
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe intriguing journey of gH625-dendrimers
A. Falanga a,
L. Lombardib,
R. Taralloc,
G. Francib,
E. Perilloa,
L. Palombab,
M. Galdierob,
D. Pontoni*d,
G. Fragneto*e,
M. Weck*c and
S. Galdiero*a
a,
L. Lombardib,
R. Taralloc,
G. Francib,
E. Perilloa,
L. Palombab,
M. Galdierob,
D. Pontoni*d,
G. Fragneto*e,
M. Weck*c and
S. Galdiero*a
aDepartment of Pharmacy, CIRPEB, University of Naples “Federico II”, Via Mezzocannone 16, Naples 80134, Italy. E-mail: stefania.galdiero@unina.it
bDepartment of Experimental Medicine, Second University of Naples, Via de Crecchio 7, Naples 80138, Italy
cMolecular Design Institute, Department of Chemistry, New York University, New York 10003, USA. E-mail: marcus.weck@nyu.edu
dESRF – The European Synchrotron, 71 avenue des Martyrs, Grenoble, France. E-mail: pontoni@esrf.fr
eInstitut Laue-Langevin, 71 avenue des Martyrs, BP 156, 38000 Grenoble Cedex, France. E-mail: fragneto@ill.eu
First published on 30th January 2017
Abstract
The knowledge of the mechanism used by vectors to gain access to cell interiors is key to the development of effective drug delivery tools for different pathologies. The role of the initial interaction with the membrane bilayer is widely recognized, although not fully understood. We use neutron reflectivity experiments and internalization studies with cells to reveal the extent of interaction of dendrimers functionalized with the peptide gH625 with biomimetic membranes. We further investigate the internalization by use of Caco-2 cells for assessing the membrane permeability properties of the peptide–dendrimer construct. Neutron reflectivity allowed for the hypothesis that the peptide–dendrimer is able to pass across the bilayer which was confirmed via permeability studies. We find that gH625-dendrimers interact more strongly with cholesterol containing membranes. The advances in our understanding of the mechanism of drug uptake are extremely useful to push further the design of new drug delivery systems.
Introduction
Membranes surround all living cells and cell organelles and play an essential role in maintaining and protecting the cell and its compartments. Being selectively permeable, membranes allow the penetration of some substances but not others; thus, non-specific and non-disruptive penetration across the membrane is only applicable to the adsorption of small molecules, less than approximately 500 Da, while larger molecules can be adsorbed with low efficiency and usually cause significant modifications of the lipid bilayer structure.1 Endocytosis (pinocytosis and phagocytosis) offers a route to large macromolecules, which are otherwise incapable of crossing the membrane, to be taken up by cells. Endosomes fuse with lysosomes to form secondary lysosomes and, the macromolecule is degraded by lysosomal enzymes. Sometimes, following uptake of a drug via receptor-mediated endocytosis, only some endosomes carrying the drug can bypass the lysosomes and release the un-degraded drug into the intracellular space.Thus, poor cellular delivery and low bioavailability of drugs compel to develop novel approaches aimed to transport molecules across the membrane without permanently damaging it and without involving endocytic mechanisms of uptake.2 Peptide supported transport of bioactive molecules into cells is an emerging field and constitutes a promising strategy. Moreover, a variety of drug delivery carriers (polymer nanoparticles, liposomes, dendrimers, metal nanoparticles) are in preclinical and clinical development to improve drug bioavailability.3 Among these, dendrimers are nanocontainers able to encapsulate or conjugate therapeutic drugs or imaging moieties and offering the potential for orthogonal multi-functionalization, a requirement for theranostic platforms. Dendrimers are a class of well-defined synthetic nanostructured macromolecules with a high number of active termini, a polydispersity of one, a globular shape at higher generations and the potential for a multivalent display, i.e. the introduction of several functionalities (such as drugs, labelling agents, targeting groups, cell-penetrating peptides, and/or solubilizing groups) into the dendrimer structures.4
Despite these advantages, dendrimers usually exploit endocytosis to cross cell barriers;5,6 thus dendrimers are mainly entrapped in endosomes with only a small amount finding their way to the intracellular target. The combination of dendrimers with cell penetrating peptides (CPPs) significantly helps in solving this problem. In particular, membranotropic viral peptides, able to efficiently cross biological membranes, to promote lipid-membrane restructuring processes and cause temporary membrane destabilization and reorganization,7–15 represent a challenging opportunity to modify the internalization mechanism of dendrimers. The peptide gH625 is derived from herpes simplex virus 1 and was developed in our research group as the first membranotropic sequence able to enhance drug delivery.16–19 We also previously reported on a poly(amide)-based dendrimer20–22 coupled to gH625, showing its ability to cross the membrane bilayer through an energy-independent process, without evidence of endocytosis, poration, or cytotoxicity.
The gH625-dendrimer presents a high affinity for the membrane bilayer and deeply penetrates the bilayer where it does not form holes.20 Our previous data support the hypothesis that the interaction with the bilayer can be splitted into a first adsorption step that leads to an increased macromolecule concentration on the membrane and a second insertion step which involves the penetration into the lipid bilayer and likely a conformational change of the peptide bound to the dendrimer which assumes a helical conformation when in a membrane mimetic environment. Clearly, the mechanism of lipid association is key in the translocation process and thus in cellular uptake. Further studies, however, are indispensable to fully clarify the mechanism of translocation and to design and develop new and effective drug delivery tools.
Liposomes are remarkable model systems because of their simple and membrane-like composition, their easiness to prepare, biodegradability, biocompatibility and sufficient stability over time.23 From the analysis of liposome and dendrimer interactions, we can draw conclusions about transport pathways implicated in drug delivery.
Quite a few modes of interaction between dendrimers and liposomes have been reported.24–35 As examples, dendrimers can pass through the lipid bilayer or dendrimer–lipid micelles can be generated.36 Hydrophobic forces may be involved in the interaction between the hydrophobic dendrimer interior and lipid acyl chains.37,38 The strength of the interaction will depend on the size and charge of the molecule39–41 and the phase of lipids.42
This work describes the mechanism by which gH625-dendrimer gains access to the cell interior as determined by neutron reflectivity (NR) measurements, using model system membranes composed of cholesterol (Chol) and neutral phospholipids such as phosphatidylcholine (PC). The results described herein complement previous reports supporting the view that gH625-dendrimers are able to pass through and do not disrupt the biological membrane.
