
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of hydroxyapatite particle size on the formation of chloropyromorphite in anglesite–hydroxyapatite suspensions†
Yu Wanga,
Xiaoqing Dong *a,
Jing Cuib,
Zhenggui Wei*b and
Xiaohong Wanga
*a,
Jing Cuib,
Zhenggui Wei*b and
Xiaohong Wanga
aAnhui Key Laboratory of Functional Coordination Compounds, School of Resources and Environments, Anqing Normal University, Anqing, Anhui 246013, P. R. China. E-mail: dongxiaoqing@aqnu.edu.cn; Tel: + 86 556 5526325
bDepartment of Environmental Science, Nanjing Normal University, Nanjing, Jiangsu 210097, P. R. China. E-mail: weizhenggui@gmail.com
First published on 17th February 2017
Abstract
Anglesite (PbSO4), a widely available form of Pb whose bioavailability has been the focus of a few studies, was selected in the present study and reacted with synthetic hydroxyapatites (Ca5(PO4)3OH, HAPs) of different sizes (approximately 20.08 nm, 60.24 nm, and micron-sized) at various P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb molar ratios and under various constant pH conditions. Particle size was discovered to strongly affect the immobilisation of Pb and the transformation from anglesite to chloropyromorphite (Pb5(PO4)3Cl) by the HAPs, in addition to the dissolution properties of HAP. The complete transformation from anglesite to chloropyromorphite was achieved at a pH of 4–5 for 20.08 nm-sized HAPs, whereas incomplete transformations of the 60.24 nm and micro-HAPs were obtained at these pH. At pHs of ≥6 (HAPs of size 20.08 nm) and ≥5 (60.24 nm), the smaller-particle HAPs were incompletely dissolved, and their surfaces were coated by newly formed chloropyromorphite, which was in contrast to the micro-HAPs, which were at pH ≥ 3. These results demonstrated that the surface coating rate decreased when the HAP particle size was decreased; thus, smaller-particle HAPs completely immobilised anglesite by forming chloropyromorphite at higher pH, particularly when the P:Pb molar ratio was high. This study demonstrated that nano-HAPs have potential for use in the situ immobilisation of Pb in low-acidity soil and waste without requiring the P solubility enhancement through the use of acidic conditions or an increase in the number of HAPs.
:Pb molar ratios and under various constant pH conditions. Particle size was discovered to strongly affect the immobilisation of Pb and the transformation from anglesite to chloropyromorphite (Pb5(PO4)3Cl) by the HAPs, in addition to the dissolution properties of HAP. The complete transformation from anglesite to chloropyromorphite was achieved at a pH of 4–5 for 20.08 nm-sized HAPs, whereas incomplete transformations of the 60.24 nm and micro-HAPs were obtained at these pH. At pHs of ≥6 (HAPs of size 20.08 nm) and ≥5 (60.24 nm), the smaller-particle HAPs were incompletely dissolved, and their surfaces were coated by newly formed chloropyromorphite, which was in contrast to the micro-HAPs, which were at pH ≥ 3. These results demonstrated that the surface coating rate decreased when the HAP particle size was decreased; thus, smaller-particle HAPs completely immobilised anglesite by forming chloropyromorphite at higher pH, particularly when the P:Pb molar ratio was high. This study demonstrated that nano-HAPs have potential for use in the situ immobilisation of Pb in low-acidity soil and waste without requiring the P solubility enhancement through the use of acidic conditions or an increase in the number of HAPs.
Introduction
Lead (Pb) contamination is a major and worldwide public health issue because Pb is toxic to humans and animals.1,2 In most cases, Pb is present in Pb-contaminated media in the form of highly soluble minerals such as cerussite (PbCO3), hydrocerussite (Pb3(CO3)2(OH)2), anglesite (PbSO4), and litharge (PbO).3,4 Mao et al.5 estimated the potential toxicity of lead emissions in tailings from 52 countries and eight regions, discovering that the main lead-bearing constituents of tailings are fine particles of the lead minerals galena, cerussite, and anglesite. Liang and Mao6 estimated lead emissions and temporal accumulation according to the evolution of the lead cycle over 70 years. The main species identified were anglesite, litharge, and galena, which together accounted for 61.2% of the total emissions. To remove them, these Pb species must be dissolved and released as Pb(II) depending on their source and the prevailing environmental conditions.3 Such factors may crucially affect Pb exposure and bioavailability. For example, anglesite is commonly present in mine waste and soil, and its Pb bioavailability is determined by kinetic constraints.3,7,8 Furthermore, highly bioavailable Pb species are a primary source of young children's Pb exposure.2 Thus, they are a form of Pb that has substantial control over Pb mobility and reactivity in soil; and if they can be immobilised, their bioavailability can be reduced.Various types of remediation technology have been developed to remediate Pb-contaminated soil, such as excavation, solidification, and chelation/extraction.3,9,10 However, these methods are often ineffective at removing Pb or reducing its bioavailability. Furthermore, they are dangerous and costly. Using apatite materials (AMs) to immobilised Pb(II) has been considered as an alternative and promising method of pollution control because of its cost-effectiveness and less disruptive nature.11–13 The main mechanism of Pb immobilisation using AMs is apatite dissolution and the subsequent precipitation of sparingly soluble pyromorphite-type minerals (PY, Pb5(PO4)3X, where X = F, Cl, Br, or OH).3,8,11 In this dissolution–precipitation mechanism, the pH affecting apatite dissolution, PY precipitation, and particularly the soluble P concentration affect the Pb(II) immobilisation using AMs. Zhang and Ryan used various lead minerals to study the formation of pyromorphite in aqueous systems in the presence of synthetic apatite under varying pH conditions and proposed that at a low pH (<4), the cerussite dissolution rate and cerussite-to-pyromorphite conversion rate are high.14 A high pH (>5) is favourable for the dissolution of galena (PbS) and the transformation of galena into pyromorphite.15 Cao et al.16 reported induced acidic soil conditions that can promote the transformation of Pb(II) into insoluble phosphates. The study also demonstrated the importance of soil pH in determining the effectiveness of Pb immobilisation and revealed that acidic conditions (pH ≤ 5) are favourable for transforming Pb(II) into insoluble PYs. This is because acidic conditions facilitate the dissolution of Pb(II) compounds and the release of Pb(II) from Pb minerals. Acidic conditions also promote the liberation of P from AMs and enhance the formation of PYs. However, the eutrophication of surface water and the possibility of soil acidification remain a concern.3,11,16 Therefore, researchers have mainly focused on Pb(II) immobilisation that utilizes the various physical and chemical properties of AMs, such as particle size, and remarkable Pb(II) immobilisation results have been obtained.
