
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ preparation of oxygen-deficient TiO2 microspheres with modified {001} facets for enhanced photocatalytic activity†
Xiaogang Liuab and
Yingpu Bi *a
*a
aState Key Laboratory for Oxo Synthesis & Selective Oxidation, National Engineering Research Center for Fine Petrochemical Intermediates, Lanzhou Institute of Chemical Physics, CAS, Lanzhou 730000, China. E-mail: yingpubi@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 3rd February 2017
Abstract
A facile in situ synthetic strategy has been developed to prepare highly active oxygen-deficient anatase TiO2 microsphere single crystals with modified {001} facets by simply controlling the hydrothermal reaction time. More importantly, on investigating the performance of photocatalytic hydrogen evolution, TiO2 microspheres consisting of micro-decahedrons with etched {001} facets show superior photoreactivity compared with TiO2 microspheres wrapped with intact-{001} faceted anatase crystals. As determined by electron spin resonance (ESR) measurement, short hydrothermal reaction time facilitates the formation of oxygen vacancies, whereas longer hydrothermal reaction time contributes to the decrease of oxygen vacancies and formation of subsurface Ti3+ defect states. Based on the PL emission spectra obtained at different excited wavelengths, a possible mechanism of charge separation and transport resulting from changing of bulk/surface trapping sites on the building blocks of TiO2 microspheres is proposed, and the enhanced photocatalytic efficiency of the as-prepared TiO2 microsphere is largely attributed to the efficient separation and transport of photogenerated charge carriers caused by rational surface modulation and modification.
Titanium dioxide (TiO2) is well known as one of the most widely used materials in the scope of photocatalysis and solar energy conversion.1–3 In particular, TiO2 crystals with tailored facets have been one of the most interesting research topics among those of various metal oxides in the past few years.4–9 Triggered by Lu's first study of the preparation of TiO2 crystals enclosed by 47% {001} facets,3 great attempts have been made to obtain a higher exposure percentage area than the initial value of 47%. To date, the percentage area of {001} facets has already increased up to nearly 100%.10 In spite of the promising properties associated with {001} facets, such as dissociative adsorption of water molecules and unique anchoring of dye molecules, its synergistic effect with other facets is not quite clear and remains controversial. For example, Han11 et al. showed that the photodegradation percentage of methylene blue increases from 46% to 59%, 73%, and 98% with increasing percentage area of anatase {001} facets from nearly 0% to 5%, 20%, and 60%. This means that the anatase (001) surface is indeed more reactive in decomposing organic molecules than that of anatase (101). Some other researchers, however, hold the opposite viewpoint. One remarkable example reported by Pan12 et al. was that the micrometer-sized anatase crystals with 24% {001} and 76% {101} facet areas exhibit higher activities in both hydrogen and OH radical generation than those with 40% {001} and 60% {101} facet areas, and crystals with 14% {001}, 33% {101} and 53% {010} facet areas have the highest activities. Recently, Yu13 et al. proposed a new “surface heterojunction” concept to explain the differences in the photocatalytic activity of TiO2 with co-exposed {001} and {101} facets, finding that an optimal ratio of the exposed {101} and {001} facets leads to the highest photocatalytic activity toward reduction of CO2 to CH4.
Tremendous efforts have been put towards facet engineering, and extensive studies have also been devoted to investigate spherical titania structures, including mesoporous spheres,14 spherical flaky assemblies,15 and dendritic particles that are monodisperse and of variable diameter,16 due to their realized and potential applications in the areas of chromatographic separation,17 lithium-ion batteries,18 dye-sensitized solar cells,19 and photocatalytic water splitting.20 For example, Chen et al. synthesized hierarchical anatase microspheres assembled from the high surface area nanosheets with nearly 100% {101} facets.21 Micrometer-sized anatase spheres with variable pore sizes have shown excellent performance as packing materials for chromatographic separation and selective phosphopeptide enrichment, and thus provide instructive strategies to prepare more novel and efficient spherical-based titania photocatalysts.
Recently, reduced TiO2 containing the Ti3+ or oxygen vacancy has been demonstrated to extend the light absorption spectrum and enhance the electron mobility as well as electron–hole separation efficiency.22–24 Different strategies, such as H2/Ar treatment25 and laser irradiation,26 as well as metal reduction methods using Al,27 Zn28 and Mg,29 were used to prepare Ti3+-doped or oxygen-vacancy-rich TiO2. Although progress has been achieved using these methods, they are rather time-consuming, not environmentally friendly and are energy wasteful. Exploring more mild and cost-effective preparation methods to achieve oxygen-deficient TiO2 without introducing any impurity element is therefore highly desired and necessary.
