
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePodophyllotoxin–pterostilbene fused conjugates as potential multifunctional antineoplastic agents against human uveal melanoma cells
Lei Zhang *a,
Jing Wang*a,
Lai Liua,
Chengyue Zhenga,
Yang Wanga,
Yongzheng Chena and
Gang Weib
*a,
Jing Wang*a,
Lai Liua,
Chengyue Zhenga,
Yang Wanga,
Yongzheng Chena and
Gang Weib
aSchool of Pharmacy, Zunyi Medical University, 201 Dalian Road, Zunyi 563003, P. R. China. E-mail: lzhang@zmc.edu.cn; wangjing@zmc.edu.cn
bCSIRO Manufacturing Flagship, PO Box 218, Lindfield, NSW 2070, Australia
First published on 7th February 2017
Abstract
Uveal melanoma is the most common primary intraocular malignancy with a high tendency for early metastasis. There is an urgent need for novel anticancer agents for the therapy of uveal melanoma. In this paper, two novel conjugates of podophyllotoxin–pterostilbene were prepared and evaluated for their cytotoxicity against human uveal melanoma cells (MUM-2B and C918) by the CCK-8 assay. Conjugate B1 exhibited a significant IC50 value of 0.081 ± 0.004 μM against MUM-2B cells. Treatment of MUM-2B cells with B1 caused S cell cycle arrest through reductions in CyclinB1, CDK1 and CDK2 levels. In addition, B1 showed antimigratory activity by down-regulating the expression of VEGFR-2 and MMP-2, and up-regulating the level of E-cadherin. Furthermore, B1 treatment resulted in the induction of apoptosis as characterized by Hoechst 33342 staining, flow cytometry and cleavage of procaspases-3, -8, and -9. Finally, B1 significantly inhibited TOPOIIα and TOPOIIβ expression, simultaneously suppressing the ERK1/2 and AKT pathways in MUM-2B cells.
Introduction
Uveal melanoma, a rare form of melanoma, is the most frequent primary intraocular malignancy in adults.1 Unfortunately, uveal melanoma has a high tendency for early metastasis, especially spreading hematogenously to the liver.2 Meanwhile, uveal melanoma cells are highly resistant to antineoplastic drugs.3 Untill now, there are lack of effective treatments for uveal melanoma. Therefore, there is an urgent need for novel anticancer agents for the therapy of uveal melanoma patients.4Natural products have been widely and successfully used in treating multiple diseases, such as carcinoma.5,6 Podophyllotoxin (PPT, 1, Fig. 1), a well-known non-alkaloid toxin lignan exhibiting great anti-tumor activity by inhibiting tubulin polymerization,7 is isolated from Podophyllum hexandrum.8,9 However, due to unacceptable toxic side effects (e.g., myelosuppression, liver injury and neutropenia), it is unable to be applied in cancer treatment.10 To overcome the faults, numerous structural modifications of podophyllotoxin have been undertaken to generate some analogs with better pharmacological characters,11 such as etoposide (2, Fig. 1) and teniposide (3, Fig. 1), which are used for the treatment of small-cell lung cancer, leukaemia, neuroblastoma and non-Hodgkin's lymphoma.12 In recent years, hundreds of derivatives have been synthesized from the skeleton of PPT, with higher antineoplastic profiles, such as GL-331, QS-ZYX-1-61 and Tafluposide.13 Notably, several researchers showed that PPT and its derivatives presented cytotoxicity in skin melanoma. For instance, Kamal et al. reported that PPT and its derivatives displayed significant antiproliferative activity against A375 and B16 skin melanoma cells.14,15 However, whether PPT and its derivatives exert antitumor effect in uveal melanoma remains unclear.
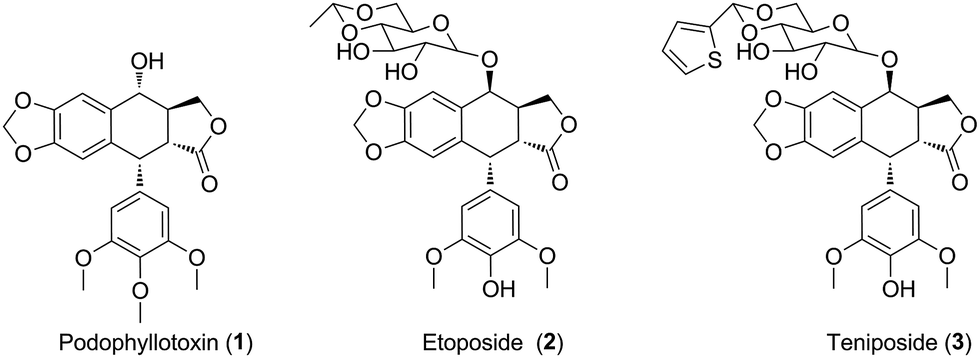 | ||
| Fig. 1 The structures of podophyllotoxin and its analogues. | ||