Several studies have suggested that a decrease in AMs particle sizes could substantially enhance Pb(II) immobilisation. Chen et al.17 used a pot experiment to study how the particle size of rock phosphate affected its immobilisation of Pb(II) in Pb-contaminated soil. The results indicated that rock phosphate with a small grain size was superior to that with a large grain size for in situ immobilisation of Pb. This finding was consistent with the literature, in which previous studies have reported that rock phosphate can significantly reduce the phytoavailability of Pb, Zn, Cu, and Cd in soil through immobilisation, and that these effects are strengthened as the particle size of the rock phosphate decreases.18 Da Silva et al.19 investigated the efficiency and mechanisms of lead adsorption by calcium phosphate of two particle sizes from an aqueous solution. Decreasing the biosorbent particle size was discovered to increase the adsorption rate at low initial metal ion concentrations. Similar findings were reported by Simonescu et al.,20 who demonstrated that the highest Pb(II) adsorption was achieved for nanometre-sized rather than micrometre-sized hydroxyapatite (HAP).
The adsorption capacity of AMs is also greatly affected by the AMs particle size for other ions. Da Rocha et al.21 reported that Cd(II) adsorption is controlled by HAP crystallite dimensions and that a decrease in adsorption capacity is induced by an increase in crystal size. The adsorption of copper ions from a Cu solution onto HAP particles with sizes 1323.2 and 82.3 nm at three initial pHs of 4.50, 6.00, and 7.50 was studied by Bazargan-Lari et al.22 The results determined natural HAP's maximum adsorption capacity to be 1.631 mmol g−1 at a pH of 7.5 and with a small particle size. Gao et al.23 identified the size-dependent defluoridation properties of HAPs. HAPs with a small particle size and high adsorption efficiency performed favourably. In our previous study, AMs were used for the adsorption of oxalic acid and fluoride.24–26 However, despite numerous investigations into the effect of AMs particle size, the effect of the particle size of AMs on the formation of pyromorphite at different pH in aqueous solutions using nano-HAPs remains unclear.
Anglesite, a widely available form of Pb – and the bioavailability of which has been the focus of a few studies, was selected in the present study – and reacted with different nano-HAP particle sizes at pH 2–7. The objectives of this study were to investigate the effect of particle size on the immobilisation of Pb(II) using nano-HAPs and the mechanism through which it occurs. The findings of this study may be crucial to understanding the effects of particle size on immobilising Pb(II) from anglesite using nano-HAPs.
Experimental
Sample preparation
All chemicals were analytical grade and purchased from Nanjing Chemical Reagent Co., Ltd. (China). The deionized water used in all experiments was obtained from Milli-Q water purification systems.The nano-HAPs used were synthesized using a precipitation method and a sol–gel method. The precipitation was performed according to the method proposed in ref. 27. Briefly, (NH4)2HPO4 was add to a Ca(NO3)2·4H2O solution at a stoichiometric ratio of Ca:P ratio of 1.67, while the pH was maintained at approximately 11 using an occasional addition of ammonia. The resulting suspension was washed with distilled water and then dried at 110 °C for 24 h. This synthesized sample was denoted Sample A. The nano-HAP was synthesized using a sol–gel method with Ca(NO3)2·4H2O and NH4H2PO4 as the starting materials, ammonia solution to adjust the pH, and EDTA as a complex reagent.28 Aliquots of these complex precursors were decomposed at 800 °C for 4 h. This sample was named Sample B. To provide comparison, micro-HAP was obtained using direct solid-state reactions in the CaCO3 and CaHPO4·2H2O mixture maintained at 1100 °C for 4 h in air. This sample was named Sample C.
The synthetic HAPs were characterised using X-ray diffraction (XRD, Rigaku D/max-2000) and transmission electron microscopy (TEM, Hitachi H-7650) (ESI†). Both TEM and XRD (Scherrer formula) results indicated that the mean particle size in Sample A, B and C was 20.08 nm, 60.24 nm and micrometre, respectively.29
Dissolution of HAPs and anglesite
In each run, 1.0 g of HAP/anglesite was placed in a glass reactor together with 1 L of solution, under a static pH condition (pH 2–7). A 1 L solution containing 0.1 mol L−1 NaNO3 and 0.001 mol L−1 NaCl was adjusted to the desired pH prior to the addition of HAP or anglesite. The suspension maintained at a constant pH was adjusted by adding 0.1 mol L−1 HNO3 and/or NaOH using an automatic titrator. The contents were shaken thoroughly with a stirring paddle at 37 °C for 2 h, which was considered to be the time required for the solution to equilibrate. The suspension was sampled periodically using a 5 mL injector during 2 h reaction period.Reactions between HAPs and anglesite
The reactions between the anglesite (0.454 g) and HAPs occurred at a range of pHs from 2 to 7 and with three P:Pb molar ratios (3:5, 6:5, and 9:5), which provide 1, 2, and 3 times the stoichiometric P/Pb ratio of chloropyromorphite. After adding the anglesite–HAP mixture to 1.0 L solution containing 0.1 mol L−1 NaNO3 and 0.001 mol L−1 NaCl, the contents were continuously stirred for 2 h at a constant pH. The suspension was periodically sampled during the 2 h reaction period, which was determined by preliminary dissolution experiments.