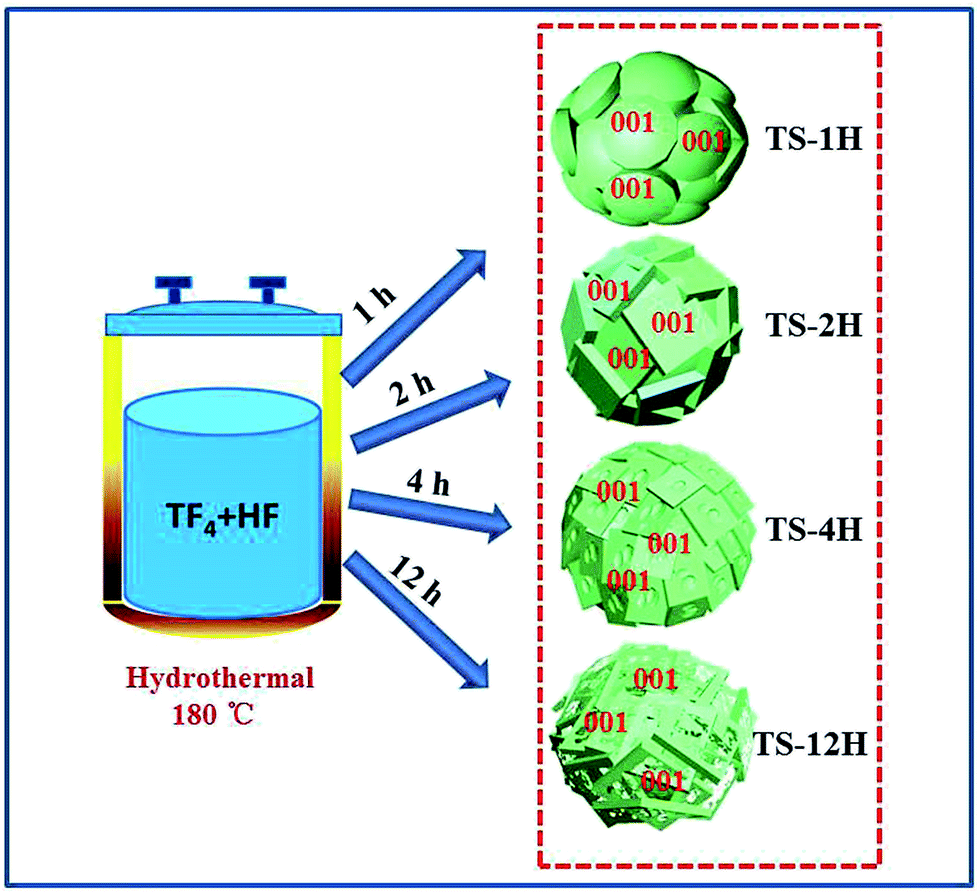
Herein, we demonstrate a facile method for fabricating Ti3+-doped anatase TiO2 microspheres (denoted TS-xH, where x represents the hydrothermal reaction time) consisting of etched- or intact-{001} faceted truncated bipyramidal TiO2 (the building units of microspheres are referred as etched- and intact-TiO2). Approximately 3.6 and 8 times higher hydrogen evolution rates (255 and 92.95 μmol h−1 for microspherical TiO2 consisted of etched- and intact-(001) faceted TiO2 anatase crystals, respectively) than that of their building blocks (69.25 μmol h−1 and 11.4 μmol h−1 for etched- and intact-TiO2, respectively) have been achieved under the same conditions. The unique microspherical structures based on specific facet moderation play a key role in the transfer processes of photo-excited charge carriers and therefore their photocatalytic activities. These findings may provide a new strategy for designing more efficient TiO2-based photocatalysts for solar energy conversion.
Fig. 1 shows typical scanning electron microscope (SEM) images of the prepared TiO2 microsphere single crystals synthesized under different hydrothermal times. As can be seen from Fig. 1A, the TS-1H obtained from a reaction medium containing 5 wt% HF and 0.04 M TiF4 under 1 h has already shown spherical morphology, indicating that the formation rate of spherical crystals is quite rapid. By prolonging the reaction time to 2 h, the outer exposed surface of TS gradually exhibits squared crystal facets consisting of well-faceted truncated octahedra (Fig. 1B), in which the two square surfaces and the other eight isosceles triangular, surfaces have been confirmed to be {001} and {101} facets, respectively.30 Furthermore, the percentage of exposed {001} facets is calculated to be 60.69% (the detailed measuring method is presented in the ESI and eqn (1)†). Moreover, all of the crystal surfaces are extremely smooth without any trace of corrosion. However, the TS-4H sample partially preserves the growth of the {001} facet along with slight corrosion phenomenon observed in the center of the {001} facet (Fig. 1C), which can be attributed to the dual role of HF for controlling crystal facet growth of TiO2.31,32 Furthermore, the facial etching gradually intensifies as the reaction time increases from 4 h to 12 h, which can be observed from the SEM images shown in Fig. 1D and E. In addition, as can be seen in Fig. 1F, TS-12 h exhibits very uniform etching activity on the {001} facet with several newly-formed tiny pits on the outer exposed surface. In order to observe the TiO2 microspheres more clearly, the transmission electron microscope (TEM) was further used. As shown in Fig. S1A, 2A and 3A,† the average diameter of the prepared TiO2 microspheres is ca. 2 μm. The high-resolution (TEM) images (Fig. S1B, C, S2B, S3B and C†) show a distinct lattice spacing of 0.19 nm and 0.35 nm, which can be ascribed to the (010) and (101) atomic planes of anatase TiO2, respectively.
 | ||
| Fig. 1 SEM images of the prepared anatase TiO2 microspheres at different hydrothermal reaction times. (A) 1 h, (B) 2 h, (C) 4 h, (D) 6 h, (E) 9 h, (F) 12 h. | ||
The dual role of HF in controlling crystal facet growth has been experimentally demonstrated and theoretically confirmed. HF stabilizes the growth of {001} facets at low concentration but selectively destroys the growth of {001} facets at high concentration. As for the prepared TiO2 microspheres, its formation is a result of secondary nucleation, growth and further etching, in which F elements play a vital role in the formation process. Herein, a schematic of morphology evolution of micro-spherical TiO2 at different reaction states is proposed, as shown in Fig. 2. At the beginning of the hydrothermal reaction, TiF4 was readily hydrolysed in an aqueous solution, followed by nucleation and then budding to a spherical morphology (Fig. 1A). As the reaction time prolonged to 2 h, more F atoms were adsorbed on both {001} and {101} faceted surfaces, leading to a higher fluorinated surface. At this stage, HF preserved the growth of the {001} facet to form TiO2 microspheres consisting of well faceted, truncated octahedra (Fig. 1B). Further prolonged reaction time only affected the fraction of available adsorption sites occupied by F atoms. At this stage, the {001} faceted surface was only selectively etched (as shown in Fig. 1C) due to the different geometrical arrangements of oxygen and titanium on the {001} and {101} faceted surfaces. When the reaction time prolonged from 6 h to 12 h, the etching degree was gradually intensified on the {001} faceted surface, as shown in the SEM images of TS-6, 9, 12 h (Fig. 1D–F).
 | ||
| Fig. 2 Schematic of morphology evolution for the prepared anatase TiO2 microsphere single crystals. | ||
The XRD patterns of the prepared TS samples before and after calcination at 600 °C in air are shown in Fig. S4 and S5A,† respectively. The XRD profiles of all the prepared TiO2 samples can be indexed to pure anatase TiO2 with tetragonal structure (space group I41/amd, a = b = 3.782 Å, c = 9.502 Å, JCPDS card 73-1764). The UV-visible absorption spectra of the as-prepared TiO2 microspheres are shown in Fig. S5B.† It can be seen that the microsphere sample has stronger absorption in both the visible and ultraviolet range along with extended reaction time due to the controllable surface carving in the {001} facets and the unique spherical geometry morphology. Among them, the TS-9H sample shows the strongest absorption in both the visible and ultraviolet range with a red shift in the absorption edge. On the basis of the formula a = Bi(hν − Eg)2/hν (where a is the absorption coefficient, hν is the incident photon energy, and Bi is the absorption constant for indirect transitions), the band gaps are deduced to be 3.15 and 3.19 eV for TS-9H and TS-1H, respectively (see inset of Fig. S4B†).
In order to get a clean surface, all the samples were treated by an annealing method. Fig. S6† presents the high resolution XPS survey of F element before and after calcination. As shown in Fig. S6A,† the F 1s peak at 684.8 eV in TS-6H is consistent with fluorine bound to the surface of TiO2. However, the F 1s peaks after calcination at 600 °C for 120 min in TS-1H, 2H, 4H, 6H, 9H, 12H samples have disappeared (Fig. S6B†), indicating the surface F atoms of the as-prepared TiO2 microspheres have been completely removed. The Brunauer–Emmett–Teller (BET) specific surface area (SBET) and pore structure of the prepared samples were investigated using nitrogen adsorption–desorption measurements (see Table S1 and Fig. S6 in the ESI†). The nitrogen adsorption–desorption isotherm, shown in Fig. S7A,† can be attributed to type IV, according to IUPAC classification. The corresponding Barrett–Joyner–Halenda (BJH) method pore size distribution plot, shown in Fig. S7B,† indicates the presence of mesopores (2–50 nm) between the building blocks of the prepared TiO2 microspheres.
A Xe lamp (300 W) was used to evaluate the H2 production under UV-visible light irradiation, and the results are shown in Fig. 3. With the increase of hydrothermal reaction time, the H2 production is first increased and then decreased, as shown in Fig. 3A. As shown in Fig. 3B, the TS-6 h sample shows the highest hydrogen evolution rates (denoted as HERs) of 255 μmol h−1 g−1, which is nearly 3.7 times and 1.8 times higher than that of TS-1H (68.65 μmol h−1 g−1) and TS-12 h (136.66 μmol h−1 g−1), respectively. Above all, it can be found that once the {001} facet of the spherical microcrystal was corroded, its photocatalytic H2 production is enhanced ultimately compared with that of non-etched {001} facet ones. However, a too strong etching phenomenon on the {001} facet could lead to decreased photocatalytic performance (see the reduced HERs of TS-9H and TS-12H compared with that of TS-6 h). Moreover, the building blocks (denoted as intact-, etched-TiO2) of the as-prepared TS samples are also introduced to investigate the difference in HERs and the typical SEM images are shown in Fig. S8A and B.† It is found that the HERs of TiO2 microspheres are far more than those of their single TiO2 building block microstructures (Fig. 3C). To be specific, the calculated HERs of TS-2H and TS-6H are about 8 and 3.6 times that of their basic building blocks (11.53 μmol h−1 g−1 for intact- and 92.95 μmol h−1 g−1 for etched-TiO2), respectively (Fig. 3D). Moreover, the higher charge carriers separation efficiency and photocatalytic activity can be further confirmed by the measurement of the hydroxyl (OH) radical (shown in Fig. S9†). It can be clearly seen that the TS-6H sample possesses the highest fluorescence intensity of OH radicals among these samples, which further confirms the effective separation of photo-induced charge carriers. Moreover, the results of OH radicals were in good agreement with those of the photocatalytic H2 production.
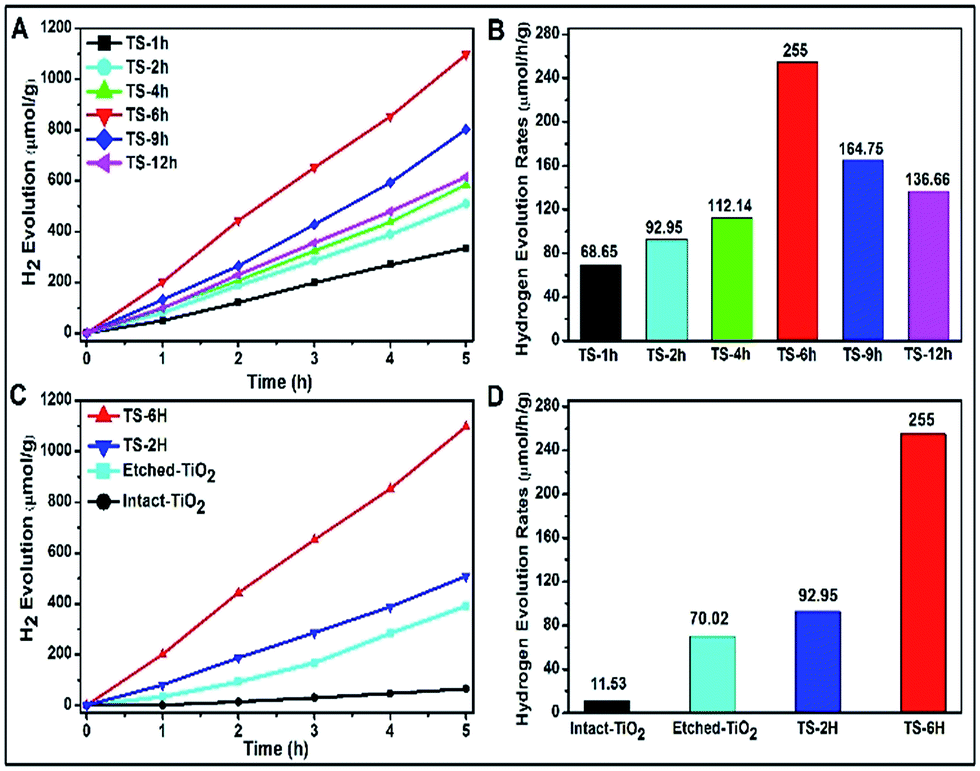 | ||
| Fig. 3 (A) H2 evolution vs. UV-vis light exposure time for TiO2 microspheres prepared under different reaction times and (B) the corresponding H2 evolution rates. (C) A comparative result of H2 evolution vs. UV-vis light exposure times for different TiO2 samples (intact-TiO2, etched-TiO2, TS-2H, TS-6H) and (D) their corresponding H2 evolution rates. | ||
Owing to its high sensitivity and non-destructive character, the photoluminescence (PL) technique has been widely used to investigate the electronic structure, optical and photochemical properties of semiconductor materials. Using this information, surface oxygen vacancies and defects, as well as the efficiency of charge carrier immigration and transfer can be obtained.33–35 Based on the above discussion, the photoluminescence