
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew quinacridone derivatives with π-extended conjugation in central core†
Piotr Kurzepa,
Łukasz Skórkaa,
Malgorzata Zagorskaa,
Piotr A. Guńka a,
Marzena Banasiewiczb,
Bolesław Kozankiewiczb and
Irena Kulszewicz-Bajer*a
a,
Marzena Banasiewiczb,
Bolesław Kozankiewiczb and
Irena Kulszewicz-Bajer*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: ikulsz@ch.pw.edu.pl; Tel: +48 22 2345584
bInstitute of Physics, Polish Academy of Science, Al. Lotników 32/46, 02-668 Warsaw, Poland
First published on 30th January 2017
Abstract
A novel synthetic strategy for the preparation of quinacridone derivatives with extended conjugation in central aromatic core is proposed allowing for fine tuning of their electronic properties. Two new quinacridone derivatives with carbazole (1) and naphthalene (2) core are described, which are excellent candidates for optoelectronic applications as evidenced by their electrochemically determined redox properties combined with strong photoluminescence (the fluorescence quantum yield of 0.83 and the radiative rate constant of 90.2 × 107 s−1 in the case of 2).
Introduction
Quinacridone, QA, is a well-known industrial dye. Due to its excellent thermal and environmental stabilities it is commonly used for fabrication of paints and inkjet toners. However, recently QA has also attracted considerable attention as an organic semiconductor exhibiting interesting luminescent,1 transport1d,2 and photovoltaic3 properties. QA and its derivatives have been successfully used as efficient electron transporting layers4 or organic emitters in OLEDs with low driving voltage and high power efficiency.1b,1e Copolymers of QA and fluorene yield white electroluminescence.5 The planar structure of QA and its strong H-bonding properties favor its charge transport abilities. As a consequence QA and its polymeric derivatives exhibit high charge carrier mobility in organic field-effect transistors.2Results and discussion
Synthetic strategy
Physicochemical properties of QA can be easily tuned by modification of its chemical structure. Thus, the incorporation of new functional groups on amine1a,6 or carbonyl sites7 changes the solubility, planarity and the electronic structures of these derivatives. However, the most significant modification of the physicochemical properties of this semiconductor can be obtained by the elongation of its π-conjugated structure. Till now the π-conjugation has been extended by the addition of aromatic moieties to lateral phenylene rings of pristine QA. Thus, quinacridone derivatives containing indole,1d indene8 and dithianaphthene2d rings fused to QA have been synthesized. On the other hand, the electronic structure can be also changed by the modification of the central aromatic core of quinacridone. However, the classical method of QA preparation is not compatible with this type of modification.1a,9 Here, we present a new strategy for tuning the properties of QA-type derivatives by the incorporation of different aromatic cores into the center of the QA molecule (Scheme 1).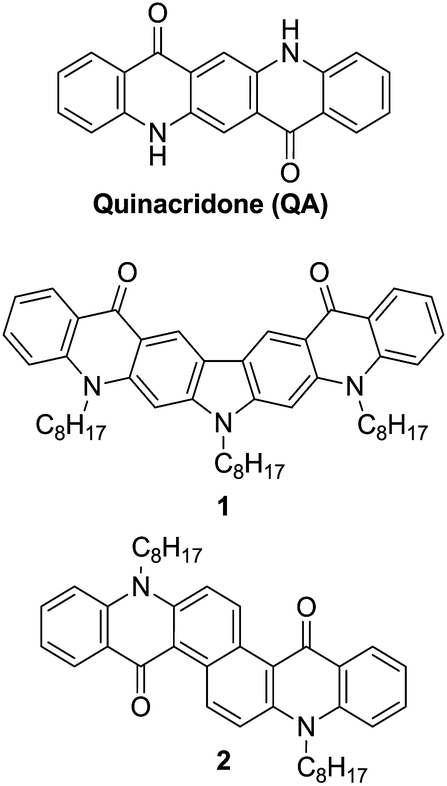 | ||
| Scheme 1 Structures of quinacridone and its derivatives 1 and 2. | ||
The synthesis of the new compounds (1 and 2) is based on the C–N coupling reaction. It was shown earlier that the Ullmann condensation of anthranilic acid with dihalogenobenzene in the presence of the Cu catalyst requires the use of diiodo-derivatives and leads to a mixture of mono- and disubstituted products, obtained with low to medium yields.10 Dibromoderivatives are even less reactive in this reaction.10 However, the coupling in the presence of the palladium catalyst is significantly more efficient, but it largely depends on the reaction conditions. Thus, the amination reactions of 1,3-dibromobenzene with anthranilic acid in the presence of Pd(OAc)2/BINAP10b or 1,2-dibromobenzene with methyl anthranilate using Pd(OAc)2/t-Bu3P catalytic system proceeds in sufficiently good yields of 70–80%.11
Taking into consideration these results we have applied the palladium catalyzed C–N coupling for the amination of para-substituted dibromoarenes (Scheme 2). The reactions of methyl anthranilate with 2,7-dibromocarbazole using catalytic system Pddba2/t-Bu3P or with 2,6-dibromonaphthalene in the presence of Pd(OAc)2/t-Bu3P led to the intermediate products 3a and 4a with 53% and 90% yields, respectively. Diesters 3a and 4a were easily hydrolized in NaOH/EtOH solution to the corresponding diacids 3b and 4b. Then, diacids were condensed in standard conditions using polyphosphoric acid/P2O5 mixture. Since these condensation products were sparingly soluble they were directly alkylated with n-octyl bromide. The final yields were 30% and 56% for carbazole and naphthalene derivatives, respectively. In the case of the derivative 2 the cyclization reaction proceeded only at α positions of naphthalene. No other isomers corresponding to ring-closure at β position were detected. It should be emphasized that this simple route is a rather general method, and should allow to synthesize a new class of quinacridone derivatives, in which π-conjugation is extended by the incorporation of different aromatic cores in the center of the QA molecule.
 | ||
| Scheme 2 Synthetic pathways leading to 1 and 2. | ||
Electrochemical studies
The electrochemical properties of the compounds 1 and 2 were studied by cyclic voltammetry (CV) (see ESI†) and differential pulse voltammetry (DPV) and compared to that of N,N′-dioctylquinacridone, QA-C8. The electrochemical behavior of the studied compounds in the positive potential range strongly depended on the nature of the central aromatic core. Thus, the differential pulse voltammogram of 1 exhibited three reversible redox couples at 0.55 V, 0.66 V and 0.99 V (vs. Fc/Fc+) (Fig. 1a). The first two peaks overlapped and were typical of the carbazole moiety oxidation.12a The third peak was ascribed to the oxidation of acridone unit and its potential was shifted to higher value than that measured for QA-C8, due to the positive charge imposed on the molecule in the process of the carbazole core oxidation. Similar oxidation behavior was observed by Wang et al. for QA substituted at 2,9 positions with carbazole dendrimers.1c | ||
| Fig. 1 Differential pulse voltammograms – oxidation mode: (a) 1, (b) 2, (c) QA-C8 in CH2Cl2 solution (the concentration of the compounds was c = 10−3 M) containing an electrolyte – 0.1 M Bu4NBF4. | ||
The differential pulse voltammogram of 2, registered at positive potentials, showed two reversible redox couples at 0.80 V and 1.16 V vs. Fc/Fc+ (Fig. 1b), which could be attributed to consecutive oxidations of two acridone moieties present in the molecule.1c,12b It should be noted that the oxidation potential of 2 increases as compared to that of QA-C8 (Fig. 1c), despite its extended conjugation. This effect is attributed to nonlinearity of the molecule's conjugated core which leads to more difficult oxidation as observed earlier for similar compounds.13
The reduction processes of 1 and 2 were significantly different. The CV of 1 (ESI, Fig. S6a†) displayed an irreversible process and its DPV curve could not be properly registered at such negative potentials. In contrast, the differential pulse voltammogram of 2 showed a reversible reduction peak at −2.08 V and an additional quasi-reversible one at −2.30 V vs. Fc/Fc+ (Fig. 2a). These reduction peaks were shifted to lower potential values as compared to the corresponding peaks in the voltammogram of QA-C8 registered in the same experimental conditions, i.e. −1.82 V and −2.17 V vs. Fc/Fc+ (Fig. 2b).
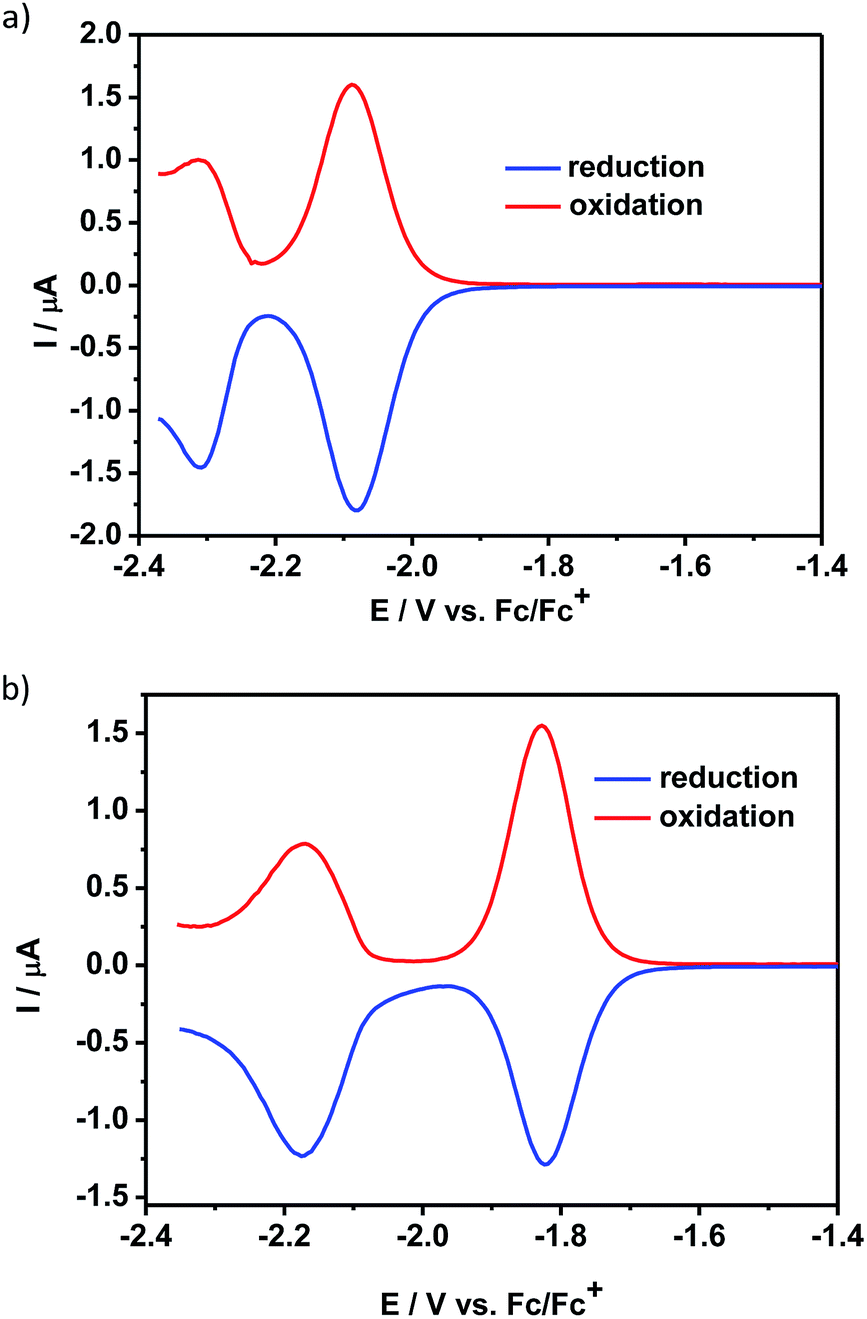 | ||
| Fig. 2 Differential pulse voltammograms – reduction mode: (a) 2, (b) QA-C8 in CH2Cl2 solution (the concentration of the compounds was c = 10−3 M) containing an electrolyte – 0.1 M Bu4NBF4. | ||
From the formal potential of the first oxidation process it is possible to calculate the ionization potentials, IP, of 1 and 2 expressing their values with respect to a vacuum level, whereas from the formal potential of the first reduction process – the electron affinity, EA could be calculated.14 The IP value of QA-C8 is intermediate between those calculated for 1 and 2 (Table 1). The EA value of 2 (Table 1) is higher by ca. 0.25 eV than that for QA-C8. This trend is clearly reflected in quantum chemical calculations (compare Table 2).
| Compound | Conditions | HOMO [eV] | LUMO [eV] | Eg [eV] | IP [eV] | EA [eV] |
|---|---|---|---|---|---|---|
| a Calculated energies of HOMO and LUMO orbitals, the band gap, Eg, ionization potential, IP, and electron affinity, EA, (calculated at the B3LYP/6-31G(d,p) level of theory in vacuum and in DCM solvent for quinacridone derivatives). | ||||||
| 1 | vac. | −5.08 | −1.40 | 3.68 | 6.05 | −0.32 |
| DCM | −5.21 | −1.63 | 3.59 | 5.15 | −1.70 | |
| 2 | vac. | −5.20 | −1.84 | 3.37 | 6.32 | −0.73 |
| DCM | −5.39 | −2.07 | 3.32 | 5.35 | −2.17 | |
| QA-C8 | vac. | −5.17 | −2.08 | 3.09 | 6.35 | −0.90 |
| DCM | −5.29 | −2.29 | 3.00 | 5.25 | −2.36 | |
DFT calculations
The electronic structures of the new quinacridone derivatives were calculated using the Gaussian09 Revision E.01 package and employing hybrid B3LYP exchange correlation potential combined with 6-31G(d,p) basis set.Fig. 3 shows that both HOMO and LUMO were located on whole π-systems. The calculated HOMO energy levels of 1 and 2 were very close to that of QA-C8 (Table 2). However, the LUMO level of 1 was far above the LUMO levels calculated for 2 and QA-C8, which is in agreement with the electrochemical measurements. The DFT calculations demonstrated also that the solvation changes the HOMO/LUMO levels, dropping down both values by ca. 0.2 eV. The calculated IP values were ca. 0.2 eV smaller than those derived from the cyclic voltammetry investigations, however, the trend remains the same.
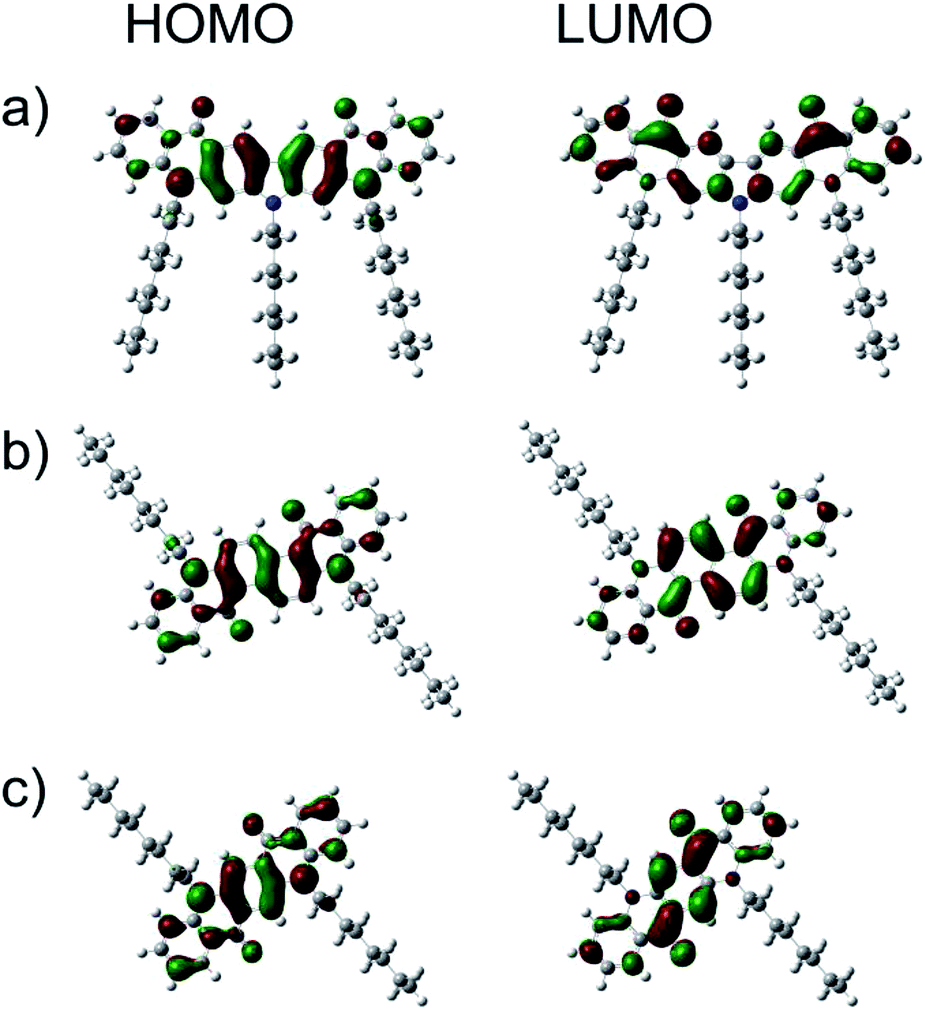 | ||
| Fig. 3 HOMO and LUMO contours for compounds 1 (a), 2 (b) and QA-C8 (c) calculated at B3LYP/6-31G(d,p) level of theory (isosurface value: 0.03 green and red stand for negative and positive value, respectively). | ||
X-ray diffraction studies
The crystal structure of 2 was determined at RT by X-ray diffraction (ESI, Table S1†).15 2 crystallizes in the P21/c space group with half a molecule in the asymmetric unit.The molecules of 2 (the structure in Fig. 4) are centrosymmetric (Ci point group) and consist of a flat aromatic core and n-octyl groups with disordered terminal carbon atoms (C18, C19 and C20). There are two intramolecular C–H⋯O hydrogen bonds between oxygen atoms of carbonyl groups and hydrogen atoms of the aromatic core (d(C2⋯O1i) = 2.770(2) Å, d(H2⋯O1i) = 2.09 Å, C2–H2⋯O1i = 129°, ii = −x, −y, −z). Molecules are arranged by π stacking into columns running along the crystallographic Y axis. The distance between aromatic core planes of adjacent molecules in columns is 3.4793(12) Å. The columns are held together in the 3D crystal structure by dispersion interactions between interdigitated alkyl chains and by C–H⋯O interactions (d(C3⋯O1ii) = 3.352(2) Å, d(H3⋯O1ii) = 2.56 Å, C3–H3⋯O1ii = 144°, ii = x, 1/2 − y, 1/2 + z).
 | ||
| Fig. 4 Molecular structure (a) and ball-and-stick model of crystal packing (b) of compound 2. Only the major part of the disordered n-octyl groups is shown. Thermal ellipsoids drawn at 50% level (a) and hydrogen atoms omitted for clarity. i = −x, −y, −z. | ||
Optical studies
Modification of the conjugated core in 1 and 2 led to profound changes in their optical properties as compared to the case of QA-C8 The UV-Vis absorption and fluorescence spectra of these three compounds are compared in Fig. 5. A clear hypsochromic shift of the absorption and the emission bands of 1 and 2 with respect to the corresponding bands of QA-C8 is observed (by 102 nm and 91 nm for 1 and 71 nm and 79 nm for 2) (ESI, Table S2†) again consistent with quantum chemical calculations. These significant hypsochromic shifts reflect the influence of the modification of the central aromatic core on optical properties of new derivatives and allow to displace the emission towards the blue-yellow range.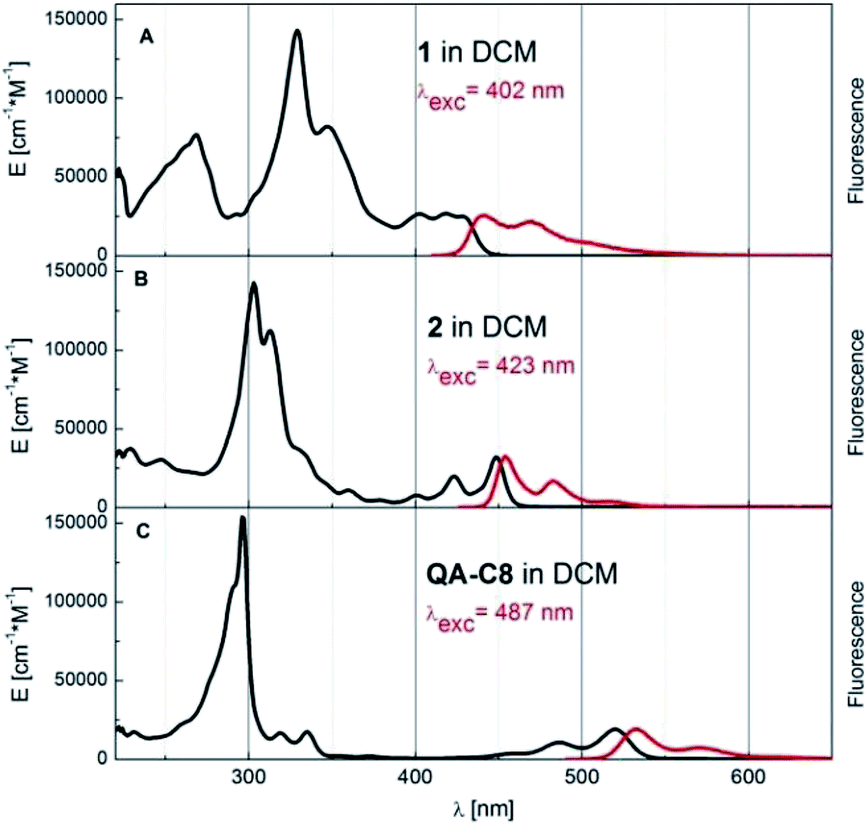 | ||
| Fig. 5 Absorption (black curve, given in the extinction units) and fluorescence (red, arbitrary units) spectra of compounds 1, 2 and QA-C8 in dichloromethane (DCM) at room temperature. | ||
The naphthalene derivative 2 is especially interesting for potential optoelectronic applications, for which a very small Stokes shift between the absorption and fluorescence bands maxima was observed. Furthermore, this compound is a very good emitter of fluorescence photons, much better than QA-C8 (see ESI Table S2† and the spectroscopic data of quinacridone reported previously1a,16,17). The fluorescence quantum yield of 2 is pretty high 0.83 and the lifetime as short as 0.92 ns, which results in very high radiative rate constant kr = 90.2 × 107 s−1.
Conclusions
In summary, we have demonstrated that the palladium catalyzed C–N coupling reaction can be applied for the synthesis of new quinacridone derivatives and allows the incorporation of moieties other than phenylene ring into the central core of the molecule, which can influence the electronic, optical and electrochemical properties of these new compounds. Two new quinacridone derivatives were prepared and characterized by electrochemical and optical studies. Furthermore, the compound 2 with the naphthalene central core is a very good emitter of fluorescence photons, much better than classical quinacridone. The synthesized new molecules are promising candidates for optoelectronics.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
PK, LS, MZ and IKB wish to acknowledge financial support from National Centre of Science in Poland, NCN, Grant No. 2015/17/B/ST5/00179. PAG thanks Warsaw University of Technology for financial support. The Gaussian09 calculations were carried out in the Wroclaw Centre for Networking and Supercomputing, Poland, http://www.wcss.wroc.pl, under calculation Grant No. 283. The authors thank Professor Adam Proń for his helpful and critical review of the manuscript.References
- (a) K. Ye, J. Wang, H. Sun, Y. Liu, Z. Mu, F. Li, S. Jiang, J. Zhang, H. Zhang, Y. Wang and C. M. Che, J. Phys. Chem. B, 2005, 109, 8008 CrossRef CAS PubMed; (b) H. Bi, K. Ye, Y. Zhao, Y. Yang, Y. Liu and Y. Wang, Org. Electron., 2010, 11, 1180 CrossRef CAS; (c) C. Wang, S. Chen, K. Wang, S. Zhao, J. Zhang and Y. Wang, J. Phys. Chem. C, 2012, 116, 17796 CrossRef CAS; (d) C. Wang, D. Chen, W. Chen, S. Chen, K. Ye, H. Zhang, J. Zhang and Y. Wang, J. Mater. Chem. C, 2013, 1, 5548 RSC; (e) S. Wang, Y. Zhang, W. Chen, J. Wei, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 11972 RSC; (f) C. Wang, S. Wang, W. Chen, Z. Zhang, H. Zhang and Y. Wang, RSC Adv., 2016, 6, 19308 RSC.
- (a) E. D. Głowacki, L. Leonat, M. Irimia-Vladu, R. Schwodiauer, M. Ullah, H. Sitter, S. Bauer and N. S. Sariciftci, Appl. Phys. Lett., 2012, 101, 023305 CrossRef; (b) I. Osaka, M. Akita, T. Koganezawa and K. Takimiya, Chem. Mater., 2012, 24, 1235 CrossRef CAS; (c) H. Li, C. Gu, L. Jiang, L. Wei, W. Hu and H. Fu, J. Mater. Chem. C, 2013, 1, 2021 RSC; (d) W. Chen, K. Tian, X. Song, Z. Zhang, K. Ye, G. Yu and Y. Wang, Org. Lett., 2015, 17, 6146 CrossRef CAS PubMed.
- (a) H. Li, X. Wang, F. Liu and H. Fu, Polym. Chem., 2015, 6, 3283 RSC; (b) T. L. Chen, J. Jun-An, L. Catane and B. Ma, Org. Electron., 2011, 12, 1126 CrossRef CAS; (c) C. Wang, W. Chen, S. Chen, S. Zhao, J. Zhang, D. Qiu and Y. Wang, New J. Chem., 2012, 36, 1788 RSC; (d) S. Mattiello, A. Sanzone, P. Brazzo, M. Sassi and L. Beverina, Eur. J. Org. Chem., 2015, 5723 CrossRef CAS.
- T. V. Pho, P. Zalar, A. Garcia, T. Q. Nguyen and F. Wudl, Chem. Commun., 2010, 46, 8210 RSC.
- J. Liu, B. Gao, Y. Cheng, Z. Xie, Y. Geng, L. Wang, X. Jing and F. Wang, Macromolecules, 2008, 41, 1162 CrossRef CAS.
- (a) J. Wang, Y. Zhao, J. Zhang, J. Zhang, B. Yang, Y. Wang, D. Zhang, H. You and D. Ma, J. Phys. Chem. C, 2007, 111, 9177 CrossRef CAS; (b) A. Ortiz, W. H. Flora, G. D. D'Ambruso, N. R. Armstrong and D. V. McGrath, Chem. Commun., 2005, 444 RSC.
- (a) T. Takeda, H. Sugihara, Y. Suzuki, J. Kawamata and T. Akutagawa, J. Org. Chem., 2014, 79, 9669 CrossRef CAS PubMed; (b) W. Chen, J. Lv, J. Han, Y. Chen, T. Jia, F. Li and Y. Wang, J. Mater. Chem. A, 2016, 4, 2169 RSC.
- Y. Zou, T. Yuan, H. Yao, D. J. Frazier, D. J. Stanton, H. J. Sue and L. Fang, Org. Lett., 2015, 17, 3146 CrossRef CAS PubMed.
- U. Keller, K. Mullen, S. De Feyter and F. C. De Schryver, Adv. Mater., 1996, 8, 490 CrossRef CAS.
- (a) O. Baudoin, M. P. Teulade-Fichou, J. P. Vigneron and J. M. Lehn, J. Org. Chem., 1997, 62, 5458 CrossRef CAS; (b) C. Jacquelin, N. Saettel, C. Hounsou and M. P. Teulade-Fichou, Tetrahedron Lett., 2005, 46, 2589 CrossRef CAS.
- R. Lartia, H. Bertrand and M. P. Teulade-Fichou, Synlett, 2006, 4, 610 Search PubMed.
- (a) K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601 CrossRef CAS; (b) R. E. Sioda and B. Frankowska, J. Electroanal. Chem., 2008, 612, 147 CrossRef CAS.
- V. D. Parker, J. Am. Chem. Soc., 1978, 98, 98 CrossRef.
- (a) S. Trasatti, Pure Appl. Chem., 1986, 58, 955 CAS; (b) T. van der Poll, J. A. Love, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646 CrossRef CAS PubMed; (c) J. Sworakowski, J. Lipiński and K. Janus, Org. Electron., 2016, 33, 300 CrossRef CAS.
- CCDC 1515700 contains the supplementary crystallographic data for this paper.†.
- P. H. Liu, H. Tian and C. P. Chang, Photochem. Photobiol., 2000, 137, 99 CrossRef CAS.
- S. N. Shcherbo, G. A. Val'kova, D. N. Shigorin, Z. Z. Moiseeva and E. S. Bir, Zh. Fiz. Khim., 1978, 53, 566 (Russ. J. Phys. Chem., 1978, 53, 320) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic procedure and spectroscopic characterization of all compounds, electrochemical measurements (CV and DPV), X-ray diffraction, optical absorption and fluorescence studies. X-ray crystallographic data for compound 2 (CIF). CCDC 1515700. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28567h |
| This journal is © The Royal Society of Chemistry 2017 |