Analytical procedures
The suspension samples were filtered with a 0.22 μm membrane and analysed for soluble Ca, PO4, Pb, and SO4 concentrations using an ICP-AES (Perkin-Elmer Optimal 2100 DV). Solution pH was measured using a pH meter (Thermo 868). After these experiments were finished, the residues were collected and then examined using X-ray diffraction (XRD) with a step-scanning rate of 0.02° 2θ s−1 using Cu Ka (λ = 1.5405 Å) radiation. The morphology and size of the HAPs were characterised using Hitachi Model H-7650 transmission electron microscope (TEM); and scanning electron microscoy (SEM, Hitachi S-3000N), which was equipped with an energy dispersive X-ray (EDX) detector.Results and discussion
Dissolution of HAPs and anglesite
A comparison of the dissolution rates among the HAPs is presented in Fig. 1. Our results indicated that a constant concentration was reached after approximately 1 h (Fig. 1c). The dissolution was inversely related to the pH, and below a pH of 4, the dissolution occurred too rapidly in the first 5 min to be accurately determined under the experimental conditions. At a low pH (2, 3, and 4), the concentrations of Ca and PO4 after 1 h indicated that the added Sample A had nearly dissolved completely, but only 45.16%, 12.52%, and 3.07% of these HAPs had dissolved after 2 h when pHs of 5, 6, and 7 were used, respectively (Fig. 1a and b). The dissolution of Sample B exhibited the same trends as that of Sample A. Sample B was dissolved completely at pHs of 2 and 3, but, it was only 57.61%, 21.67%, 9.66%, and 1.72% dissolved at pHs of 4, 5, 6, and 7, respectively (Fig. 1a and b). Sample C did not completely dissolve at any pH from 2-to-7, but rather 80.50%, 57.62%, 22.24%, 5.67%, 1.76%, and 0.82% was dissolved at pHs of 2–7, respectively (Fig. 1a and b). Notably, the micro-HAPs were significantly less soluble than the nano-HAPs, with Sample A exhibiting the maximal solubility. The notable difference among the three samples was the large particle sizes of Sample B and C. Overall, the solubility of the HAPs increased as the HAP particle size was decreased. The small sized HAPs were generally considered to have stronger structures than the large-sized HAPs, which lead to the higher solubility of Sample A than Sample B and C. Similar results were obtained by Wei et al.30 and Kede et al.,31 who reported that poorly crystallized and small-sized HAPs release more Ca and P than well-crystallized and large-sized HAPs. This result was consistent with a study by Xie et al.,32 which demonstrated that the dissolution of pyromorphite was affected by its particle size.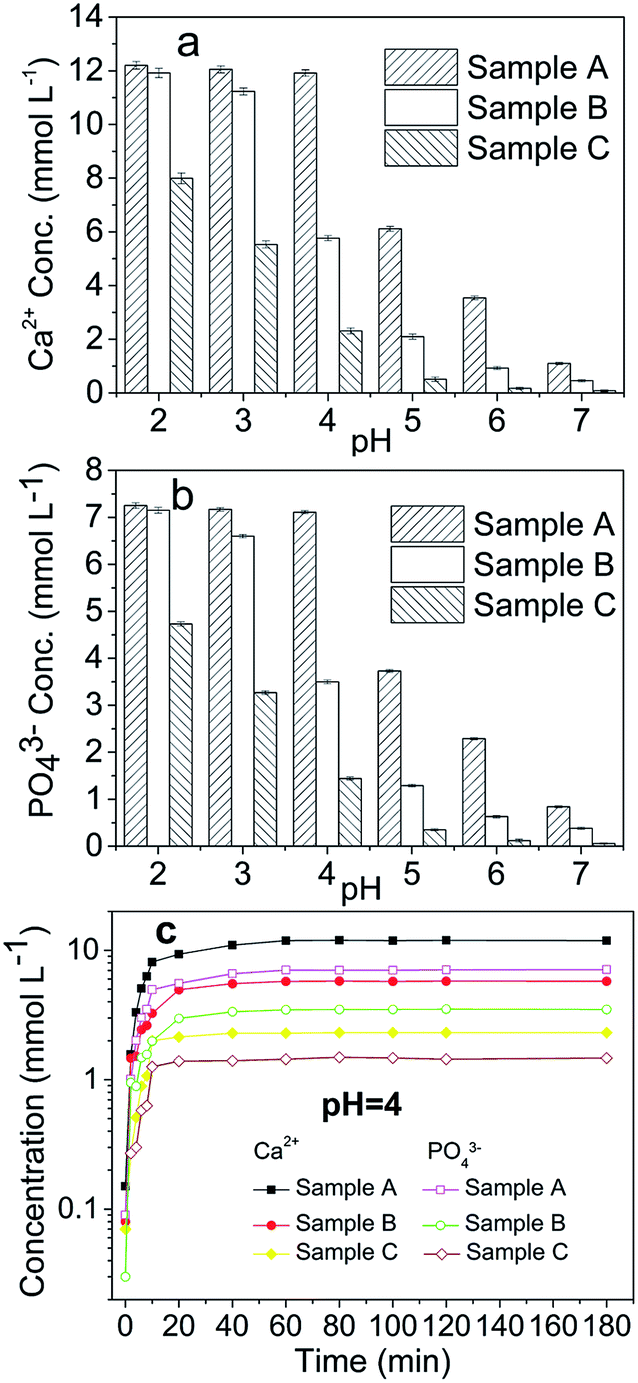 | ||
| Fig. 1 Dissolution of the HAPs with different particle sizes. Samples A, B, and C had average particle sizes of approximately 20.08 nm, 60.24 nm and micrometres-HAP, respectively. | ||
In the present study, the dissolution of anglesite was expressed as a function of the soluble Pb and SO4 concentrations versus time for each pH. The results revealed that dissolution was pH-independent within the pH range 2–7. The concentration of soluble Pb and SO4 in the anglesite suspension was approximately 0.5 mmol L−1 after 60 min of reaction. The anglesite dissolution findings were consistent with those of a study by Zhang.8
Effect of HAP particle size
When phosphate and Pb were mixed at stoichiometric concentrations, the newly formed chloropyromorphite rapidly precipitated in the presence of Cl ions. Therefore, the overall reaction that occurs in the anglesite–HAP system was kinetically controlled by the dissolution of HAPs and/or anglesite. Previous studies have indicated that no other processes, such as adsorption or ion exchange, remove soluble Ca and SO4 in anglesite–HAP suspensions.8 Thus, in the present study, the dissolution of the HAPs and anglesite was determined through the total concentrations of soluble Ca and SO4 in the solution, respectively. Differences between the dissolved and measured PO4 and Pb were attributed to the formation of chloropyromorphite.No formation of chloropyromorphite
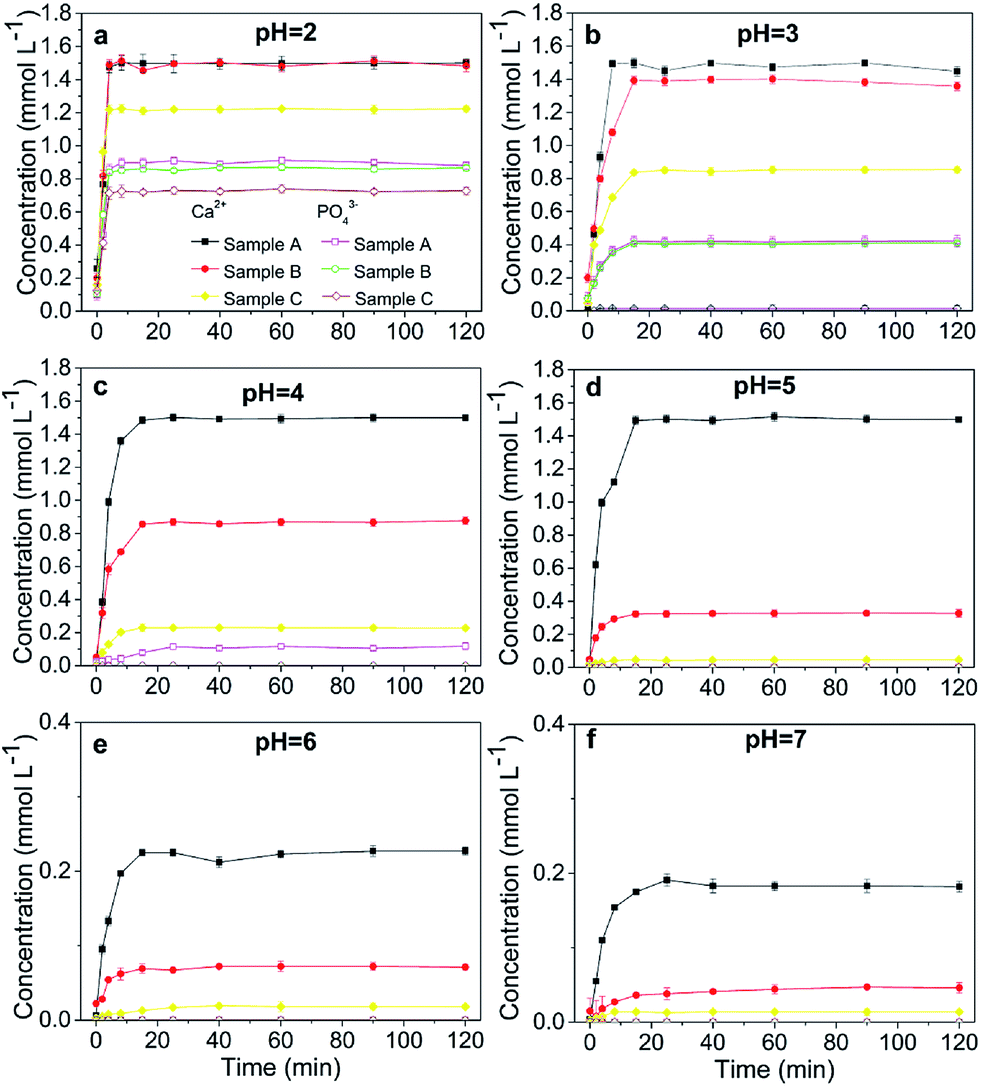
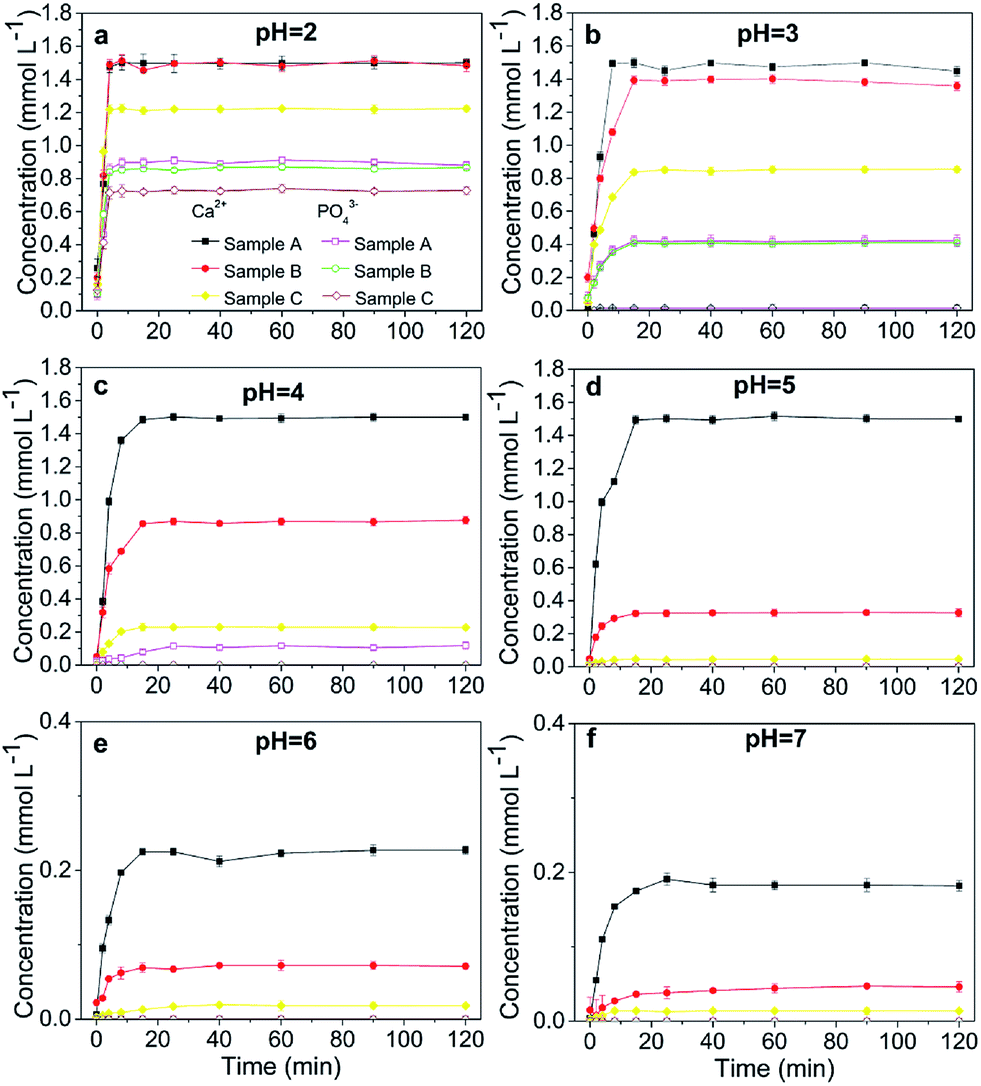
At pH 2, the concentrations of soluble Ca and PO4 varied depending on the HAP particle size. Smaller-particle HAPs exhibited higher solubility (Fig. 2a). However, the solubility of chloropyromorphite was higher at relatively low pH, and only limited chloropyromorphite formed under these conditions. Therefore, no new phase was formed in the solid collected after the reaction of HAPs and anglesite with a P:Pb molar ratio of 3:5, regardless of HAP particle size. These results indicated that only anglesite–Pb was present, which was consistent with the results of XRD (Fig. 4) and SEM (Fig. 5a) analysis of the solids. The concentrations of Ca and PO43−, were approximately 1.40 and 0.9 mmol L−1, respectively, at pH 2 after 10 min reaction for all solutions, which indicated the complete dissolution of Sample A and B (Fig. 2a). Sample C, however, was not completely dissolved after even 120 min (Fig. 2a). The soluble Pb and SO42− concentrations increased with time at nearly equivalent rates, implying that no dissolved Pb was immobilised by HAPs (Fig. 3a). According to the solubility product constant of chloropyromorphite (Ksp = 10−84.4), a small amount of chloropyromorphite should have formed. But none was detected using XRD and SEM. No structural changes in Sample A were detected using XRD analysis of the solid residue, which was obtained after the reaction of Sample A with anglesite at pH 2 (Fig. 4). Lead, phosphorous, chlorine, and calcium were the only elements detected by SEM/EDX elemental microanalysis. SEM/EDX analysis of the solids did also not detect chloropyromorphite after 120 min of reaction, with the predominant mineral was the added anglesite (Fig. 4 and 5a). This may have been due to the strong acidity conditions (pH 2). Although chloropyromorphite was formed, it gradually dissolved. The formed chloropyromorphite may also have been too small in amount to be detected.
 | ||
| Fig. 2 Concentration of the soluble Ca and PO4 in the anglesite–HAP suspension as a function of time with P:Pb = 3:5. Samples A, B, and C had average particle sizes of approximately 20.08 nm, 60.24 nm and micrometres-HAP, respectively. | ||
 | ||
| Fig. 3 Concentration of the soluble Pb and SO4 in the anglesite–HAP suspension as a function of time with P:Pb = 3:5. Samples A, B, and C had average particle sizes of approximately 20.08 nm, 60.24 nm and micrometres-HAP, respectively. | ||
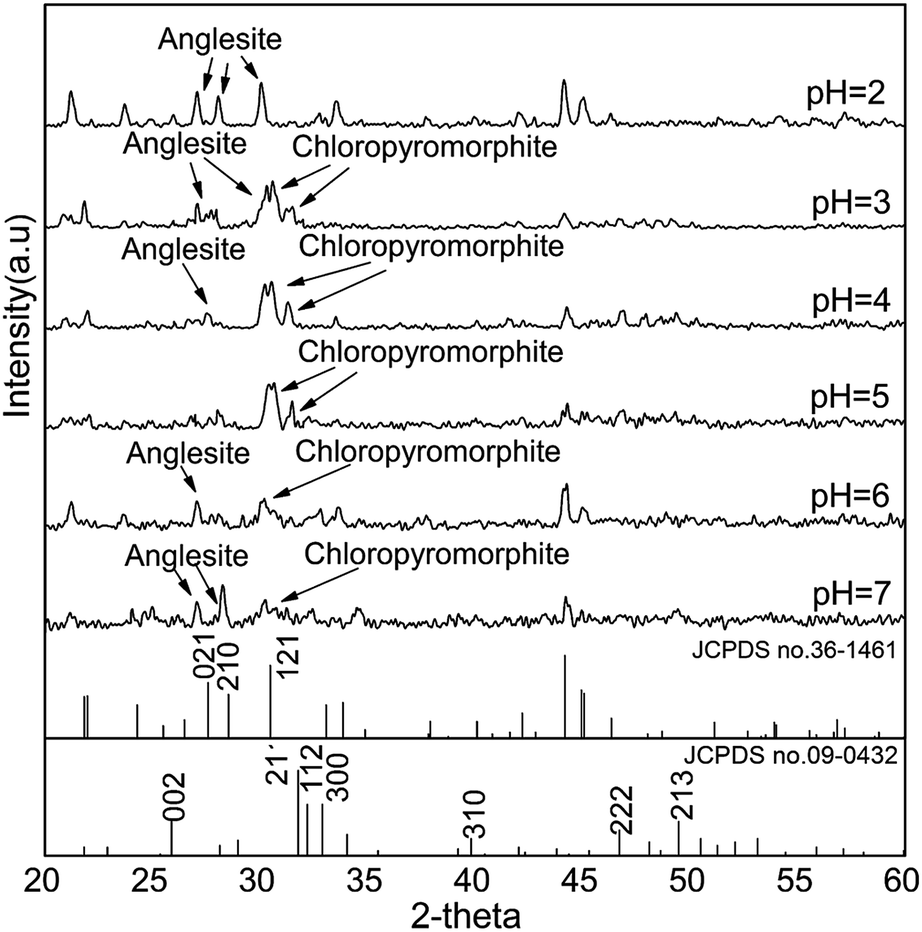 | ||
| Fig. 4 XRD patterns of solids collected from sample A after 120 min of reaction with P:Pb = 3:5. Sample A had average particle size of approximately 20.08 nm. | ||
 | ||
| Fig. 5 SEM images of the solids collected from sample A at pHs of 2 (a), 3 (b), 4 (c), 5 (d), 6 (e), 7 (f). Sample A had average particle size of approximately 20.08 nm. | ||
Formation of chloropyromorphite
The rate of transforming anglesite into chloropyromorphite decreased as the HAP particle size was increased, and a complete transformation was observed at pHs 4–5 in Sample A. The transformation was incomplete in Sample B and C, however, and for these samples the highest transformation rate was obtained at pH levels of 4 and 3, respectively. The transformation rates of the sample for pHs 2–5 were ordered as follows: Sample A (20.08 nm) > Sample B (60.24 nm) > Sample C (micrometres), indicating that anglesite was transformed into chloropyromorphite at the highest rate at low pH and with small HAP particle size.The variation in transformation efficiency was due to the individual dissolution of the HAP samples, and the availability of the soluble P was critical to forming chloropyromorphite (Fig. 1). The micro-HAPs were less soluble than the nano-HAPs; consequently, the micro-HAPs were less reactive in their supply of P for immobilising Pb(II) than the nano-HAPs were in forming chloropyromorphite. The primary mechanism of Pb(II) immobilisation by HAPs was the dissolution of HAPs, which liberated phosphate for the subsequent precipitation of Pb(II) into chloropyromorphite. The precipitation into chloropyromorphite was dependent on the HAP dissolution, a process that released large amounts of P supported the formation of a solid solution, and preceded chloropyromorphite precipitation. This result confirmed those of previous study, in which the effectiveness of rock phosphate at remediating Pb-contaminated soil was affected by the rock phosphate's grain size.17 The present results were also consistent with the findings of Gao et al., which demonstrated that HAPs with a smaller particle size exhibited higher adsorption efficiency.23
Our results indicated that chloropyromorphite formed at pHs higher than 2 for all the HAP samples. However, the chloropyromorphite content in the collected solids differed, with that in Sample A more substantial than that in Sample C. The concentrations of PO4 were 0.42, 0.12, and 10−4 mmol L−1 at pHs of 3, 4, and 5 for Sample A, respectively (Fig. 2). Furthermore, the differences in Ca and PO4 concentration were equivalent to the amount required to precipitate 0.8 (pH 3), 1.3 (pH 4), and 1.5 (pH 5) mmol L−1 dissolved Pb to then form chloropyromorphite. The estimated Pb concentrations were in consistent with the measured concentrations of 0.063, 0.01, and 0.001 mmol L−1 at pHs of 3, 4, and 5, respectively (Fig. 3). Therefore, the proportion of chloropyromorphite in the collected solids increased from 85.5% to 99% when the pH increased from 3 to 5. The conversion rates of Sample B were lower than those of Sample A, which were 85.12% and 96.54% at pHs of 3 and 4, respectively (Fig. 2 and 3); for Sample C, the transformation rate was 62.71% at a pH 3 (Fig. 2 and 3). The excellent Pb(II) immobilisation performance of Sample A was mainly dependent on its special properties. Compared with Sample B and C, Sample A had a smaller particle size, more favourably dispersed morphology, which gave Sample A much higher immobilisation capacity than Sample B and C.
SEM (Fig. 5) and XRD (Fig. 4) both determined that only chloropyromorphite was formed from the reaction of anglesite with the HAPs. The absence of other solid phases was consistent with the results of the study by Ma et al.,33 which reported that Pb immobilisation occurred because of HAP dissolution and PY precipitation. In the present study, the formation of chloropyromorphite was confirmed though analyse of the XRD patterns of the collected solids after anglesite reacted with HAPs at pHs of 3–5 (Fig. 4). The chloropyromorphite phase was easily identified in the spectrum of Sample A by comparing it with that in the JCPDS database (pyromorphite, JCPDS no. 19-0701). The presence of chloropyromorphite (three strongest peaks at 2.89, 2.96, and 2.99 Å) was detected at pHs 3–7. The three strongest peaks (d: 2.07, 3.01, and 3.33 Å) corresponding to anglesite were only clearly observed at a pH of 2. The three strongest peak of hydroxyapatite at 2.72, 2.78, 2.81 Å. As the pH was increased, the XRD peaks for chloropyromorphite became sharper and narrow, indicating that these formed precipitates had a higher degree of crystallinity. XRD demonstrated that the smaller-particle-sized Sample A was almost completely converted into chloropyromorphite at pH 5, suggesting that the immobilisation efficiency of Pb(II) by Sample A was much higher than that of Sample B or C. At pH 3, micrometres needle-shaped particles were observed in addition to the dominant anglesite particles (Fig. 5b). TEM micrographs (Fig. 6a) showed that the original HAP was irregular and needle like particles for Sample A. The average particle size of Sample A was approximately 20 nm. SEM micrographs (Fig. 6b) showed that the original anglesite was spherical particles. The EDX spectrum (Fig. 7a), indicated that the new needle-shaped particles were mostly composed of chloropyromorphite. More needle-shaped particles were produced at pH 4, and only minerals were identified in the reacted solid after 120 min of reaction at pH 5 (Fig. 5c and d). These results indicated that as the pH was increased, the amount of chloropyromorphite produced also increased because Sample A dissolved rapidly at high pH (<6). Some previous studies have also demonstrated that chloropyromorphite formation is favoured when PO43− is present at pH 3–5.8,16 Chloropyromorphite was discovered to be the most stable form of Pb(II).3 The formation of chloropyromorphite substantially decreases Pb bioavailability and improves ecosystem health.16
 | ||
| Fig. 6 TEM images of original HAP (a) and SEM images of original anglesite (b). | ||
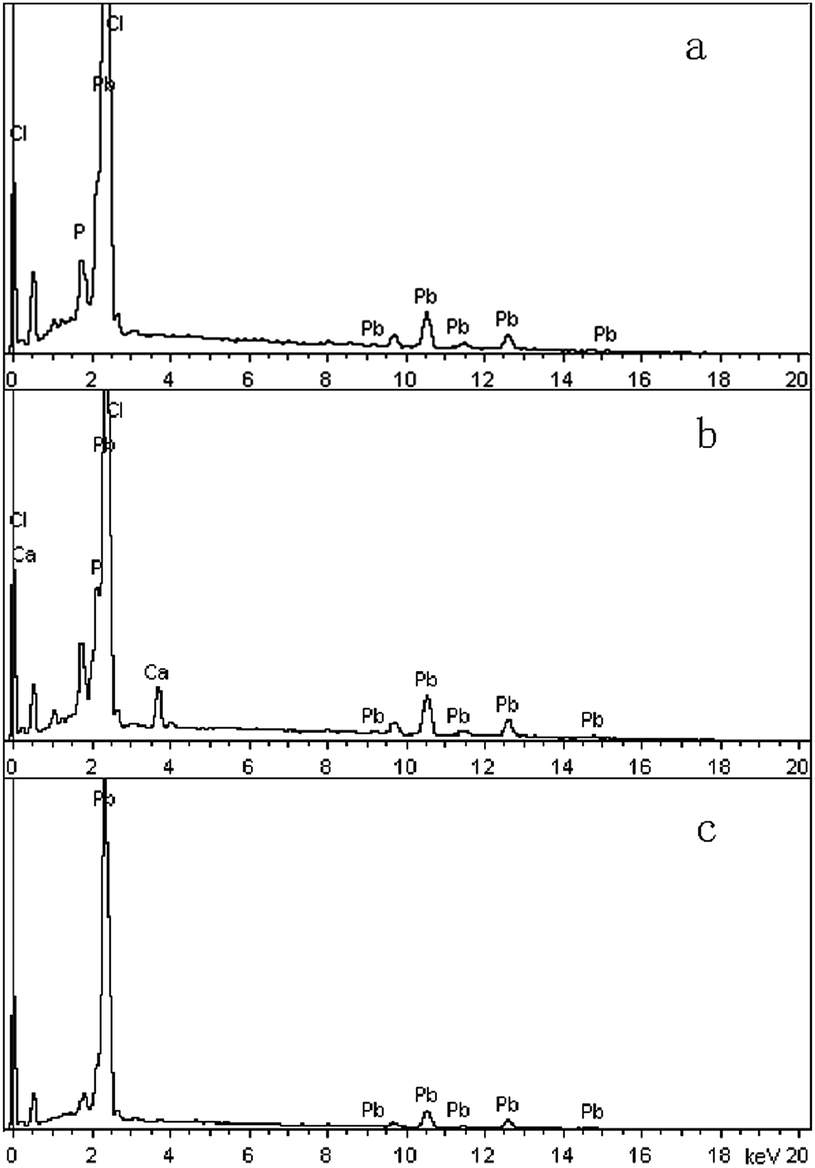 | ||
| Fig. 7 EDX spectra of new solid phase (sample A) at pH 3 (a) and 6 (b), and original anglesite (c). Sample A had average particle size of approximately 20.08 nm. | ||
Coated HAPs
The dissolution of HAPs decreased as the pH was increased, and only an incomplete transformation from anglesite into chloropyromorphite occurred at higher pH; because of insufficient soluble PO4 concentration. The surfaces of the undissolved HAPs were coated by newly formed crystals (Fig. 5e and f). These crystals are attributed to chloropyromorphite, as confirmed by XRD analyses (Fig. 4). Fig. 5 presents the SEM image of sample A after 120 min for immobilisation of Pb(II). The SEM image shows crystals with needle-like morphologies attributed to the new chloropyromorphite phase, which has the same hexagonal symmetry as hydroxyapatite P63/m (Fig. 5). According to Prado da Silva et al.,34 the continuous process of monetite dissolution and HAP reprecipitation maintains the original morphology of the monetite crystals. This topotaxial process was probably favored because of the gradual nanometric HAP crystal formation on the micrometric monetite crystals.34 The EDX spectrum revealed that the composites comprised Ca elements, in addition to P, Cl and Pb (Fig. 6b). The high Ca (weight% 6.95, atomic% 28.56) and Pb (weight% 93.62, atomic% 74.47) content confirmed that the HAP particles were partly coated by chloropyromorphite. Furthermore, the HAP surfaces served as site for chloropyromorphite nucleation,8,35 which was concluded to be rapid and usually homogeneous.36–38 The surface coating might have further decreased the HAP dissolution rate and thus the rate of transformation of anglesite into chloropyromorphite by the HAPs. As the HAP particle size was decreased, precipitation of chloropyromorphite on the surfaces of the undissolved HAPs was achieved at higher pH. A surface coating was observed at pH > 5, pH > 4, and pH > 2 for Samples A, B, and C, respectively. The XRD peaks corresponding to chloropyromorphite became weaker and broader as the pH of the solution was increased from 5-to-7 for Sample A, suggesting that the quantity and crystallinity of the chloropyromorphite decreased as the pH was increased (Fig. 4).Effect of P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Pb ratio
:Pb ratio
Similar results were obtained for reactions involving P:Pb ratios 6:5 and 9:5, as were obtained for P:Pb ratio of 3:5, regardless of the HAP particle size (Fig. 8). When the pH was increased from 2 to 7, the rate of transformation from anglesite into chloropyromorphite first increased and then decreased, which may have been due to the pH-dependent rate of HAP dissolution. Compared with its the complete transformation at pH 4–5 that was achieved with a P:Pb ratio of 3:5, Sample A was completely transformed at higher pHs 4–6 and 4–7 when the P:Pb ratio was 6:5 and 9:5, respectively. Sample B was also completely transformed at pH 4–5; however, Sample C did not completely transform at any P:Pb ratios. In addition, the surfaces of the undissolved HAPs were coated by chloropyromorphite at a pH of ≥7, ≥6, and >2 for Samples A, B, and C, respectively, with P:Pb ratios of either 6:5 or 9:5. This suggested that the surface was coated at higher pH when the P:Pb ratio was increased and that anglesite was completely immobilised under neutral/or alkaline pH conditions. As the P:Pb ratio was increased, the concentration of soluble Pb decreased remarkably, and the rate of transformation from anglesite into chloropyromorphite increased, implying that the soluble PO4 concentration was a limiting factor. The solubility of the HAPs was controlled by their particle size and determined the soluble PO4 concentration. Thus, HAP particle size was the primary rate-limiting factor.
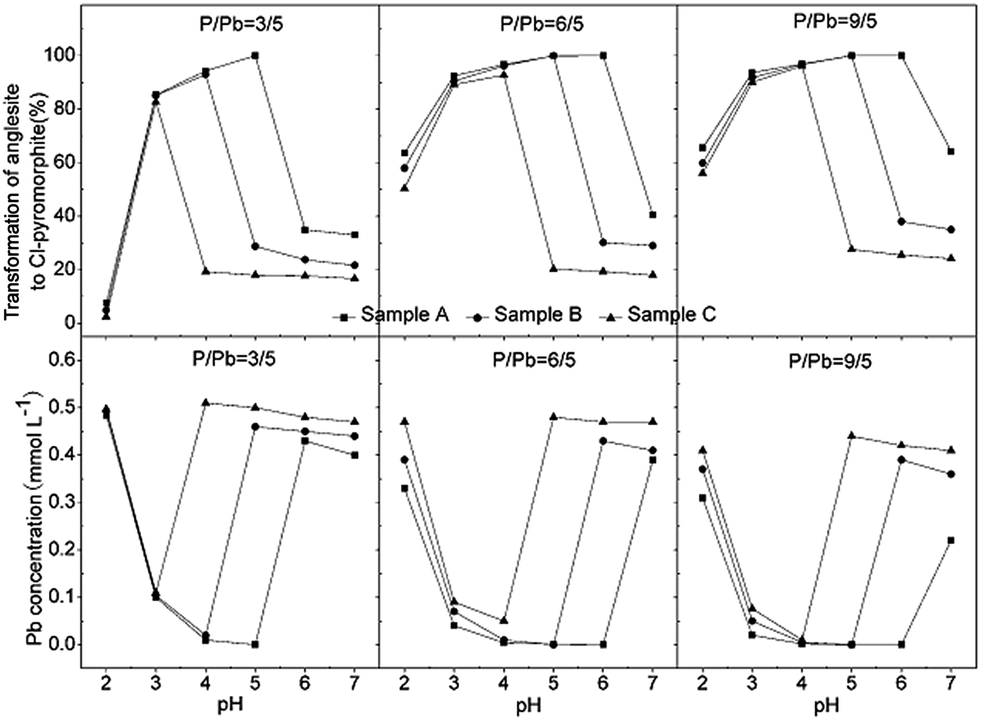 | ||
| Fig. 8 Effect of HAP particle size on the rate of transformation from anglesite into chloropyromorphite and on the soluble Pb concentration in the anglesite–hydroxyapatite suspension after 120 min of reaction for each of the three P:Pb ratios. Samples A, B, and C had average particle sizes of approximately 20.08 nm, 60.24 nm and micrometres-HAP, respectively. | ||
Conclusions
The rates of transformation from anglesite into chloropyromorphite using the three particle-sized HAPs was ordered Sample A (20.08 nm) > Sample B (60.24 nm) > Sample C (micro-HAPs), indicating that the immobilisation efficiency of the nano-HAPs was superior to that of the micro-HAPs. As the HAP particle size was decreased, the surfaces of the undissolved HAPs were coated by newly formed chloropyromorphite at a higher pH value, and particularly at a high P:Pb ratio, indicating that HAP particle size was a rate-limiting factor. The results of this study revealed that smaller particle size of HAPs, particularly nano-HAPs, may be for effective Pb immobilisation, with this process forming chloropyromorphite under neutral/and alkaline pH conditions, which expands the applicability of HAPs to a broader pH range.
Acknowledgements
This work is supported by the National Programs for High Technology Research and Development of China (no. 2007AA10Z406), the National Natural Science Foundation of China (no. 21307002), the Foundation of Anhui Educational Committee (no. KJ2013B120), the Doctoral Program of Higher Education (no. 044-K05000000290).Notes and references
- J. Zheng, T. Huynh, M. Gasparon, J. Ng and B. Noller, Environ. Sci. Pollut. Res., 2013, 20, 8404 CrossRef CAS PubMed.
- E. Deshommes and M. Prevost, Environ. Sci. Technol., 2012, 46, 6269 CrossRef CAS PubMed.
- G. M. Hettiarachchi and G. M. Pierzynski, Environ. Prog., 2004, 23, 78 CrossRef CAS.
- H. Henry, M. F. Naujokas, C. Attanayake, N. T. Basta, Z. Cheng, G. M. Hettiarachchi, M. Maddaloni, C. Schadt and K. G. Scheckel, Environ. Sci. Technol., 2015, 49, 8948 CrossRef CAS PubMed.
- J. S. Mao, J. Cao and T. E. Graedel, Environ. Pollut., 2009, 157, 2670 CrossRef CAS PubMed.
- J. Liang and J. Mao, Environ. Sci. Pollut. Res., 2015, 22, 7129 CrossRef CAS PubMed.
- M. V. Ruby, A. Davis, J. H. Kempton, J. W. Drexler and P. D. Bergstrom, Environ. Sci. Technol., 1992, 26, 1242 CrossRef CAS.
- P. Zhang, Environ. Sci. Technol., 1998, 32, 3318 CrossRef CAS.
- Y. Song, M. Ammami, A. Benamar, S. Mezazigh and H. Wang, Environ. Sci. Pollut. Res., 2016, 11, 10577 CrossRef PubMed.
- D. Wu, S. Wen and J. Deng, New J. Chem., 2015, 1922 RSC.
- P. Miretzky and A. Fernandez-Cirelli, Environ. Chem. Lett., 2008, 6, 121 CrossRef CAS.
- C. Zhang, M. Zhu, G. Zeng, Z. Yu, F. Cui, Z. Yang and L. Shen, Environ. Sci. Pollut. Res., 2016, 23, 4370 CrossRef PubMed.
- H. E. Khal and N. H. Batis, New J. Chem., 2015, 3597 RSC.
- P. Zhang and J. A. Ryan, Environ. Sci. Technol., 1999, 33, 625 CrossRef CAS.
- P. Zhang and J. A. Ryan, Environ. Sci. Technol., 1999, 33, 618 CrossRef CAS.
- X. Cao, L. Q. Ma, S. P. Singh and Q. Zhou, Environ. Pollut., 2008, 152, 184 CrossRef CAS PubMed.
- S. B. Chen, Y. G. Zhu and Y. B. Ma, J. Hazard. Mater., 2006, 134, 74 CrossRef CAS PubMed.
- Z. Zhao, G. Jiang and R. Mao, J. Soil Sci. Plant Nutr., 2014, 14, 258 Search PubMed.
- E. A. Borges Da Silva, C. A. E. Costa, V. J. P. Vilar, C. M. S. Botelho, M. B. Larosi, J. M. P. Saracho and R. A. R. Boaventura, Water, Air, Soil Pollut., 2012, 223, 989 CrossRef.
- C. M. Simonescu, A. Tatarus, C. Tardei, D. Patroi, M. Dragne, D. C. Culita, R. Patescu, L. T. Busuioc and I. Melinte, Rev. Chim., 2015, 66, 732 CAS.
- N. C. C. Da Rocha, R. C. De Campos, A. M. Rossi, E. L. Moreira, A. D. F. Barbosa and G. T. Moure, Environ. Sci. Technol., 2002, 36, 1630 CrossRef CAS PubMed.
- R. Bazargan-Lari, M. E. Bahrololoom, A. Nemati and Z. Salehi, J. Food, Agric. Environ., 2011, 9, 652 Search PubMed.
- S. Gao, R. Sun, Z. Wei, H. Zhao, H. Li and F. Hu, J. Fluorine Chem., 2009, 130, 550 CrossRef CAS.
- Y. Wang, N. Chen, W. Wei, J. Cui and Z. Wei, Desalination, 2011, 276, 161 CrossRef CAS.
- Y. Wang, X. Q. Dong, B. H. Zhou and Z. B. Xu, Adv. Mater. Res., 2012, 610, 390 Search PubMed.
- W. Wei, X. Wang, Y. Wang, M. Xu, J. Cui and Z. Wei, Desalin. Water Treat., 2014, 52, 6219 CrossRef CAS.
- I. Mobasherpour, M. S. Heshajin, A. Kazemzadeh and M. Zakeri, J. Alloys Compd., 2007, 430, 330 CrossRef CAS.
- M. Long, F. Hong, W. Li, F. Li, H. Zhao, Y. Lv, H. Li, F. Hu, L. Sun, C. Yan and Z. Wei, J. Lumin., 2008, 128, 428 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- W. Wei, J. Cui and Z. Wei, Chemosphere, 2014, 105, 14 CrossRef CAS PubMed.
- M. L. F. M. Kede, E. Mavropoulos, N. C. C. D. Rocha, A. M. Costa, M. H. P. D. Silva, J. C. Moreira and A. M. Rossi, Surf. Coat. Technol., 2012, 206, 2810 CrossRef CAS.
- L. Xie and D. E. Giammar, Environ. Sci. Technol., 2007, 41, 8050 CrossRef CAS PubMed.
- Q. Y. Ma, S. J. Traina, T. J. Logan and J. A. Ryan, Environ. Sci. Technol., 1993, 27, 1803 CrossRef CAS.
- M. H. Prado da Silva, J. H. C. Lima, G. A. Soares, C. N. Elias, M. C. de Andrade, S. M. Best and I. R. Gibson, Surf. Coat. Technol., 2001, 137, 270 CrossRef CAS.
- L. Wang, C. V. Putnis, E. Ruiz-Agudo, H. E. King and A. Putnis, Environ. Sci. Technol., 2013, 47, 13502 CrossRef CAS PubMed.
- S. K. Lower, P. A. Maurice, S. J. Traina and E. H. Carlson, Am. Mineral., 1998, 83, 147 CrossRef CAS.
- S. K. Lower, P. A. Maurice and S. J. Traina, Geochim. Cosmochim. Acta, 1998, 62, 1773 CrossRef CAS.
- M. Manecki, P. A. Maurice and S. J. Traina, Am. Mineral., 2000, 85, 932 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28770k |
| This journal is © The Royal Society of Chemistry 2017 |