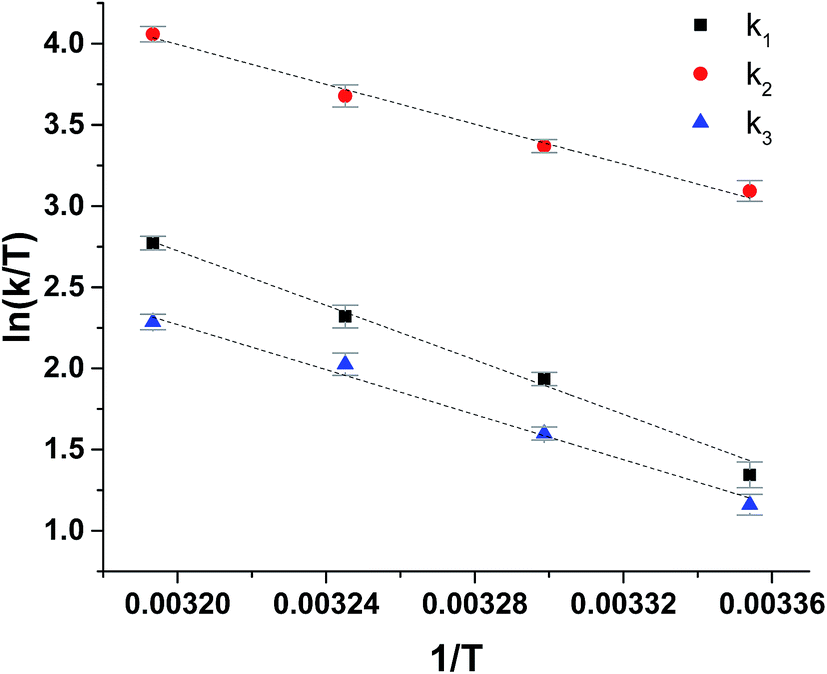 | ||
| Fig. 5 Eyring plots for reduction of ferrylmyoglobin by uric acid/urate for each possible reaction path, see Scheme 1. | ||
The reaction between the protonated ferrylmyoglobin and uric acid dominating at low pH has a relative large value for the enthalpy of activation, which is larger than the value obtained for reduction by ascorbate and by chlorogenate, as will be discussed below and, notably, is also larger than the value for autoreduction. The activation is largely entropy-controlled for the reduction by uric acid. Autoreduction seems to include an initial intermolecular acid/base equilibrium involving a protein radical cation, see eqn (3), leading to reduction of iron(IV)-oxo to iron(III) and to protein oxidation.
For uric acid reduction, proton coupled electron transfer may be important for the formation of the radical cation form of ferrylmyoglobin. Proton-coupled electron transfer seems even more important for the reduction of ferrylmyoglobin at intermediate pH. An initial proton-transfer to ferrylmyoglobin by uric acid as depicted in Fig. 6 will lead to a protonated ferrylmyoglobin for which electron transfer becomes facilitated. This process involves proton transfer from uric acid followed by electron transfer from urate and corresponds to the reaction of eqn (6). However, a transition state with the same composition arises from the reaction of eqn (5) for which, however, the proton is transferred from protonated ferrylmyoglobin to urate followed by electron transfer in the opposite direction. The proton-coupled electron transfer, i.e. the reaction of eqn (6) seems a more feasible reaction path and activation parameters for this reaction will be used for comparison with reduction of ferrylmyoglobin by other reactants. The important role of a proton for electron transfer is evident, since the reaction between the non-protonated ferrylmyoglobin and urate becomes non-significant and a limiting value for the rate constant for eqn (6) may be calculated from eqn (9). A suggested mechanism for the proton-coupled electron transfer is shown in Fig. 6.