Inspired by the molecules hybridization concept,16 in this study, we described the design of two novel podophyllotoxin conjugates, with the natural product pterostilbene (trans-3,5-dimethoxy-4′-hydroxystilbene, PTS, 4, Fig. 2),17,18 a dimethylated analog of resveratrol (5, Fig. 2). PTS, existed in many fruits (such as blueberries and vaccinium berries), showed diverse pharmacological properties,19–24 including anticancer, antioxidant, anti-diabetic, hypolipidemic activities and anti-inflammation. It was further confirmed that PTS had no toxic effect on mice at high does.25 Additionally, previous studies reported that PTS possessed anticarcinogenic activity against skin melanoma cells.26,27
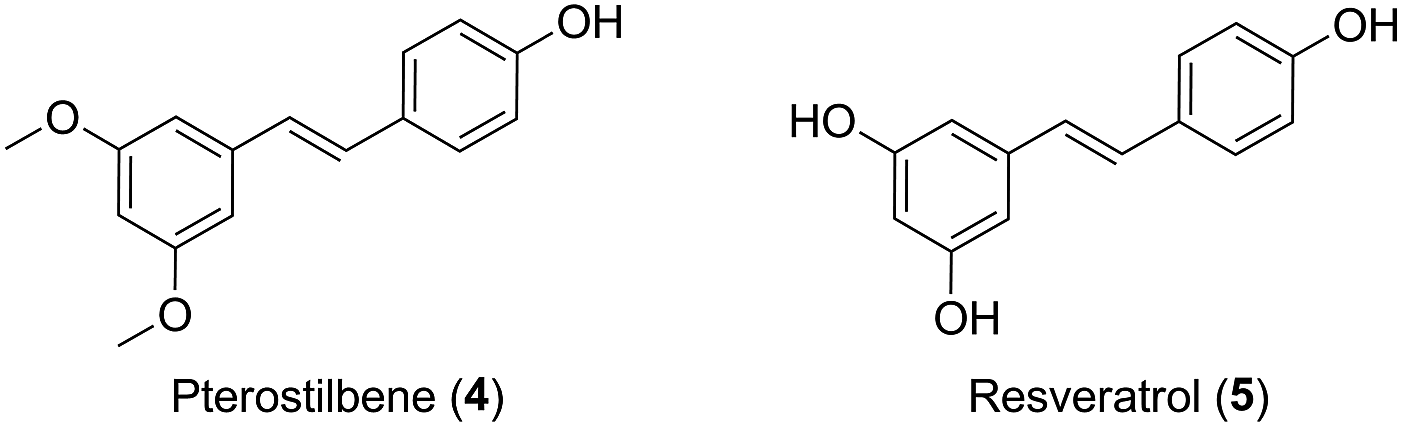 | ||
| Fig. 2 Examples of pterostilbene and resveratrol. | ||
It's well known that PPT shows significant cytotoxicity in both of cancer and normal cells. Therefore, it is an urgent need to solve the toxicity issue of PPT. To reduce the toxicity of PPT and find new multifunctional antineoplastic agents based on the PPT skeleton in our group,28–30 here we reported the design, synthesis and antiproliferative activity of novel podophyllotoxin–pterostilbene conjugates using molecules hybridization (Fig. 3) in human uveal melanoma cells (MUM-2B and C918), simultaneously, the underlying molecular mechanisms were also investigated.
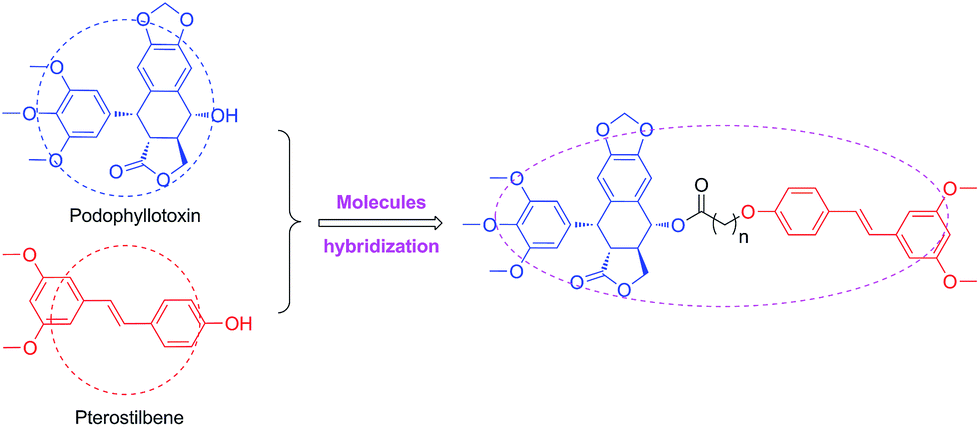 | ||
| Fig. 3 Design of targeted molecules. | ||
Results and discussion
The hybrid compounds (B1 and B2) were synthesized as shown in Scheme 1. Firstly, PPT was reacted with chloroacetyl chloride or 3-chloropropionyl chloride to provide the intermediates 6 or 7. Then, compounds 6 or 7 were reacted with PTS in the presence of potassium carbonate and potassium iodide to give the target molecules B1 or B2. The structures of podophyllotoxin–pterostilbene conjugates were confirmed by 1H NMR, 13C NMR and HR-MS.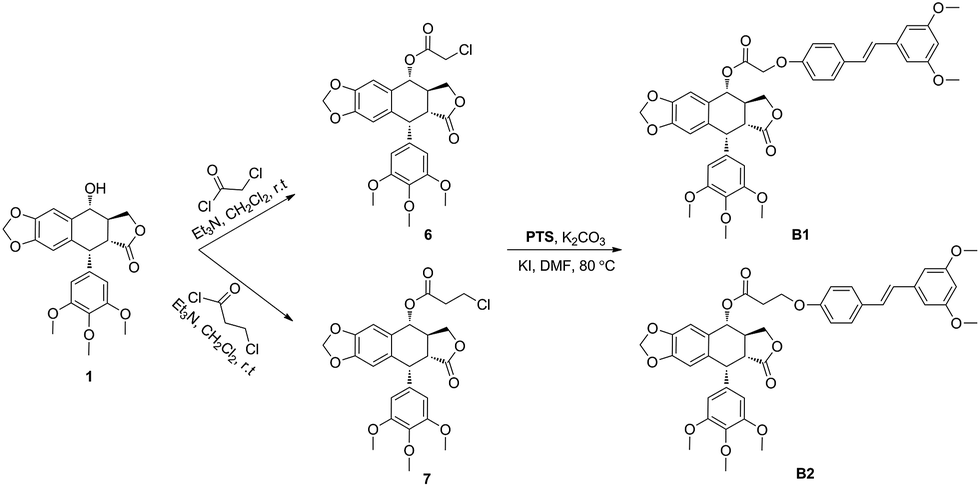 | ||
| Scheme 1 Synthesis of podophyllotoxin–pterostilbene conjugates. | ||
The novel podophyllotoxin–pterostilbene conjugates B1 and B2 were tested by CCK-8 assay for their in vitro antineoplastic activity against human uveal melanoma cells (MUM-2B and C918) and human umbilical vein endothelial cells (HUVEC). PPT, PTS and clinical drug etoposide were used as positive compounds. The results were summarized in Table 1. All tested molecules displayed potent cytotoxicity. Positive compound PPT exhibited nonspecific cytotoxicity against MUM-2B, C919 and HUVEC cells with IC50 values of 0.016 ± 0.006, 0.012 ± 0.002 and 0.018 ± 0.008 μM, respectively, which showed that PPT not only had strong antiproliferative activity, but also possessed high toxicity. In addition, PTS showed weaker cytotoxic activity than etoposide against above three cells with IC50 values ranging from 40.314 ± 9.862 to 80.809 ± 15.742 μM. Interesting, hybrid compounds B1 and B2 showed significant antiproliferative activity compared with PTS. Our data showed that conjugates B1 and B2 displayed less cytotoxic activity than PPT, which may be involved with the solubility property. In addition, B1 showed better anticancer effect against MUM-2B (0.081 ± 0.004 μM) than C918 cells (0.347 ± 0.054 μM), showing 497- and 196-fold more cytotoxic than that of PTS (40.314 ± 9.862 and 68.124 ± 7.422 μM), respectively. Moreover, the IC50 value of B1 against human normal HUVEC cells was 0.362 ± 0.109 μM which was less cytotoxicity than that of PPT, indicating that conjugate had less toxicity. From the data of B1 and B2, conjugate displayed less anticancer activity with longer linker, at the same time, exhibited less toxicity. It was found that MUM-2B cells were more sensitive to conjugate as well as PTS than C918 cells. Furthermore, the solubility property and more details in the structure–activity relationship need further study.