Results and discussion
Synthesis of the gH625-dendrimer
The octadecaazide dendrimer was synthetized as previously reported.43 As shown in Fig. 1 we obtained a symmetrical dendrimer with 18 azido-termini.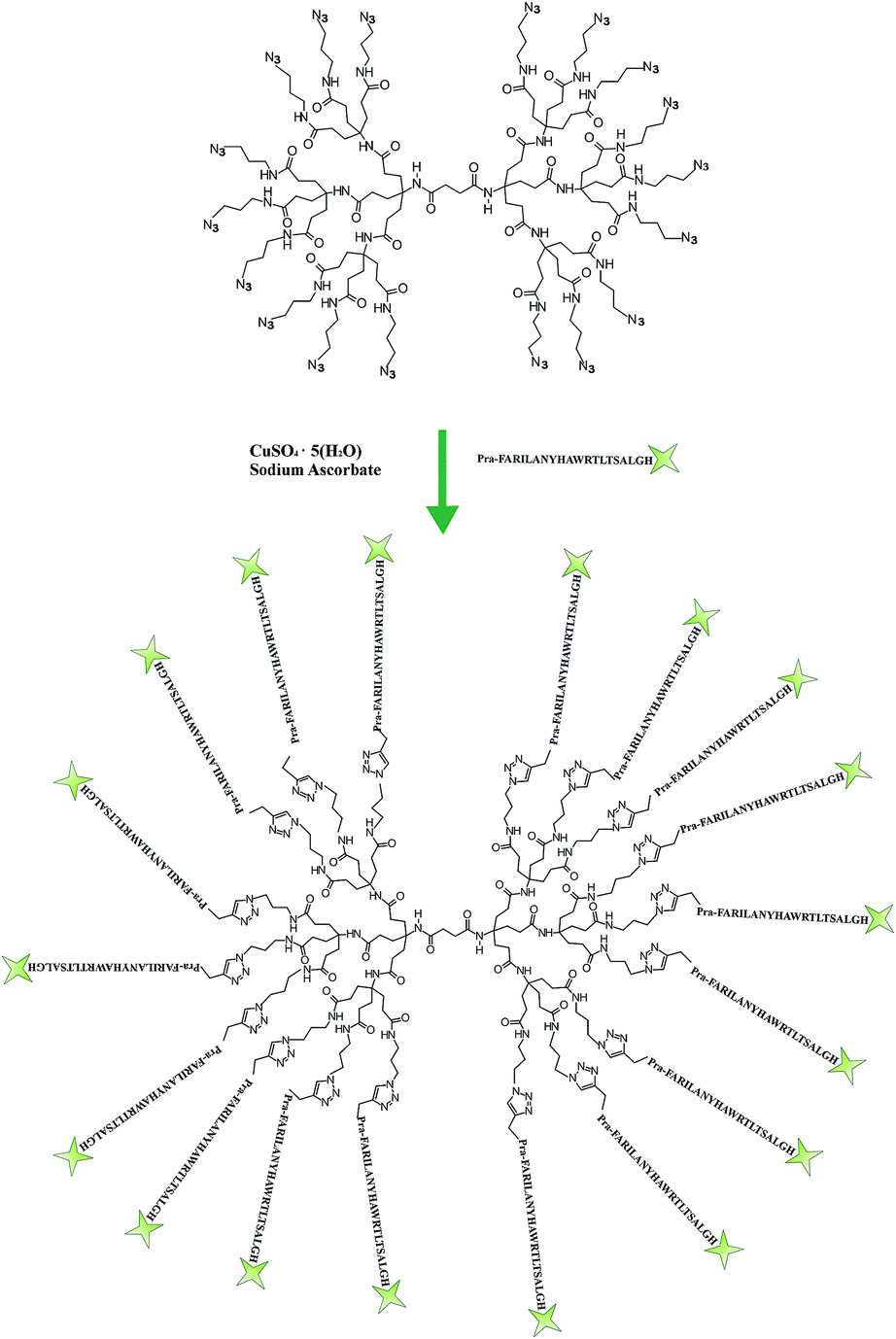 | ||
| Fig. 1 Schematic representation of the functionalization step to yield the target peptide–dendrimer. | ||
The gH625 peptide sequence (Table 1) was synthesized with a propargylglycine residue (PrA) at the C terminus to perform the copper-catalyzed azide–alkyne cycloaddition (CuAAC). The functionalization of the dendrimer was achieved in a water/methanol solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) using CuSO4·5H2O as catalyst and sodium ascorbate as reducing agent. The obtained peptide–dendrimer was purified by HPLC as reported in the Experimental section. The amount of peptide functionalization was determined to be 71% by UV analysis (εgH625 = 6990 M−1 cm−1 at λ = 280 nm).
:1 v/v) using CuSO4·5H2O as catalyst and sodium ascorbate as reducing agent. The obtained peptide–dendrimer was purified by HPLC as reported in the Experimental section. The amount of peptide functionalization was determined to be 71% by UV analysis (εgH625 = 6990 M−1 cm−1 at λ = 280 nm).
| Peptide | Sequence |
|---|---|
| gH625 | NH2-HGLASTLTRWAHYNALIRAF-CONH2 |
| NBD-gH625-PrA | NBD-HGLASTLTRWAHYNALIRAF-PrA-CONH2 |
| gH625-PrA | NH2-HGL-Ad3-STLTRW-Ad3-HYN-Ad3-LIR-Ad3-F-PrA-CONH2 |
Neutron reflectivity (NR)
Neutron reflectometry (NR) allows the study of the interaction between the peptide–dendrimer and the lipids. NR was chosen not only because it allows deuteration-based contrast variation approaches, but also because it is exempt from substrate charging effects encountered when applying synchrotron X-ray reflectivity techniques.44,45 Despite such charging effects do not induce detectable structural changes in e.g. lipid multilayers deposited on silicon substrates,46 they may alter the interactions between the single lipid bilayers and the dendrimers studied here, thus requiring detailed investigations of possible irradiation-induced alterations of the dendrimer/lipid-bilayer system. NR makes such control experiments superfluous, and despite the available beam flux is considerably lower than at synchrotrons, the attainable spatial resolution is still sufficient to detect alterations of the bilayer structure due to the insertion of the peptide–dendrimer constructs into the lipid bilayers.Measurements were performed on the lipid bilayers alone and after their interaction with gH625-PrA-dendrimer. In detail, two lipid compositions were chosen: in the first experiment the lipid bilayer was made only of one-chain-deuterated POPC (d-POPC) while in the second experiment the bilayer comprised a mixture of fully hydrogenated POPC (h-POPC) with 20% hydrogenated cholesterol (h-Chol).
As neutron reflectivity is sensitive to isotopic substitution, not only different isotopic lipid compositions were used, but also different water contrasts (light water, deuterated water or a mix of both) were exchanged for the same bilayers both before and after addition of the peptide–dendrimer. The simultaneous fitting of NR profiles pertaining to multiple water contrasts for the same system enhances considerably the reliability of the results. For all systems, a five-layer model produced the best fits to the data (Fig. 2) yielding meaningful surface-normal scattering length density profiles for the bare bilayers (Fig. 2A and C) and after their interaction with the peptide–dendrimer (Fig. 2B and D). The five layers are representative of (1) the native silicon oxide sub-nanometre thick layer present on the silicon crystals, (2) a thin water layer separating the lipid bilayer from the silicon oxide surface, (3) the polar heads of the inner bilayer leaflet adjacent to the silicon block, (4) the hydrophobic tails of both leaflets, and (5) the polar heads exposed to the bulk solution and belonging to the outer leaflet of the lipid bilayer. Each layer of the model is characterized by four parameters: (a) thickness, (b) scattering length density ρ, (c) percentage of water present in the layer, (d) roughness of its interface with the subsequent layer. The best fit parameters for all five layers in each of the four experiments presented here are summarized in Table 2.