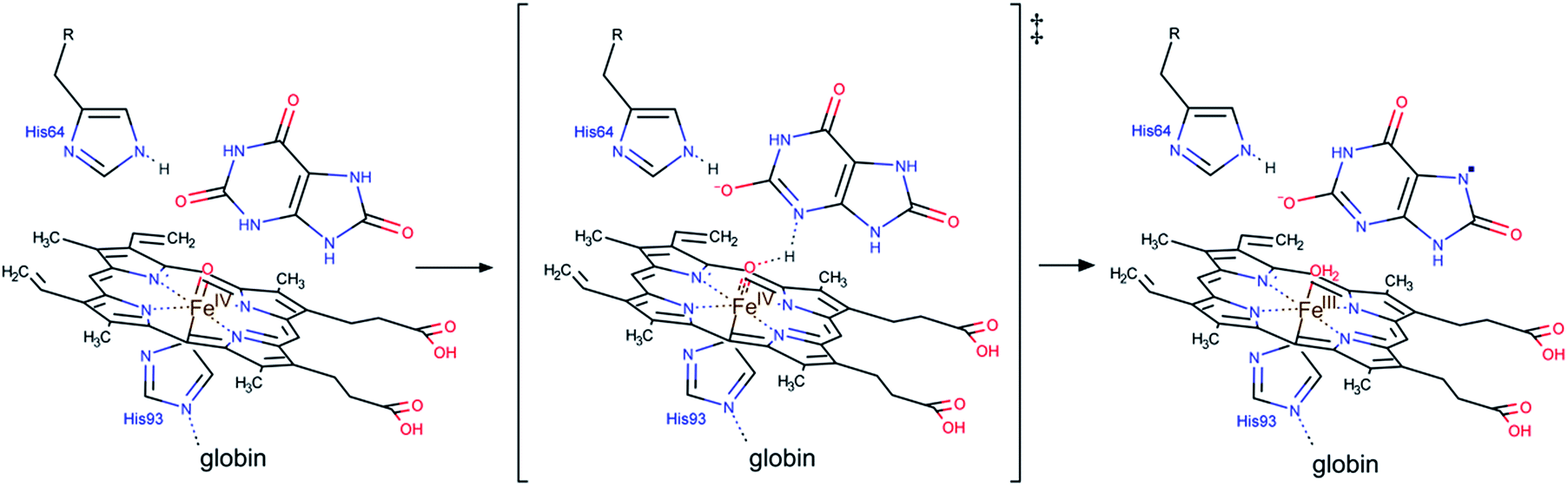 | ||
| Fig. 6 A possible mechanism for the proton coupled electron transfer from uric acid to non-protonated ferrylmyoglobin. | ||
Deactivation of ferrylmyoglobin is important in muscles during oxidative stress and decreasing pH as for anaerobic metabolism. Uric acid seems to be important for such conditions rather than for normal pH-conditions. Ascorbate has a similar pH-profile and is also faster as a reductant at lower pH, as may be seen from the comparison of rate constants and activation parameters in Table 2. Hydrogen sulfide (H2S) seems to have a special role in deactivating hypervalent muscle pigments, since cysteine as another sulfur based reductant, is less efficient than hydrogen sulfide as reductant and even less efficient than oxygen-based reductants like ascorbate and chlorogenic acid.27–29 Uric acid is a nitrogen-based reductant and apparently the nitrogen based anion does not allow transfer of an electron without the assistance of a proton, see Scheme 2. Hydrogen atom transfer from uric acid is kinetically hampered due to a strong hydrogen bonding pattern involving the nitrogen–hydrogen bonds.41 However, the nitrogen hydrogen are slightly acidic corresponding to a pKa = 5.75 and may initiate reduction through a proton transfer. Urate is as reductant different from cysteine, as the anion of cysteine reduces ferrylmyoglobin without the assistance of a proton in contrast to urate.
| k (L mol−1 s−1) | ΔH‡ (kJ mol−1) | ΔS‡ (J mol−1 K−1) | |
|---|---|---|---|
| a From Carlsen et al., 2000,27 determined in aqueous solution with ionic strength 0.16 mol L−1.b From Libardi et al., 2013,28 apparent values as determined at pH 6.8.c From Libardi et al., 2014,29 apparent values as determined at pH 6.8.d From Mikkelsen et al., 1995 (ref. 26).e From Østdal et al., 1996;42 apparent values as determined at pH 7.0.f From Jórgensen et al., 1997;43 apparent values as determined at pH 7.0. | |||
MbFe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O,H+ reduction by O,H+ reduction by |
|||
| Ascorbatea | 1.7 × 103 | 62 | 24 |
| Chlorogenatea | 2.7 × 103 | 59 | 15 |
| Uric acid | 1.1 × 103 | 70 | 58 |
| H2Sb | 2.5 × 106 | ||
| Cysteinec | 5.1 | ||
| Autoreductiond | 7.9 × 102 | 59 | 3 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| MbFe(IV)O reduction by |
|||
| β-Lactoglobuline | 2.4 | 45 | −93 |
| Ascorbatea | 2.9 | 51 | −63 |
| Chlorogenatea | 2.2 × 102 | 73 | 41 |
| Uric acid | 9.5 × 102 | 59 | 12 |
| Urate | 0 | ||
| H2Sb | 4.5 × 105 | ||
| Cysteine thiolatec | 3.1 × 10−1 | ||
| Crocinf | 1.8 × 102 | 55 | −17 |
| Autoreductiond | 1.6 × 10−4 | ||
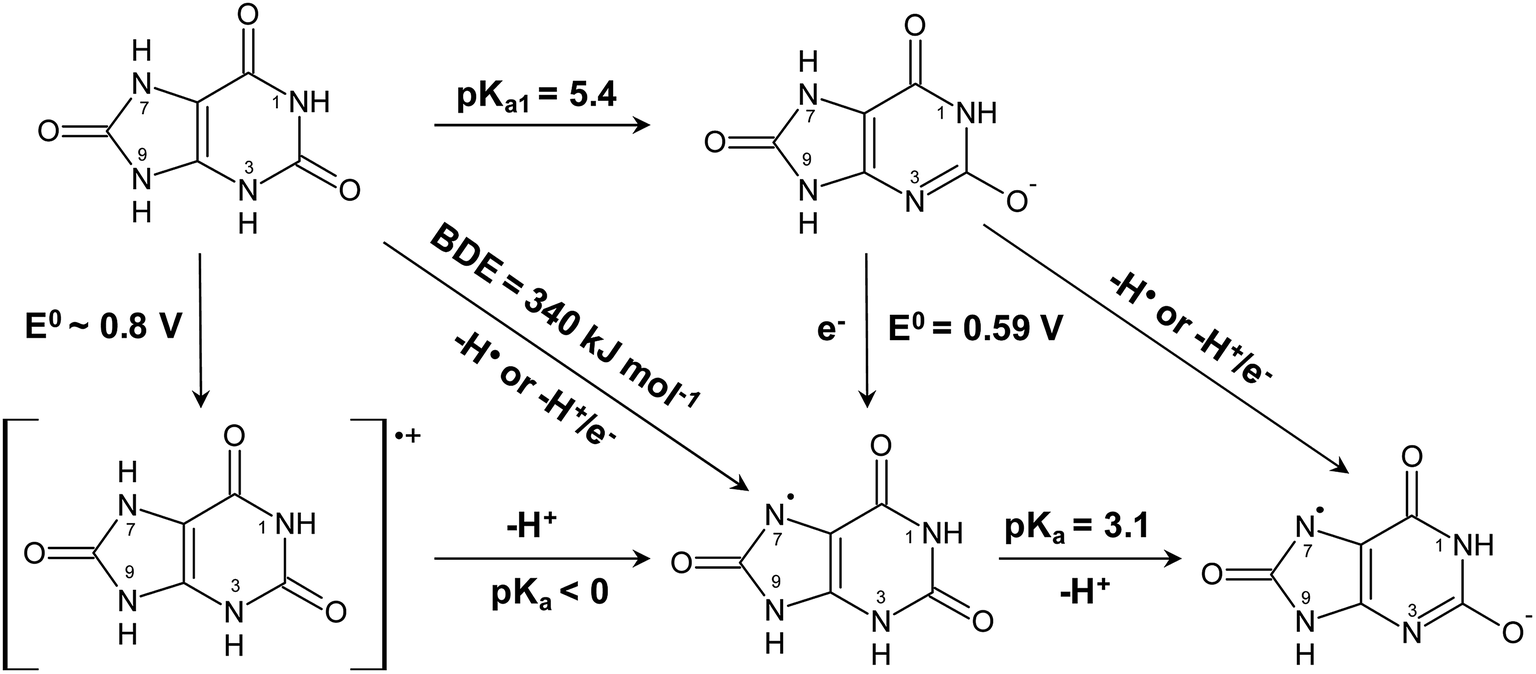 | ||
| Scheme 2 Aqueous thermochemistry of uric acid used to identify the actual reactant for reduction of ferrylmyoglobin. | ||
Reduction of ferrylmyoglobin by various physiological relevant reductants has a moderately high enthalpy of activation as may be seen from Table 2, where oxygen and sulfur based reductants are compared with uric acid/urate as a nitrogen-based reductant. In general, protonization of MbFe(IV)O to yield MbFe(IV)O,H+ accelerates the reduction due to entropy effects as are typically for outer-sphere electron transfer, which has a positive value for entropy of activation. Such outer-sphere electron transfer may occur for a protein radical cation, see eqn (3). For the non-protonated MbFe(IV)O, entropy of activation is negative or close to zero, in effect counteracting the moderate enthalpy of activation suggesting an inner-sphere electron transfer mechanism. This compensation effect is especially evident for the sulfur-based reductants hydrogen sulfide, cysteine and β-lactoglobulin.42 For the oxygen-based reductant ascorbate, a similar effect is rather seen, when reduction of the protonated MbFe(IV)O,H+ is compared with the non-protonated MbFe(IV)O.27 The more negative entropy of activation for the reduction of the non-protonated MbFe(IV)O by the sulfur-based reductant or by ascorbate may reflect the high organization required in the transition state with a proton partly transferred prior to electron transfer to the iron center of ferrylmyoglobin. For the water-soluble carotenoid crocin, a negative entropy of activation is likewise counteracting the moderate enthalpy of activation and probably relates to direct electron transfer from the conjugated double bond.43 A similar effect seems evident when reduction of MbFe(IV)O,H+ and MbFe(IV)O by uric acid is compared. For the reduction of non-protonated MbFe(IV)O by uric acid, the initial proton transfer entails high organization lowering the entropy of activation from 35 J mol−1 K−1 to 12 J mol−1 K−1 when compared to MbFe(IV)O,H+. For urate as a possible reductant of non-protonated MbFe(IV)O, a further increase in activation entropy makes this reaction of no importance kinetically when compared to ferrylmyoglobin autoreduction.
Uric acid is accordingly considered to be the active reductant for reaction at acidic conditions and for intermediate pH. For intermediate pH, proton coupled electron transfer is equivalent to donation of a hydrogen atom, as shown in Scheme 2, to MbFe(IV)O, where the oxidized uric acid is seen subsequently to lose an additional proton from the uric acid neutral radical. The urate ion is concluded not to be a reductant for ferrylmyoglobin.
Sulfur is concluded to be a better electron donor for reduction of ferrylmyoglobin compared to oxygen as evident from lower enthalpies of activation. Uric acid/urate as a N–H purine base reductant is less efficient and comparable to the carotenoid crocin as reductant. For uric acid/urate initial proton transfer from N–H bond becomes of even more importance.
The prooxidative activity of hypervalent heme iron as formed during digestion of red meat and meat products is getting increasing attention as one possible explanation of the negative health effect of a high red meat intake.16–18 A balanced diet with plant based antioxidants like polyphenols and carotenoids have been found to protect against radical formation as the primary oxidation step through efficient reduction of perferrylmyoglobin and ferrylmyoglobin under the acidic conditions of the stomach. Endogenous factors in the form of metabolites like carbon monoxide, hydrogen sulfide and nitric oxide may, however, may also be important for the protection against toxic effects of hypervalent iron. Uric acid may now be added to the list of small molecules with protective effect against radical formation during digestion of meat and, in muscles, during oxidative stress. The level of uric acid depends on dietary factors. However, an important observation seems to be that urate despite its strong reducing capacity, only becomes active against hypervalent heme iron through proton coupled electron transfer as for the decreasing pH of muscles during anaerobic conditions or under the acidic conditions of the stomach.
Acknowledgements
This research is part of the bilateral Brazilian/Danish Food Science Research Program “BEAM – Bread and Meat for the Future” supported by FAPESP (Grant 2011/51555-7) and by the Danish Research Council for Strategic Research (Grant 11-116064). D. R. C. thanks the Brazilian National Research Council – CNPq for the research Grant (396491/2015-0), and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) for a Science without Borders fellowship (Grant 99999.008903/2014-00 to A. Z.).References
- B. F. Becker, Free Radical Biol. Med., 1993, 14, 615–631 CrossRef CAS PubMed.
- Y. Y. Sautin and R. J. Johnson, Nucleosides, Nucleotides Nucleic Acids, 2010, 27, 608–619 Search PubMed.
- M. G. Simic and S. V. Jovanovic, J. Am. Chem. Soc., 1989, 111, 5778–5782 CrossRef CAS.
- A. Amić, Z. Marković, J. M. Dimitrić, B. Lučić, V. Stepanić and D. Amić, Theor. Comput. Chem., 2016, 1077, 2–10 CrossRef.
- D. P. Chong, Theor. Comput. Sci., 2013, 1, 1–7 CrossRef.
- W. N. Kelley, F. M. Rosenbloom, J. F. Henderson and J. E. Seegmiller, Proc. Natl. Acad. Sci., 1967, 57, 1735–1739 CrossRef CAS.
- G. K. Glantzounis, E. C. Tsimoyiannis, A. M. Kappas and D. A. Galaris, Curr. Pharm. Des., 2005, 11, 4145–4151 CrossRef CAS PubMed.
- M. A. Cameron and K. Sakhaee, Urol. Clin., 2007, 34, 335–346 CrossRef PubMed.
- R. J. Johnson, D. H. Kang, D. Feig, S. Kivlighn, J. Kanellis, S. Watanabe, K. R. Tuttle, B. Rodriguez-Iturbe, J. Herrera-Acosta and M. Mazzali, Hypertension, 2003, 41, 1183–1190 CrossRef CAS PubMed.
- J. Fang and M. H. Alderman, JAMA, 2000, 283, 2404 CrossRef CAS PubMed.
- K. H. Yu, L. C. See, Y. C. Huang, C. H. Yang and J. H. Sun, Semin. Arthritis Rheum., 2008, 37, 243–250 CrossRef CAS PubMed.
- C. P. Baron and H. J. Andersen, J. Agric. Food Chem., 2002, 50, 3887–3897 CrossRef CAS PubMed.
- B. J. Reeder and M. T. Wilson, Curr. Med. Chem., 2005, 12, 2741–2751 CrossRef CAS PubMed.
- A. Sesink, D. S. Termont, J. H. Kleibeuker and R. Van Der Meer, Carcinogenesis, 2000, 21, 1909–1915 CrossRef CAS PubMed.
- S. C. Larsson, J. Rafter, L. Holmberg, L. Bergkvist and A. Wolk, Int. J. Cancer, 2005, 113, 829–834 CrossRef CAS PubMed.
- D. E. Corpet, Meat Sci., 2011, 89, 310–316 CrossRef CAS PubMed.
- B. Lorrain, O. Dangles, C. Genot and C. Dufour, J. Agric. Food Chem., 2010, 58, 676–683 CrossRef CAS PubMed.
- B. Lorrain, C. Dufour and O. Dangles, Free Radical Biol. Med., 2010, 48, 1162–1172 CrossRef CAS PubMed.
- B. J. Reeder and M. T. Wilson, Free Radical Biol. Med., 2001, 30, 1311–1318 CrossRef CAS PubMed.
- C. Giulivi and E. Cadenas, Free Radical Biol. Med., 1998, 24, 269–279 CrossRef CAS PubMed.
- J. H. Horner, X. Sheng, R. E. P. Chandrasena, R. Zhang, Q. Wan and M. Newcomb, ECS Trans., 2009, 19, 71–80 CAS.
- B. N. Ames, R. Cathcart, E. Schwiers and P. Hochstein, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 6858–6862 CrossRef CAS.
- H. K. Choi, S. Liu and G. Curhan, Arthritis Rheum., 2005, 52, 283–289 CrossRef PubMed.
- B. H. S. Pai, G. Swarnalatha, R. Ram and K. V. Dakshinamurty, Indian J. Nephrol., 2013, 23, 280–286 CrossRef PubMed.
- B. S. Rocha, J. O. Lundberg, R. Radi and J. Laranjinha, Redox Biol., 2016, 8, 407–414 CrossRef CAS PubMed.
- A. Mikkelsen and L. H. Skibsted, Journal of Food Research and Research, 1995, 200, 171–177 CAS.
- C. U. Carlsen, M. V. Kröger-Ohlsen, R. Bellio and L. H. Skibsted, J. Agric. Food Chem., 2000, 48, 204–212 CrossRef CAS PubMed.
- S. H. Libardi, H. Pindstrup, D. R. Cardoso and L. H. Skibsted, J. Agric. Food Chem., 2013, 61, 2883–2888 CrossRef CAS PubMed.
- S. H. Libardi, H. Pindstrup, J. M. Amigo, D. R. Cardoso and L. H. Skibsted, RSC Adv., 2014, 4, 60953–60958 RSC.
- P. Bugnon, J. Chott, J. Jestin, B. Jung, G. Laurenczy, M. Maeder, A. E. Merbach and D. Andreas, Anal. Chim. Acta, 1994, 298, 193–201 CrossRef CAS.
- R. G. Wilkins, Kinetics and mechanism of reactions of transition metal complexes, Weinheim, 2nd edn, 1991 Search PubMed.
- P. Faulkner, B. Y. P. George and D. H. Irvine, Biochem. J., 1955, 60(4), 596 CrossRef.
- C. Carlsen, J. Moller and L. Skibsted, Coord. Chem. Rev., 2005, 249, 485–498 CrossRef CAS.
- J. F. Taylor and V. E. Morgan, J. Biol. Chem., 1942, 144, 15–20 CAS.
- M. Tsuruga, A. Matsuoka and Y. Sugawara, J. Biol. Chem., 1998, 273, 8607–8615 CrossRef CAS PubMed.
- R. Silaghi-Dumitrescu, B. J. Reeder, P. Nicholls, C. E. Cooper and M. T. Wilson, Biochem. J., 2007, 403, 391–395 CrossRef CAS PubMed.
- F. J. Romeros, I. Ordoñez, A. Arduini and E. Cadenas, J. Biol. Chem., 1992, 267, 1680–1688 Search PubMed.
- W. R. Wilcox, A. Khalaf, A. Weinberger, I. Kippen and J. R. Klinenberg, J. Med. Biol. Eng., 1972, 10, 522–531 CrossRef CAS.
- C. Wang, R. Yuan, Y. Chai, S. Chen, F. Hu and M. Zhang, Anal. Chim. Acta, 2012, 741, 15–20 CrossRef CAS PubMed.
- F. Bergmann and S. Dikstein, J. Am. Chem. Soc., 1955, 77, 691–696 CrossRef CAS.
- G. Litwinienko and K. U. Ingold, Acc. Chem. Res., 2007, 40, 222–230 CrossRef CAS PubMed.
- H. Østdal, B. Daneshvar and L. H. Skibsted, Free Radical Res., 1996, 24, 429–438 CrossRef.
- L. V. Jørgensen, H. J. Andersen and L. H. Skibsted, Free Radical Res., 1997, 27, 73–87 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28314d |
| This journal is © The Royal Society of Chemistry 2017 |