
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc porphyrin/fullerene/block copolymer micelle for enhanced electron transfer ability and stability†
Ruolin Wang,
Rui Qu,
Chen Jing,
Yan Zhai,
Yingli An and
Linqi Shi *
*
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Functional Polymer Materials, Ministry of Education, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Institute of Polymer Chemistry, Nankai University, Tianjin 300071, China. E-mail: shilinqi@nankai.edu.cn
First published on 6th February 2017
Abstract
Inspired by the structures of antenna-reaction centers in photosynthesis, a complex micelle was prepared from zinc tetrakis(4-sulfonatophenyl) porphyrin (ZnTPPS), modified fullerene (mC60) and poly(ethylene glycol)-block-poly(L-lysine) (PEG-b-PLys) by electrostatic interactions. The core–shell structure made the donor–acceptor system work in an aqueous environment. In the micellar core, ZnTPPS and mC60 molecules were surrounded by each other which ensured effective energy migration from the donor to the acceptor. The emission of the porphyrin was quenched efficiently which was confirmed by a series of fluorescence spectra. In comparison with the ZnTPPS micelle, the interaction of the mC60 with the porphyrin inhibited the generation of singlet oxygen, which was measured by electron paramagnetic resonance (EPR) and iodide method. In addition, enhanced generation of the superoxide radical was detected by reduction of nitro blue tetrazolium (NBT) in the presence of an electron donor. What is more, the complex micelle exhibited high electron transfer performance in the photocatalytic reduction of methyl viologen. The complex micellar structure endowed the donor–acceptor system with improved stability in an acidic environment. This strategy would be helpful for designing a new electron transfer platform and artificial photosynthetic system.
1. Introduction
Photosynthesis is a fundamental process on earth, which converts light energy into chemical energy.1 The primary reaction of photosynthesis is light-driven charge separation, recognized as initial conversion of light energy into electrochemical potential, carried out in reaction centers.2 In the pursuit of artificial photosynthetic reaction centers, various donor–acceptor systems formed by porphyrins and fullerenes in both covalent and non-covalent forms displaying photoinduced energy and electron transfer processes have attracted considerable attention.3–8 However, the covalently linked systems of building artificial photosynthetic systems differ from natural photosynthesis systems where vast pigments and other active components are elegantly assembled together by non-covalent interactions.9 To date, a series of excellent intermolecular porphyrin and fullerene systems have been mainly designed via metal–ligand coordination, π–π stacking, hydrogen bonding, pseudorotaxane association, and electrostatic interaction.10–15 Recently, D'Souza et al. reported a supramolecular tetrad formed by coordinating functionalized fullerene to porphyrin triad both in o-dichlorobenzene and toluene as a charge-separating photosynthetic antenna-reaction center mimic.16 Guldi and coworkers reported a novel supramolecular electron donor–acceptor hybrid with strong binding via cooperativity between π–π stacking and hydrogen bonding in o-dichlorobenzene.17 However, in organic solvents, the energy transfer efficiency was limited due to binding equilibrium. For example, in supramolecular such as dyads formed via coordination interaction and pseudorotaxane association, dozens to hundred equivalents of acceptor were added to reach efficient fluorescence quenching of donor.15 In addition, most of them are hydrophobic and can be only worked in organic solutions. In the prosperous researches fields such as photodynamic therapy/inactivation, optical sensors and photocatalysis of contaminant, in which energy/electron transfer mechanisms usually occur, water-soluble systems are quite needed to adapt biological environment and meet requests for green chemistry.18–22 With intensive studies of water-soluble porphyrins and fullerenes, non-covalent assemblies through electrostatic interaction are allowed to work in aqueous, though less being reported.14,23 Nevertheless, regulating the ratio of donor and acceptor precisely to mimic specific ratio as nature dose is challenging.24 What is more, water-soluble metalloporphyrins bear kinds of instability, for instance, hydrolyzation and self-quenching due to aggregation.25,26 In a word, it is highly desired to find a straightforward approach to fabricate a porphyrin/fullerene donor–acceptor system which can be used directly in aqueous solution with great stability.In natural photosynthesis antenna-reaction centers, chlorophylls, carotenoids and cozymase are embedded in protein matrixes. The protein plays the role of a programmed solvent, accommodating high density of pigments, ensuring their efficiently transfer energy to the reaction centers, and simultaneously avoiding energy losses due to concentration quenching.27 Carotenoid molecules serve as both light harvesters and photo-protective agents. When exceed sun light intensity arise, carotenoids avoid the generation of harmful singlet oxygen.28 It is worth learning from nature that in vivo chlorophyll–protein interactions play important roles in the stabilization and high efficiency of chlorophyll. From another point of view, polymeric assemblies in aqueous based on block copolymers such as poly(ethylene glycol) block polyaminoacid can serve as excellent carriers because of their various and versatile blocks.29–33 Hitherto, many functional micelles containing target molecules have been constructed. The main driving forces between block copolymers and the loaded components are attributed to non-covalent interactions, such as hydrophobic/electrostatic/coordination interactions, and host–guest recognition,34–37 which are almost same with patterns of natural photosynthesis systems. Similar to the protective environment created by proteins in the case of natural photosynthetic systems, the micellar hydrophobic core can offer pigments protection from demetallation and photodegradation.38,39
Inspired by the structures of antenna-reaction centers in photosynthesis and in connection with our ongoing research programs on cooperative macromolecular self-assembly toward polymeric assemblies,40,41 we envision that micelles seem to be the most suitable and realistic platform to realize the photosynthetic pigment–protein complexes. Herein, we fabricated the complex micelle consist of PEG-b-PLys, ZnTPPS and mC60 in aqueous solution, which formed a proteins-like micelle with electron transfer activity, as shown in Scheme 1. The three components formed complex micelle through the electrostatic interaction between the amino groups on the polylysine and the carboxyl anions/sulfonic anions on the two photosensitizers, thus further chemical synthesis was needless, making this method easy to prepare and repeat. By electrostatically binding to the PLys chains, ZnTPPS distributed uniformly in the micellar core, and thus aggregation and self-quenching were inhibited. Simultaneously, ZnTPPS and mC60 were associated in the protein-like core together and surrounded by each other, affording condition for efficient interaction. With the interaction between ZnTPPS and mC60, the complex micelle performed relatively diminished singlet oxygen production and higher generation in superoxide radical in comparison with ZnTPPS micelle. The complex micelle exhibited improved electron transfer activity and enhanced stability in acid aqueous. Therefore, an efficient and stable donor–acceptor system was successfully constructed.