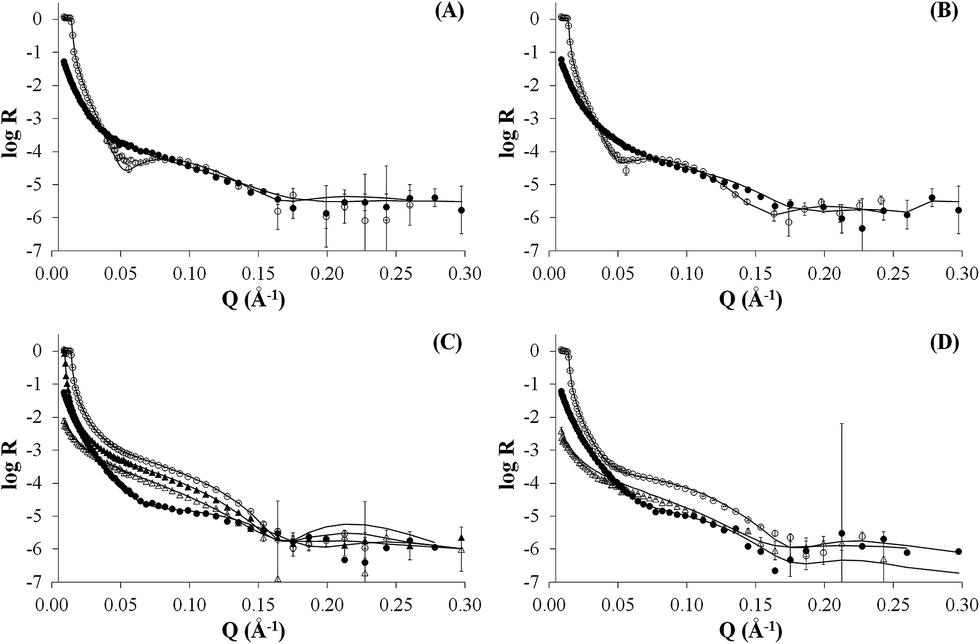 | ||
| Fig. 2 NR profiles of dPOPC bilayer (panel A), dPOPC and gH625-PrA-dendrimer (panel B), hPOPC and hChol 20% bilayer (panel C), hPOPC and hChol 20% bilayer and gH625-PrA-dendrimer (panel D). | ||
| Thickness (Å) | ρ × 10−6 (Å−2) | % water | Roughness (Å) | |
|---|---|---|---|---|
| dPOPC | ||||
| Silicon oxide | 7 ± 1 | 3.4 ± 0.1 | 0 ± 2 | 4 ± 1 |
| Water | 8 ± 1 | — | 100 | 6 ± 2 |
| Inner head | 7 ± 1 | 1.9 ± 0.1 | 15 ± 5 | 6 ± 2 |
| Tails | 26 ± 1 | 3.3 ± 0.1 | 5 ± 1 | 8 ± 2 |
| Outer head | 8 ± 1 | 1.9 ± 0.1 | 40 ± 12 | 7 ± 2 |
|
||||
| dPOPC + gH625-dendrimer | ||||
| Silicon oxide | 7 ± 1 | 3.5 ± 0.1 | 0 ± 2 | 4 ± 1 |
| Water | 5 ± 1 | — | 100 | 3 ± 2 |
| Inner head | 10 ± 1 | 1.90 ± 0.1 | 25 ± 8 | 5 ± 2 |
| Tails | 29 ± 1 | 3.1 ± 0.1 | 2.0 ± 0.2 | 7 ± 2 |
| Outer head | 8 ± 1 | 2.0 ± 0.1 | 40 ± 10 | 7 ± 2 |
|
||||
| hPOPC + hChol 20% | ||||
| Silicon oxide | 8 ± 1 | 3.4 ± 0.1 | 0 ± 2 | 2 ± 1 |
| Water | 4 ± 1 | — | 100 | 3 ± 2 |
| Inner head | 6 ± 1 | 1.7 ± 0.1 | 25 ± 8 | 2 ± 2 |
| Tails | 33 ± 1 | −0.11 ± 0.02 | 0 ± 1 | 3 ± 2 |
| Outer head | 6 ± 1 | 1.7 ± 0.1 | 25 ± 8 | 6 ± 2 |
|
||||
| hPOPC + hChol 20% + gH625-dendrimer | ||||
| Silicon oxide | 7 ± 1 | 3.4 ± 0.1 | 0 ± 2 | 3 ± 1 |
| Water | 4 ± 1 | — | 100 | 3 ± 2 |
| Inner head | 8 ± 1 | 2.0 ± 0.1 | 35 ± 10 | 4 ± 2 |
| Tails | 32 ± 1 | 0 ± 0.1 | 30 ± 5 | 5 ± 2 |
| Outer head | 8 ± 1 | 1.7 ± 0.1 | 60 ± 18 | 4 ± 2 |
The NR data measured for the bare d-POPC bilayer (Fig. 2A) yield the scattering length density profiles reported in Fig. 3A. In particular, the profile pertaining to H2O (lower curve in Fig. 3A) presents a central bump caused by the deuteration of one of the two hydrophobic chains composing the POPC molecule. This bump hides the headgroup peaks usually seen in fully hydrogenated POPC bilayers in H2O (lower curve in Fig. 3C) and makes them appear slightly thicker (7 ± 1 and 8 ± 1 Å) with respect to the fully hydrogenated case (6 ± 1 Å). Conversely, the thickness of the hydrophobic tail layer is slightly smaller (26 ± 1 Å) than the value usually found (28 ± 1 Å) in pure hydrogenated POPC bilayers.47 Although all these differences are essentially within error bars, this may suggest minor changes of the internal bilayer structure induced by the partial deuteration of POPC. However, the total fitted thickness of this d-POPC bilayer, calculated as the sum of the thicknesses of headgroup and hydrophobic tails model layers (41 Å), is in full agreement with molecular dynamics simulations of hydrogenated POPC bilayers, where the bilayer thickness is usually estimated as the average distance between the phosphorous atoms of the upper and lower bilayer leaflets.48 The ρ profile for the bare d-POPC bilayer in D2O (upper line in Fig. 3A) demonstrates that water can reach the region in between the silicon oxide surface and the headgroup of the inner bilayer leaflet, thus transforming a local minimum in the ρ profile into a local maximum. In addition, hydration of the hydrophilic headgroups by D2O makes the profile flatter in the central bilayer region. Most importantly, in this central region the two profiles in Fig. 3A never coincide, suggesting that some water can enter also in between the hydrophobic tails, as indicated by a non-zero value (5%) of water fraction in the fitted layer representing the hydrophobic chain region of the bilayer (Table 2). Finally, the solvation of this bare d-POPC bilayer appears to be asymmetrical, as demonstrated by a lower (15%) water fraction for the inner silicon-exposed headgroups with respect to the solution-exposed ones (40%). The above discussion indicates that the starting d-POPC bilayer in this experiment does not exhibit the highest possible structural quality. This may be due to partial-deuteration effects, preparation conditions for this specific sample, or combinations of both effects. A more perfect bilayer structure can be achieved using fully deuterated POPC,49 but these molecules are not commercially available yet. We therefore proceeded to study the interaction of this partially deuterated POPC bilayer with our peptide–dendrimer construct.