| Compound | IC50a (μM) | ||
|---|---|---|---|
| MUM-2B | C918 | HUVEC | |
| a Data were expressed as mean IC50 ± SD (μM) from three independent experiments. | |||
| B1 | 0.081 ± 0.004 | 0.347 ± 0.054 | 0.362 ± 0.109 |
| B2 | 0.613 ± 0.307 | 1.035 ± 0.152 | 2.265 ± 0.862 |
| PPT | 0.016 ± 0.006 | 0.012 ± 0.002 | 0.018 ± 0.008 |
| PTS | 40.314 ± 9.862 | 68.124 ± 7.422 | 80.809 ± 15.742 |
| Etoposide | 1.965 ± 0.221 | 1.279 ± 0.221 | 2.255 ± 0.224 |
To investigate the effect of B1 on the cell cycle, MUM-2B cells were treated with 0.1 μM B1 for 48 h, and then analyzed by flow cytometry. PPT and PTS were used as positive compounds. As shown in Fig. 4, after treatment with 0.1 μM B1, 77.69% of cell numbers were accumulated in S phase, compared with 16.54% cells accumulation in S phase in control group. Meanwhile, the percentages of cells in S phase were 81.42% and 74.11%, respectively, in 0.01 μM PPT and 50 μM PTS-treated groups. It was revealed that conjugate B1 could induce MUM-2B cells arrest at S phase.
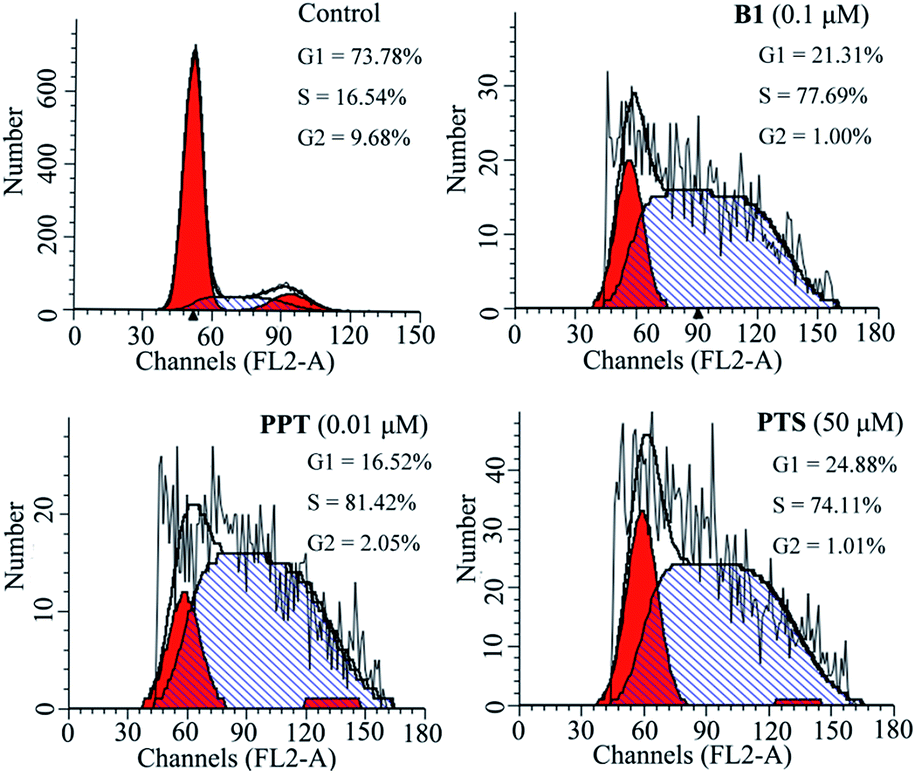 | ||
| Fig. 4 Effects of conjugate B1, PPT and PTS on the cycle of MUM-2B cells. Cells were treated with vehicle, 0.1 μM B1, 0.01 μM PPT or 50 μM PTS for 48 h. | ||
Next, to validate whether the inhibitory effect of B1 on MUM-2B cells proliferation are accompanied by apoptosis, MUM-2B cells were treated with 0.1 μM B1 for 48 h, and the apoptotic cells were analyzed by flow cytometry. PPT and PTS were used as positive compounds. We observed that the rate of apoptosis in 0.1 μM B1-treated group was 37.72% (Fig. 5), which were stronger than that of the control group (4.16%). Similarly, apoptotic cell number increased to 32.95% and 48.73% when the cells were incubated with 0.01 μM PPT and 50 μM PTS, respectively. These results indicated that conjugate B1 could induce apoptosis in MUM-2B cells.
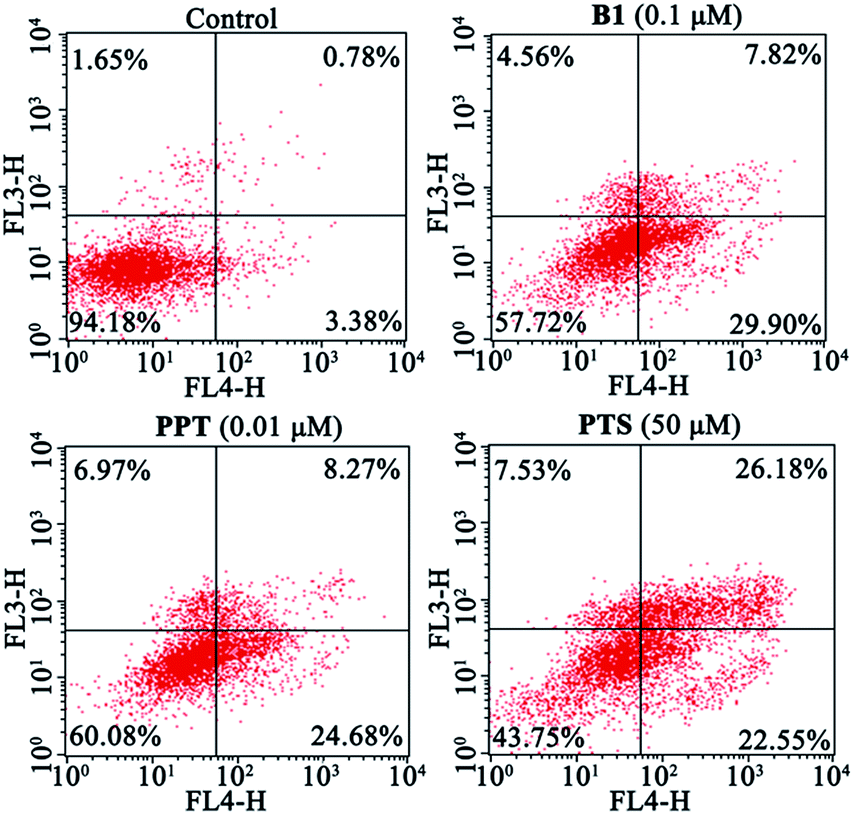 | ||
| Fig. 5 Effects of conjugate B1, PPT and PTS on the apoptosis of MUM-2B cells. Cells were treated with vehicle, 0.1 μM B1, 0.01 μM PPT or 50 μM PTS for 48 h. | ||
Furthermore, to confirm the induction of apoptosis of B1, MUM-2B cells were tested by Hoechst 33342 staining. As seen in Fig. 6, treatment with 0.1 μM B1 led to cell shrinkage and chromatin condensation. Meanwhile, bright blue fluorescent and condensed nuclei in apoptosis cells were observed by treatment with 0.1 μM B1. Similar effects were observed when cells were incubated with 0.01 μM PPT and 50 μM PTS. There were no significant characteristics of apoptosis in control group. The above results significantly proved that conjugate B1 was effective in inducing MUM-2B cells apoptosis.
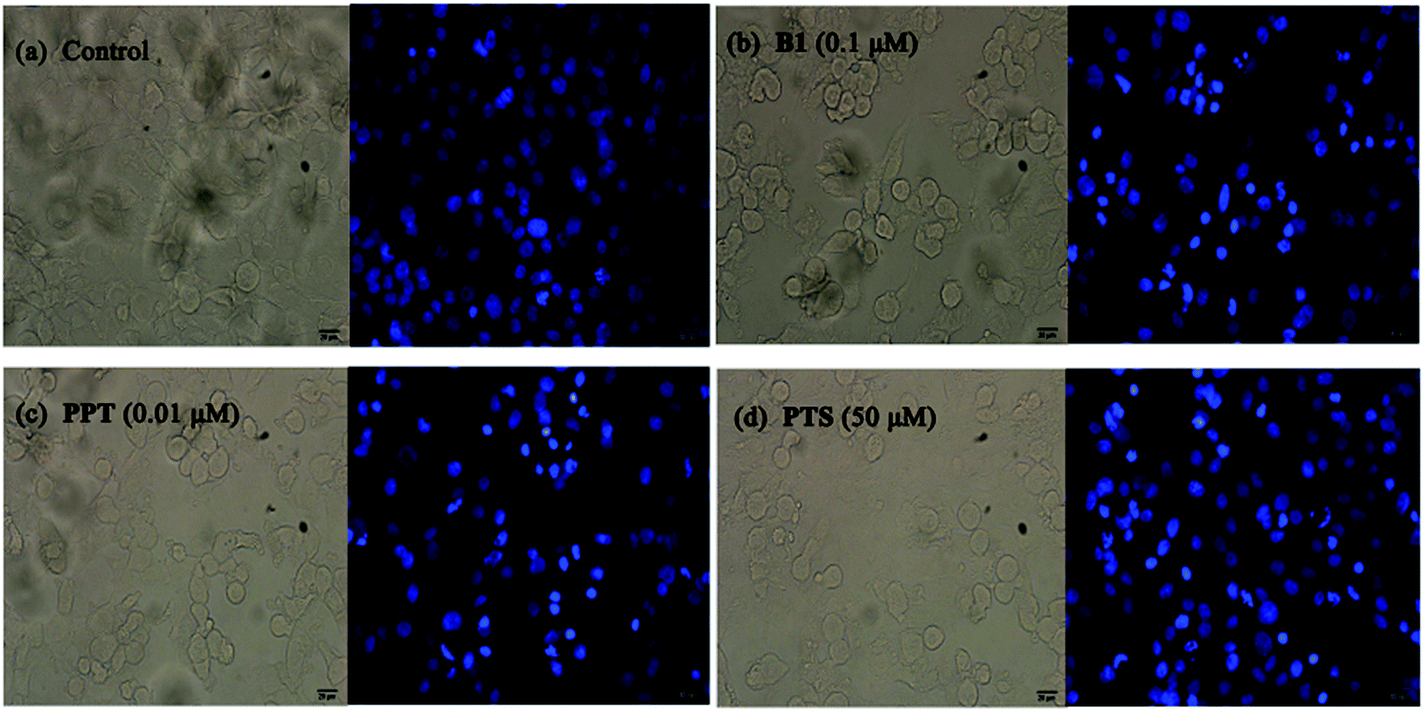 | ||
| Fig. 6 Effects of conjugate B1, PPT and PTS on the morphology of MUM-2B cells. Cells were treated with vehicle, 0.1 μM B1, 0.01 μM PPT or 50 μM PTS for 24 h, then stained with Hoechst 33342 for microscopy. (a) Control group; (b) 0.1 μM B1; (c) 0.01 μM PPT; (d) 50 μM PTS. Magnification: 200×, scale 20 μm. | ||
Recently, some studies reported that PPT and PTS both had the potential to suppress tumor migratory and metastasis.31–33 To further test the effect of conjugate B1 on the migration potential of cancer cells, wound healing assay was conducted on MUM-2B cells in vitro. As shown in Fig. 7, after treatment with 0.1 μM B1, 0.01 μM PPT or 50 μM PTS for 24 h, the migration of MUM-2B cells was suppressed obviously, compared with the control group. These data indicated that conjugate B1 might have the potential to inhibit the migratory and metastasis of MUM-2B cells.
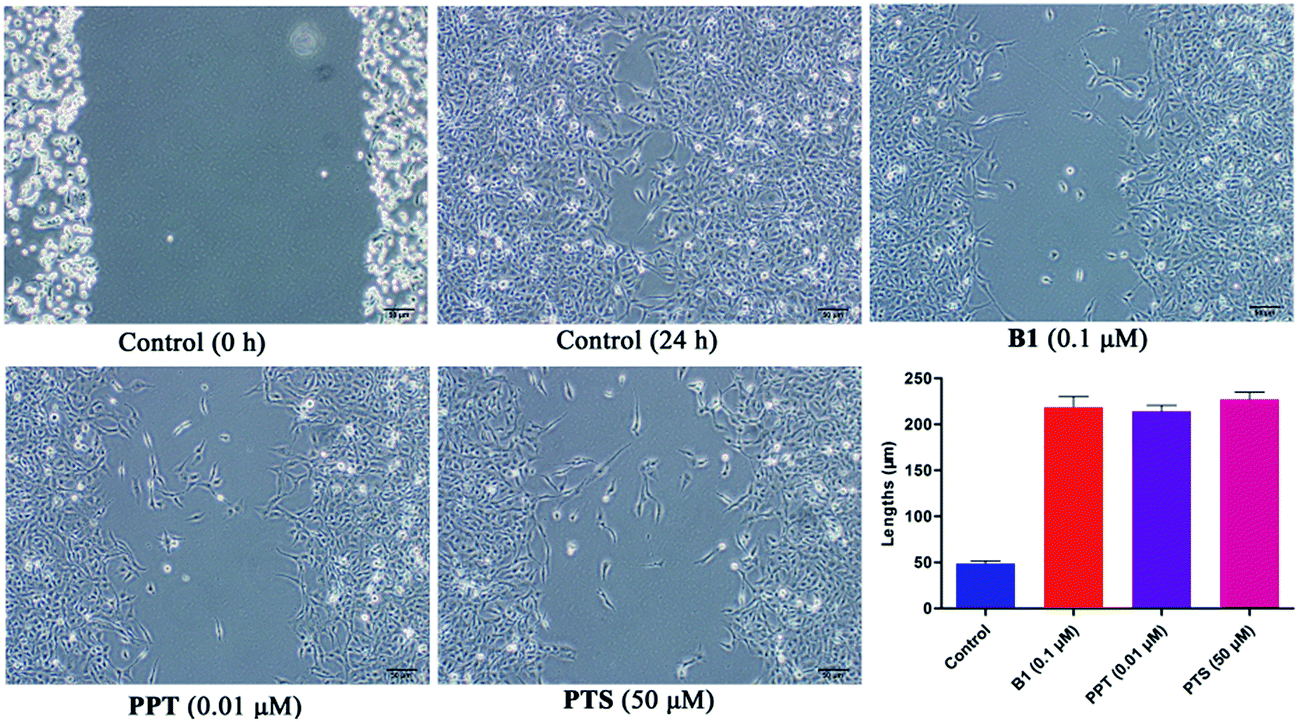 | ||
| Fig. 7 Effects of conjugate B1, PPT and PTS on the migration of MUM-2B cells in vitro. Cells were treated with vehicle, 0.1 μM B1, 0.01 μM PPT or 50 μM PTS for 24 h. Columns mean and bars given here follow ±SD of three independent experiments. Magnification: 100×, scale 50 μm. | ||
To evaluate the mechanisms involved in S cell cycle arrest in MUM-2B cells, the effects of B1 on expression of cell cycle-related proteins were examined by western blotting using PPT and PTS as positive compounds. As shown in Fig. 8A, 0.1 μM B1 significantly decreased the expression levels of CDK1, CDK2 and CyclinB1, compared with the vehicle-treated control, however, CyclinA level in MUM-2B cells was not changed. Similar effects were observed when cells were incubated with 0.01 μM PPT and 50 μM PTS. Taken together, these data proved that conjugate B1 blocked MUM-2B cells in the S phase by down-regulation of CDK1, CDK2 and CyclinB1.
 | ||
| Fig. 8 Effects of conjugate B1, PPT and PTS on the cell cycle- and apoptosis-related proteins expression, respectively, in MUM-2B cells by western blotting using β-actin as a control. (A) Control MUM-2B cells; (B) MUM-2B cells treated with 0.1 μM B1; (C) MUM-2B cells treated with 0.01 μM PPT; (D) MUM-2B cells treated with 50 μM PTS. | ||
In order to further investigate the mechanisms involved in apoptosis induced by B1, the expression of apoptotic proteins (the cleavage states of caspase 3, 8 and 9) in MUM-2B cells was examined by western blotting (Fig. 8B). PPT and PTS were used as positive compounds. The data revealed that 0.1 μM B1-treatment dramatically increased the relative levels of cleaved caspase 3, 8 and 9 in MUM-2B cells. Similar effects were observed when cells were incubated with 0.01 μM PPT and 50 μM PTS. Collectively, these findings strongly demonstrated that conjugate B1-induced apoptosis of MUN-2B cells was mediated by signaling cascade of apoptosis.
Our above findings showed that conjugate B1 possessed antimigratory activity in MUM-2B cells. Furthermore, Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2), Matrix Metalloproteinase-2 (MMP-2) and E-cadherin are crucial events of invasion and metastasis in malignant cancer cells.34–36 Thus, we next further assess the roles of above metastasis-related proteins in B1-treated MUM-2B cells by western blotting. As shown in Fig. 9A, treatment with 0.1 μM B1 significantly increased the expression level of E-cadherin, simultaneously, down-regulated the levels of VEGFR-2 and MMP-2 in MUM-2B cells, compared with the control group. Interestingly, the levels of VEGFR-2 and E-cadherin were not altered by 0.01 μM PPT, whereas, 50 μM PTS did, suggesting that the antimigratory activity of B1 was related with the structure of PTS. These findings suggested that the antimigratory effect of conjugate B1 was mediated by VEGFR-2, MMP-2 and E-cadherin.
 | ||
| Fig. 9 Effects of conjugate B1, PPT and PTS on the metastasis-, TOPO- and inflammatory-related proteins expression, respectively, in MUM-2B cells by western blotting using β-actin as a control. (A) Control MUM-2B cells; (B) MUM-2B cells treated with 0.1 μM B1; (C) MUM-2B cells treated with 0.01 μM PPT; (D) MUM-2B cells treated with 50 μM PTS. | ||
Ren et al. demonstrated that PPT derivative could inhibit the expression of DNA topoisomerase IIα in HepG2 cells.37 Consequently, we next investigated the effects of B1 on the DNA topoisomerase IIα (TOPOIIα) and DNA topoisomerase IIβ (TOPOIIβ) proteins expression by western blotting. As shown in Fig. 9B, 0.1 μM B1 demonstrated the ability to decrease the expression of TOPOIIα and TOPOIIβ in MUM-2B cells. Similar effect was observed when cells were incubated with 0.01 μM PPT. Interestingly, 50 μM PTS could down-regulate the TOPOIIα expression level, while, increase the expression of TOPOIIβ. Collectively, the results indicated that conjugate B1 could lower the expression levels of TOPOIIα and TOPOIIβ in MUM-2B cells.
It was reported that PTS showed anti-inflammatory and anticarcinogenic activity by inhibition of COX-2 and iNOS.38–40 Thus, the expression of inflammatory-related proteins in B1-treated MUM-2B cells was performed by western blotting. As seen in Fig. 9C, treatment with 0.01 μM B1 could not alter the expression levels of COX-2 and iNOS in MUM-2B cells. Similar effect was observed in 0.01 μM PPT group. However, 50 μM PTS could down-regulate the COX-2 level. The results confirmed that the cytotoxic activity of conjugate B1 against MUM-2Bcells might not be mediated by COX-2 and iNOS.
More recently, we showed that PPT and its analogue could stimulate the ERK1/2 pathway in K562/adr cells.41 To further clarify the molecular mechanisms underlying the activity of B1, we finally investigated the effects of B1 on the ERK1/2, AKT and STAT3 signaling in MUM-2B cells. Cells were treated with vehicle, 0.1 μM B1, 0.01 μM PPT or 50 μM PTS for 48 h. Phosphorylation of ERK1/2, AKT and STAT3 was determined by western blotting. Fig. 10 showed that treatment with 0.1 μM B1 significantly suppressed the phosphorylation of ERK1/2 and AKT in MUM-2B cells, however, the phosphorylation of STAT3 was not altered, compared with the control group. Similar effects were observed when cells were treated with 0.01 μM PPT and 50 μM PTS. These results suggested that conjugate B1 might inhibit the activation of the ERK1/2 and AKT pathways, which contributed to its antineoplastic activity in MUM-2B cells.
 | ||
| Fig. 10 Effects of conjugate B1, PPT and PTS on the ERK1/2, AKT and STAT3 signaling in MUM-2B cells by western blotting using β-actin as a control. (A) Control MUM-2B cells; (B) MUM-2B cells treated with 0.1 μM B1; (C) MUM-2B cells treated with 0.01 μM PPT; (D) MUM-2B cells treated with 50 μM PTS. | ||
Experimental
General
Melting points were taken on a SGWX-4 meltingpoint apparatus. High-resolution mass spectra (HR-MS) were performed on an Agilent Accurate-Mass-Q-TOF-MS 6520. The 1H and 13C NMR spectra were recorded on an Agilent-NMR-vnmrs (400 MHz) instrument using TMS as internal standard.Preparation of intermediates 6 and 7
To a solution of PPT (1.9 mmol, 1 eq.) and triethylamine (4 eq.) in dry dichloromethane (15 mL) for at 0 °C, chloroacetyl chloride or 3-chloropropionyl chloride (2.5 eq.) was added. The mixture was stirred under argon at room temperature for 1 h. After that, the mixture was quenched with NH4Cl (5 mL) for 10 min and extracted with dichloromethane (3 × 10 mL). The combined organic phase was washed with brine, then dried over anhydrous MgSO4 and evaporated under reduced pressure. The crude was further purified by column chromatography (dichloromethane/methanol = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give the intermediate.
:1) to give the intermediate.
Preparation of target compounds B1 and B2
To a solution of 6 or 7 (0.25 mmol, 1 eq.), potassium iodide (0.01 eq.), potassium carbonate (1 eq.) in DMF (4 mL), PTS (1.1 eq.) was added, and the reaction mixture was stirred under argon at 80 °C for 1 h. The reaction mixture was cooled, added to water, and extracted with dichloromethane (3 × 10 mL). The combined organic phase was washed with brine, then dried over anhydrous MgSO4 and evaporated under reduced pressure. The crude was further purified by column chromatography (ethyl acetate/petroleum ether = 1:2–1:4) to give targeted molecule.
Pharmacology
000g at 4 °C for 10 min, and then determined using BCA Protein Assay kit. The proteins were separated by 10% SDS-PAGE, and then transferred to a nitrocellulose membrane, which was blocked with 5% skim milk for 2 h and incubated with primary antibodies: CDK1, CDK2, CyclinA, CyclinB1, cleaved caspase 3, cleaved caspase 8, cleaved caspase 9, VEGFR-2, MMP-2, E-cadherin, TopoIIα, TopoIIβ, COX-2, iNOS, p-ERK1/2, p-AKT, p-STAT3 and β-actin overnight at 4 °C. After washing, membranes were then treated with secondary antibodies at room temperature for 2 h. The immunoreactive bands were visualized using enhanced chemiluminescence detection system and quantitated with using Gel-Pro32 software.Conclusions
In summary, two novel conjugates of podophyllotoxin–pterostilbene were synthesized and evaluated against human uveal melanoma cell lines in vitro. Compound B1 exhibited a significant IC50 value of 0.081 ± 0.004 μM against MUM-2B cells. Treatment with B1 induced MUM-2B cell cycle arrest at S phase and apoptosis, as well as showed antimigratory activity. Furthermore, B1 significantly inhibited TOPOIIα and TOPOIIβ expression, simultaneously, suppressed the ERK1/2 and AKT pathways in MUM-2B cells. The antitumor mechanisms of B1 were presented in Fig. 11. Overall, our findings indicated that conjugate B1 might be a potent antineoplastic agent for uveal melanoma chemotherapy, however, metabolic stability evaluation and anticancer efficacy in vivo of conjugates still need our further studies.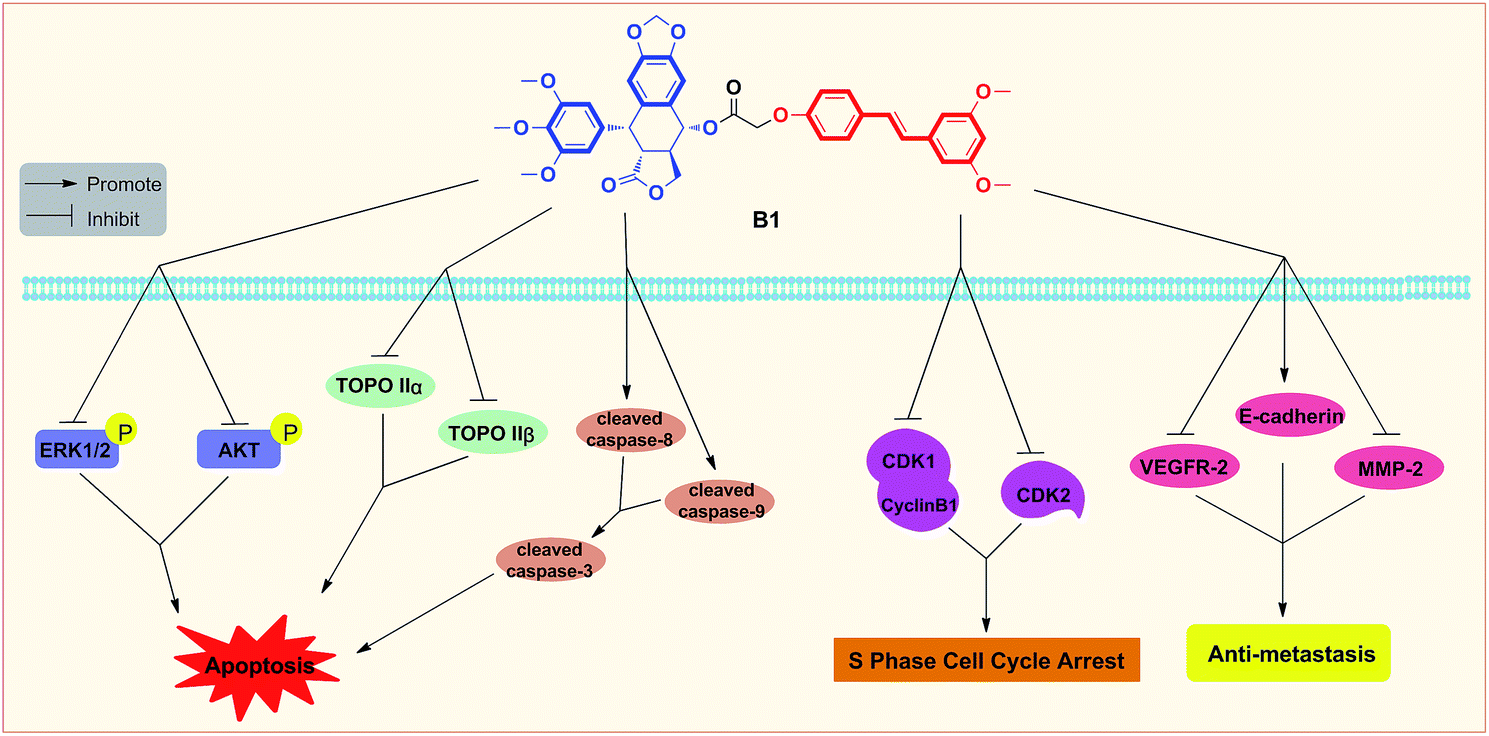 | ||
| Fig. 11 Proposed diagrams of antineoplastic activity of conjugate B1 against human uveal melanoma MUM-2B cells. | ||
Acknowledgements
This work was financially supported by the Department of Science and Technology of Guizhou Province (No. [2014]7565, [2014]7557, [2015]6010), Ministry of Education “Chunhui Project” Foundation of China (No. Z2015008) and Discipline Construction Funding of Zunyi Medical University.Notes and references
- A. D. Singh, M. E. Turell and A. K. Topham, Ophthalmology, 2011, 118, 1881–1885 CrossRef PubMed.
- S. E. Coupland, S. L. Lake, M. Zeschnigk and B. E. Damato, Eye, 2013, 27, 230–242 CrossRef CAS PubMed.
- P. L. Blanco, L. A. Lim, C. Miyamoto and M. N. Burnier, Melanoma Res., 2012, 22, 334–340 CrossRef PubMed.
- P. A. O'Neill, M. Butt, C. V. Eswar, P. Gillis and E. Marshall, Melanoma Res., 2006, 16, 245–248 CrossRef PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- Y. Damayanthi and J. W. Lown, Curr. Med. Chem., 1998, 5, 205–252 CAS.
- M. Gordaliza, P. A. García, J. M. Miguel del Corral, M. A. Castro and M. A. Gómez-Zurita, Toxicon, 2004, 44, 441–459 CrossRef CAS PubMed.
- H. Xu, M. Lv and X. Tian, Curr. Med. Chem., 2009, 16, 327–349 CrossRef CAS PubMed.
- L. Bohlin and B. Rosen, Drug Discovery Today, 1996, 1, 343–351 CrossRef CAS.
- Y. Q. Liu, J. Tian, K. Qian, X. B. Zhao, S. L. Morris-Natschke, Y. Liu, X. Nan, X. Tian and K. H. Lee, Med. Res. Rev., 2015, 35, 1–62 CrossRef PubMed.
- Y. You, Curr. Pharm. Des., 2005, 11, 1695–1717 CrossRef CAS PubMed.
- A. Kamal, S. M. Ali Hussaini, A. Rahim and S. Riyaz, Expert Opin. Ther. Pat., 2015, 25, 1025–1034 CrossRef CAS PubMed.
- A. Kamal, A. Mallareddy, P. Suresh, V. L. Nayak, R. V. C. R. N. C. Shetti, N. S. Rao, J. R. Tamboli, T. B. Shaik, M. V. P. S. Vishnuvardhan and S. Ramakrishna, Eur. J. Med. Chem., 2012, 47, 530–545 CrossRef CAS PubMed.
- A. Kamal, J. R. Tamboli, M. V. P. S. Vishnuvardhan, S. F. Adil, V. L. Nayak and S. Ramakrishna, Bioorg. Med. Chem. Lett., 2013, 23, 273–280 CrossRef CAS PubMed.
- B. Meunier, Acc. Chem. Res., 2008, 41, 69–77 CrossRef CAS PubMed.
- A. M. Rimando, M. Cuendet, C. Desmarchelier, R. G. Mehta, J. M. Pezzuto and S. O. Duke, J. Agric. Food Chem., 2002, 50, 33453–33457 CrossRef.
- E. S. Park, Y. Lim, J. T. Hong, H. S. Yoo, C. K. Lee, M. Y. Pyo and Y. P. Yun, Vasc. Pharmacol., 2010, 53, 61–67 CrossRef CAS PubMed.
- C. Stockley, P. L. Teissedre, M. Boban, C. Di Lorenzo and P. Restani, Food Funct., 2012, 10, 995–1007 Search PubMed.
- L. Pari and M. A. Satheesh, Life Sci., 2006, 79, 641–645 CrossRef CAS PubMed.
- A. M. Rimando, R. Nagmani, D. R. Feller and W. Yokoyama, J. Agric. Food Chem., 2005, 53, 3403–3407 CrossRef CAS PubMed.
- W. Nutakul, H. S. Sobers, P. Qiu, P. Dong, E. A. Decker, D. J. McClements and H. Xiao, J. Agric. Food Chem., 2011, 59, 10964–10970 CrossRef CAS PubMed.
- C. P. Ko, C. W. Lin, M. K. Chen, S. F. Yang, H. L. Chiou and M. J. Hsieh, Oral Oncol., 2015, 51, 593–601 CrossRef CAS PubMed.
- M. H. Pan, Y. S. Chiou, W. J. Chen, J. M. Wang, V. Badmaev and C. T. Ho, Carcinogenesis, 2009, 30, 1234–1242 CrossRef CAS PubMed.
- M. J. Ruiz, M. Fernández, Y. Picó, J. Mañes, M. Asensi, C. Carda, G. Asensio and J. M. Estrela, J. Agric. Food Chem., 2009, 57, 3180–3186 CrossRef CAS PubMed.
- P. Ferrer, M. Asensi, R. Segarra, A. Ortega, M. Benlloch, E. Obrador, M. T. Varea, G. Asensio, L. Jordá and J. M. Estrela, Neoplasia, 2005, 7, 37–47 CrossRef CAS PubMed.
- C. M. Remsberg, J. A. Yanez, Y. Ohgami, K. R. Vega-Villa, A. M. Rimando and N. M. Davies, Phytother. Res., 2008, 22, 169–179 CrossRef CAS PubMed.
- L. Zhang, F. Chen, J. Wang, Y. Chen, Z. Zhang, Y. Lin and X. Zhu, RSC Adv., 2015, 5, 97816–97823 RSC.
- L. Zhang, Z. Zhang, J. Wang, Y. Chen, F. Chen, Y. Lin and X. Zhu, RSC Adv., 2016, 6, 2895–2903 RSC.
- L. Zhang, F. Chen, Z. Zhang, Y. Chen and J. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 38–42 CrossRef CAS PubMed.
- J. H. Cho, W. G. Hong, Y. J. Jung, J. Lee, E. Lee, S. G. Hwang, H. D. Um and J. K. Park, Tumor Biol., 2015, 37, 1–11 Search PubMed.
- K. K. Mak, A. T. Wu, W. H. Lee, T. C. Chang, J. F. Chiou, L. S. Wang, C. H. Wu, C. Y. F. Huang, Y. S. Shieh, T. Y. Chao, C. T. Ho, G. C. Yen and C. T. Yeh, Mol. Nutr. Food Res., 2013, 57, 1123–1134 CAS.
- K. Li, S. J. Dias, A. M. Rimando, S. Dhar, C. S. Mizuno, A. D. Penman, J. R. Lewin and A. S. Levenson, PLoS One, 2013, 8, e57542 CAS.
- K. J. Huang and L. H. Sui, Med. Oncol., 2012, 29, 318–323 CrossRef CAS PubMed.
- S. Valastyan and R. A. Weinberg, Cell, 2011, 147, 275–292 CrossRef CAS PubMed.
- N. Khan and H. Mukhtar, Cancer Metastasis Rev., 2010, 29, 435–445 CrossRef PubMed.
- J. Ren, L. Wu, W. Q. Xin, X. Chen and K. Hu, Bioorg. Med. Chem. Lett., 2012, 22, 4778–4782 CrossRef CAS PubMed.
- C. S. Lai, J. H. Lee, C. T. Ho, C. B. Liu, J. M. Wang, Y. J. Wang and M. H. Pan, J. Agric. Food Chem., 2009, 57, 10990–10998 CrossRef CAS PubMed.
- Y. S. Chiou, M. L. Tsai, K. Nagabhushanam, Y. J. Wang, C. H. Wu, C. T. Ho and M. H. Pan, J. Agric. Food Chem., 2011, 59, 2725–2733 CrossRef CAS PubMed.
- S. Paul, A. J. DeCastro, H. J. Lee, A. K. Smolarek, J. Y. So, B. Simi, C. X. Wang, R. Zhou, A. M. Rimando and N. Suh, Carcinogenesis, 2010, 31, 1272–1278 CrossRef CAS PubMed.
- L. Zhang, F. Chen, Z. Zhang, Y. Chen, Y. Lin and J. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 4466–4471 CrossRef CAS PubMed.
- L. Zhang, Z. Zhang, F. Chen, Y. Chen, Y. Lin and J. Wang, Eur. J. Med. Chem., 2016, 123, 226–235 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |