
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of thermochemically stable tetraphenyladamantane-based microporous polymers as gas storage materials†
Xiong Li a,
Jianwei Guo*a,
Hangbo Yuea,
Jiawei Wangb and
Paul D. Topham*b
a,
Jianwei Guo*a,
Hangbo Yuea,
Jiawei Wangb and
Paul D. Topham*b
aGuangdong University of Technology, School of Chemical Engineering and Light Industry, China. E-mail: guojw@gdut.edu.cn
bChemical Engineering & Applied Chemistry, Aston University, UK. E-mail: p.d.topham@aston.ac.uk
First published on 14th March 2017
Abstract
In view of environmental pollution control and purification of natural gases, developing ideal porous materials for small gas molecule (hydrogen, methane and carbon dioxide) capture is an important, pressing challenge. Accordingly, herein, three microporous organic polymers (MOP-Ad) have been synthesized by Suzuki coupling polymerization of 1,3,5,7-tetrakis(4-bromophenyl)adamantane “knots” with three phenylboronic acid-type “rods”. Gas adsorption studies of the MOP-Ad materials demonstrated their permanent porosity and good gas storage capabilities (1.07 wt% H2 at 77.3 K and 1.13 bar, 10.3 wt% CO2 and 2.4 wt% CH4 at 273.1 K and 1.13 bar), as well as moderate CO2/CH4 adsorption selectivity. Moreover, high thermal stability (up to 520 °C) and remarkable chemical resistance to strong acids and bases were found in these polymers, making them suitable candidates as gas storage materials in harsh chemical environments.
1. Introduction
Advanced porous organic materials could play a pivotal role in next-generation clean energy technologies to compete with established materials such as zeolites, activated carbons and porous polymeric materials in the field of gas storage/separation, heterogeneous catalysis and chemical sensors, etc.1,2 In view of environmental pollution control and purification of natural gases for fuels, developing ideal porous materials for small gas molecule (hydrogen, methane and carbon dioxide) capture has been a hot topic for some time. Metal–organic frameworks (MOFs),3 covalent–organic frameworks (COFs)4 and microporous organic polymers (MOPs)5 have been reported as the most promising materials for gas capture owing to their large specific surface area, controllable pore structure, low mass density and high flexibility in structural design. For instance, Hong et al. reported that magnesium-based metal–organic frameworks mmen-Mg2(dobpdc), synthesized via solvothermal and microwave methods, displayed excellent capacity for CO2 adsorption (169.84 mg g−1 at 298 K and 1 bar) in combination with CO2 over N2 selectivity of over 96%.6 Yaghi and Furukawa showed that COF-102 and COF-103 materials, prepared by self-condensation reactions of tetra(4-(dihydroxy)borylphenyl)methane and tetra(4-(dihydroxy)borylphenyl)silane, exhibited remarkable gas uptake (1200 mg g−1 and 1190 mg g−1 for CO2 at 298 K and 55 bar),7,8 whilst Han and co-workers designed tetraphenylethylene-based microporous organic polymers, which also gave high CO2/CH4 selectivity (5.4–13.4 at 273 K and 1 bar),9 Zou et al. reported work on a novel charged MOF with a nitrogen-containing tetracarboxylate ligand and indium(III) ions, which exhibited significant CO2 adsorption capacity as well as high CO2 adsorption selectivity over N2 (up to 106 at 273 K and 1 bar) and CH4 (up to 25 at 273 K and 1 bar).10 Despite promising gas absorption capabilities, these porous materials suffer from various structural instabilities under certain conditions, a limiting factor that has not yet been fully addressed. The presence of moisture and other harsh components in mixed gases lead to the degradation of most MOF materials via the hydrolysis of coordinate bonds.11 Likewise, some COF materials suffer from chemical/hydrothermal instability owing to the breakage of reversibly formed bonds, such as boroxines or imines in the structure.7 During the past decade, much attention has also focused on the functionalization of MOP materials for gas separation. For instance, Li and Wang reported a series of nitrogen-rich microporous poly(Schiff-base)s, which exhibited excellent CO2/CH4 selectivity (14, 273 K and 1 bar).12 Senker et al. synthesized series of microporous polyimides (MOPIs), via polycondensation of amine and anhydride linker molecules, which showed high CO2/CH4 selectivity (13, 273 K and 1 bar).13 Nevertheless, the presence of heteroatoms (such as B, N, O, S, etc.) in the MOP backbones, diminished their structural stability,14–16 compared to their non-functionalized counterparts.17,18 Therefore, it is remains essential to search for, and develop porous organic materials that can maintain physicochemical stability especially for adsorption over long periods of time or repeated uses in harsh environments.Adamantane and its derivatives, a class of unique compounds with a relatively rigid three-dimensional framework and high physicochemical stability,19,20 have been employed as suitable building blocks in the preparation of thermostable microporous polymers.21,22 For instance, Han and co-workers15 reported networks of hexakis(4-bromophenyl)benzene and diethynylbenzene (HPOP-2), synthesized via Sonogashira coupling, possessed a high Brunauer–Emmett–Teller (BET) surface area of 742 m2 g−1 but degraded around 280 °C. In contrast, Chang et al.18 designed and synthesized novel MOP materials in the same way and all reagents except that 1,3,5,7-tetrakis(4-iodophenyl)adamantane were used in place of hexakis(4-bromophenyl)benzene, showing a high BET surface area (up to 665 m2 g−1) and a degradation temperature around 340 °C. They attributed the enhanced thermal stability to the introduction of adamantane into the molecular structure.
Substantially building on this pioneering work, thermochemically stable MOP based on adamantane (termed MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3) with three different “rod” lengths have been successfully synthesized via Suzuki coupling polymerization, as reported herein. The synthesized materials possess a crosslinked network comprising solely C–C bonds, which significantly contribute to the chemical and hydrothermal stability of the resulting polymers, and this structural design could also lead to an increase in CO2 capture capacity as well as CO2/CH4 adsorption selectivity.
2. Experimental
2.1 Materials
Adamantane, benzene-1,4-diboronic acid, 4,4′-dibromobiphenyl, p-terphenyl, tetrakis(triphenylphosphine) palladium(0), were purchased from Shanghai Macklin Biochemical Co., Ltd, Sigma Aldrich and TCI and used as received. 1,3,5,7-Tetrakis(4-bromophenyl)adamantane (TBPA),20 biphenyl-4,4′-diboronic acid22 and 4,4′′-dibromo-p-terphenyl23 were prepared according to the procedures described in the literature.2.2 Synthesis of MOP-Ad networks
Synthesis of the MOP-Ad series is outlined in Scheme 1. For MOP-Ad-1, to a mixture of TBPA (0.30 g, 0.40 mmol, 1.0 equiv.), benzene-1,4-diboronic acid (0.15 g, 0.91 mmol) in DMF (100 ml), tetrakis(triphenylphosphine) palladium(0) (45.85 mg, 39.68 μg, 10.0 mol%) and an aqueous solution of K2CO3 (2.0 M, 16.0 ml) were added. The resulting mixture was degassed via freeze–pump–thaw cycles, purging with Ar, and stirring at 150 °C for 72 h. After cooling to room temperature, the mixture was poured into deionized water. The precipitate was collected by filtration, and washed with water to remove the inorganic salts. Further purification of the precipitate was carried out by Soxhlet extracted with water, methanol, acetone, tetrahydrofuran and chloroform (sequentially), each of for 24 h. The polymers were dried under vacuum at 200 °C for 24 h, to give an off-white powder (0.23 g, 97.7% yield). Anal. calcd for C46H36: C, 93.88; H, 6.12. Found: C, 92.54; H, 5.43.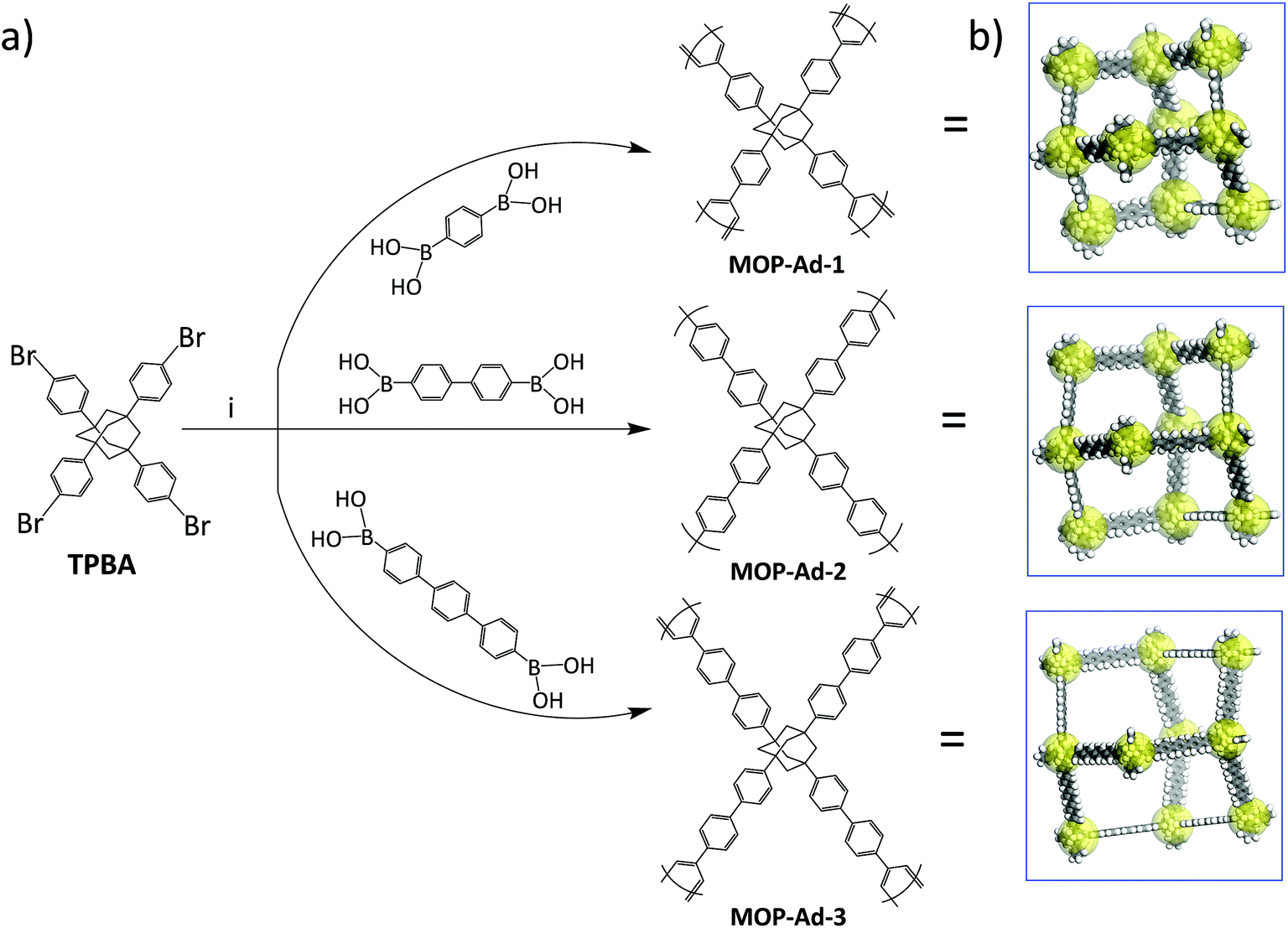 | ||
| Scheme 1 (a) Synthetic routes to the MOP-Ad networks and the notional idealized polymer structures with (b) cartoon 3D representations of the networks in each case. | ||
The synthetic procedure for MOP-Ad-2 and MOP-Ad-3 were similar to that of MOP-Ad-1 except that the “rod” used was 4,4′′-dibromo-p-terphenyl for MOP-Ad-2 and p-terphenyl-4,4′′-diboronic acid for MOP-Ad-3 instead of benzene-1,4-diboronic acid. Yields were 90.2% (anal. calcd for C58H44: C, 94.05; H, 5.95. Found: C, 91.87; H, 6.15 for MOP-Ad-2) and 65.7% (anal. calcd for C70H52: C, 94.17; H, 5.83. Found: C, 91.99; H, 6.58 for MOP-Ad-3) based on hypothetical 100% polycondensation.
2.3 Characterization
1H and 13C NMR spectra were recorded on a Bruker AVANCE III 400 MHz Superconducting Fourier in dimethyl sulfoxide-d6 (DMSO-d6) or deuterated chloroform (CDCl3) and were referenced to external tetramethylsilane (TMS). Solid-state cross polarization magic angle spinning (CP/MAS) NMR spectra were recorded on a Bruker Avance III 400 NMR spectrometer. Fourier transform infrared (FTIR) spectra were obtained using a Thermo Electron Nicolet-6700 spectrometer. Target polymers were pressed into KBr pellets for the spectral measurements. Powder X-ray diffraction (XRD) data were collected on a Bruker X'pertpro multipurpose diffractometer (MPD). Samples were mounted on a sample holder and measured using Cu Kα radiation with a 2θ range of 5° to 80°. Small-angle X-ray scattering (SAXS) was performed with a rotating anode generator (Bruker Nanostar, turbospeed solution) equipped with a pinhole camera, and using monochromatized CuKα (λ = 0.154 nm) radiation collimated with crossed Goebel mirrors. The SAXS profiles for the MOP-Ad networks were collected in the q range from 0.3 nm−1 to 3.5 nm−1. The nitrogen adsorption–desorption isotherms were measured on 3H-2000PM2 analyzer and the adsorption of hydrogen, methane and carbon dioxide analyses was measured on 3H-2000PS2 apparatus at 77 K/1 bar (H2) and 273 K/1 bar (CH4 and CO2). Thermogravimetric analysis (TGA) was performed using a NETZSCH STA 409 PC thermal gravimetric analyzer at a heating rate of 10 °C min−1 in nitrogen. SEM analysis was performed on a Hitachi S-3400N scanning electron microscope to investigate the surface morphologies of the polymers. Elemental analysis was performed with a Perkin Elmer Series II 2400 elemental analyzer. All the samples were dried at 200 °C for 24 h under vacuum prior to measurement.3. Results and discussion
Three MOP-Ad networks were synthesized via Suzuki coupling polymerization, using TPBA as a so-called “knots” to connect benzene-1,4-diboronic acid, 4,4′′-dibromo-p-terphenyl, or p-terphenyl-4,4′′-diboronic acid (TPDBA) “rods”, resulting in MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3, respectively, as summarized in Scheme 1. The moderate yield of MOP-Ad-3 (65.7%) was due to the fact that TPDBA was practically a suspension (insoluble) in solvents because of its “long” rod length and thus its participation in the reaction is somewhat limited in contrast to the synthesis of MOP-Ad-1 (97.7%) and MOP-Ad-2 (90.2%). Nevertheless, this synthetic approach provides relatively simple, targeted structural diversity and selectivity, as compared with Friedel–Crafts coupling,17 with moderate to high yield.3.1 Characterization of MOP-Ad networks
FTIR spectroscopy has been widely used in probing the structure of microporous organic polymers. Fig. 1 shows the FTIR spectra of TBPA and the corresponding three MOP-Ad networks, after subtraction of the KBr background. When comparing the building molecule TBPA with its resulting polymers, one of the biggest differences in the spectra is the disappearance of the band at 1070 cm−1, which arises from the C–Br oscillation.24,25 Furthermore, new bands appeared near 3020 and 1600 cm−1 after the coupling polymerization, which are ascribed to the characteristic adsorption of the conjugated system of biphenyl groups.9,25 The band that appeared at 3430 cm−1 is characteristic of the B–OH end-group vibrations in MOP-Ad samples25 and was higher for MOP-Ad-3, indicative of a higher number of unreacted termini within the network. Overall, the spectral assignments indicate that the designed MOP networks were successfully synthesized, which is further corroborated by nuclear magnetic resonance (NMR) results (Fig. 2).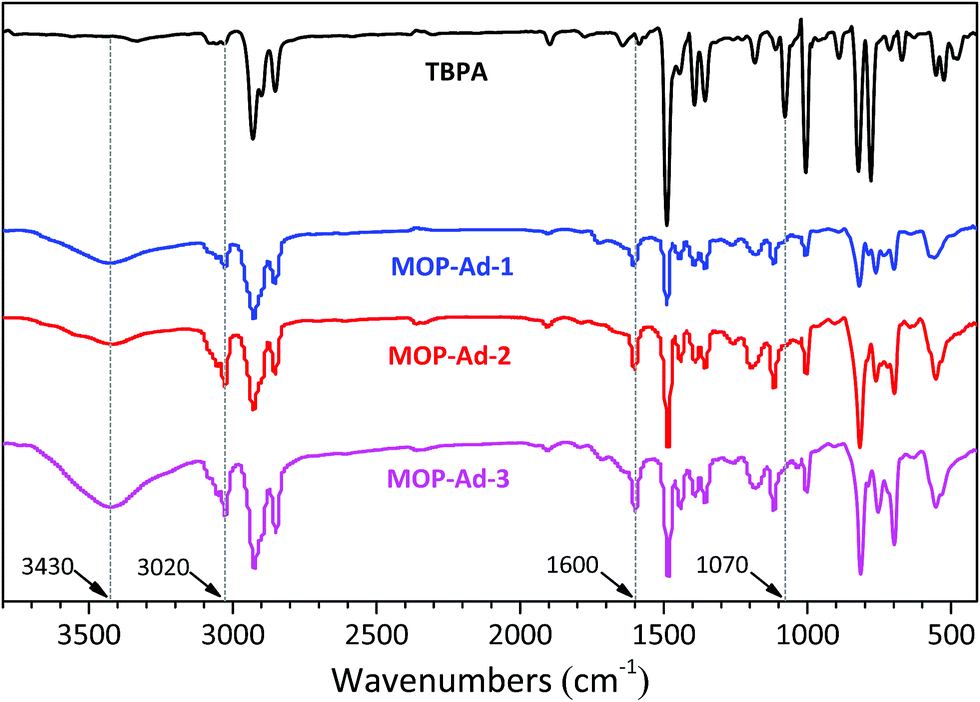 | ||
| Fig. 1 FTIR spectra for TBPA and target polymers, MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3. | ||
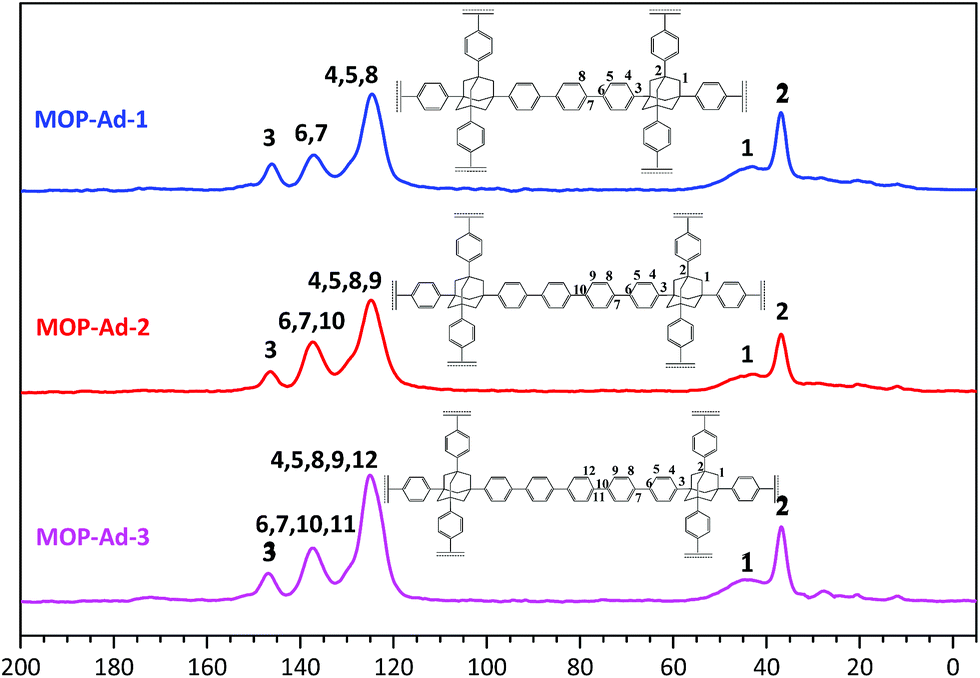 | ||
| Fig. 2 13C CP/MAS NMR spectra of MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3. | ||
13C cross-polarization magic-angle spinning (CP/MAS) NMR spectra with resonance assignment for MOP-Ad networks are shown in Fig. 2. There are five main pronounced peaks. For MOP-Ad-1, the single peaks at 146.1, 137.1 and 124.6 ppm correspond to the substituted phenyl carbons (C3 at 146.1 ppm and C6 & C7 at 137.1 ppm) and unsubstituted phenyl carbons (C4, C5 & C8 at 124.6 ppm). Additionally, peaks at 43.0 and 36.7 ppm correspond to the methylene (C1) and quaternary bridgehead carbon (C2) from adamantane, respectively. For MOP-Ad-2 and MOP-Ad-3, all of these peaks can also be observed in the corresponding spectra in Fig. 2. In addition, no clear, sharp diffraction peaks could be observed in the XRD profiles (see ESI Fig. S2†), indicating that the MOP-Ad networks are amorphous in nature.
3.2 Stability and porosity of MOP-Ad networks
In comparison with most tetrahedral benzene-based microporous organic polymers, such as those from biphenyl monomer PPN-10 (ref. 26) (stable up to 240 °C), HPOP-2 (ref. 15) (stable up to 280 °C) and triptycene-based STP-4 (ref. 14) (stable up to 300 °C), the three MOP-Ad networks in our work do not show a dramatic weight-loss until up to 520 °C (Fig. 3), and exhibit decomposition temperatures over 600 °C. Clearly, the introduction of the rigid adamantane unit has enhanced the thermal stability of our target polymers.19,22 Moreover, the higher the adamantane content, the better the thermal stability. For example, the molar ratio of adamantane between MOP-Ad-1 and MOP-Ad-3 is 22.45% and 14.80%, and we found that MOP-Ad-1 was more stable (up to 520 °C) than MOP-Ad-3 (up to 480 °C). This excellent thermal stability is a consequence of the presence of exclusively C–C bonds in the skeletal structure of MOP-Ad networks.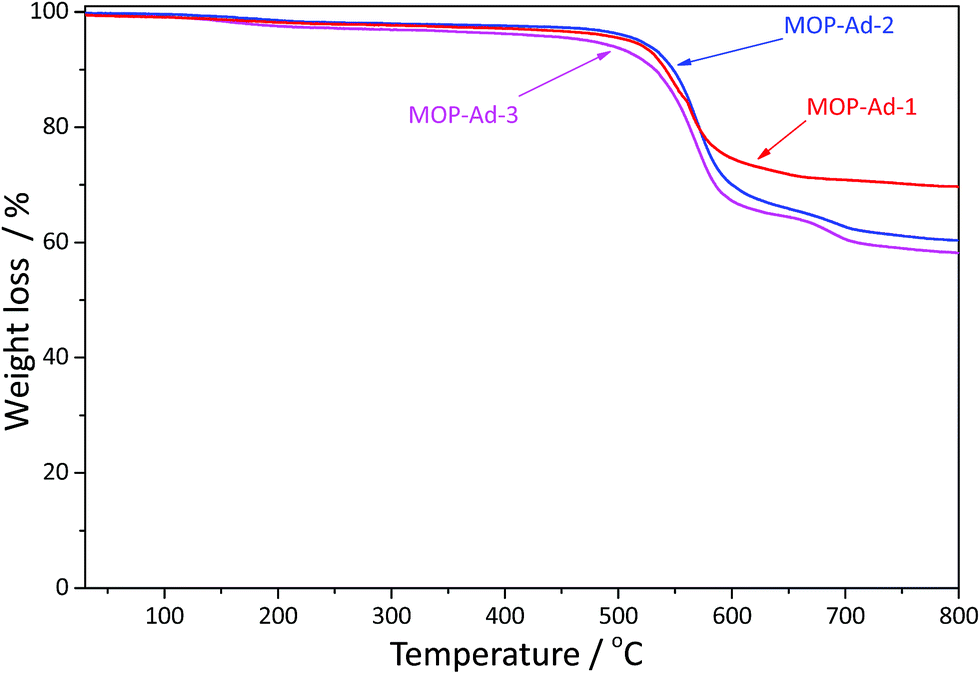 | ||
| Fig. 3 TGA curves of MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3. | ||
To further investigate the stability of MOP-Ad networks, the three materials were rigorously stirred at 1500 rpm in hydrochloric acid (2.0 M) and then sodium hydroxide (2.0 M), both for 72 h. Following such harsh treatments, the FTIR spectra (see ESI, Fig. S3†) showed similar bands to the untreated polymers (Fig. 1), demonstrating that MOP-Ad materials are structurally stable both in strong acid and base. Similar results were also obtained in the 13C CP/MAS NMR spectra (see ESI Fig. S4†) of the MOP-Ad networks before and after acid/base treatment. Moreover, these MOP-Ad networks, like many other MOP materials,18,24 also exhibited excellent chemical stability to common organic solvents such as methanol, acetone, tetrahydrofuran and chloroform.
Gas physisorption measurements were conducted to examine the porous nature of the MOP-Ad networks. Fig. 4 shows the N2 adsorption/desorption isotherms and the pore-size distributions (PSD) of the MOP-Ad networks, and Table 1 summarizes the porous properties of these polymers at 77.3 K and 1.13 bar. All of the polymers gave rise to type II isotherms (Fig. 4(a)) according to the IUPAC classification.15 The isotherms clearly displayed a sharp uptake at low relative pressure (p/p0 < 0.001), especially for MOP-Ad-1 and MOP-Ad-2, indicating substantial microporous structure in these networks.16,27 The upward deviation at higher p/p0 values for all three MOP-Ad networks was also observed, indicating the presence of mesopores in these networks, due to inter-grain porosity or voids.28 All of the desorption isotherms for the three polymers exhibited hysteresis to some extent, also indicating that these three polymers possess micro- and mesoporous structures. Interestingly, the hysteresis could also indicate a swelling of the polymer matrix or the limited release of trapped nitrogen molecules from the narrow pore channels, which is particularly prevalent for non-ordered nanoporous materials, as described elsewhere.7,29
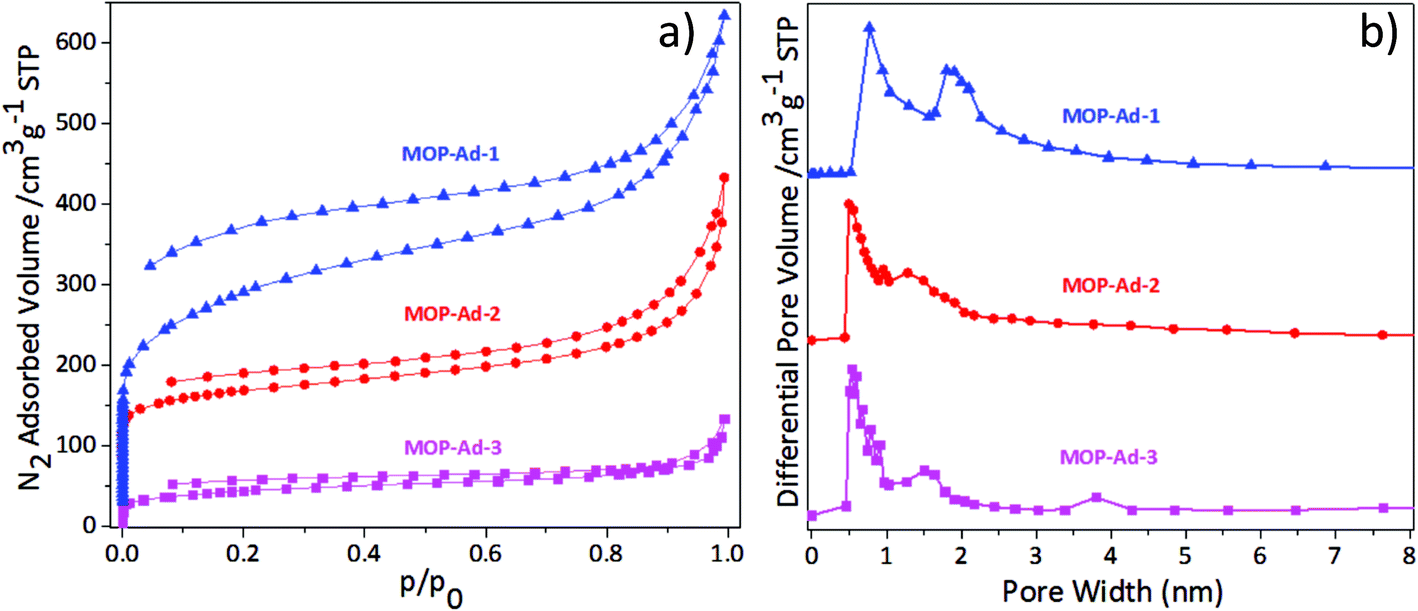 | ||
| Fig. 4 (a) Nitrogen sorption isotherms of MOP-Ad-1, 2 and 3 at 77.3 K and (b) pore size distribution (PSD) for MOP-Ad-1, 2 and 3. | ||
Fig. 4(b) shows the PSD curves for the three MOP-Ad networks as calculated using non-local density functional theory (NLDFT).21 p-Terphenyl-4,4′′-diboronic acid possesses a larger aspect ratio than both benzene-1,4-diboronic acid and biphenyl-4,4′-diboronic acid. Therefore, after cross-linking, it is logical to anticipate that the longer rod in MOP-Ad-3 would generate larger pores compared with MOP-Ad-2 and MOP-Ad-1. However, according to Fig. 4(b), the pore diameter of MOP-Ad-2 and MOP-Ad-3 appeared at approximately 0.55 nm, and much lower than that of MOP-Ad-1 (0.74 nm). In particular, with an increased degree of conformational freedom in the longer rod, greater intermolecular and intramolecular intercalation can be obtained in the process of polymerizing, as well as more efficient space filling in the structure.28,30 Actually, MOP-Ad-3 possessed a distributed pore diameter ranging from 0.49 to 1.91, owing to the intermolecular and intramolecular intercalation in the network, with SAXS (see ESI Fig. S5†) displaying a less ordered network overall. Field-emission scanning electron microscopy (FE-SEM) (see ESI Fig. S6†) was used to observe the surface morphology of the MOP-Ad networks, showing that MOP-Ad-3 has a certain degree of intercalation while MOP-Ad-1 and MOP-Ad-2 exhibit typical agglomerating surfaces.2,25
Concomitantly, the surface areas of the MOP-Ad networks were shown to be dependent on the length of the rods. Thus, by using different length rods, the BET surface area can be tuned. In the present study, the ranking of the length of rods from the largest to the smallest is p-terphenyl-4,4′′-diboronic acid, 4,4′′-dibromo-p-terphenyl and benzene-1,4-diboronic acid. Consequently, the shortest rods between the adamantane rings (MOP-Ad-1) exhibited the highest surface areas of 974 m2 g−1, followed by MOP-Ad-2 (653 m2 g−1) and MOP-Ad-3 (282 m2 g−1). The same trend in surface area for materials of similar kind was found by Lim et al.17 However, compared with their material (adamantane-3), synthesized from 1-bromoadamantane with terphenyl, via Friedel–Crafts reaction, our MOP-Ad-1 network exhibited a higher BET surface area (974 m2 g−1 against 720 m2 g−1). Presumably, this was attributed to the Suzuki coupling strategy processing higher selectivity and target activity in the terminal groups (–Br in the knots and –B(OH)2 in the rods) than the Friedel–Crafts reaction (–Br in 1-bromoadamantane and a large number of –H in terphenyl), resulting a large amount of side reactions occurring in the Friedel–Crafts reactions (the possible mechanisms were shown in Scheme S3†).31–33
3.3 Gas uptake capabilities
The gas (H2, CO2 and CH4) adsorption performance of all MOP-Ad networks were studied to evaluate their applicability in gas storage/purification. In accordance with the results obtained from gas physisorption (Fig. 5 and Table 2) with pressure up to 1.13 bar, one can see that MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3 exhibited moderate uptake capacities for H2 (1.07, 0.92 and 0.49 wt%) and CO2 (10.3, 7.5 and 5.2 wt%), respectively. This gas adsorption performance is comparable to currently favorable microporous organic polymers under the same condition, such as networks based on spirocyclic tetraether (SPOP-8, 0.67 wt% for H2, 4.2 wt% for CO2),34 and triptycene (STP-3 and STP-4, 0.5 and 0.9 wt% for H2, 6.2 and 7.5 wt% for CO2, respectively).14 Nevertheless, the MOP-Ad networks were not as effective as PBI-Ad-1 and PBI-Ad-2 (1.6 and 1.3 wt% for H2, 17.3 and 13.7 wt% for CO2), which connected 1,3,5,7-tetrakis(4-formylphenyl)adamantane with 3,3′-diaminobenzidine or 1,2,4,5-tetraaminobenzene, respectively.35 In addition, the physisorption isotherms had not reached saturation state at pressure 1.13 bar for both H2 and CO2 uptakes by MOP-Ad networks, implying that higher capacities can be expected at increased pressures. Moreover, the H2 and CO2 uptake properties of the MOP-Ad networks can be directly correlated with the surface area and microporous volume. As expected, the polymers with the highest surface areas and microporous volumes gave rise to the highest H2 and CO2 uptakes under the same testing conditions.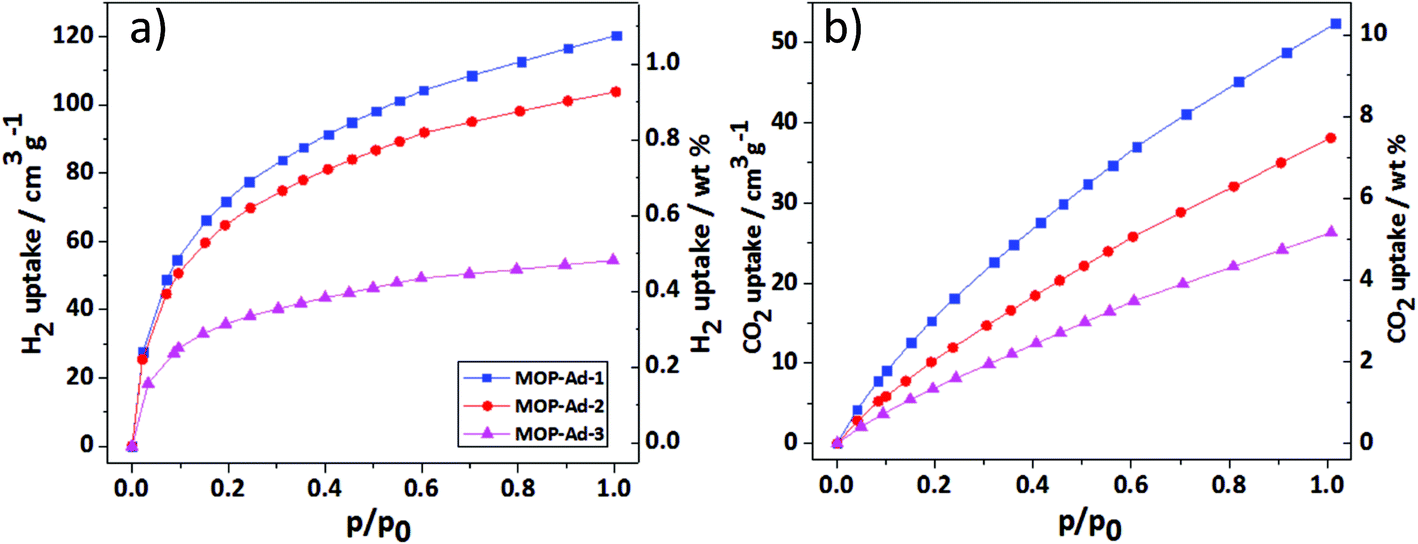 | ||
| Fig. 5 Gas sorption isotherms (a) H2 at 77.3 K, (b) CO2 at 273.1 K. | ||
CH4 adsorption of the MOP-Ad networks was also investigated; MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3 (Table 2 and ESI Fig. S7†) exhibited better CH4 uptake capacities (2.4, 1.6 and 1.0 wt%, respectively) than most non-functionalized porous polymer networks, such as tetrahedral porous frameworks based on 2,2′,7,7′-tetraethynyl-9,9′-spirobifluorene POP-1 (0.8 wt%),2 nanoporous covalent triazine-based frameworks with adamantane PCTF-2 (1.0 wt%),36 but lower than the fluorinated porous poly(arylene-ethynylenes) FPOP-1 and FPOP-2 (2.7 and 3.3 wt%, respectively)37 due to the fluorine heteroatoms that enhance interactions between the pore walls and CH4 molecules.21
For CO2 capture and separation applications, high selectivity is also essential. We estimated the selectivity of CO2 over CH4 (i.e., CO2/CH4 selectivity) for MOP-Ad-1, MOP-Ad-2 and MOP-Ad-3 networks by applying Henry's law to the adsorption data at 273.1 K and low pressure (less than 0.15 bar),16,38 which is typical partial pressure of CO2 in flue gas. As shown in Table 2, the CO2/CH4 selectivities of MOP-Ad networks are calculated to be 3.8, 5.3, and 5.9, respectively (see ESI Fig. S8†).
For the adsorption and selectivity performance of MOP-Ad networks, MOP-Ad-1 possesses relatively higher CO2 and CH4 uptakes, but MOP-Ad-3 possesses higher CO2/CH4 selectivities. The higher CO2 and CH4 uptake is attributed to the higher surface areas and microporous volumes (Table 1). Additional, the narrow pore size distribution also leads to fine selectivity.39 Although MOP-Ad-1 showed the highest CO2 uptake, lower selectivity was obtained due to a higher content of mesoporous surface area and wider pore size distribution in the structure, which apperas to be disadvantageous for the recognition of small CO2 molecules (3.30 Å) over the large CH4 (3.82 Å) molecule. Additionally, it was found that the three MOP-Ad networks exhibited a slightly lower CO2/CH4 selectivity than some functionalized 3D microporous organic polymers, such as tetraphenylethylene-based microporous organic polymers (MTPOPs, 5.4–7.2 at 273 K),9 and tetraphenyladamantane-based microporous polybenzimidazoles (PBI-Ads, 9.0–11.0 at 273 K),35 N-doped porous carbons (NAHA, 7.9–10.7 at 273 K),40 benzothiazole- and benzoxazole-linked porous polymers (6.9–9.4 at 273 K),38 and the mesoporous polymeric organic frameworks (mesoPOF, 10–15 at 273 K).41 Again, this difference was attributed to the presence of polarized sites or the utilization of N-rich or O-rich building blocks, which can increase the gas–framework interactions via hydrogen bonding or dipole–quadrupole interactions.42 Care must be taken with such strategies, however, as the introduction of heteroatoms (such as N or O) into networks, as previously reported,12 reduces the thermal stability of the structure, thus limiting their application. Nevertheless, the CO2/CH4 selectivity of the MOP-Ad networks was still superior to many other 2D functional polymers, such as carbazole–spacer–carbazole type conjugate microporous networks (3.0–4.0 at 273 K),43 conjugated microporous copolymers based on 1,3,6,8-tetrabromocarbazole CP-CMP (3.4–4.2 at 273 K),44 even some 3D functional polymers, such as N-functionalized microporous organic polymer based on tetraethynyl monomer TEPS-TPA (3.9 at 273 K),45 tetraethynylspirobifluorene-containing porous organic polymers (POP, 3.4–4.8 at 298 K),2 the tetra-armed triphenylamine-containing MOPs (3.4–4.3 at 273 K),46 tetrakis(4-ethynylphenyl)methane-based porous materials (PAF-26-COOH, 4.0 at 298 K).21 Additionally, the adsorption and selectivity performance of MOP-Ad networks after immersing in hydrochloric acid and sodium hydroxide were also investigated in the same conditions. As expected, we found no significant changes in the CO2 and CH4 uptake capacities (see ESI Fig. S9 and Table S1†), as well as the CO2/CH4 selectivities, which reflects their high chemical stability.
4. Conclusions
In summary, three microporous organic polymers (MOP-Ad) have been synthesized by Suzuki coupling and a rich structural diversity of them was achieved by using tetraphenyl adamantane “knots” and different phenylboronic acid “rods” as modular building blocks. Importantly, owing to the rational design of the molecular skeleton, these MOP-Ad networks not only exhibited extremely high thermal stability (stable up to 520 °C), but were also stable in common organic solvents, and strong acid and base. For potential use as gas storage materials, they showed desirable adsorption ability over small gas molecules. For example, MOP-Ad-1, the network with the shortest rod linkers, can reversibly absorb 1.07 wt% H2 (77.3 K and 1.13 bar), 10.3 wt% CO2 and 2.4 wt% CH4 (273.1 K and 1.13 bar). Moreover, the three porous networks showed good CO2/CH4 selectivity without any post modification, highlighting them as promising candidates for post-combustion CO2 capture and separation.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21476051), Science and Technology Program of Guangdong Province (No. 2016A050502057) and Natural Science Foundation of Guangdong Province (No. 2016A030310349). H. Yue is grateful to China Postdoctoral Science Foundation. PDT thanks the State Administration for Foreign Experts Affairs and the Royal Society of Chemistry for a Visiting Researcher Programme grant to China.References
- B. Coasne, A. Galarneau, R. J. M. Pellenq and F. D. Renzo, Chem. Soc. Rev., 2013, 42, 4141–4171 RSC.
- Q. Y. Ma, B. X. Yang and J. Q. Li, RSC Adv., 2015, 5, 64163–64169 RSC.
- B. Wang, A. P. Côté, H. Furukawa, M. O'keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- Z. Xiang and D. Cao, J. Mater. Chem. A., 2013, 1, 2691–2718 CAS.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- H. Li, X. Ding and B. H. Han, RSC Adv., 2016, 6, 51411–51418 RSC.
- R. Q. Zhong, Z. L. Xu, W. Z. Bi, S. B. Han, X. F. Yu and R. Q. Zou, Inorg. Chim. Acta, 2016, 443, 299–303 CrossRef CAS.
- K. V. Rao, R. Haldar, T. K. Maji and S. J. George, Polymer, 2014, 55, 1452–1458 CrossRef CAS.
- G. Li, B. Zhang, J. Yan and Z. Wang, J. Mater. Chem. A., 2014, 2, 18881–18888 CAS.
- C. Klumpen, M. Breunig, T. Homburg, N. Stock and J. Senker, Chem. Mater., 2016, 28, 5461–5470 CrossRef CAS.
- C. Zhang, Z. Wang, J. J. Wang, L. Tan, J. M. Liu, B. Tan, X. L. Yang and H. B. Xu, Polymer, 2013, 54, 6942–6946 CrossRef CAS.
- Q. Chen, M. Luo, T. Wang, J. X. Wang, D. Zhou, Y. Han, C. S. Zhang, C. G. Yan and B. H. Han, Macromolecules, 2011, 44, 5573–5577 CrossRef CAS.
- C. Zhang, X. Yang, Y. Zhao, X. Wang, M. Yu and J. X. Jiang, Polymer, 2015, 61, 36–41 CrossRef CAS.
- H. Lim, M. C. Cha and J. Y. Chang, Polym. Chem., 2012, 3, 868–870 RSC.
- H. Lim and J. Y. Chang, Macromolecules, 2010, 43, 6943–6945 CrossRef CAS.
- S. Q. Fu, J. W. Guo, D. Y. Zhu, Z. Yang, C. F. Yang, J. X. Xian and X. Li, RSC Adv., 2015, 5, 67054–67065 RSC.
- R. K. Totten, M. H. Weston, J. K. Park, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2013, 3, 1454–1459 CrossRef CAS.
- H. Ma, H. Ren, X. Zou, S. Meng, F. Sun and G. Zhu, Polym. Chem., 2014, 5, 144–152 RSC.
- J. W. Guo, X. F. Lai, S. Q. Fu, H. B. Yue, J. W. Wang and P. D. Topham, Mater. Lett., 2017, 187, 76–79 CrossRef CAS.
- B. J. Dahl and N. S. Mills, Org. Lett., 2008, 10, 5605–5608 CrossRef CAS PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H. C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498–13502 RSC.
- W. Lu, Z. Wei, D. Yuan, J. Tian, S. Fordham and H. C. Zhou, Chem. Mater., 2014, 26, 4589–4597 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- J. Weber, J. Schmidt, A. Thomas and W. Böhlmann, Langmuir, 2010, 26, 15650–15656 CrossRef CAS PubMed.
- J. X. Jiang, A. Trewin, F. Su, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658–2666 CrossRef CAS.
- Z. Tian, J. Huang, Z. Zhang, G. Shao, A. Liu and S. Yuan, Microporous Mesoporous Mater., 2016, 234, 130–136 CrossRef CAS.
- Y. Yang, Q. Zhang, S. Zhang and S. Li, Polymer, 2013, 54, 5698–5702 CrossRef CAS.
- J. H. Zhu, Q. Chen, Z. Y. Sui and B. H. Han, J. Mater. Chem. A., 2014, 2, 16181–16189 CAS.
- M. Y. Jiang, Q. Wang, Q. Chen, X. M. Hu, X. L. Ren, Z. H. Li and B. H. Han, Polymer, 2013, 54, 2952–2957 CrossRef CAS.
- B. Zhang, G. Li, J. Yan and Z. Wang, J. Phys. Chem. C, 2015, 119, 13080–13087 CAS.
- A. Bhunia, I. Boldog, A. Möller and C. Janiak, J. Mater. Chem. A., 2013, 1, 14990–14999 CAS.
- D. P. Liu, Q. Chen, Y. C. Zhao, L. M. Zhang, A. D. Qi and B. H. Han, ACS Macro Lett., 2013, 2, 522–526 CrossRef CAS.
- M. G. Rabbani, T. Islamoglu and H. M. El-Kaderi, J. Mater. Chem. A., 2017, 5, 258–265 CAS.
- K. Y. Yuan, C. Liu, J. H. Han, G. P. Yu, J. Y. Wang, H. M. Duan, Z. G. Wang and X. G. Jian, RSC Adv., 2016, 6, 12009–12020 RSC.
- B. Yuan, J. Wang, Y. X. Chen, X. F. Wu, H. M. Luo and S. G. Deng, J. Mater. Chem. A., 2016, 4, 2263–2276 CAS.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2012, 24, 471–479 CrossRef CAS.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C. G. Yan and B. H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- S. Qiao, Z. Du and R. Yang, J. Mater. Chem. A., 2014, 2, 1877–1885 CAS.
- M. Yu, X. Wang, X. Yang, Y. Zhao and J. X. Jiang, Polym. Chem., 2015, 6, 3217–3223 RSC.
- H. J. Zhang, C. Zhang, X. C. Wang, Z. X. Ze, X. M. Liang, B. Chen, J. W. Xu, J. X. Jiang, Y. D. Li, H. Li and F. Wang, RSC Adv., 2016, 6, 113826–113833 RSC.
- X. Yang, S. W. Yao, M. Yu and J. X. Jiang, Macromol. Rapid Commun., 2014, 35, 834–839 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28833b |
| This journal is © The Royal Society of Chemistry 2017 |