properties were adopted to better understand the surface processes of the as prepared TiO2 microspheres. Typical PL spectra with excitation wavelengths of 380 nm and 253 nm are shown in Fig. 4. As can be seen from Fig. 4A, the TS samples exhibit a relatively strong and wide PL signal at 570 nm with excitation light of energy (the excitation wavelength is 380 nm) higher than the band gap energy of TiO2. The PL signal is generally attributed to excitonic PL,36 which mainly results from surface charge carrier trapping and defects of TiO2 microspheres. Specifically, the excitonic PL intensity of the as-prepared TiO2 initially decreases with increasing reaction time, and then increases along with increasing reaction time. Among the PL spectra, the TS-6H sample has the weakest PL intensity in the range of 550 nm to 590 nm. Moreover, the PL spectrum (shown in Fig. 4B) was also performed with the excitation wavelength of 253 nm. It can be seen that strong peaks at 392 nm were observed, which can be ascribed to band–band PL.37 According to PL attributes, the band–band PL spectrum can directly reflect the separation of photo-induced charge carriers, viz. the stronger the band–band PL signal, the higher the recombination rate of photo-induced carriers.38 Moreover, the PL intensity order excited with the excitation wavelength of 253 nm is TS-1H > TS-2H > TS-12H > TS-9H > TS-4H > TS-6H, which is in good agreement with the order of photocatalytic H2 production. In addition, regardless of the different line shape of the PL spectrum excited with a different wavelength, the intensity order of PL spectra at a different wavelength range is in good accord with the photocatalytic H2 production rates.
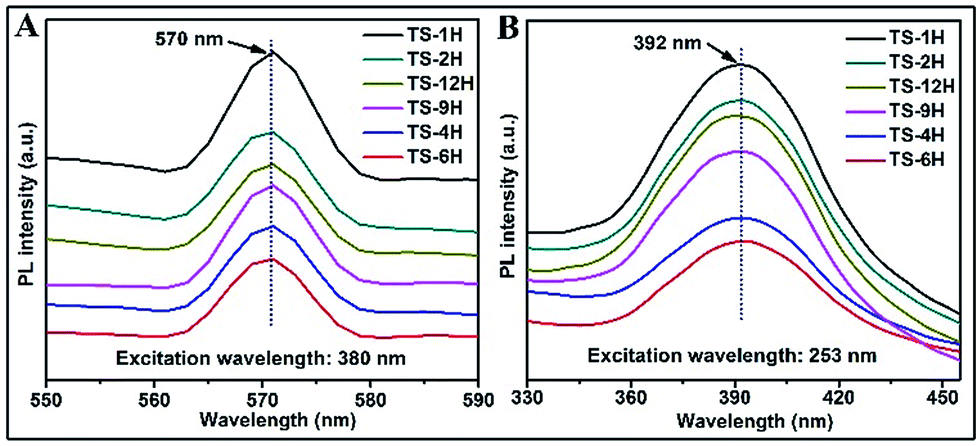 | ||
| Fig. 4 PL spectra of prepared microspherical TiO2 with different reaction times under different excitation wavelengths: (A) 380 nm and (B) 253 nm. | ||
Electron paramagnetic resonance (EPR) spectra were recorded at room temperature to investigate the presence of Ti3+ in the prepared TiO2 microspheres, and the results are shown in Fig. 5. TS-2H has a very strong EPR signal at g = 2.003, which could be assigned to the oxygen vacancy species.39 Furthermore, new paramagnetic signal peaks centered at g = 1.967 (the signal can be ascribed to the subsurface paramagnetic Ti3+ center40) appear for TS-6H and TS-12H compared with that of TS-2H. In addition, the signal peak intensity of subsurface paramagnetic Ti3+ for TS-6H is sharper and stronger than that of TS-12H. The intensity variation of both the signals at g = 2.003 and g = 1.967 indicates that the quantity of both the surface and bulk trapping sites (here referring to Ti3+ ions and oxygen vacancies) have been changed with extension of the hydrothermal reaction time.
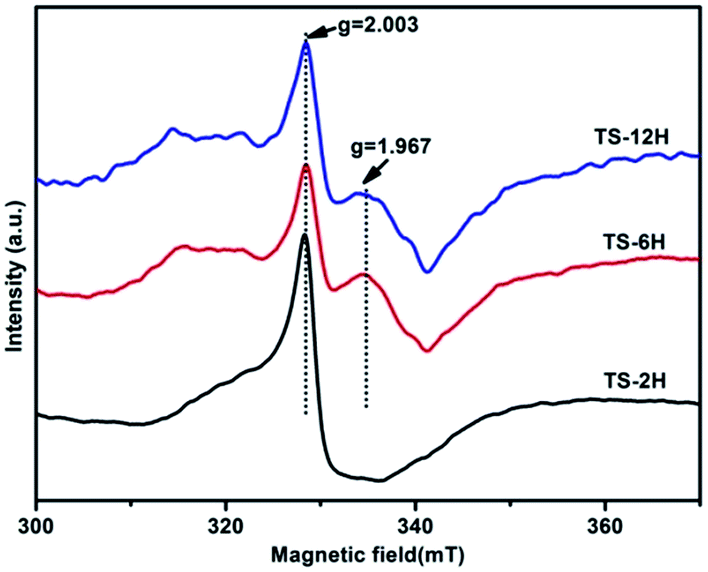 | ||
| Fig. 5 EPR spectra of the TiO2 samples obtained after different hydrothermal reaction times. | ||
Furthermore, X-ray photoelectron spectroscopy (XPS) measurement was performed to explore the chemical state of titanium and oxygen elements in the TS samples. Typical high-resolution XPS spectra of the TS samples are shown in Fig. S10.† The XPS spectrum of Ti 2p for all the samples exhibits two typical peaks with a spin–orbital doublet splitting (Ti 2p3/2 and Ti 2p1/2), which can be assigned to oxidation state of titanium being Ti4+.41 No peak corresponding to Ti3+ around 458.1 eV was detected. For one thing, the surface of TS was dominated by Ti4+, which rules out the presence of Ti3+ on the sample surface.42 For another, the radius of the prepared TiO2 microspheres (ca. 2 μm) is larger than the detecting depth of XPS since the XPS technique monitors the electron binding energy of sites within a few nanometers of the particle surface. To be specific, as for TS-1H, the peaks at 458.68 eV (Fig. S10A†) and 529.97 eV (Fig. S10B†) could be indexed to Ti 2p3/2 and O 1s, respectively. However, prolonged reaction time leads to a slight shift to lower binding energy for both Ti 2p and O 1s core level spectra, as observed in the XPS result of TS-12H (the binding energy positions of Ti 2p3/2 and O 1s shift to 458.60 eV and 529.88 eV, respectively). The subtle variations in XPS result may result from Ti3+ doping-induced changes in the electronic properties and surface structure of the prepared TS samples.
The widely accepted photoactivation mechanism is as follows: (a) upon excitation by UV light absorption, electrons are excited from the valence band to the conduction band; (b) the photogenerated electrons (e−) and holes (h+) migrate from bulk to surface, where the electrons reduce adsorbed electron acceptors (e.g., O2) and holes oxidize adsorbed donor species (e.g., organic species or hydroxyl); (c) the e−–h+ recombination occurs in bulk trapping sites and on surface trapping sites. The overall moderate photocatalytic efficiency is mainly ascribed to the competition between recombination of photogenerated charge carriers and charge transfer to adsorbates. As for the prepared TiO2 microspheres, the (001) faceted surface is changed with extension of the reaction time. Thus, the quantity of both bulk trapping sites and surface trapping sites has changed to result in optimum values. In other words, the relative ratio of surface/bulk trapping sites plays a decisive role in adsorption and photoreactivity of TiO2 crystals. Based on the above discussion and PL spectra results, a surface/bulk trapping sites-mediated photo-excitation mechanism is proposed, as shown in Fig. 6. The corrosive effect generated at {001} facets has a meaningful influence on the amount of both bulk trapped and surface trapped sites. Since different degrees of corrosion have been observed, compared with TiO2 with intact {001} facets, there exists a relatively appropriate ratio of surface defects to bulk defects. It is concluded that a relatively suitable corrosion phenomenon in {001} facets of anatase TiO2 microcrystals leads to the production of more reactive under-coordinated Ti 5c atoms, but a relatively reduced concentration ratio of bulk defects to surface defects in TiO2 microcrystals. An optimum concentration of bulk/surface defects can improve the charge separation of photogenerated charge carriers, and thus provide a maximum enhancement of photocatalytic performance. However, an excess concentration of surface/bulk defects results in a decrease of photocatalytic performance.
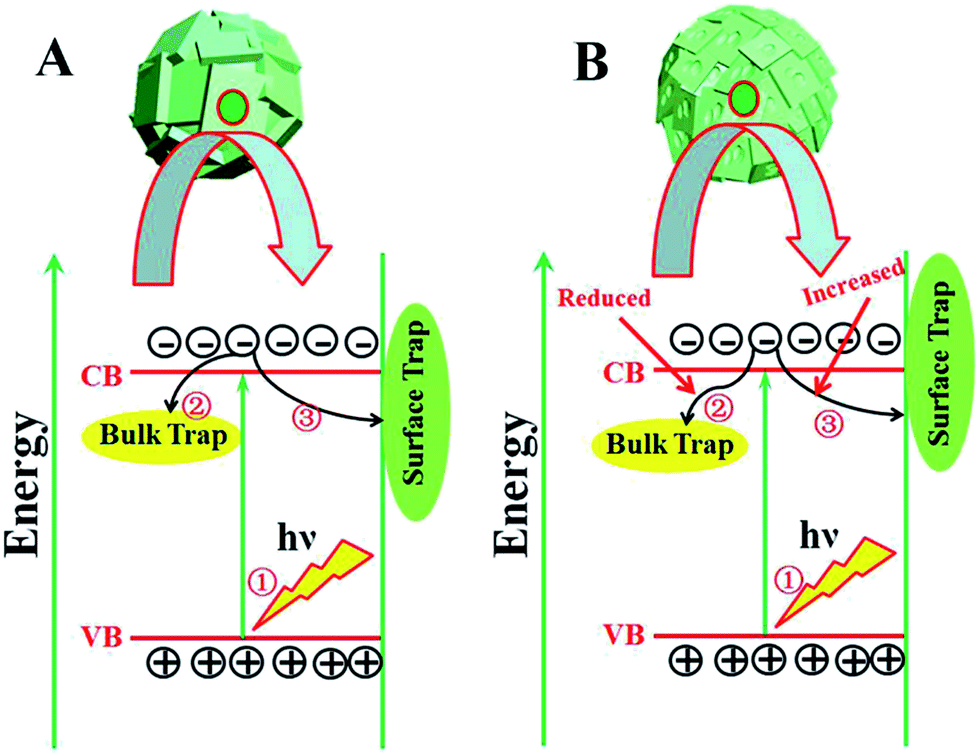 | ||
| Fig. 6 Surface/bulk trapping sites-mediated photo-excitation process for TiO2 microspheres consisting of (A) intact and (B) etched {001} faceted truncated octahedra. | ||
In summary, a facile hydrothermal method has been applied to fabricate oxygen-deficient anatase TiO2 microspheres consisting of intact or etched {001}-faceted TiO2 by simply changing the hydrothermal reaction time. Such prepared TiO2 microspheres consisting of etched {001}-faceted surfaces show excellent photocatalytic hydrogen production, far exceeding than that of microspherical TiO2 consisting of intact {001}-faceted surfaces. Moreover, short hydrothermal reaction time facilitates the formation of oxygen vacancies, whereas prolonged hydrothermal reaction time contributes to the decrease of oxygen vacancies and formation of subsurface Ti3+ defect states. Moderate etching effects occurring on the {001} facets of prepared TiO2 microspheres bring out an optimum surface/bulk trapping ratio and thus the most excellent photocatalytic performance.
Acknowledgements
This study was supported by the “Hundred Talents Program” of the Chinese Academy of Science and the National Natural Science Foundation of China (21622310, 21573264, 21603247).Notes and references
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- X. Chen, L. Liu, Y. Y. Peter and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- E. J. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander-Webber and H. J. Snaith, Nature, 2013, 495, 215–219 CrossRef CAS PubMed.
- H. B. Jiang, Q. Cuan, C. Z. Wen, J. Xing, D. Wu, X. Q. Gong, C. Li and H. G. Yang, Angew. Chem., Int. Ed., 2011, 50, 3764–3768 CrossRef CAS PubMed.
- Y.-F. Li, Z.-P. Liu, L. Liu and W. Gao, J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS PubMed.
- S. Yang, B. X. Yang, L. Wu, Y. H. Li, P. Liu, H. Zhao, Y. Y. Yu, X. Q. Gong and H. G. Yang, Nat. Commun., 2014, 5, 5355 CrossRef CAS PubMed.
- A. S. Ichimura, B. M. Mack, S. M. Usmani and D. G. Mars, Chem. Mater., 2012, 24, 2324–2329 CrossRef CAS PubMed.
- X. Han, X. Wang, S. Xie, Q. Kuang, J. Ouyang, Z. Xie and L. Zheng, RSC Adv., 2012, 2, 3251–3253 RSC.
- J. Pan, G. Liu, G. Q. M. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- J. Wang, Y. Zhou, Y. Hu, R. O'Hayre and Z. Shao, J. Phys. Chem. C, 2011, 115, 2529–2536 CAS.
- Y. Mao, M. Kanungo, T. Hemraj-Benny and S. S. Wong, J. Phys. Chem. B, 2006, 110, 702–710 CrossRef CAS PubMed.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y.-M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- J. Konishi, K. Fujita, K. Nakanishi, K. Hirao, K. Morisato, S. Miyazaki and M. Ohira, J. Chromatogr. A, 2009, 1216, 7375–7383 CrossRef CAS PubMed.
- J. Chen, W. Song, H. Hou, Y. Zhang, M. Jing, X. Jia and X. Ji, Adv. Funct. Mater., 2015, 25, 6793–6801 CrossRef CAS.
- P. Cheng, S. Du, Y. Cai, F. Liu, P. Sun, J. Zheng and G. Lu, J. Phys. Chem. C, 2013, 117, 24150–24156 CAS.
- Y. Zhang, B. Wu, Y. Tang, D. Qi, N. Wang, X. Wang, X. Ma, T. C. Sum and X. Chen, Small, 2016, 12, 2291–2299 CrossRef CAS PubMed.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS PubMed.
- Z. Wang, B. Wen, Q. Hao, L.-M. Liu, C. Zhou, X. Mao, X. Lang, W.-J. Yin, D. Dai, A. Selloni and X. Yang, J. Am. Chem. Soc., 2015, 137, 9146–9152 CrossRef CAS PubMed.
- L. Liu, Y. Jiang, H. Zhao, J. Chen, J. Cheng, K. Yang and Y. Li, ACS Catal., 2016, 6, 1097–1108 CrossRef CAS.
- R. Kumar, S. Govindarajan, R. K. Siri Kiran Janardhana, T. N. Rao, S. V. Joshi and S. Anandan, ACS Appl. Mater. Interfaces, 2016, 8, 27642–27653 CAS.
- T. Leshuk, R. Parviz, P. Everett, H. Krishnakumar, R. A. Varin and F. Gu, ACS Appl. Mater. Interfaces, 2013, 5, 1892–1895 CAS.
- J. M. Coronado, A. J. Maira, J. C. Conesa, K. L. Yeung, V. Augugliaro and J. Soria, Langmuir, 2001, 17, 5368–5374 CrossRef CAS.
- C. Yang, Z. Wang, T. Lin, H. Yin, X. Lü, D. Wan, T. Xu, C. Zheng, J. Lin, F. Huang, X. Xie and M. Jiang, J. Am. Chem. Soc., 2013, 135, 17831–17838 CrossRef CAS PubMed.
- Z. Zheng, B. Huang, X. Meng, J. Wang, S. Wang, Z. Lou, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2013, 49, 868–870 RSC.
- A. Sinhamahapatra, J.-P. Jeon and J.-S. Yu, Energy Environ. Sci., 2015, 8, 3539–3544 CAS.
- X. Y. Ma, Z. G. Chen, S. B. Hartono, H. B. Jiang, J. Zou, S. Z. Qiao and H. G. Yang, Chem. Commun., 2010, 46, 6608–6610 RSC.
- Y. Wang, H. Zhang, Y. Han, P. Liu, X. Yao and H. Zhao, Chem. Commun., 2011, 47, 2829–2831 RSC.
- X. H. Yang, H. G. Yang and C. Li, Chem.–Eur. J., 2011, 17, 6615–6619 CrossRef CAS PubMed.
- J. Liqiang, S. Xiaojun, X. Baifu, W. Baiqi, C. Weimin and F. Honggang, J. Solid State Chem., 2004, 177, 3375–3382 CrossRef.
- J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- T. Toyoda, T. Hayakawa, K. Abe, T. Shigenari and Q. Shen, J. Lumin., 2000, 87, 1237–1239 CrossRef.
- L.-Q. Jing, B.-F. Xin, D.-J. Wang and F.-l. Yuan, Chem. J. Chin. Univ., 2005, 26, 111–115 CAS.
- X. Li, F. Li, C. Yang and W. Ge, J. Photochem. Photobiol., A, 2001, 141, 209–217 CrossRef CAS.
- J. Liqiang, Q. Yichun, W. Baiqi, L. Shudan, J. Baojiang, Y. Libin, F. Wei, F. Honggang and S. Jiazhong, Sol. Energy Mater. Sol. Cells, 2006, 90, 1773–1787 CrossRef.
- I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara, S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem., 2000, 161, 205–212 CrossRef CAS.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- J. G. Li, R. Büchel, M. Isobe, T. Mori and T. Ishigaki, J. Phys. Chem. C, 2009, 113, 8009–8015 CAS.
- F. Zuo, K. Bozhilov, R. J. Dillon, L. Wang, P. Smith, X. Zhao, C. Bardeen and P. Feng, Angew. Chem., Int. Ed., 2012, 51, 6223–6226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28533c |
| This journal is © The Royal Society of Chemistry 2017 |