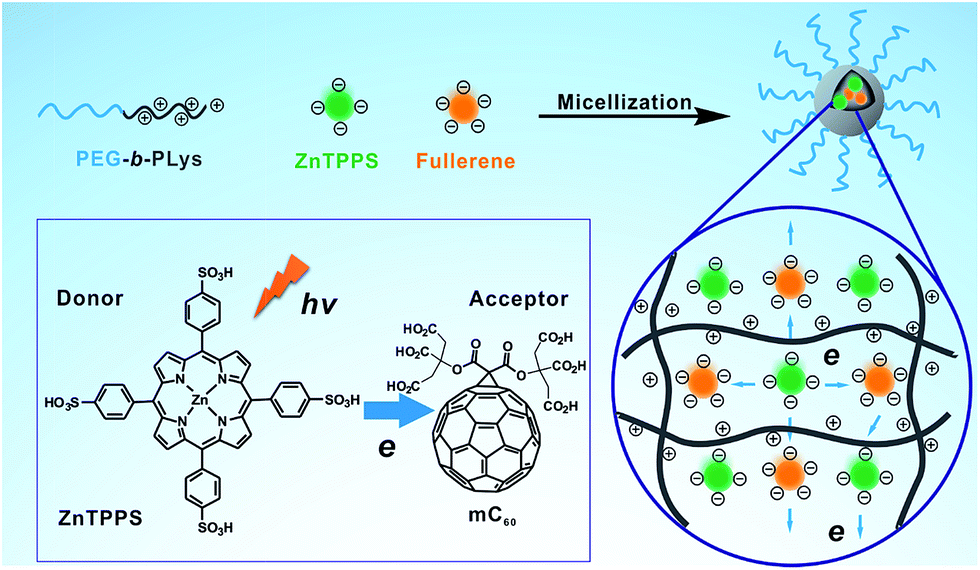 | ||
| Scheme 1 Schematic illustration of the formation of the complex micelle and its photoinduced property. | ||
2. Experimental
2.1. Synthesis of the block copolymer
Poly(ethylene glycol)-b-poly(L-lysine) (PEG-b-PLys) was prepared as described previously.42 Briefly, Lys(Z)-NCA (0.98 g, 3.2 mmol) was dissolved in 30 mL of DMF and polymerized by addition of MeO-PEG113-NH2 (2.0 g, 0.4 mmol) with the terminal primary amino group as the initiator. The reaction mixture was stirred for 3 days at r.t. under a dry argon atmosphere. Then solvent was evaporated under reduced pressure. The resulting product was dissolved in 15 mL of CHCl3 and then precipitated into excessive diethyl ether to obtain PEG-b-PLys(Z) (yield 90%). Deprotection of Z group in PEG-b-PLys(Z) was carried out by addition of HBr (33 wt% in HOAc, 2 mL) to the solution of PEG-b-PLys(Z) (2.0 g) in 20 mL CF3COOH for 2 h at 0 °C. The reaction mixture was precipitated into excessive cold diethyl ether. The precipitation was dissolved in DMF and purified by filtering through a 0.22 μm Millipore filter. The filtrate was precipitated in excessive diethyl ether to remove the residual CF3COOH and obtain PEG-b-PLys (yield 60%). Then the product was dried at room temperature under vacuum.2.2. Synthesis of water-soluble fullerene (mC60)
The triethyl citrate (TEC) (2.76 g, 10.0 mmol) and the malonyl dichloride (0.5 mL, 5.0 mmol) were mixed in a 100 mL round-bottom flask with a calcium chloride tube at the top of condenser and toluene (30 mL) was used as solvent. After 5.0 mol% of DMAP·HCl (0.078 mg, 0.5 mmol) was added, the reaction mixture was stirred at 110 °C. The reaction was stopped according to thin layer chromatography (TLC). Then the mixture was allowed to cool to room temperature and the solvent was removed by rotary evaporation to afford the crude product, which was purified by the flash column chromatography (PE–EtOAc = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give the product TEC malonate as colourless oil.43
:1) to give the product TEC malonate as colourless oil.43
To a solution of fullerene C60 (103 mg, 0.14 mmol) in toluene (85 mL) were sequentially added I2 (35 mg, 0.14 mmol), TEC malonate (86 mg, 0.14 mmol) and DBU (23 mg, 0.15 mmol). After being stirred for 4 h at room temperature, the reaction mixture was concentrated by rotary evaporation. TEC malonate fullerene was isolated by column chromatography on silica gel with the yield 40% using the eluent petroleum ether/ethyl acetate (3:1) as brown-black solid (compound 1, see Scheme S1 in the ESI†).44
To a stirred solution of compound 1 (35 mg, 0.026 mmol) in EtOH (5 mL) and THF (5 mL) was added 1 M aqueous LiOH (15 mL) and the reaction mixture was stirred at room temperature for 12 h. The reaction was stopped when started material was disappeared and turned into large-polar compounds according to the TLC. The mixture was neutralized by conc. HCl and dialyzed against distilled water using a dialysis membrane (molecular weight cutoff = 500). The aqueous solution of purified product was lyophilized.
2.3. Preparation of polymeric micelles
Complex micellization was carried out by adding the stock ZnTPPS, mC60, or ZnTPPS/mC60 (1:0.25, 1:0.5, 1:1, 1:1.5, 1:2 molar ratio) solution dropwise into PEG-b-PLys solutions under stirring at room temperature. The obtained complex solutions had the same polymer concentration of 0.3 mg mL−1 and ZnTPPS concentration of 20 μM.
2.4. Detection of singlet oxygen via EPR measurement
ESR electronic spinning resonance measurements were performed on a JEOL JES-FA200 apparatus (JEOL Ltd., Tokyo), using TEMP as a spin trap for 1O2. To a micelle solution, distilled water and TEMP in ethanol were added and mixed well under an aerobic condition (the final concentration of TEMP was 7 mg mL−1). The mixed solution was collected in a 1 cm path length quartz cell, irradiated with a red lamp (635 nm) at the distance of 15 cm (20 mW cm−2) in 30 min, and immediately subjected to EPR measurement.2.5. Detection of singlet oxygen via iodide method
5 mL of a detection solution (0.1 M) potassium iodide (KI), containing 5 μM ZnTPPS and/or mC60 in the micelles, was prepared. The solutions were stirred and irradiated using a red lamp at room temperature. The reaction was monitored by recording the increase in the intensity of the 351 nm absorption peak attributed I3− ion as a function of their radiation time.2.6. Detection of super radical oxygen (NBT method)
The nitro blue tetrazolium (NBT) method was used to detect superoxide anion radical (O2˙−) formation in water. This procedure was carried out using 0.2 mM NBT, 0.5 mM reduced nicotinamide adenine dinucleotide (NADH), and different micelles. Samples (3 mL in total) were irradiated in 1 cm path length quartz cells under aerobic condition with red lamp (635 nm) at the distance of 15 cm (20 mW cm−2). The progress of the reaction was observed by following the increase of the absorbance at λ = 560 nm.2.7. Photosensitized reduction of MV2+
A sealed square quartz cuvette (4 mL) which contained water solution of micelles, MV2+ (8.0 × 10−4 M) and TEOA (0.1 M) was irradiated with white light from a 150 W high-pressure Xe lamp with a cut-off filter of >360 nm. The photochemical reaction was monitored by measuring the absorption spectra at different irradiation times. The mixture was degassed by three repeated freeze–evacuate–thaw cycles with argon before the irradiation.3. Results and discussion
3.1. Characterization of water-soluble fullerene
In order to get water-solution fullerene with negative charges, the mC60 moiety was synthesized from malonyl chloride, triethyl citrate (TEC) and C60 (see Scheme S1 in the ESI†). The hydrolysis from the obtained compound 1 was confirmed by FT-IR, 1H NMR and ESI-MS. The FT-IR spectrum (Fig. S7†) of compound 1 exhibited the characteristic absorption band at 2900 cm−1 and 1450 cm−1 due to the C–H stretching vibration of the ethyl group on the esters. After being hydrolyzed to acid form, the hydrolysis products showed a broad peak at 3200–2500 cm−1, which did not exist in compound 1. The strong absorbance at ∼1770 cm−1 and 920 cm−1 was due to the –OH group (in carboxylic groups) in hydrolysis products. The 1H NMR of mC60 moiety in D2O also showed residual ethyl signals (Fig. S5†). The EIS-mass confirmed the compositions of the hydrolysis products. ESI-mass analysis of the reaction mixture showed peaks at m/z = 459.2 and 649.0, which were assigned to be penta-acid compound (compound 2, Scheme S1†) and hexa-acid compound (compound 3, Scheme S1†), respectively (see Fig. S8†). That is to say, there was averagely one ethyl group left on the mC60 molecule after hydrolysis. It is worth emphasizing that though with much carboxyl group, mC60 was inclined to form aggregations, familiar with compounds reported by both Guldi and Hirsch.45,46 This provided us a direct measure of the cluster size by dynamic light scattering (DLS), transmission electron microscopy (TEM). Fig. 1A showed an average hydrodynamic diameter (Dh) of 25 ± 1.0 nm in solution, which was also supported by TEM analysis (Fig. S9A†) showing an average diameter of about 20 nm as aggregate particles. Absorption spectroscopic of the mC60 solution (Fig. S10†) showed absorption at 200–400 nm region. Zeta potential of mC60 solution in pure water was intensively negative (Table 1). This result confirmed that there were massive carboxyl groups on the surface of the particles. These results above fully proved that water-soluble fullerene with negative charges was successfully prepared.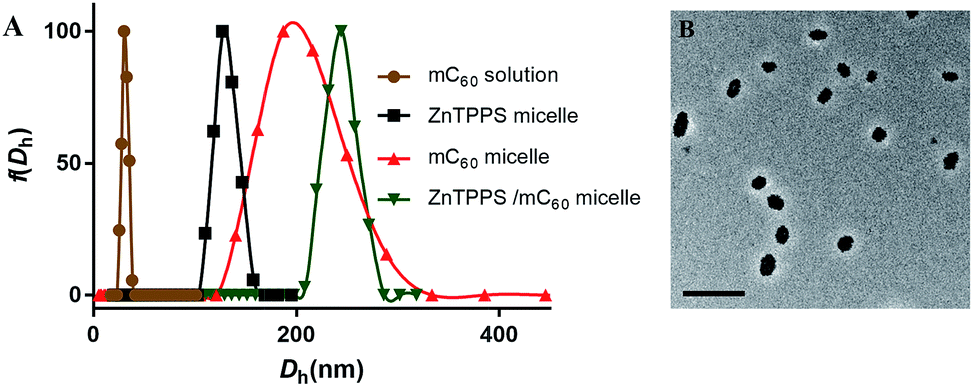 | ||
| Fig. 1 (A) DLS profile of water-soluble fullerene solution and three kinds of micelles under pH = 6.5. (B) TEM micrograph of ZnTPPS/mC60 (1:1 in molar ratio) complex micelle. The scale bar was 500 nm. | ||
| Sample | ζ-Potential (mV) |
|---|---|
| a The samples were conducted in pure water (pH 6.5) at 25 °C. | |
| ZnTPPS micelle | 7.94 ± 1.02 |
| mC60 micelle | 7.06 ± 0.57 |
| ZnTPPS/mC60 micelle | 3.44 ± 1.91 |
| mC60 solution | −19.05 ± 0.55 |
3.2. Morphology characterization of micelles
In the present study, a series of polymeric micelles with ZnTPPS and/or mC60 in cores were fabricated through the straightforward self-assembly. These micelles were prepared by adding the photosensitizers stock solutions dropwise into PEG-b-PLys solutions under stirring. They had electrostatic cross-linked cores consist of PLys chains and photosensitizers. The samples were named as single micelles (ZnTPPS or mC60 alone in core) and complex micelles (both ZnTPPS and mC60 in core with a series of molar ratio). Dynamic light scattering with the scattering angle of 90° was exploited to measure the hydrodynamic diameter distribution of three types of micelles. As demonstrated in Fig. 1A, the average hydrodynamic diameter (Dh) of ZnTPPS micelle, mC60 micelle and complex micelle were 122 ± 1.6 nm, 200 ± 4.5 nm, and 241 ± 3.8 nm, respectively. The TEM image (Fig. 1B) showed a spherical morphology of the complex micelle.The surface charge properties of these three kinds of micelles were confirmed by the zeta potential measure. As shown in Table 1, zeta potentials of micelles were all positive which was due to the excess of lysine units. The complex micelle with both components in core had less positive zeta potentials than either of the single micelle, under pH = 6.5, illustrating increase of negative charge units in the complex micellar core.
3.3. Spectroscopic properties of micelles
Absorption spectroscopic properties of the micelles were investigated. As observed in Fig. S10,† the absorption spectra of mixed solution containing ZnTPPS and mC60 was approximately identical with the superposition of the absorptions of the two components, indicating that there was little interaction between the moieties in the ground state and the two chromophores retained their individual identities.As Fig. 2A showed, for ZnTPPS micelle, compared with ZnTPPS solution, the Soret band showed a blue shift of ∼7 nm. For complex micelles, Soret bands showed gradual bathochromic shift and the peak intensity was diminished greatly with the increasing content of mC60 in the micelles. Appearance of isosbestic point was found at about 430 nm. The maximum shift of the Soret band of ZnTPPS/mC60 complex micelles showed a bathochromic shift of ∼22 nm with respect to ZnTPPS micelle, suggesting appreciable interaction between ZnTPPS and mC60 in the core.
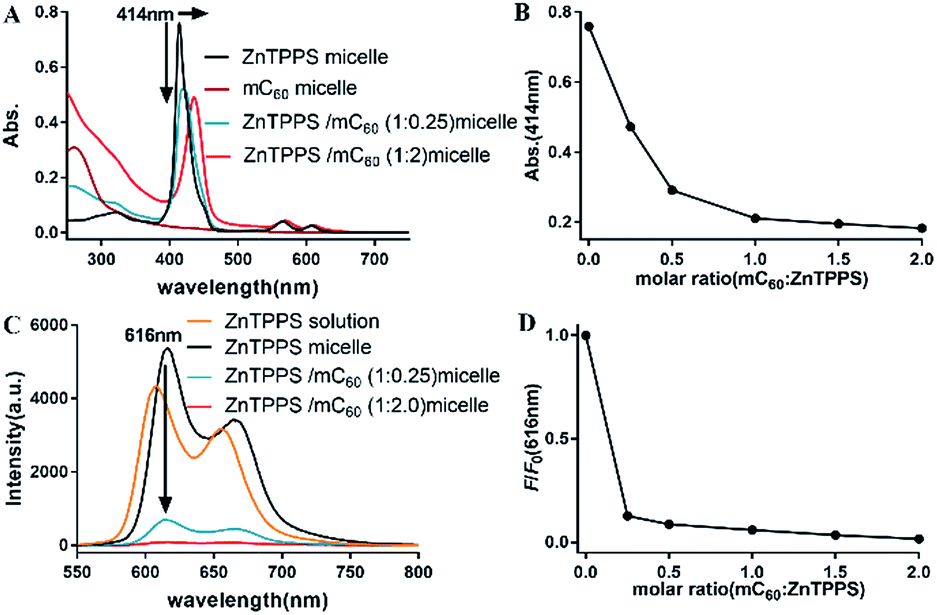 | ||
| Fig. 2 Absorption spectra (A) and fluorescence emission spectra changes (C) of micelles upon varying the molar ratios of ZnTPPS:mC60 from 1:0 to 1:2. Plot of absorptions (B) at 414 nm and plot of the relative fluorescence intensities (D) at 616 nm as a function of molar ratio of ZnTPPS:mC60 in micelles. Arrows indicate the progression of the addition of mC60. λexc = 430 nm. | ||
To shed light onto the interaction, the fluorescence quenching property of the as prepared micelles with different donor–acceptor molar ratio was investigated via comparing the steady-state fluorescence emission spectra of them. Though the maximum absorption peaks varied along with ZnTPPS/mC60 ratio, the excitation wavelength was set at 430 nm for the measurements, which was about the average maximum absorption wavelength of the complex micelles. The ZnTPPS micelle displayed two bands at 616 nm and 665 nm in the red spectral region when exciting the sample at 430 nm. However, the fluorescence emission of the complex micelles weakened successively with the increase of mC60/ZnTPPS molar ratio, indicating strong quenching of the excited porphyrin by nearby fullerene derivatives (Fig. 2C). Since there was no appreciable emitting band from fullerene (∼720 nm), this result indicated that an electron transfer process might occur from the porphyrin to the fullerene moiety.47 Quenching efficiencies (according to fluorescence intensity at 616 nm) of these donor–acceptor systems were calculated in plot of F/F0 (Fig. 2D). F0 was the fluorescence intensity of ZnTPPS micelle and F was the fluorescence intensity of complex micelle in the presence of mC60 in core. The maximum quenching fell by 98.3% in the complex micelle with a ZnTPPS:mC60 molar ratio of 10:20. The fluorescence quantum yield of ZnTPPS/mC60 micelle was calculated to be 2% (see ESI†). Similar quenching results have been reported in both coordination supermolecule and porphyrin derivatives attached to a fullerene structure in organic solvents, while in such supramolecular formed via coordination interaction and pseudorotaxane association, dozens to hundred equivalents of fullerene derivative were needed to reach a majority of quenching of donor fluorescence.15,48
In the absence of block copolymer, the fluorescence titration of ZnTPPS solution on increasing addition of mC60 in water was also investigated (Fig. S11†). The result revealed no significant decrease of ZnTPPS emission intensity as compared to the spectrum of the complex micelle, indicating an absence of interaction between the fullerene derivatives with ZnTPPS molecules in aqueous. This can be attributed to the charge repulsions between ZnTPPS and mC60. All these spectrum results fully proved that block copolymer could sever as an effective frame in associating ZnTPPS with mC60 together and constructing a donor–acceptor model.
3.4. Fluorescence lifetimes of micelles
Photoinduced charge-separation and charge-recombination processes were examined in the micelles by means of time-resolved fluorescence lifetime measurements (Fig. 3). The spectral features of the micelles were essentially the same as those observed by steady-state measurements. The time profile of the fluorescence of ZnTPPS micelle showed mono exponential decay in H2O. The lifetime of ZnTPPS micelle was evaluated as τf = 0.388 μs. Upon forming the complex micelle with mC60 in side, the fluorescence lifetime became shorter to 0.134 μs with a ZnTPPS/mC60 ratio of 1:2, suggesting that the efficient charge-separation originated from the excited ZnPPPS moiety to the C60 moiety took place, generating mC60˙− → ZnPPPS˙+ with high quantum efficiency. In addition, in the complex micellar system with a ZnTPPS/mC60 ratio of 1:2, the energy-transfer rate constant kET between ZnTPPS donor and mC60 acceptor was calculated to be 1.26 × 108 s−1 (see the ESI†).
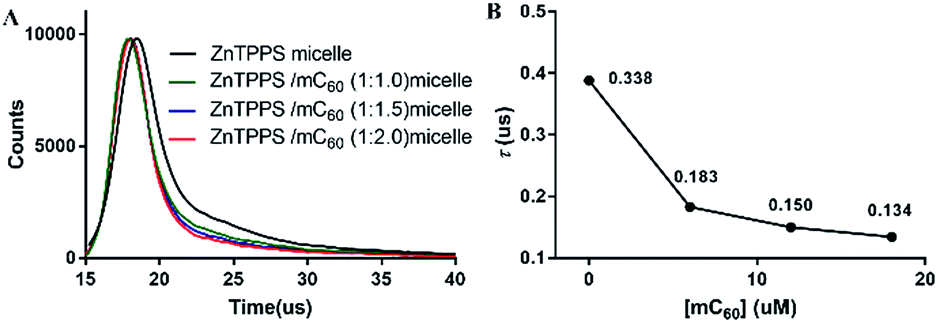 | ||
| Fig. 3 (A) Fluorescence decay profiles of ZnTPPS micelle and ZnTPPS/mC60 micelle in water. λex = 430 nm, λem = 616 nm. (B) Fluorescence lifetime of complex micelles. The molar ratio of ZnTPPS to mC60 in complex micelles were 1:1, 1:1.5, 1:2, respectively. ZnTPPS in these samples had the same concentration of 10 μM. | ||
The high efficiency of energy migration can be attributed to the structure of the micellar core. In there, by electrostatically binding to PLys chains, ZnTPPS and mC60 distributed uniformly in the micellar core, were surrounded by each other nearby. This formation made the interaction between excited ZnTPPS and the mC60 moiety convenient and efficient.
3.5. Detection of singlet oxygen via EPR and KI methods
It is known that singlet oxygen (1O2) and superoxide radical (O2˙−) are most common reactive oxygen species (ROS). 1O2 can be generated by energy transfer from a triplet photosensitizer (Type II mechanism) and O2˙− is produced by electron transfer from a radical anion (Type I mechanism).49 Both ZnTPPS and fullerene are effective photosensitizers in 1O2 generation. To further evaluate the interaction between these two in the micellar core, we used EPR spectroscopy to detect short-lived 1O2 upon photo-irradiation. A trace amount of 2,2,6,6-tetramethylpiperidine (TEMP), a typical trapping molecule, was used to capture 1O2 by yielding a paramagnetic product TEMPO.50EPR spectra intensities were originated from Fig. S12.† As shown in Fig. 4A the micelle with ZnTPPS inside had higher production of singlet oxygen than that of mC60 single micelle and complex micelle. The lower 1O2 production in complex micelle compared to ZnTPPS micelle indicated that the formation of charge-transfer state took place, decreasing the efficiency of triplet-state formation, and finally, decreasing the production of 1O2. The mixed micelles consist of both ZnTPPS micelle and mC60 micelle, containing equal amount of photosensitizers to the complex micelle, performed an approximate sum 1O2 production of the two single micelles produced, which clearly indicated that the ZnTPPS and mC60 in two separated micelles acting no interaction when mixed together.
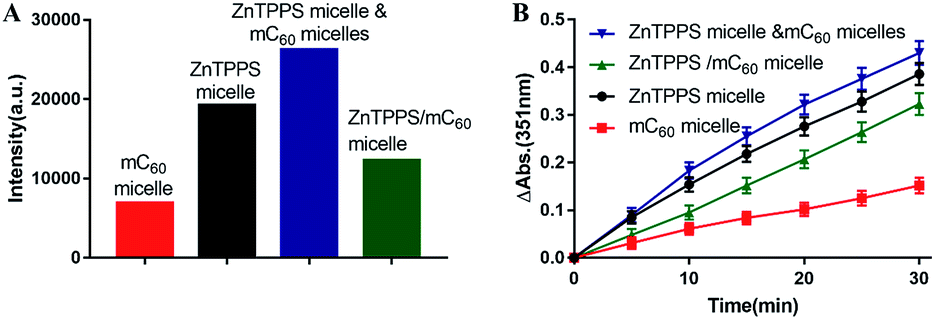 | ||
| Fig. 4 (A) EPR spectra intensities of the mC60 micelle, ZnTPPS micelle, complex micelle, and mixed micelles under irradiation. (B) Kinetics of tri-iodide (I3−) formation at λ = 351 nm as a function of time. | ||
The iodide method was used as an alternative method for the detection of 1O2. Potassium iodide served as reactant. The concentration of photoproduced tri-iodide ion (I3−) was proportional to the concentration of 1O2.51 As showed in Fig. S13,† where reaction conducted by complex micelle was shown as typical example. Increasing absorption at 351 nm indicated the I3− ion formation. The intensity increased with time indicated that the concentration of the 1O2 molecules increased with time in the aqueous solution. Then the absorption intensities of the I3− ion peak at 351 nm in the reactions conducted by different samples were recorded and were plotted against the photoirradiation time. As shown in Fig. 4B, the amount of 1O2 production in complex micelle was lower than that in ZnTPPS micelle. These results are in accordance with the EPR measurement of the micelles, thus confirming the aforementioned assumption.
It is well known that in photosystem II, the singlet oxygen generated by triplet chlorophyll attacks lipids and other tissues, causing photodamage. Photosynthetic organisms limit such photodamage by using carotenoid as photoprotective agents to quench neighboring chlorophyll. In this work, accompanying with ZnTPPS in core, mC60 served similar effect with carotenoid offering photoprotection.
3.6. Photosensitized reduction of NBT
Photosensitized decomposition of NBT occurred mainly through a Type I photoreaction process under aerobic conditions. In this work, the photoinduced superoxide radical oxygen generation was detected by NBT method.52 The reduction of NBT to formazan was used in the presence of reduced nicotinamide adenine dinucleotide (NADH) showed in Fig. 5F. Typical results were shown in Fig. 5A–D for reactions photosensitized by the different micelles. The increase of formazan absorption at λ = 560 nm was examined as a function of time after irradiation of the samples in water. As shown in Fig. 5E, the reductions of NBT were moderate in the reactions using the two single micelles. Compared with them, the complex micelle performed a higher photodynamic activity in the presence of the ZnTPPS and mC60 moiety in micellar core. In addition, the mixed micelles consist of both ZnTPPS micelle and mC60 micelle, containing equal amount of photosensitizers to the complex micelle, performed lower production of O2˙− than that of complex micelle. This can be interpreted as that, under irradiation, Type II route mainly occurred in ZnTPPS micelle and the production of O2˙− was low. The production enhancement effect in complex micelle could be ascribed to electron transfer from ZnTPPS to mC60 in the micellar core and to O2˙− finally.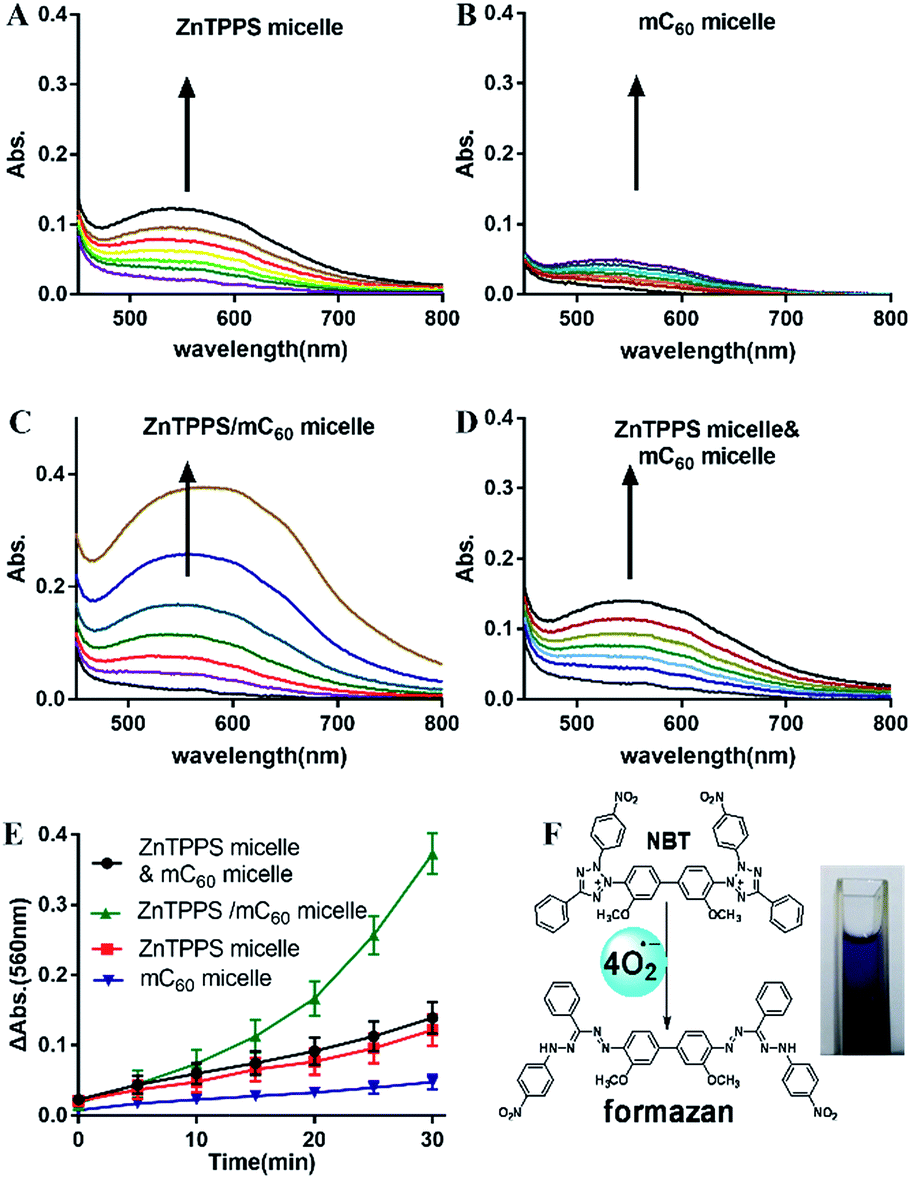 | ||
| Fig. 5 Absorption spectra changes of NBT reactions after different irradiation times (Δt = 5 min) photosensitized by different micelles and mixed micelles (A–D). Time course of O2˙− generation detected as an increase in the absorption at 560 nm by the NBT method (E). The chemical reaction of NBT method and the image of formazan generation (F). Arrows indicate the changes of spectra with time. | ||
3.7. Electron transfer ability
As reported, despite trapped inside the micellar core, metalloporphyrins in the electrostatic micelles still have the electron transfer ability.53 The photoreduction of methyl viologen (MV2+) was the most important step in a photochemical hydrogen production involved electron transfer.54 To gain more insight into the role of the fullerene derivative in complex micelles, electron transfer ability was investigated by the photoreduction of MV2+.Photosensitized reduction of MV2+ by micelles was studied in deoxygenated water. Fig. S14† showed the time dependent absorption spectral change mediated by complex micelle, and a new maximum absorbance appeared at 605 nm, which was assigned to MV+˙. The kinetics of MV+˙ formation at 605 nm as a function of time of all kinds of micelles were showed in Fig. 6A. Obviously, a higher value of photoreduction of MV2+ was found using the complex micelle as photosensitizer with respect to other micelles. This suggested that the porphyrin/fullerene system in the complex micelle possessed high efficiency in electron transfer. It can be inferred that, in electrostatic complex micelles, the back electron transfer was restrained in the presence of mC60. It was worth pointing out that the efficiency using mixed micelles was lower than that of complex micelle but higher than that of ZnTPPS micelle or mC60 micelle. It was probably because that in the mixed micelles, minority of the two type micelles had blended as complex micelle. Fig. 6B showed the schematic electron transfer route of photoreduction of MV2+ method and the final image of blue MV+˙ product.
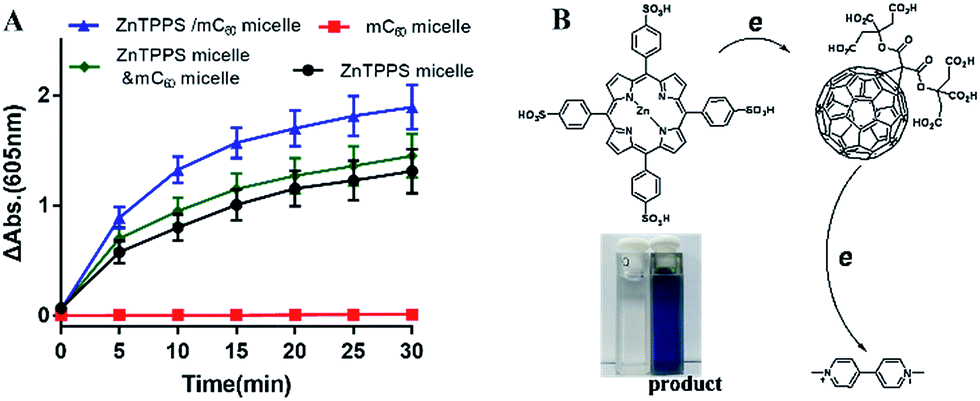 | ||
| Fig. 6 (A) Time course of the reduced MV2+ formation under various micelle solutions (MV2+, 8.0 × 10−4 M; TEOA, 0.1 M; ZnTPPS and/or mC60 were 10 μM in final mixtures) with illumination time. Samples were irradiated with white light from a 150 W high-pressure Xe lamp with a cut-off filter of >360 nm (Δt = 5 min). (B) The schematic electron transfer route of photoreduction of MV2+ method and the image of MV+˙ product solution (right quartz cuvette). | ||
It is known that C60 molecule is an efficient ROS generator. The process that ground state molecular oxygen transformed into ROS can occur by energy transfer from the triplet state fullerene (3C60*) to produce 1O2 (Type II mechanism) or by electron transfer from fullerene anion radical (C60˙−) to form O2˙− (Type I mechanism).55 Nevertheless, ultraviolet absorption limit its application in absorbing panchromatic light. On the other hand, porphyrins in the micelle core absorbed the visible light acting as antenna and electron donor. Electron transfer occurred from excited porphyrins to fullerene C60 and photoinduced charge-separated state formed. Subsequently, the produced C60˙− can transfer an electron to molecular oxygen producing O2˙−. The electron transfer reaction mainly takes place in a polar medium and in the presence of reducing substances, such as TEOA and NADH.56,57 Thus, in this work, in complex micelle core, both main photochemical reactions can be competed to produce ROS. In the presence of mC60, complex micelle was particularly effective at mediating Type I photochemical reactions as opposed to the generation of 1O2 which dominated ZnTPPS micelle (Scheme 2).
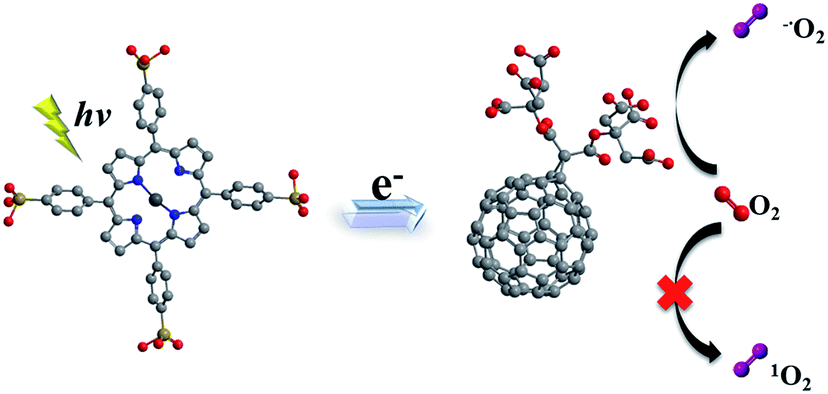 | ||
| Scheme 2 Schematic illustration of mechanism in producing ROS of the complex micelle under irradiation. | ||
3.8. Stability of micelles in acidic solution
ZnTPPS is apt to be demetallized in acidic solutions due to hydrolysis. Block copolymer poly(ethyleneglycol)-b-poly(4-vinylpyridine) (PEG-b-P4VP) can effectively protect ZnTPPS against demetallization in acidic aqueous through electrostatic interaction between positively charged pyridyls on polymer and negatively charged sulfonate groups on ZnTPPS.38 However, because of the lower hydrophobicity of PLys than P4VP, PLys micellar core might provide an inferior protective microenvironment for ZnTPPS. So it was necessary to evaluate the stability of these PEG-b-PLys micelles in acidic solution. The evolution of UV-vis absorptions of ZnTPPS solutions and micelles at pH 3.0 with time were shown in Fig. 7A–C. The intensities of the typical Soret band (421 nm) and Q bands (555 and 595 nm) in ZnTPPS solution decreased progressively indicating the hydrolysis of ZnTPPS. The decomposition was almost completed in 20 min at pH 3.0 in the absence of polymer. Whilst, the decrease in intensity at Soret band of the ZnTPPS micelle was delayed compared to that of ZnTPPS solution with time. Furthermore, complex micelle with additional mC60 in micellar core compared with ZnTPPS solution displayed more dilatory decrease in Soret band. Therefore, the result indicated that the complex micelle has a reinforced stability in acidic environment.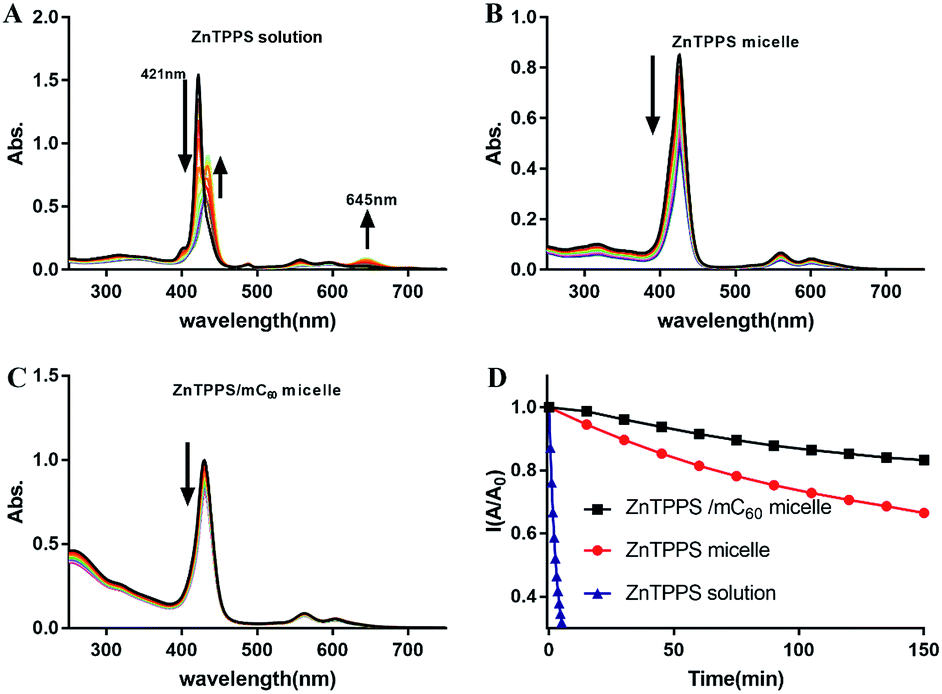 | ||
| Fig. 7 Changes in UV-vis absorption spectra of ZnTPPS solution and different micelles (the concentrations of ZnTPPS in three samples were same) in pH = 3 with time (A–C). The kinetic profiles of Soret bands absorption intensity against time for the decomposition of ZnTPPS in solution and micelles (D). Arrows showed the decrease of the Soret band intensities and the appearance of new peaks with time. | ||
This pH stability reinforce can be interpreted as: in the presence of PEG-b-PLys, the electrostatic interaction between the cationic PLys block and the anionic ZnTPPS that bears four negatively charged sulfonate groups leads to the formation of the micelle consisting of a PLys/ZnTPPS core and a PEG shell. As a result, ZnTPPS was encapsulated in the micellar core which can protect ZnTPPS from the attack of protons to some extent. In the complex micellar core with both ZnTPP and mC60, ZnTPPS and mC60 containing massive carboxy group acted as electrostatic crosslinking agents in the micellar core, which made the protonated core tighter and kept ZnTPPS from the invasion of protons. Therefore, the decomposition process was further restrained even in acidic environments. This enhanced stability extend the application fields of the nanosystem than just being dissociative molecules.
4. Conclusions
In conclusion, we have prepared an efficient donor–acceptor system through electrostatic self-assembling strategy in water. The micelle serves as an excellent carrier to associate ZnTPPS with mC60 together and a shelter to prevent proton invasion. Non-covalent interaction between ZnTPPS and mC60 mimics the inherent feature of photosynthesis reaction center. In complex micelle, the metal porphyrins act as antenna pigments to enhance the light absorption of the system. Meanwhile, it can afford efficient electron transfer from metal porphyrins donors to mC60 acceptors. The presence of fullerene derivatives endowed the system with diminished singlet oxygen production, enhanced oxygen free radical production and efficient electron transfer process under light irradiation. This construction model can be a strong guide for designing new functional materials in photocatalysis and be helpful for understanding fundamental properties of electron transfer in natural photosynthesis, and, in turn, designing new artificial photosynthetic systems.Acknowledgements
We thank the National Natural Science Foundation of China (No. 91527306, 51390483, 21620102005, 51603231) and the Natural Science Foundation of Tianjin, China (No. 16JCQNJC02500) for financial support.Notes and references
- E. Rabinowitch and Govindjee, Photosynthesis, Wiley, New York, 1969, available at, http://www.life.illinois.edu/govindjee/g/Books.html, accessed Jan 6, 2017 Search PubMed.
- J. Deisenhofer and J. Norris, Photosynthetic Reaction Center, Academic Press, 2013 Search PubMed.
- D. Gust, T. Moore and A. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed.
- D. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC.
- P. Boyd and C. Reed, Acc. Chem. Res., 2005, 38, 235 CrossRef CAS PubMed.
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453 CrossRef CAS.
- D. Lawlor and B. Ke, Ann. Bot., 2003, 91, 106 CrossRef.
- Z. Chai, R. Ma, Z. Zhang and L. Shi, Acta Polym. Sin., 2012, 10, 1108 Search PubMed.
- C. Kc and F. D'Souza, Coord. Chem. Rev., 2016, 322, 104 CrossRef CAS.
- D. Williams, E. Dolgopolova, D. Godfrey, E. Ermolaeva, P. Pellechia, A. Greytak, M. Smith, S. Avdoshenko, A. Popov and N. Shustova, Angew. Chem., Int. Ed., 2016, 55, 9070 CrossRef CAS PubMed.
- E. Dietel, A. Hirsch, E. Eichhorn, A. Rieker, S. Hackbarth and B. Röder, Chem. Commun., 1998, 1981 RSC.
- F. Wessendorf, B. Grimm, D. Guldi and A. Hirsch, J. Am. Chem. Soc., 2010, 132, 10786 CrossRef CAS PubMed.
- L. Moreira, J. Calbo, B. Illescas, J. Aragó, I. Nierengarten, B. Delavaux-Nicot, E. Ortí, N. Martín and J. Nierengarten, Angew. Chem., Int. Ed., 2015, 54, 1255 CrossRef CAS PubMed.
- D. Balbinot, S. Atalick, D. Guldi, M. Hatzimarinaki, A. Hirsch and N. Jux, J. Phys. Chem. B, 2003, 107, 13273 CrossRef CAS.
- F. D'Souza, R. Chitta, S. Gadde, M. Zandler, A. McCarty, A. Sandanayaka, Y. Araki and O. Ito, Chem.–Eur. J., 2005, 11, 4416 CrossRef PubMed.
- C. Kc, G. Lim, P. Karr and F. D'Souza, Chem.–Eur. J., 2014, 20, 7725 CrossRef CAS PubMed.
- R. Calderon, J. Valero, B. Grimm, J. de Mendoza and D. Guldi, J. Am. Chem. Soc., 2014, 136, 11436 CrossRef CAS PubMed.
- J. Worlinsky, S. Halepas and C. Bruckner, Org. Biomol. Chem., 2014, 12, 3991 CAS.
- Y. Yan, J. Tian, F. Hu, X. Wang and Z. Shen, RSC Adv., 2016, 6, 113991 RSC.
- J. Almeida, J. Tome, M. Neves, A. Tome, J. Cavaleiro, A. Cunha, L. Costa, M. Faustino and A. Almeida, Photochem. Photobiol. Sci., 2014, 13, 626 CAS.
- L. Costa, M. Faustino, J. Tomé, M. Neves, A. Tomé, J. Cavaleiro, Â. Cunha and A. Almeida, J. Photochem. Photobiol., B, 2013, 120, 10 CrossRef CAS PubMed.
- O. Kwon, J. Kim, J. Cho and J. Kim, ACS Appl. Mater. Interfaces, 2014, 7, 318 Search PubMed.
- N. Wang, X. Zhang, W. Zheng, D. Ouyang and R. Yang, Supramol. Chem., 2015, 27, 72 CrossRef CAS.
- Y. Liu, J. Jin, H. Deng, K. Li, Y. Zheng, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7952 CrossRef CAS PubMed.
- A. Farajtabar, F. Gharib, P. Jamaat and N. Safari, J. Chem. Eng. Data, 2008, 53, 350 CrossRef CAS.
- S. Gandini, V. Yushmanov and M. Tabak, J. Inorg. Biochem., 2001, 85, 263 CrossRef CAS PubMed.
- A. Ruban, M. Johnson and C. Duffy, Energy Environ. Sci., 2011, 4, 1643 CAS.
- G. Fleming, G. Schlau-Cohen, K. Amarnath and J. Zaks, Faraday Discuss., 2012, 155, 27 RSC.
- W. Jang, N. Nishiyama, G. Zhang, A. Harada, D. Jiang, S. Kawauchi, Y. Morimoto, M. Kikuchi, H. Koyama, T. Aida and K. Kataoka, Angew. Chem., Int. Ed., 2005, 44, 419 CrossRef CAS PubMed.
- D. Chen and M. Jiang, Acc. Chem. Res., 2005, 38, 494 CrossRef CAS PubMed.
- C. Giacomelli, V. Schmidt, K. Aissou and R. Borsali, Langmuir, 2010, 26, 15734 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37 CrossRef.
- Y. Cao, Y. Li, Y. Wu, W. Li, C. Yu, Y. Huang, L. Sun, Y. Bao and Y. Li, RSC Adv., 2016, 6, 105442 RSC.
- Y. Liu, H. Busscher, B. Zhao, Y. Li, Z. Zhang, H. van der Mei, Y. Ren and L. Shi, ACS Nano, 2016, 10, 4779 CrossRef CAS PubMed.
- L. Shen, R. Qu, H. Shi, F. Huang, Y. An and L. Shi, Biomater. Sci., 2016, 4, 857 RSC.
- R. Qu, L. Shen, Z. Chai, C. Jing, Y. Zhang, Y. An and L. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 19207 CAS.
- Y. Chen, D. Zhao and Y. Liu, Chem. Commun., 2015, 51, 12266 RSC.
- X. Wang, L. Zhao, R. Ma, Y. An and L. Shi, Chem. Commun., 2010, 46, 6560 RSC.
- X. Wang, Z. Chai, R. Ma, L. Zhao, Z. Zhang, Y. An and L. Shi, J. Colloid Interface Sci., 2012, 388, 80 CrossRef CAS PubMed.
- Z. Zhang, R. Ma and L. Shi, Acc. Chem. Res., 2014, 47, 1426 CrossRef CAS PubMed.
- L. Zhao, R. Qu, A. Li, R. Ma and L. Shi, Chem. Commun., 2016, 52, 13543 RSC.
- S. Motala-Timol, D. Jhurry, J. Zhou, A. Bhaw-Luximon, G. Mohun and H. Ritter, Macromolecules, 2008, 41, 5571 CrossRef CAS.
- Z. Liu, Q. Ma, Y. Liu and Q. Wang, Org. Lett., 2013, 16, 236 CrossRef PubMed.
- J. Fan, Y. Wang, A. Blake, C. Wilson and E. Davies, Angew. Chem., Int. Ed., 2007, 46, 8013 CrossRef CAS PubMed.
- D. Guldi, H. Hungerbuehler and K. Asmus, J. Phys. Chem., 1995, 99, 13487 CrossRef CAS.
- M. Brettreich and A. Hirsch, Tetrahedron Lett., 1998, 39, 2731 CrossRef CAS.
- H. Imahori, M. El-Khouly, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 325 CrossRef CAS.
- J. Costa, A. Farinha, M. Neves and A. Tomé, Dyes Pigm., 2016, 135, 163 CrossRef CAS.
- Y. Yamakoshi, N. Umezawa, A. Ryu, K. Arakane, N. Miyata, Y. Goda, T. Masumizu and T. Nagano, J. Am. Chem. Soc., 2003, 125, 12803 CrossRef CAS PubMed.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285 CrossRef CAS PubMed.
- L. Slavětínská, J. Mosinger and P. Kubát, J. Photochem. Photobiol., A, 2008, 195, 1 CrossRef.
- M. Ballatore, J. Durantini, N. Gsponer, M. Suarez, M. Gervaldo, L. Otero, M. Spesia, M. Milanesio and E. Durantini, Environ. Sci. Technol., 2015, 49, 7456 CrossRef CAS PubMed.
- Z. Chai, C. Jing, Y. Liu, Y. An and L. Shi, Colloid Polym. Sci., 2014, 292, 1329 CAS.
- Y. Amao, ChemCatChem, 2011, 3, 458 CrossRef CAS.
- D. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS PubMed.
- F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC.
- M. Milanesio, M. Alvarez, V. Rivarola, J. Silber and E. Durantini, Photochem. Photobiol., 2005, 81, 891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00196g |
| This journal is © The Royal Society of Chemistry 2017 |