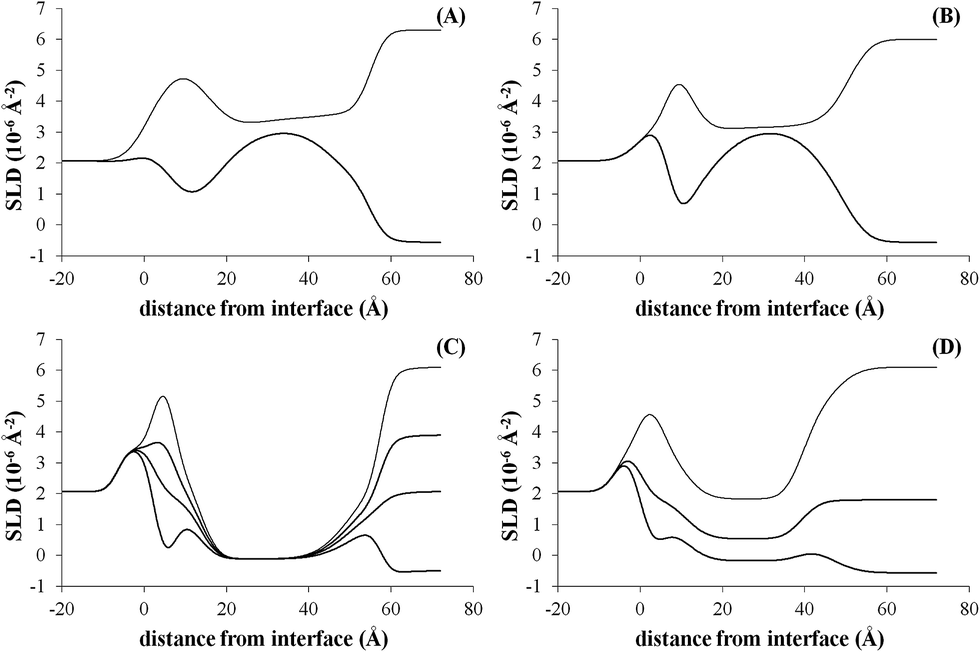 | ||
| Fig. 3 Scattering length density profiles corresponding to the reflectivity data of Fig. 2 (in the same order). | ||
The interaction between the d-POPC bilayer and the gH625-dendrimer does not produce dramatic changes in the measured NR data (Fig. 2B) and corresponding density profiles (Fig. 3B). The only qualitative changes that are easily observable in Fig. 3B are a sharper local dip/peak related to the silicon-oxide-adjacent water layer (its fitted thickness decreases from 8 to 5 Å) and a smaller separation between the profiles in the centre of the hydrophobic inner region of the bilayer, which translates into a tail solvation decrease from 5% to 2% (Table 2). Further inspection of the fitted parameters (Table 2) provides however useful insights. While the outer polar heads layer remains basically unchanged, the ρ value of the tails layer decreases from 3.3 × 10−6 to 3.1 × 10−6 Å−2, which indicates the presence of peptide–dendrimer molecules because their ρ value is estimated to be 1.29 × 10−6 Å−2 (see Experimental section). Most prominently, the tails layer thickness increases from 26 to 29 Å and the inner heads layer thickens from 7 to 10 Å, thus causing an overall bilayer thickness increase from 41 to 47 Å. Finally, the water content of the inner headgroups increases from 15% to 25%. These findings are all compatible with the deep penetration of the peptide–dendrimer construct inside the bilayer. The former would push towards the inner headgroups region some of the residual water found initially in the tails layer, thus increasing the inner heads solvation and thinning down (from 8 to 5 Å) the water layer intercalated between them and the silicon oxide surface. The above indications of a deep penetration of the peptide–dendrimer inside the lipid bilayer were cross-checked by investigating a more conventional fully hydrogenated POPC (hPOPC) bilayer containing also 20% of hydrogenated cholesterol (hChol). The latter is expected to further promote the interactions between membranotropic peptides and lipid bilayers.
The structural quality of the starting mixed hPOPC–hChol bilayer was assessed by measuring NR data (Fig. 2C) using four different water contrasts, as described in the Experimental section. All the resulting fitted density profiles (Fig. 3C) coincide in the central hydrophobic region of the bilayer, indicating that no water was present initially in between the lipid tails. Moreover, both headgroup layers are better defined (6 Å thickness) and the tails layer is considerably thicker (33 Å) with respect to the dPOPC case (26 Å). This determines an overall bilayer thickness increase from 41 Å (dPOPC case) to 45 Å, which is perfectly in line with the known bilayer thickening effects due to addition of hChol.47 Finally, the starting condition with respect to headgroup hydration is symmetrical in this bilayer (25% water for both inner and outer head layers), while in the dPOPC case the outer head layer was more solvated (40% water) than the inner one (15%). All these findings suggest that the starting sample for this second NR investigation is better suited than the dPOPC case for detecting interactions between the bilayer and the peptide–dendrimer construct.
Indeed when adding gH625-dendrimer to the hPOPC + hChol bilayer the changes in the NR data (Fig. 2D) and corresponding density profiles (Fig. 3D) are more pronounced than what found in the pure dPOPC case. This is well in line with the expectation that the presence of cholesterol further enhances the bilayer structural changes induced by its interaction with the peptide–dendrimer construct. The most striking difference that can be easily noticed in Fig. 3D is that the density profiles depart from each other when using different water contrasts, which is the signature of a dramatic bilayer structural alteration causing the increase of the water content in the tails layer from 0% to 30%. After addition of gH625-dendrimer, the water content in the inner head layer is even higher in the hPOPC + hChol bilayer (35%) than in the dPOPC case (25%). Also the outer heads become dramatically more solvated (from 25% to 60%) and the local peaks associated to both heads in the H2O contrast case (lowest curve in Fig. 3D) become visibly broader, as attested by their thickness increase from 6 to 8 Å. This translates into a total bilayer thickness increase from 45 to 48 Å after addition of the gH625-dendrimer. In view of the fact that the ρ value for the overall gH625-dendrimer molecule is estimated to be 1.29 × 10−6 Å−2 (see Experimental section), the ρ increase from −0.11 × 10−6 Å−2 to 0 is another indication of the presence of gH625-dendrimer in the tails region, while the increase of the inner heads ρ from 1.7 × 10−6 Å−2 to 2.0 × 10−6 Å−2 may indicate that the deuterated amino acids present in the peptides accumulate within the inner leaflet headgroups. This suggests that the peptides would transport the dendrimer completely across the bilayer if this process was not hindered by the presence of the hard-wall represented by the bilayer-supporting silicon block.
With the 2D detector present on the instrument and that allows to detect simultaneously specular and off-specular reflectivity during a single measurement, we did not observe off-specular signal suggesting the absence of micrometer scale inhomogeneities in the sample. Despite specular NR yields only laterally averaged surface-normal structural information, complementary investigations9 suggest that peptide–dendrimers do not cause poration in model liposomes. The NR data presented here therefore confirm that gH625-dendrimers enter POPC bilayers by altering their molecular-level structure both in the absence and presence of cholesterol. In the latter case in particular, the effects on the bilayer structure seem to be further enhanced. In both cases, NR data suggest that the peptide–dendrimer constructs may be able to fully cross the model membranes in the absence of the solid silicon support. These conclusions motivated the cell permeability studies discussed in the next section.
Cell permeability studies
Caco-2 cells, derived from a human colon adenocarcinoma and cultured on semipermeable membranes, differentiate into a highly functionalized epithelial barrier with significant morphological and biochemical similarity to small intestinal columnar epithelium.50 Fully differentiated cell monolayers can be used to determine membrane transport capabilities of novel molecules.We tested the ability of gH625 to enhance dendrimer crossing of a Caco-2 confluent monolayer (Fig. 4A). In particular, in this experiment we have determined the fraction of NBD-labelled compounds taken up into Caco-2 cells in order to probe the ability of gH625 to change the internalization of polyamidoamine (PAMAM) dendrimers. We incubated the compounds with Caco-2 cells at 37 °C at different peptide concentrations (20, 25 μM) for 30, 60 and 120 min. Data reported in Fig. 4B show that gH625-dendrimer crosses more efficiently the layer compared to the un-functionalized dendrimer.
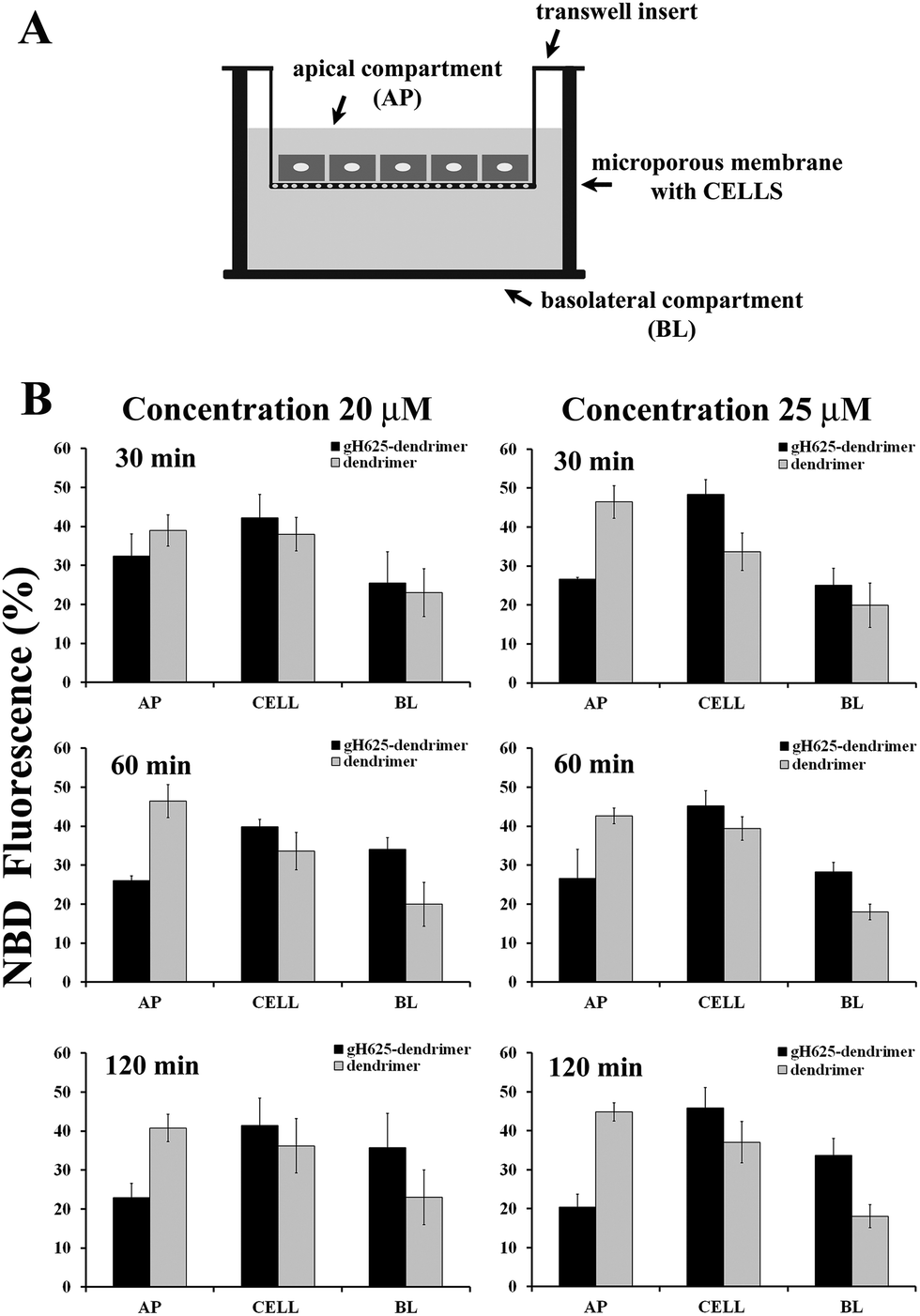 | ||
| Fig. 4 Permeability studies performed on Caco-2 cell monolayers (panel A). Apical (AP), basal (BL) and cellular (CELL) content were measured following NBD fluorescence and reported as percentages for the peptide–dendrimer and for the dendrimer (panel B). P-Value ≤ 0.05. | ||
More precisely, the permeability (P) of the monolayer calculated at 25 μM and 120 min was 8.6 (±0.3) × 10−5 cm s−1 for gH625-dendrimer, approximatively two fold higher than the P value of 4.5 (±0.4) × 10−5 cm s−1 found for the unfunctionalized dendrimer. Moreover, no changes in control permeability were observed after exposure to our compounds, indicating that they did not perturb the integrity of the cell monolayer in the experimental conditions used for the assay (data not shown).
We isolated the AP, BL and CELL content of dendrimer and peptide–dendrimer and quantified the content by fluorescence spectroscopy (Fig. 4B). To better understand the kinetics of internalization, we incubated the compounds with cells at 37 °C for various times (30, 60, 120 min) and then determined the localization of the compounds in the AP, BL and CELL compartments. Fig. 4B shows the fraction of compounds taken up by cells at different time points. The analysis of the obtained results clearly indicates that when the dendrimer is functionalized with the peptide, the percentage of compound in the BL and CELL compartments is higher. At 30 min, we observed that a high percentage of the peptide–dendrimer is located inside the CELL; at 1 h we observe an increase of the percentage located in the BL compartment while the quantity in the AP decreases significantly. In detail, we observed that when we compare the data obtained for the peptide–dendrimer and the dendrimer alone, the percentage of peptide–dendrimer located in the CELL or BL compartments is always greater than that of the dendrimer. This result clearly indicates that the peptide is enhancing the internalization of the dendrimer.
Experimental
Materials
Fmoc-protected amino acids, coupling reagents N-hydroxybenzotriazole (HOBT) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) and rink-amide p-methylbenzhydrylamine MBHA resin (0.54 mmol g−1) were purchased from Novabiochem. Fmoc-L-propargylglycine (Fmoc-PrA-OH) was purchased from polypeptide, 4-chloro-7-nitro-1,2,3-benzoxadiazole (NBD-chloride) from Sigma-Aldrich and L-alanine-N-Fmoc (3,3,3-d3 98%) from Cambridge Isotope Laboratories. All other reagents and chemicals for the solid-phase peptide synthesis (piperidine, N,N-diisopropylethylamine (DIPEA), HPLC-grade acetonitrile (ACN), trifluoroacetic acid (TFA) and dry N,N-dimethylformamide (DMF) were from Romil Delchimica and Sigma-Aldrich).The lipids, hydrogenated and deuterated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (h-POPC and d-POPC) and cholesterol, were purchased from Avanti Polar Lipids.
Analytical and preparative reverse-phase high-performance liquid chromatography (HPLC) was performed on a Shimadzu LC8 pump setup using Phenomenex Jupiter C4 5 μm 300 Å 4.6 by 150 mm and Phenomenex Jupiter C4 10 μm 300 Å 21.20 by 250 mm columns.
Liquid chromatography-mass spectrometry (HPLC-MS) analyses of the peptide were performed on a Thermo Finnigan LC-MS with an electrospray source (DECA) on a Phenomenex Jupiter 4 μm Proteo C12 90 Å 150 by 4.6 mm column. Transwell permeable supports 6.5 mm inserts, 24 well plates, 0.4 μm polyester membrane were purchased from Corning. D2O (99.7% purity) was provided by the Institut Laue-Langevin (ILL).
Peptide synthesis
Two peptides were synthesized. The peptide NBD-gH625-PrA was used to perform the experiments on transwell permeable supports while the same peptide without the NBD group, gH625-PrA, was used for NR measurements (Table 1).Furthermore, in gH625-PrA the alanine residues were deuterated. Both peptides were synthesized on a rink amide MBHA (0.54 mmol g−1) in DMF using the standard solid phase method.12 Briefly, the amide MBHA resin was deprotected (30% v/v piperidine in DMF, twice for 10 min) to remove 9-fluorenylmethoxy carbonyl (Fmoc) group, then the first Fmoc protected amino acid (Fmoc-PrA-OH, 4 equivalents) was double coupled with 4 equivalents of 0.45 M HOBT/HBTU and 8 equivalents of 2 M DIPEA (30 min for each coupling). The synthesis was performed alternating steps of Fmoc deprotection and double coupling of the amino acids. For NBD-gH625-PrA, after the whole synthesis, before the cleavage, NBD was bound at the N-terminus (4 eq. in DMF with 1% v/v DIPEA). The peptides were cleaved from the resin and deprotected by treatment with TFA/thioanisole/anisole/water/EDT 82.5/5/5/5/2.5, precipitated in ice-cold ethylic ether and freeze-dried. The peptides were analysed by electrospray ionization (ESI) LC-MS using a linear gradient of acetonitrile (0.1% TFA) in water (0.1% TFA) from 20 to 80% and purified by preparative reversed-phase high-performance liquid chromatography (RP-HPLC) using a gradient of acetonitrile (0.1% TFA) in water (0.1% TFA) from 20 to 80% in 25 min.
Synthesis of the octadecaazide dendrimer
The Newkome-style dendron was coupled to the hemisuccinate dendron by using 2-[7-aza-1H-benzotriazol-1-yl]-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) to obtain a symmetrical dendrimer. The tert-butyl groups were removed using an acidic wash to yield the carboxylic group that can be reacted with azide-terminated amine molecules. Details can be found in the literature.43 The final molecule is an octadecaazide dendrimer.Peptide–dendrimer preparation
The peptides of choice were attached to the dendrimer via the copper-catalyzed azide–alkyne cycloaddition (CuAAC) click reaction between the azide and the alkyne groups, obtaining the NBD-gH625-PrA-dendrimer for the experiments on transwell permeable supports or the gH625-PrA-dendrimer for the neutron reflectivity studies. In detail, aqueous solutions of NBD-gH625-PrA (200 μL, 21.6 equiv.), CuSO4·5H2O (50 μL, 82.8 equiv.), and sodium ascorbate (40 μL, 120.6 equiv.) were added to the dendrimer (290 μL, 1 equiv., corresponding to 18 functional groups) in methanol and the obtained mixture was stirred for one hour at 40 °C and for two days at RT. The peptide–dendrimer was purified by ultrafiltration in water:methanol:DMSO 50:48:2 versus 30000 molecular weight cut-off membranes, followed by reverse phase HPLC on a C4 column with water (0.1% TFA) and acetonitrile (0.1% TFA) from 30 to 95% over 20 min at 5 mL min−1. The functionalization of the gH625-dendrimer was confirmed by determining the amount of peptide attached via UV analysis (εgH625 = 6990 M−1 cm−1 at λ = 280 nm) and comparing the result to the initial dendrimer and peptide feed ratio. The yield of peptide functionalization was determined to be 71%. Further structural characterization to confirm the functionalization were performed as previously reported.20
Neutron reflectivity
For NR experiments, supported lipid bilayers were prepared by vesicle fusion on a solid support in a flow cell. The solid supports were 8 × 5 × 1 cm3 silicon (111) single crystals, cleaned with organic waters and UV/ozone to make the surface hydrophilic.51 Small Unilamellar Vesicles (SUVs) 0.5 mg mL−1, of 25–35 nm in diameter, were prepared from the multilamellar vesicle (MLV) suspension by 10 min vortexing and sonication. The lipid composition was POPC and POPC/Chol 80/20. The SUVs were injected into the NR flow cell and allowed to diffuse and adsorb on the silicon surfaces over a period of 30 min. After lipid the vesicle adsorption and fusion the sample cell was rinsed with appropriate buffer to remove excess lipids. Neutron reflectometry measurements were performed on the formed lipid bilayer. After the measurements meant to characterise the bilayer alone, a solution of gH625-PrA-dendrimer was injected, the flow cell was rinsed in order to remove the extra material which had not interacted with the lipids, and other measurements were carried to study the effect of the presence of peptide–dendrimer. Neutron reflectivity measurements were performed on the FIGARO reflectometer52 at the high flux reactor of the ILL, in time-of-flight mode using a spread of wavelengths, λ, between 2 and 30 Å at two incoming angles, θ, of 0.8° and 3°.In a neutron reflectivity experiment, the specular reflection, R, at the silicon/water interface, is measured as a function of the scattering vector modulus q = 4π/λsinθ, and is related to the scattering length density across the interface, ρ(z), by the equation53
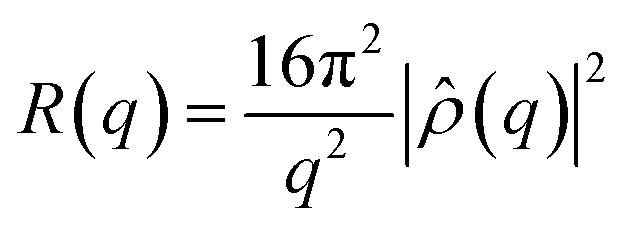
![[small rho, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b7.gif) (q) is the one dimensional Fourier transform of ρ(z), equal to:
(q) is the one dimensional Fourier transform of ρ(z), equal to: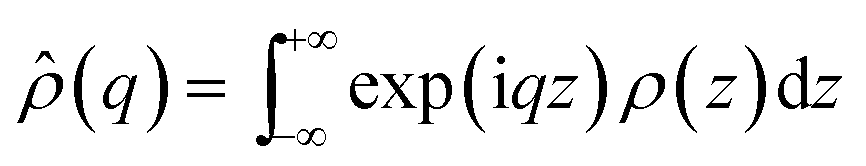
And ρ(z) is a function of the distance perpendicular to the interface and related to the composition of the adsorbed species by the equation
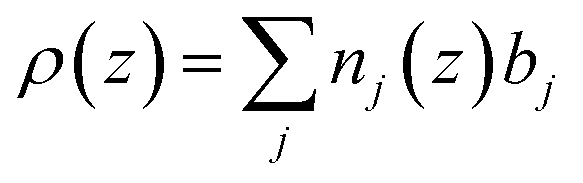
The scattering length densities of the molecules adsorbed at the surface give access to the structural information on the lipid bilayer at the nanometer level and therefore to its interfacial properties.
Measurements of a sample in different water contrasts greatly enhance the sensitivity of the method and the reliability of the results. Samples were measured at 25 °C using four water contrasts which were H2O (ρ = −0.56 × 10−6 Å−2), D2O (ρ = 6.35 × 10−6 Å−2), silicon matched water (SMW, a mixture of 38% v/v D2O and 62% H2O; ρ = 2.07 × 10−6 Å−2), and 4 matched water (4MW, 66% D2O and 34% H2O; ρ = 4 × 10−6 Å−2). The scattering length densities used for the lipids and cholesterol are as previously reported;54 that of the gH625-dendrimer was evaluated from the atom composition and volume (https://www.ncnr.nist.gov/resources/activation) to be 1.29 × 10−6 Å−2.
Neutron reflectivity profiles were analysed by using the MotoFIT piece of software.55 It allows the co-refinement of reflectivity data from the same sample in different water contrasts by defining models consisting of a series of layers. The four variable model fitting parameters related to each layer are its thickness, scattering length density, water volume fraction and interfacial roughness. Although more than one model can describe a given experimental curve, prior knowledge of the system, the definition of physically meaningful upper and lower limits for the parameters to be optimised and the use of different isotopic contrasts greatly reduce the number of possible models.
Cell cultures
Caco-2 cells (ATCC HTB-37, Virginia, US) were maintained in Dulbecco's Modified Eagle Medium (DMEM) (Sigma-Aldrich, St. Louis, US) containing 10% FBS (HyClone, Logan, US), 1% nonessential amino acids, and 2 mM L-glutamine freshly prepared. Cells were cultured at 37 °C in an atmosphere of 5% CO2 and 95% relative humidity; and were passaged at 80–90% confluence (every 3–4 days) using trypsin–EDTA solution. Culture inserts were preincubated with culture medium (1 h, 37 °C) and then seeded with 60000 cells per cm2 (0.33 cm2 per insert) on transwell permeable inserts (6.5 mm in diameter, 0.4 μm pores size; Corning Incorporated, Corning, NY, US). 100 μL and 600 μL complemented DMEM were applied in the upper and bottom part of the transwell respectively as reported in the manufacture date sheet. Caco-2 monolayers were fed with fresh medium 24 h after seeding and then 3 times per week. Caco-2 monolayers were cultured for 15 days before use. Caco-2 cells were used at passage number 25, after receipt at passage 18 from ATCC. 20 hours after feeding, monolayers were washed with PBS 1× (Euroclone #ECB4004L). The degree of monolayer confluence and tight junction development was 100%.
Permeability assays
Permeabilization assay donor solutions consisted of 20, and 25 μM peptide, dendrimer and peptide–dendrimer in transport medium containing 100 μM lucifer yellow (molecular probes) and 1% DMSO (pH 7.4). 1% DMSO was used to enhance solubility of our compounds and was demonstrated to have no effect on Caco-2 permeability (data not shown). In every test, we used lucifer yellow, a fluorescent marker for the paracellular pathway, as an internal control to confirm tight junction integrity during the assay; in fact, changes in tight junctions may significantly influence permeability results. For transport assays we used 100 μL of apical (AP) donor solution and 600 μL of basolateral (BL) acceptor solution (transport medium, pH 7.4). All experiments were performed in three replicate monolayers and statistical filter was applied. The p values of t-test was <0.05 (n = 3). Monolayers were incubated with donor and acceptor solutions at 37 °C for 30, 60 and 120 min; then BL compartments, AP compartments and cell lysate (CELL) were sampled.Compounds were quantified exploiting the fluorescence of the NBD group (λex = 460 nm; λem = 520 nm). In detail the BL, AP and CELL samples were dried and then resuspended in 200 μL of water. The fluorescence was determined. No fluorescence interference was present.
The permeability P of the monolayer was calculated according to the following equation,
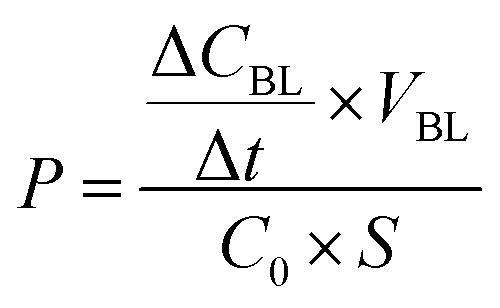
Conclusions
Dendrimers are well-defined highly branched structures which are continuously focusing much biomedical research interest for applications in drug delivery. The internalization mechanism has straight impacts on the design of drug delivery, cell transfection and gene therapy agents and in fact many reports in order to obtain a better understanding of the uptake mechanism and to increase the targeting efficiency investigate the interactions between biological membranes and dendrimers. The ideal drug delivery vehicle should be able to cross membranes without causing leakage. Data reported in the literature, corroborate the idea that the type and strength of the interaction is dependent on charge and size of the molecule. Data indicate that dendrimers either create holes in a bilayer or can be incorporated into lipid structures. In this work, we try to dissect the mechanism of interaction with membranes of a dendrimer with the external termini modified by a peptide (gH625), which has previously proved to enhance cell membrane crossing.20,43 Moreover, previous characterization of the peptide gH625 indicated that it is able to establish a strong interaction with large unilamellar vesicles (LUVs) without the formation of pores inside the bilayer. The ability to interact with membranes without the formation of pores has been considered a mean to avoid/reduce toxicity problems.11–15 We have previously shown that the peptide–dendrimer is more effective than the peptide alone at interacting and fusing with lipid membranes, demonstrating its effectiveness as a membrane-perturbing molecule and its ability to translocate across the bilayer essentially without involving the endocytic mechanism.20,43Previous results from complementary techniques such as fluorescence spectroscopy, surface plasmon resonance, isothermal calorimetry, and confocal microscopy, indicated that also the gH625-dendrimer has a high affinity for the membrane bilayer and is deeply inserted inside the bilayer.20,43 The peptide–dendrimer is able to cause membrane fusion but not holes in the bilayer, differently from the most commonly used cell penetrating peptides (such as the HIV derived TAT peptide) and from PAMAM dendrimers, which do not translocate spontaneously across bilayers but rather are taken up by cells via endocytosis.
The neutron reflectivity data allowed to obtain further information on the mechanism of uptake. We were able to better localize the peptide–dendrimer inside bilayers of different compositions and to compare the results with the dendrimer alone case. In particular, the gH625-dendrimer interacts with both lipid compositions (POPC and POPC/Chol), although the interaction is stronger with cholesterol containing liposomes. In the presence of cholesterol, the compound can penetrate deeply. From neutron reflectivity data, we can hypothesize that the peptide–dendrimer is able to pass through the lipid bilayer; but, these measurements do not allow to obtain a clear answer because the lipid bilayers are adsorbed directly on the surface of a solid substrate representing a kind of obstacle for the complete crossing.
With the aim to investigate in greater details the translocation of the peptide–dendrimer, we measured the permeability of a monolayer of Caco-2 cells with transwell inserts. This experiment allowed us to discriminate between the unbound compounds, and those able to internalize into cells or to translocate on the other side of the monolayer. The advances in our understanding of gH625-dendrimer mechanism of uptake are extremely useful to push further the design of new drug delivery systems that consist of dendrimers incorporating bioactive molecules.
Acknowledgements
The authors thank Luca De Luca for excellent technical assistance, the ILL for provision of beam-time and sample preparation facilities within the Partnership for Soft Condensed Matter (PSCM). (expt #8-02-678 “Membranotropic peptides: mechanism of membrane interaction for biological applications”).References
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- V. P. Torchilin, in Annual Review of Biomedical Engineering, Annual Reviews, Palo Alto, 2006, vol. 8, pp. 343–375 Search PubMed.
- V. Sanna, N. Pala and M. Sechi, Int. J. Nanomed., 2014, 9, 467–483 CAS.
- R. M. Pearson, S. Sunoqrot, H. J. Hsu, J. W. Bae and S. Hong, Ther. Delivery, 2012, 3, 941–959 CrossRef CAS.
- F. P. Seib, A. T. Jones and R. Duncan, J. Controlled Release, 2007, 117, 291–300 CrossRef CAS PubMed.
- A. Saovapakhiran, A. D'Emanuele, D. Attwood and J. Penny, Bioconjugate Chem., 2009, 20, 693–701 CrossRef CAS PubMed.
- R. Tarallo, A. Accardo, A. Falanga, D. Guarnieri, G. Vitiello, P. Netti, G. D'Errico, G. Morelli and S. Galdiero, Chem.–Eur. J., 2011, 17, 12659–12668 CrossRef CAS PubMed.
- A. Falanga, M. Galdiero and S. Galdiero, Int. J. Mol. Sci., 2015, 16, 25323–25337 CrossRef CAS PubMed.
- A. Falanga, R. Tarallo, G. Vitiello, M. Vitiello, E. Perillo, M. Cantisani, G. D'Errico, M. Galdiero and S. Galdiero, PLoS One, 2012, 7, e32186 CAS.
- A. Falanga, M. Vitiello, M. Cantisani, R. Tarallo, D. Guarnieri, E. Mignogna, P. Netti, C. Pedone, M. Galdiero and S. Galdiero, Nanomedicine, 2011, 7, 925–934 CAS.
- S. Galdiero, A. Falanga, G. Vitiello, M. Vitiello, C. Pedone, G. D'Errico and M. Galdiero, Biochim. Biophys. Acta, 2010, 1798, 579–591 CrossRef CAS PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, H. Browne, C. Pedone and M. Galdiero, J. Biol. Chem., 2005, 280, 28632–28643 CrossRef CAS PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, L. Raiola, R. Fattorusso, H. Browne, C. Pedone, C. Isernia and M. Galdiero, J. Biol. Chem., 2008, 283, 29993–30009 CrossRef CAS PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, L. Raiola, L. Russo, C. Pedone, C. Isernia and M. Galdiero, J. Biol. Chem., 2010, 285, 17123–17136 CrossRef CAS PubMed.
- S. Galdiero, L. Russo, A. Falanga, M. Cantisani, M. Vitiello, R. Fattorusso, G. Malgieri, M. Galdiero and C. Isernia, Biochemistry, 2012, 51, 3121–3128 CrossRef CAS PubMed.
- S. Galdiero, A. Falanga, G. Morelli and M. Galdiero, Biochim. Biophys. Acta, 2015, 1848, 16–25 CrossRef CAS PubMed.
- D. Guarnieri, A. Falanga, O. Muscetti, R. Tarallo, S. Fusco, M. Galdiero, S. Galdiero and P. A. Netti, Small, 2013, 9, 853–862 CrossRef CAS PubMed.
- E. Perillo, E. Allard-Vannier, A. Falanga, P. Stiuso, M. T. Vitiello, M. Galdiero, S. Galdiero and I. Chourpa, Int. J. Pharm., 2015, 488, 59–66 CrossRef CAS PubMed.
- S. Valiante, A. Falanga, L. Cigliano, G. Iachetta, R. A. Busiello, V. La Marca, M. Galdiero, A. Lombardi and S. Galdiero, Int. J. Nanomed., 2015, 10, 1885–1898 CAS.
- A. Falanga, R. Tarallo, T. Carberry, M. Galdiero, M. Weck and S. Galdiero, PLoS One, 2014, 9, e112128 Search PubMed.
- J. C. Roberts, M. K. Bhalgat and R. T. Zera, J. Biomed. Mater. Res., 1996, 30, 53–65 CrossRef CAS PubMed.
- C. Ornelas, R. Pennell, L. F. Liebes and M. Weck, Org. Lett., 2011, 13, 967–979 CrossRef PubMed.
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 10, 975–999 CrossRef CAS PubMed.
- J. D. Castile, K. M. G. Taylor and G. Buckton, Int. J. Pharm., 1999, 182, 101–110 CrossRef CAS PubMed.
- G. Purohit, T. Sakthivel and A. T. Florence, Int. J. Pharm., 2001, 214, 71–76 CrossRef CAS PubMed.
- S. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M. M. Kober, M. T. Islam, B. G. Orr, J. R. Baker Jr and M. M. Banaszak Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS PubMed.
- A. Mecke, S. Uppuluri, T. M. Sassanella, D. K. Lee, A. Ramamoorthy, J. R. Baker Jr, B. G. Orr and M. M. Banaszak Holl, Chem. Phys. Lipids, 2004, 132, 3–14 CrossRef CAS PubMed.
- S. Hong, A. U. Bielinska, A. Mecke, B. Keszler, J. L. Beals, X. Shi, L. Balogh, B. G. Orr, J. R. Baker Jr and M. M. Banaszak Holl, Bioconjugate Chem., 2004, 15, 774–782 CrossRef CAS PubMed.
- A. Mecke, D. K. Lee, A. Ramamoorthy, B. G. Orr and M. M. B. Holl, Langmuir, 2005, 21, 8588–8590 CrossRef CAS PubMed.
- A. Mecke, I. J. Majoros, A. K. Patri, J. R. Baker Jr, M. M. Banaszak Holl and B. G. Orr, Langmuir, 2005, 21, 10348–10354 CrossRef CAS PubMed.
- B. Klajnert and R. M. Epand, Int. J. Pharm., 2005, 305, 154–166 CrossRef CAS PubMed.
- B. Klajnert, J. Janiszewska, Z. Urbanczyk-Lipkowska, M. Bryszewska and R. M. Epand, Int. J. Pharm., 2006, 327, 145–152 CrossRef CAS PubMed.
- K. Gardikis, S. Hatziantoniou, K. Viras, M. Wagner and C. Demetzos, Int. J. Pharm., 2006, 318, 118–123 CrossRef CAS PubMed.
- M. Ionov, Z. Garaiova, D. Wróbel, I. Waczulikova, R. Gomez-Ramirez, J. De La Mata, B. Klajnert, M. Bryszewska and T. Hianik, Acta Phys. Univ. Comenianae, 2011, 52, 33–39 CAS.
- D. Wrobel, A. Kłys, M. Ionov, P. Vitovic, I. Waczulikowa, T. Hianik, R. Gomez-Ramirez, J. de la Mata, B. Klajnert and M. Bryszewska, Chem. Phys. Lipids, 2012, 165, 401–407 CrossRef CAS PubMed.
- H. Lee and R. G. Larson, J. Phys. Chem. B, 2008, 112, 7778–7784 CrossRef CAS PubMed.
- P. E. S. Smith, J. R. Brender, U. H. N. Dürr, J. Xu, D. G. Mullen, M. M. Banaszak Holl and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 8087–8097 CrossRef CAS PubMed.
- C. V. Kelly, M. G. Liroff, L. D. Triplett, P. R. Leroueil, D. G. Mullen, J. M. Wallace, S. Meshinchi, J. R. Baker, B. G. Orr and M. M. B. Holl, ACS Nano, 2009, 3, 1886–1896 CrossRef CAS PubMed.
- V. Tiriveedhi, K. M. Kitchens, K. J. Nevels, H. Ghandehari and P. Butko, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 209–218 CrossRef CAS PubMed.
- M. F. Ottaviani, R. Daddi, M. Brustolon, N. J. Turro and D. A. Tomalia, Langmuir, 1999, 15, 1973–1980 CrossRef CAS.
- M. F. Ottaviani, P. Matteini, M. Brustolon, N. J. Turro, S. Jockusch and D. A. Tomalia, J. Phys. Chem. B, 1998, 102, 6029–6039 CrossRef CAS.
- D. Wrobel, M. Ionov, K. Gardikis, C. Demetzos, J. P. Majoral, B. Palecz, B. Klajnert and M. Bryszewska, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2011, 1811, 221–226 CrossRef CAS PubMed.
- T. P. Carberry, R. Tarallo, A. Falanga, E. Finamore, M. Galdiero, M. Weck and S. Galdiero, Chemistry, 2012, 18, 13678–13685 CrossRef CAS PubMed.
- B. Salgin, D. Vogel, D. Pontoni, H. Schroder, B. Schonberger, M. Stratmann, H. Reichert and M. Rohwerder, J. Synchrotron Radiat., 2012, 19, 48–53 CrossRef PubMed.
- B. Salgin, D. Pontoni, D. Vogel, H. Schroder, P. Keil, M. Stratmann, H. Reichert and M. Rohwerder, Phys. Chem. Chem. Phys., 2014, 16, 22255–22261 RSC.
- S. K. Ghosh, B. Salgin, D. Pontoni, T. Reusch, P. Keil, D. Vogel, M. Rohwerder, H. Reichert and T. Salditt, Langmuir, 2013, 29, 815–824 CrossRef CAS PubMed.
- G. Vitiello, G. Fragneto, A. A. Petruk, A. Falanga, S. Galdiero, A. M. D'Ursi, A. Merlino and G. D'Errico, Soft Matter, 2013, 9, 6442–6456 RSC.
- A. Merlino, G. Vitiello, M. Grimaldi, F. Sica, E. Busi, R. Basosi, A. M. D'Ursi, G. Fragneto, L. Paduano and G. D'Errico, J. Phys. Chem. B, 2012, 116, 401–412 CrossRef CAS PubMed.
- N. R. Yepuri, T. A. Darwish, A. M. Krause-Heuer, A. E. Leung, R. Delhom, H. P. Wacklin and P. J. Holden, ChemPlusChem, 2016, 81, 315–321 CrossRef CAS.
- J. D. Irvine, L. Takahashi, K. Lockhart, J. Cheong, J. W. Tolan, H. E. Selick and J. R. Grove, J. Pharm. Sci., 1999, 88, 28–33 CrossRef CAS PubMed.
- J. R. Vig, J. Vac. Sci. Technol., A, 1985, 3, 1027–1034 CAS.
- R. A. Campbell, H. P. Wacklin, I. Sutton, R. Cubitt and G. Fragneto, Eur. Phys. J. Plus, 2011, 126, 1–22 Search PubMed.
- J. B. Hayter, R. R. Highfield, B. J. Pullman, R. K. Thomas, A. I. McMullen and J. Penfold, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1437–1448 RSC.
- G. Vitiello, A. Falanga, A. A. Petruk, A. Merlino, G. Fragneto, L. Paduano, S. Galdiero and G. D'Errico, Soft Matter, 2015, 11, 3003–3016 RSC.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |