
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inclusion complexes of organic salts with β-cyclodextrin as organocatalysts for CO2 cycloaddition with epoxides†
Kun Li,
Xiaohui Wu,
Qingwen Gu,
Xiuge Zhao,
Mingming Yuan,
Wenbao Ma,
Wenxiu Ni and
Zhenshan Hou *
*
Key Laboratory for Advanced Materials, Research Institute of Industrial Catalysis, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: houzhenshan@ecust.edu.cn
First published on 7th March 2017
Abstract
The inclusion complexes between β-cyclodextrin (β-CD) and the organic bases 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU)-based phenolates have been prepared by a simple method and also characterized by FT-IR, 1H NMR, 19F NMR, TGA etc. Among these inclusion complexes, DBU-based 2,3,4,5,6-pentafluorophenolate as a guest compound bound by β-CD ([DBUH][PFPhO]/β-CD) has been employed as an easily-separable organocatalyst for the cycloaddition of CO2 into cyclic carbonate and exhibited the best catalytic performance. High conversion of epoxides and excellent selectivity to carbonates could be achieved at 130 °C and under the 3.0 MPa CO2 without additional organic solvents or additives. Additionally, the organocatalyst [DBUH][PFPhO]/β-CD exhibited the better recycability in consecutive catalytic recycles, as compared with that of corresponding DBU-based phenolates. The β-CD played a crucial role in immobilizing catalytically active species and thus improving the recyclability of the present organocatalysts. The detailed characterization indicated that phenolate anions had been bound inside the cavity of β-CD, while the [DBUH]+ cation was located outside of β-CD. More interestingly, it was observed that phenolate anions could dissociate from the β-CD cavity under the reaction temperature, but the inclusion compound could form on cooling the reaction mixture after reaction, which was extremely attractive for separation and recycling of the supramolecular organocatalysts. Finally, on the basis of the characterization above, a reaction mechanism for the present organocatalysts has been proposed.
Introduction
Carbon dioxide (CO2) is a greenhouse gas that induces global warming and climate change. Meanwhile, it always can be regarded as an easily available and renewable carbon resource, which is mainly due to its advantages of being abundant, cheap, non-toxic, and nonflammable in CO2 capture and conversion.1 Transformation of CO2 into highly valuable chemicals is of great importance and has attracted increasing attention. One of the most attractive synthetic goals starting from carbon dioxide is the chemical fixation of CO2 onto epoxide to afford the five-membered cyclic carbonates,2 such products are among the most important feedstocks which have been extensively used as aprotic polar solvents, electrolyte components in lithium batteries, precursors for polycarbonates, and intermediates in organic synthesis.3A broad variety of homogeneous catalysts for the synthesis of cyclic carbonates have been developed and reported so far, such as quaternary ammonium and phosphonium salts,4 bromine,5 ionic liquid,6 triphenylphosphine-phenol,7 metalloporphyrins8 and polyoxometalate.9 However, these catalysts generally suffer from low to moderate catalytic activity, selectivity and poor recyclability. Besides, a co-solvent/additive and harsh reaction conditions such as high pressure and/or high temperature are required in some cases. Meanwhile, the inherent corrosion, toxicity and environmental problems associated with metallic cations and halide anions are also concerned from the viewpoint of green chemistry. Thus, the development of a highly efficient, metal and halide ions-free environment-friendly catalytic system for the cycloaddition of carbon dioxide with epoxide are highly desirable.10 Meanwhile, the recovery of homogeneous catalyst from products could also be a non-ignorable defect which may result in the difficulty for further product purification and catalyst reuses.11
By contrast, heterogeneous catalysts, due to its convenience of separation and recovery, have received more attention and extensive application than homogeneous analogues in the catalytic synthesis of cyclic carbonates. From this point, a series of inorganic oxides and organic polymers supported catalysts have been developed. For examples, these supports include mesoporous silica,12 polymers,13 graphene oxide,14 carbon nanotubes,15 chitosan (CS)16 etc. The synergistic effect of hydroxyl groups of solid materials on promoting the reaction has also been unveiled.
The supramolecular chemistry played a significant role in many fields, while the supramolecular catalysis has recently emerged and been well developed.17 The main strategy is to assemble building blocks by weak interactions to elaborate new catalytic systems, which could avoid the modification of the catalyst backbone by means of multistep synthesis as usually required to improve the catalytic performance of covalent catalysts. Especially, β-cyclodextrin (β-CD) has been used as a host molecule to construct the supramolecular catalysts with the aid of the interactions of the host–guest type. β-CD are cyclic oligosaccharides connected by α-1,4 linkages (Scheme 1), frequently characterized as a doughnut-shaped truncated cone.18 The primary hydroxy groups (OHs) of glucose units are located on the narrower rim of the cone, and the secondary OHs are on the wide one. The β-CD's external surface is hydrophilic to make them be soluble in water, while their hydrophobic cavity can host a variety of guest organo-molecules leading to the formation of guest/β-CD inclusion complexes. Actually, inclusion complexes between β-CD and various guest molecules have been employed as efficient catalysts and studied extensively.19
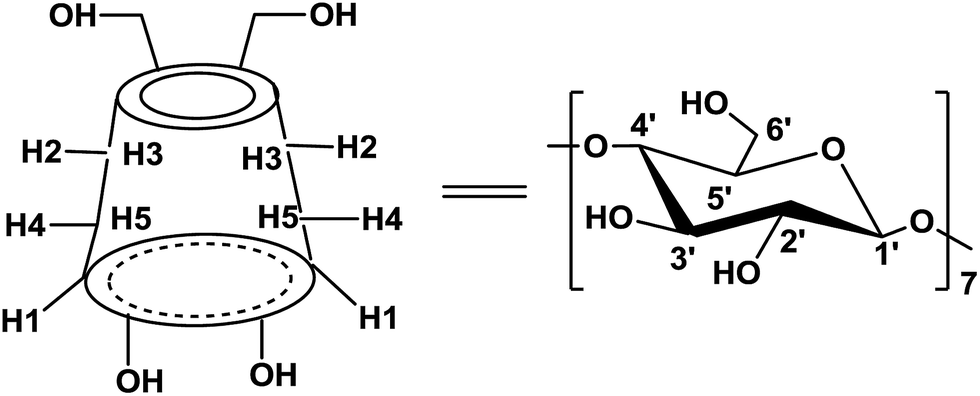 | ||
| Scheme 1 Chemical structures of β-CD. | ||
On the other hand, organic Lewis base like 1,8-diazabicyclo-[5.4.0]-undec-7-ene (DBU), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 1,4-diazabicyclo[2.2.2]octane (DABCO), etc., showed some activity for chemical fixation of CO2 into cyclic carbonates and were regarded as an environmental-friendly catalysts due to the absence of halogen ions.20,21 However, these organic base-derived materials normally acted as homogeneous catalysts, resulting in difficult recovery of the catalysts. Taking into account of the drawbacks of the homogeneous base catalysts, in our previous work, we have found that niobate salts of organic base were highly efficient catalysts for CO2 cycloaddition, and the catalyst was stable in the consecutive recycles.22 As far as we have known, the efficient synthesis of cyclic carbonate by using the metal-free heterogeneous catalysts still remains a huge challenge and is highly promising. Herewith, we attempted to prepare inclusion complexes of DBU-based phenolates with β-CD by a simple method and to use as heterogeneous organocatalysts for the cycloaddition of carbon dioxide with epoxide without additional solvents and additives. It was found that the β-CD played a crucial role in immobilizing catalytically active species and thus improving the recyclability of the supramolecular organocatalysts. Furthermore, the reaction mechanism over the present supramolecular catalysts has been proposed on the basis of the detailed characterization.
Results and discussion
Characterizations of catalysts
First, the different kinds of salts were synthesized by simply neutralizing phenol derivatives with DBU in tetrahydrofuran (THF). The β-CD was chosen as host molecules because it is highly thermally stable and owns abundant hydroxyl groups. Through the hydrogen bond interaction, DBU-based phenolates were thus included by β-CD in aqueous solution (Scheme 2).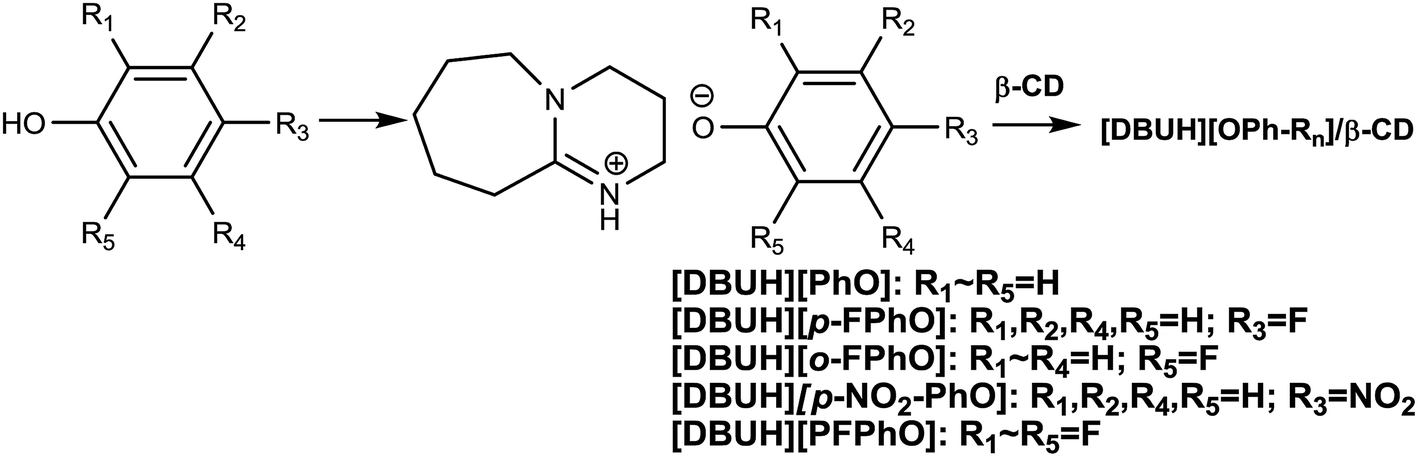 | ||
| Scheme 2 The synthetic route of β-CD inclusion complexes. | ||
In the first step, a series of DBU-based phenolate salts were firstly characterized by FT-IR spectra. As shown in Fig. 1b–f, after the neutralization reaction, the characteristic peaks at 3600 cm−1 of hydroxyl groups on phenol and other phenol derivatives disappeared, and the new peaks at 2935 cm−1 can be assigned to the stretching vibration of N+–H.22 The peaks at 1646 cm−1 and 2854 cm−1 could be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+ and C–H vibration on protonated DBU, respectively. The peak at 1499 cm−1, 1593 cm−1 could be assigned to CC vibration on benzene ring.22–24 1H NMR spectra of DBU-based phenolates have been depicted in Fig. S1,† the signal of hydroxyl hydrogen in phenol moved to downfield after the salts were formed (9–12 ppm). Additionally, the amidinium carbon in the DBU structure moved to downfield from 161.8 ppm to 178.5 ppm (13C NMR spectra shown in Fig. S2†), as compared with that of free DBU (δ = 161.5 ppm),25 revealing that DBU has been protonated after neutralization.23 All these results proved that the different kinds of DBU-based salts have been attained unambiguously.
N+ and C–H vibration on protonated DBU, respectively. The peak at 1499 cm−1, 1593 cm−1 could be assigned to CC vibration on benzene ring.22–24 1H NMR spectra of DBU-based phenolates have been depicted in Fig. S1,† the signal of hydroxyl hydrogen in phenol moved to downfield after the salts were formed (9–12 ppm). Additionally, the amidinium carbon in the DBU structure moved to downfield from 161.8 ppm to 178.5 ppm (13C NMR spectra shown in Fig. S2†), as compared with that of free DBU (δ = 161.5 ppm),25 revealing that DBU has been protonated after neutralization.23 All these results proved that the different kinds of DBU-based salts have been attained unambiguously.
 | ||
| Fig. 1 The FT-IR spectra of (a) β-CD; (b) [DBUH][PhO]; (c) [DBUH][p-FPhO]; (d) [DBUH][o-FPhO]; (e) [DBUH][p-NO2-PhO]; (f) [DBUH][PFPhO]; (g) [DBUH][PFPhO]/β-CD; (h) the recovered [DBUH][PFPhO]/β-CD organocatalyst after recycling. | ||
Because all these DBU-based phenolates served as homogeneous catalysts in cycloaddition reaction of CO2 as shown from our experimental observation, we next have prepared the inclusion complexes of DBU-based salts and β-CD with the aid of the interactions of the host–guest type. The 1H NMR spectra of all inclusion complexes was shown in Fig. S3.† Before the inclusion, the chemical shifts of H-3′ and H-5′ protons of β-CD were 3.60 and 3.56, respectively. Having included the DBU-based phenolates, the chemical shifts of H-3′ and H-5′ moved to 3.59 and 3.55. As shown in Scheme 1, the H-3′ and H-5′ were located inside the cavity of β-CD and formed the inner wall in it, when the guest molecules or the groups such as benzene rings on guest molecules were included in the cavity of β-CD, the shielding effect of these molecules could changed the chemical shifts of H-3′ and H-5′,26 but the H-2′, H-4′ and H-6′ out of the cavity still remained unchangeable. It was worth noticing that the chemical shifts of all protons on [DBUH]+ cation in all salts did not undergo any obvious changes, while the resonance signal from H6 protons in phenolates anions disappeared after inclusion (Fig. S1 vs. S3†). Moreover, the NMR signal from the protons of OH-2′ and OH-3′ in β-CD moved to higher field and became boarder after inclusion (Fig. S3a–S3f†), which might result from the hydrogen bonding interaction between the OH-2′ and OH-3′ protons and [DBUH]+ cation. From these results above, it suggested that the inclusion complexes between β-CD and DBU-based phenolates have been formed.
Sequentially, taking inclusion complexes formed between [DBUH][PFPhO] with β-CD as an example, FT-IR, 19F NMR and 13C NMR provided an additional evidence of inclusion compound formation. As shown in Fig. 1a, β-CD exhibited the characteristic peaks at 3373 cm−1 (–OH), 2926 cm−1 (C–H stretch) (too closing to overlapped with the peaks of N+–H on protonated DBU), 1160 cm−1 (C–O–C stretch/O–H bend), 1250–1370 cm−1 (C–H bend), 1029 cm−1 (C–O stretch). After inclusion (Fig. 1f and g), the catalyst still remains the characteristic peaks of β-CD, which indicated that the skeleton structure of β-CD remained untouched. The peaks at 2935 cm−1 (N+–H stretch) and 1646 cm−1 (CN+ vibration) on DBU seems unchanged after inclusion. It should be noted that the peak at 1593 cm−1 (CC on benzene ring) disappeared and the peak at 1499 cm−1 (CC on benzene ring) became weaker after inclusion. Meanwhile, 19F NMR spectra (500 MHz) of [DBUH][PFPhO]/β-CD was shown in Fig. 2. It was found that the chemical shifts of fluorine atoms on the para, ortho and meta position changed from −192.67, −172.08 and −171.31 to −192.27, −171.98 and −170.86 respectively after inclusion, which suggested the deshielding effects of β-CD on [DBUH][PFPhO].19b What's more, from the 13C NMR spectra (Fig. S4b and S4c†), after inclusion the 13C NMR signals of guest anion [PFPhO]− increased varying from 0.3 ppm to 0.8 ppm. Furthermore, the para carbon signals of phenolate anions disappeared nearly (Fig. S4c†), while all protons in [DBUH]+ did not show any obvious changes in chemical shifts after inclusion (Fig. S4b and S4c†). This had been explained in previous work that carbon atoms of guest molecules which are deeper in the cavity seem to undergo shielding effects with β-CD.26 Additionally, if the concentration of [DBUH][PFPhO]/β-CD increased further, the 13C NMR signals did not showed much change (Fig. S4d and S4e†), but the 19F NMR signals of guest anion [PFPhO]− changed considerably (Fig. S5a and S5b†). All these results also suggested that the β-CD had included the [DBUH][PFPhO] successfully. Most likely, [DBUH]+ cation was located outside of β-CD, while the hydrophobic phenol derivatives as the guest molecules was bound inside the cavity of β-CD.
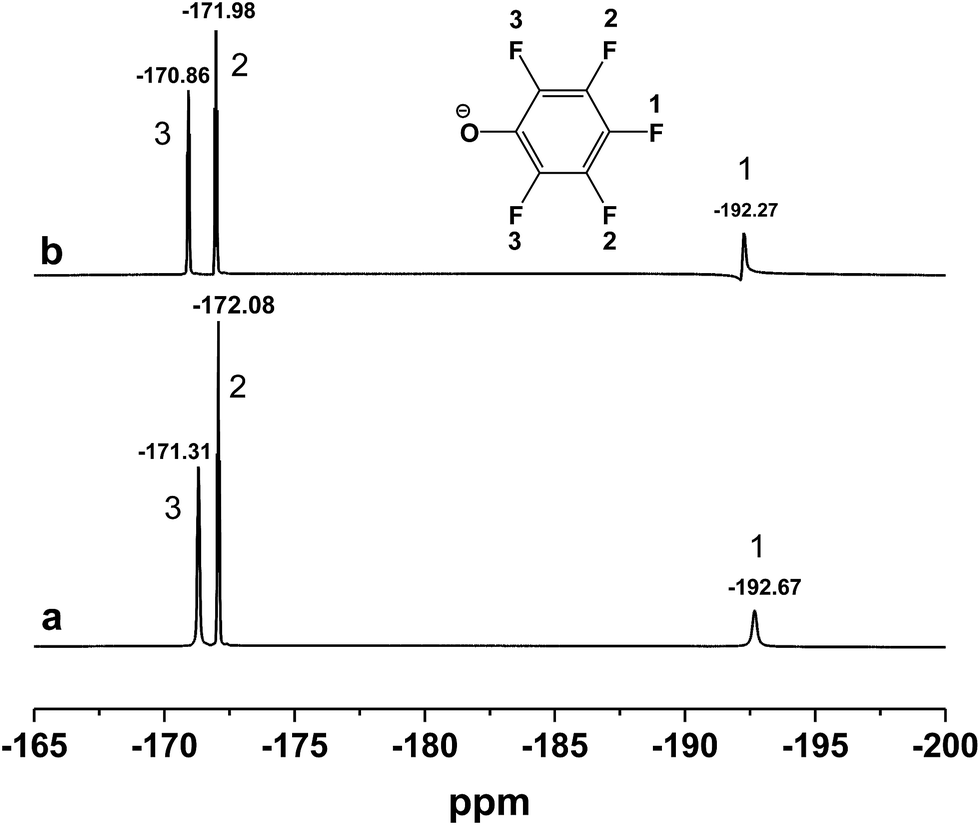 | ||
| Fig. 2 19F NMR spectra of (a) [DBUH][PFPhO]; (b) [DBUH][PFPhO]/β-CD. The concentration of [DBUH][PFPhO] in d6-DMSO was 0.054 mmol mL−1. | ||
The thermal stability of the β-CD and the inclusion complex catalysts [DBUH][PFPhO]/β-CD were investigated by TGA. As shown in Fig. S6,† β-CD showed a slightly weight loss below 100 °C, which could be attributed to the removal of the adsorbed H2O on the surface of β-CD. The β-CD started to decompose after 300 °C, which indicated its highly thermal stability. The salt [DBUH][PFPhO] appeared an obviously weight loss at 150 °C, after inclusion with β-CD, the decomposition onset temperature was around 200 °C, indicating that the thermal stability of [DBUH][PFPhO] was improved due to the formation of inclusion complexes. Scanning electron microscopy (SEM) was also employed to observe the morphology of β-CD, [DBUH][PFPhO] and [DBUH][PFPhO]/β-CD, and their SEM images were shown in Fig. S7†. Compared with pure β-CD, the inclusion compounds have a lower dispersity (Fig. S7a and 7d vs. S7c and 7f†). Modification of crystals can be added as a proof of the formation of a solid inclusion complexes with hydrophobic cavity of β-CD.
Catalyst performance
In the previous work, DBU has been proved as a well homogeneous catalyst for the cycloaddition reaction of CO2 with epoxides.20 In this work, for the sake of comparison, we synthesized a serial of salts containing DBU and different kinds of phenol derivatives, and their inclusion complexes of β-CD, which all have been employed as our catalysts. The cycloaddition reaction of propylene oxide (PO) and CO2 to propylene carbonate (PC) was chosen as a model reaction to evaluate the catalytic performance under the optimized reaction conditions. The catalytic activities over these catalysts have been displayed in Fig. 3. When only use the β-CD as catalyst, it can be seen that only PO conversion of 29.8% was attained and 1,2-propanediol was formed as a main product without any formation of cyclic carbonates. When DBU and phenol were used as homogeneous catalysts, the conversion of PO was 80% and 8.5%, respectively. However, it was found that the catalytic activity was improved considerably by using different DBU-based phenolates. Interestingly, with the decrease of pKa value of the phenolate anions within catalysts (Table S1†), the catalytic activity was increased (Fig. 3). It indicated that the stronger acidity of phenol derivatives really benefits the reaction. Previous reports have suggested that the parallel requirement of both Lewis base and Lewis acid in the fixation of CO2.27 As a result, the increasing in acidity may promote the activation of CO2.28 From this standpoint, the salt [DBUH][PFPhO] formed with DBU and 2,3,4,5,6-pentafluorophenol owning the lowest pKa value of 5.50 showed the highest catalytic activity under the present condition. On the other hand, it was worth noticing that 2,3,4,5,6-pentafluorophenol had CO2-philic properties, which presumably facilitated CO2 conversion with increasing activity.29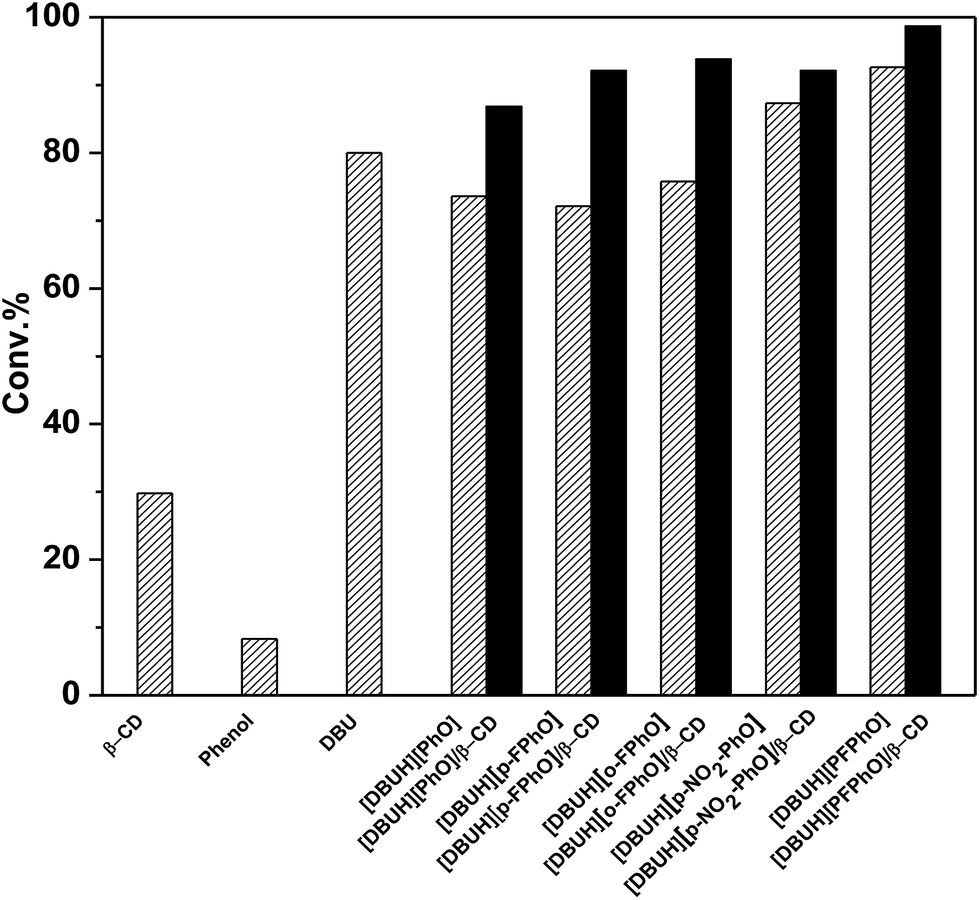 | ||
| Fig. 3 The effect of different catalysts on the conversions of cycloaddition reaction. Reaction conditions: PO 0.7 mL (10 mmol), organocatalyst (1.5 mmol%), 130 °C, 3 MPa. All catalysts showed over 98% selectivities toward cyclic carbonates except that β-CD afford 1,2-propanediol as a main by-product. | ||
Since the DBU-based phenolate catalyst was miscible with cyclic carbonate after reaction, leading to the deficient recovery of catalyst. In contrast, it was found that the inclusion complexes as organocatalysts not only were separated easily only by standing and filtration after reaction but also even afforded higher catalytic activity, in comparison with that of the corresponding DBU-based phenolate catalysts (Fig. 3), which was probably due to abundant hydroxyl on β-CD's surface.30 Moreover, it has been reported that the acidity of guest molecules (here are phenol derivatives) bound to β-CD was considerably higher than that of the corresponding free one,31 which could result in stronger acidity after inclusion in the present situation and thus benefits the reaction as well. Among all these organocatalysts, [DBUH][PFPhO]/β-CD was chosen as a main catalyst in the following investigation owing to its excellent catalytic performance.
Influence of reaction parameters
All probable effects of reaction condition (temperature, CO2 pressure and reaction time and catalyst amount) were considered and investigated over the [DBUH][PFPhO]/β-CD catalyst. Firstly, the effect of the reaction temperature has been examined and the results were shown in Fig. 4a. With the increment of temperature from 90 °C to 130 °C, the conversion of PO increased significantly from 20% to 96%. However, the PO conversion leveled off under higher reaction temperature. Consequently, it was suggested that 130 °C was the appropriate temperature for the cycloaddition reaction.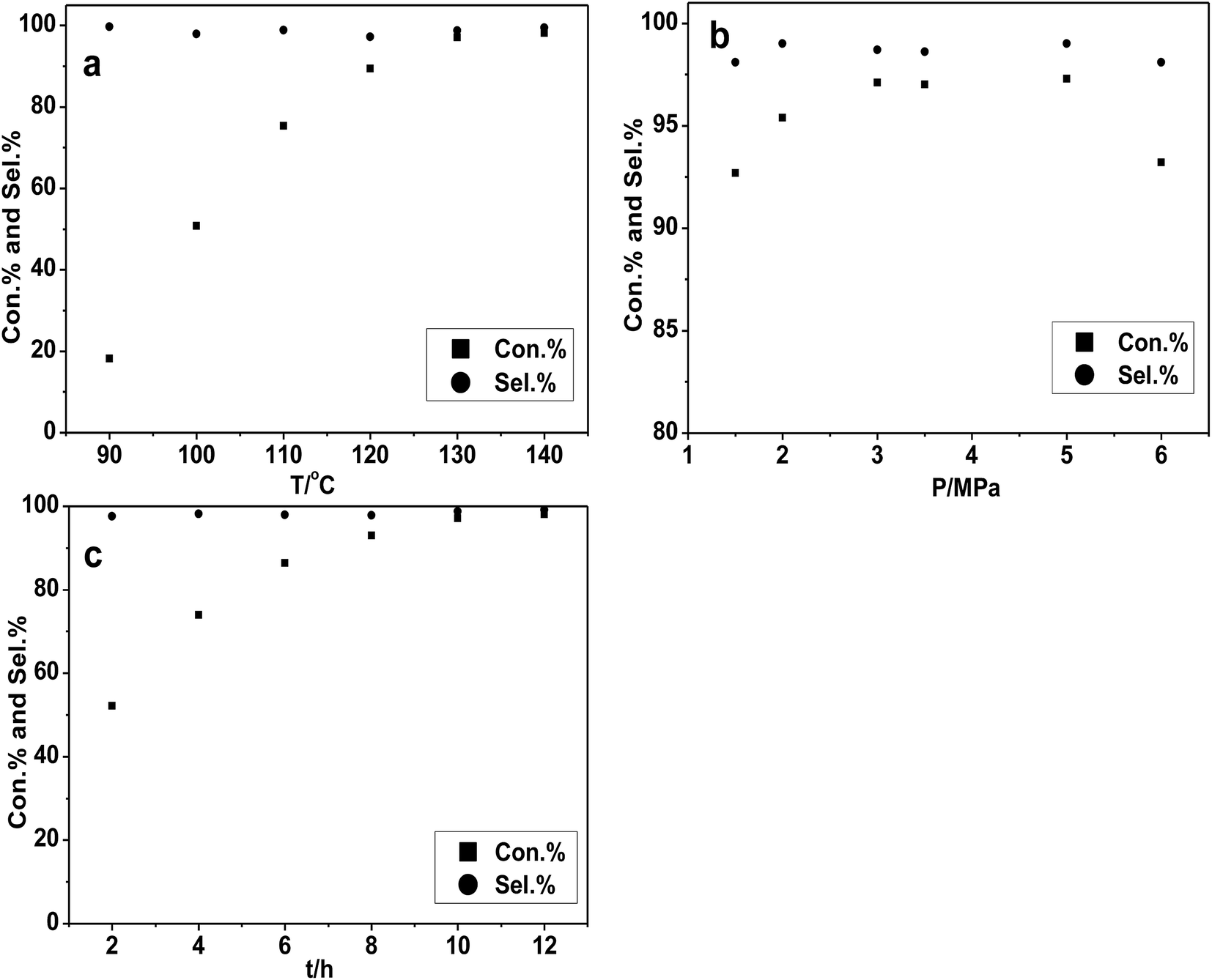 | ||
| Fig. 4 The effects of reaction temperature, pressure and time on conversion and selectivity of cycloaddition reaction. Reaction condition: PO 0.7 mL (10 mmol); organocatalyst 1.5 mmol%. (a) CO2 pressure 3 MPa; time 10 h; (b) temperature 130 °C; time 10 h; (c) temperature 130 °C; CO2 pressure 3 MPa. | ||
Then the impact of the CO2 pressure on cycloaddition reaction of PO was studied and depicted in Fig. 4b. The conversion of PO was increased in some extent with the reaction pressure changing from 1.5–3.0 MPa. It could possibly be explained that more CO2 can be dissolved in PO as increasing reaction pressure,6e which facilitated the CO2 cyclic addition reaction. However, when the reaction pressure continued to rise to 6.0 MPa, the conversion of PO has a slight decrease while the selectivity has no obvious change. This might be related to a dilution effect occurring that the excess CO2 reduced the concentration of the catalyst in the vicinity or hinder the active mass transport, which was also be observed in our group's previous studies.22,32,33 The impact of reaction time under the condition of 130 °C and 3.0 MPa can also be obtained in Fig. 4c. The conversion of PO had a gradual increasing from 2 to 10 h and then remained almost unchanged with high selectivity all time. The substrate was almost completely converted into PC within 10 h with high selectivity.
At last, we investigated the effect of the catalyst amount to the reaction and the results were shown in Table S2.† With the increasing of the catalyst amount from 1 mmol% to 1.5 mmol%, the conversion of PO had an obvious improvement. The possible explanation is that increasing the catalytic activity center indeed benefits a lot to the reaction. However, the further increasing of catalyst amount leads to a sharp descent. Due to the significant molecular weight of β-CD, the excessive amount of catalyst may be harmful to the mass transfer of the reactant and then decrease the conversion of PO. Thus, it was apparent that the appropriate catalyst amount was 1.5 mmol%.
Reaction kinetics
Sequentially, the reaction kinetics of catalysts [DBUH][PFPhO]/β-CD and [DBUH][PFPhO] was investigated respectively. The procedure for the studies of the kinetic parameters was similar to our group's previous work.33 PO (0.7 mL, 10 mmol), catalysts (1.5 mmol%) was loaded into autoclave, CO2 was introduced to the reaction and kept at 3.0 MPa under the reaction temperature. The rate constant was determined from the experimental data assuming pseudo-first order reaction kinetics. The kinetic parameters were studied over temperature ranged from 110 to 140 °C. The relationship of −ln(1 − Con.) and PO remaining with reaction time at different temperature was shown in Fig. S8.† Finally, as shown in Fig. 5, the activation energy (Ea) for the process was calculated using the Arrhenius equation, where Ea1 of [DBUH][PFPhO]/β-CD was 43.1 kJ mol−1 while the Ea2 of [DBUH][PFPhO] was 59.9 kJ mol−1. It can be seen that from this results that the inclusion of β-CD indeed owned lower Ea for the process and really benefited the cycloaddition reaction of CO2 with epoxides. The activation energy under the present conditions is consistent with that of the other many reports, in which Ea was in the range of about 35–70 kJ mol−1.34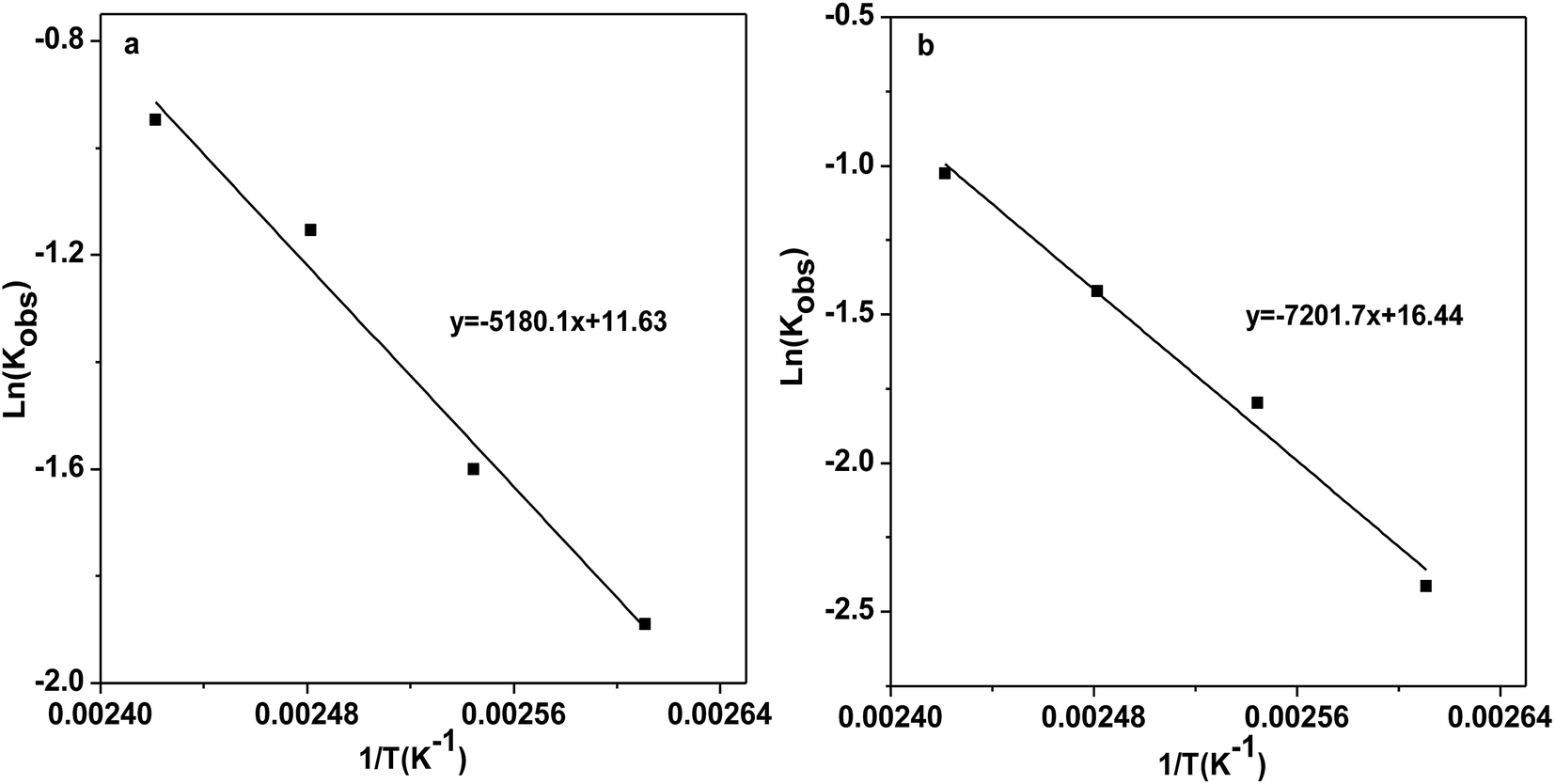 | ||
| Fig. 5 Arrhenius plots for the CO2 cycloaddition to PC catalyzed by (a) [DBUH][PFPhO]/β-CD; (b) [DBUH][PFPhO]. | ||
The substrate scope of the catalyst
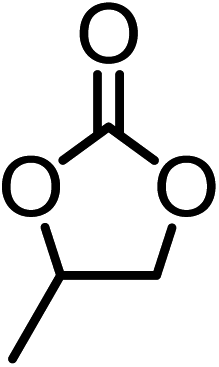
The results above demonstrated that the inclusion complexes of organic salt [DBUH][PFPhO] with β-CD was an highly efficient organocatalyst for the cycloaddition of CO2 and PO without any organic solvents and additives. Thus, a series of epoxides were tested to synthesize the corresponding carbonates under optimized conditions to check the scope of the substrates over the present catalyst and the results were shown in Table 1. It was found that the organocatalyst was applicable to producing cyclic carbonates from CO2 and various epoxides with high conversions and selectivities (Table 1, entries 1–6). Compared with other epoxides, 1,2-epoxyhexane and 1,2-epoxycyclohexane needed a longer reaction time to produce corresponding cyclic carbonates because of the higher steric hindrance (Table 1, entries 5 and 6).| Entry | Substrates | Products | Con. (%) | Sel. (%) |
|---|---|---|---|---|
| a Reaction conditions: PO 0.7 mL (10 mmol), organocatalyst (1.5 mmol%), 130 °C, 3 MPa, 10 h.b Reaction time: 30 h.c Reaction time: 45 h.d The main by-product was corresponding diol. | ||||
| 1 | 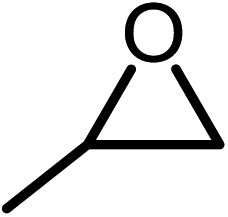 |
 |
98.7 | 99.1 |
| 2 |  |
 |
98.8 | 97.9 |
| 3 |  |
 |
95.6 | 99.8 |
| 4 | 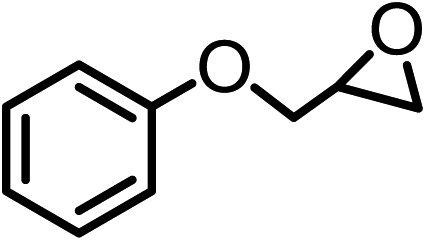 |
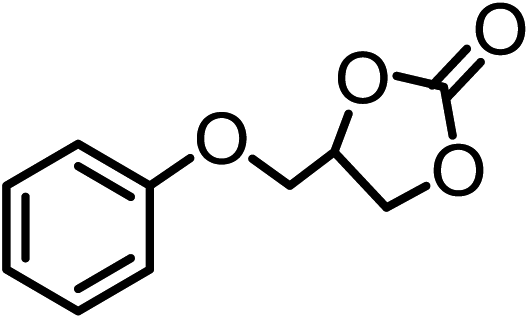 |
99.5 | 99.2 |
| 5b | 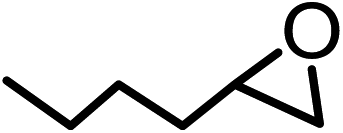 |
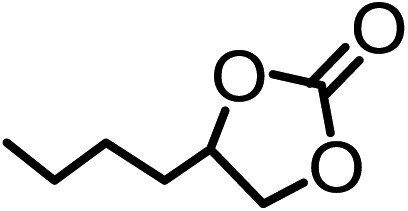 |
94.8 | 99.1 |
| 6c | 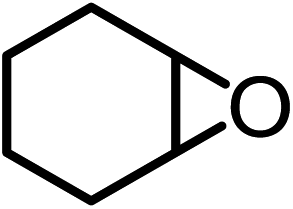 |
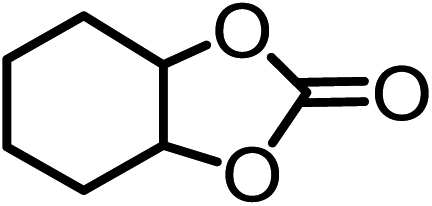 |
81.7 | 76.8d |
Catalyst stability
In the next step, the reusability of catalyst [DBUH][PFPhO]/β-CD was examined under the optimal conditions. After the reaction, the anhydrous ether was added to the reaction mixture. The catalyst was recovered by a simple standing and filtration, followed by washing with ethyl ether in 3 times and vacuum drying at 60 °C. Then the recovered catalyst can be reused for the next recycle. The reusability of [DBUH][PFPhO]/β-CD catalyst was shown in Fig. 6. It was found that the catalyst could be reusable for at least up to four times with high product selectivity, while the conversion of PO had a slight decrease in the consecutive recycles. Subsequently, the FT-IR of the reused [DBUH][PFPhO]/β-CD catalysts was displayed in Fig. 1h. Compared with the fresh catalyst, after recycling, the catalyst still remains the characteristic peaks of β-CD, which indicated that the skeleton structure of β-CD remained unchangeable. The peak at 1646 cm−1 (CN+ vibration) and the peak at 1499 cm−1 (CC on benzene ring) were still visible in the spite of the intensity was decreased, which may caused by slightly leaching of [DBUH][PFPhO] from the cavity of β-CD. Additionally, the new band at 1792 cm−1 were assigned to the CO stretching of the residual propylene carbonate over catalyst surface.22,35
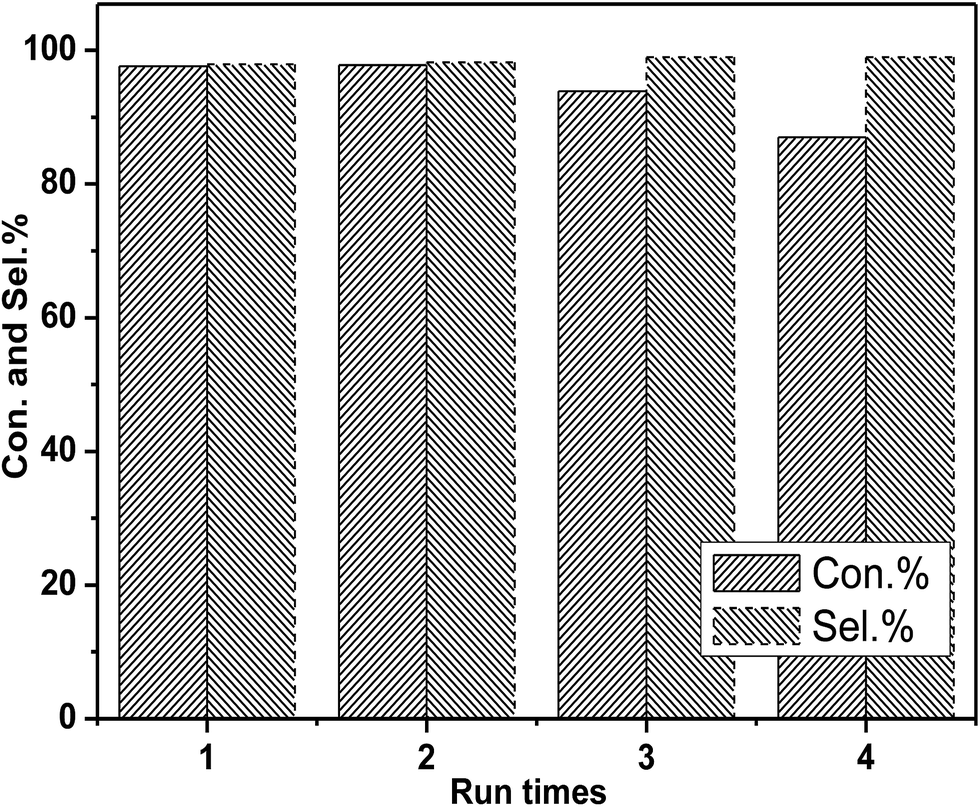 | ||
| Fig. 6 Recyclability of [DBUH][PFPhO]/β-CD. Reaction conditions: PO 0.7 mL (10 mmol), organocatalyst (1.5 mmol%), 130 °C, 3 MPa. | ||
Reaction mechanism
As we have mentioned above, the [DBUH][PFPhO] and β-CD can form the inclusion complexes by means of the host–guest interaction. In order to further investigate the chemical state of [DBUH][PFPhO] bound in the inclusion complexes during the reaction, the 19F NMR spectra (500 MHz) of the [DBUH][PFPhO]/β-CD had been determined at elevated temperature by simulating reaction condition. The organocatalyst was dissolved in d6-DMSO, and then the 19F NMR spectra was recorded in 25 °C, 50 °C and 70 °C in turn. After that, the solution was cooled down to 25 °C and recorded the spectra of the catalyst. The results were shown in Fig. 7. It was found that at 25 °C, the chemical shifts of fluorine atom on para, ortho and meta position was −192.27, −171.98 and −170.86, respectively (Fig. 7a). While the temperature was raised to 50 °C and 70 °C, the chemical shifts of all fluorine atoms moved slightly to the downfield, where was −192.94, −172.21, −171.17 and −192.98, −172.24 and −171.17, respectively (Fig. 7b and c). Increasing the concentration [DBUH][PFPhO]/β-CD in d6-DMSO, the chemical shifts of all fluorine atoms moved to the downfield more obviously (Fig. S5a vs. S5c†). When the solution cooled down back to 25 °C, the spectra of catalyst remain unchangeable, compared with its initial spectra in 25 °C (Fig. 7d). The change of chemical shifts in 50 °C and 70 °C may caused by the overflowing of guest [DBUH][PFPhO] from the β-CD's cavity. After cooled down the solution back to 25 °C, the [DBUH][PFPhO] moved into cavity and kept its initial state. This revealed that organic salt [DBUH][PFPhO] might dissociate from cavity of β-CD under the reaction temperature, but inclusion compound formed as cooling the reaction mixture after reaction, which was extremely attractive for separation and recycling of the supramolecular organocatalysts.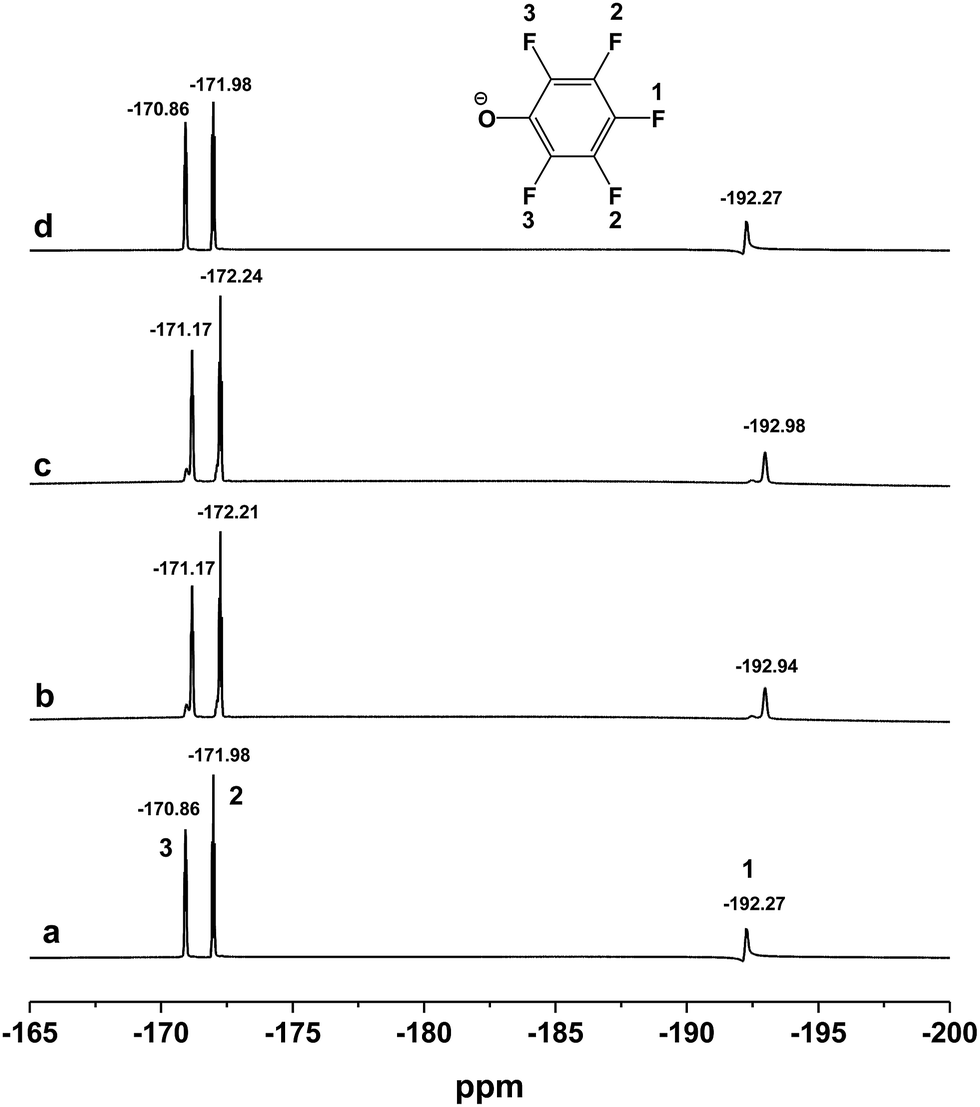 | ||
| Fig. 7 The 19F NMR spectra (500 MHz) of the [DBUH][PFPhO]/β-CD (0.054 mmol mL−1 in d6-DMSO) at different temperature. (a) 25 °C; (b) 50 °C; (c) 70 °C; (d) cooled down back to 25 °C. | ||
In order to further gain a deeper insight into the reaction mechanism, DRIFTS of in situ PO adsorption was performed over the [DBUH][PFPhO]/β-CD in 298 K. As shown in Fig. 8a, the peaks at 1423, 1409 and 1399 cm−1 can be assigned to C–H bending vibration of gas-phase PO molecules.33 After PO was introduced and purging with Ar, it was observed that the peaks of gas-phase PO molecules disappeared, and the new peaks at 1458 and 1452 cm−1 were formed (Fig. 8b and c). But these peaks were disappeared with keeping purging with Ar more longer time (Fig. 8d), indicating weak interaction of PO molecules with organocatalyst at room temperature. The new peaks could be assigned to C–O bending vibration of a probable intermediate species forming between PO and [DBUH][PFPhO] by hydrogen bond (species 2 in Scheme 3). This result suggested that the [DBUH][PFPhO] acted a crucial role in activating PO molecules. Next, the DRIFT of in situ CO2 adsorption over the [DBUH][PFPhO]/β-CD was performed and the results have been listed in Fig. 9. The gas CO2 exhibited its anti symmetric stretching vibration peaks at 2367 and 2344 cm−1 (Fig. 9a and b), respectively.36 When the CO2 was introduced on [DBUH][PFPhO]/β-CD, the peak intensity was increased. But with purging with Ar, no absorption peaks can be detected (Fig. 9h), which suggested that CO2 could only physically absorbed on the [DBUH][PFPhO]/β-CD and showed weak absorption ability on catalyst at room temperature. Meanwhile, it should be noticed that when we introduced the PO and CO2 on [DBUH][PFPhO]/β-CD simultaneously, only CO2 peaks could be observed and the peak from PO adsorption was too weak to be observable. Furthermore, we can easily found that the peak intensity became much stronger (Fig. 9d and e vs. g). From these results, it can be deduced that the existence of the adsorbed PO can promote the CO2 absorption on [DBUH][PFPhO]/β-CD organocatalyst.
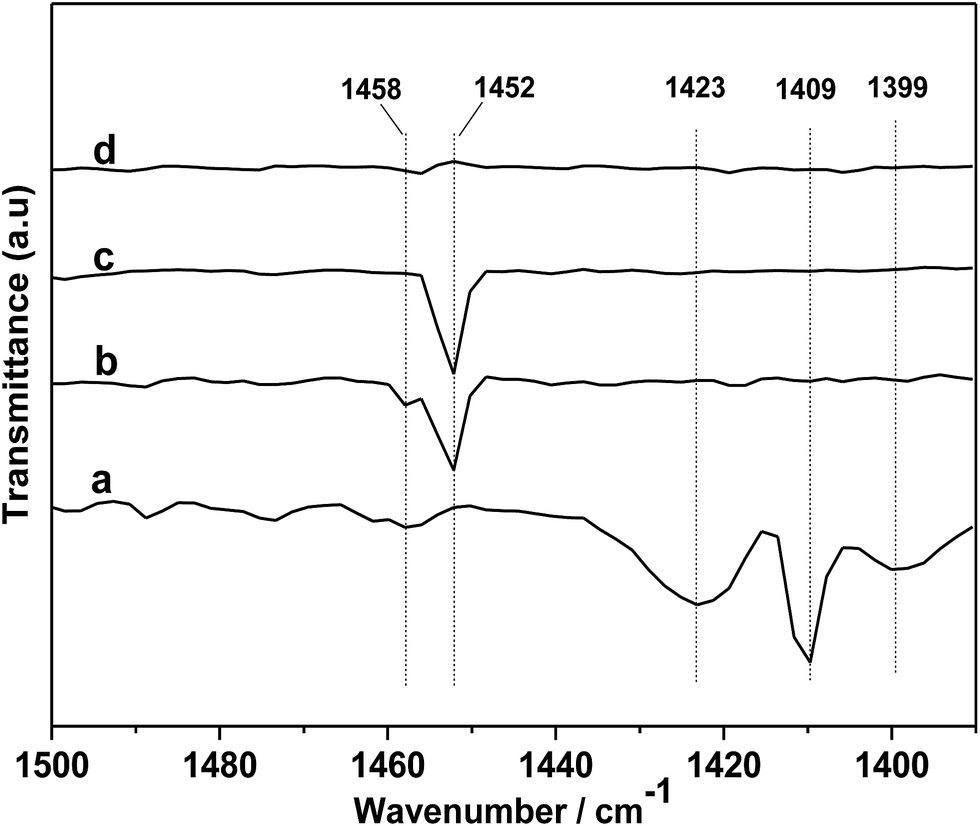 | ||
| Fig. 8 DRIFT spectra of gas-phase PO (a) and in situ PO adsorbed on [DBUH][PFPhO]/β-CD samples followed purging with the flow of Ar (50 mL min−1), (b) 3 min; (c) 6 min; (d) 9 min. | ||
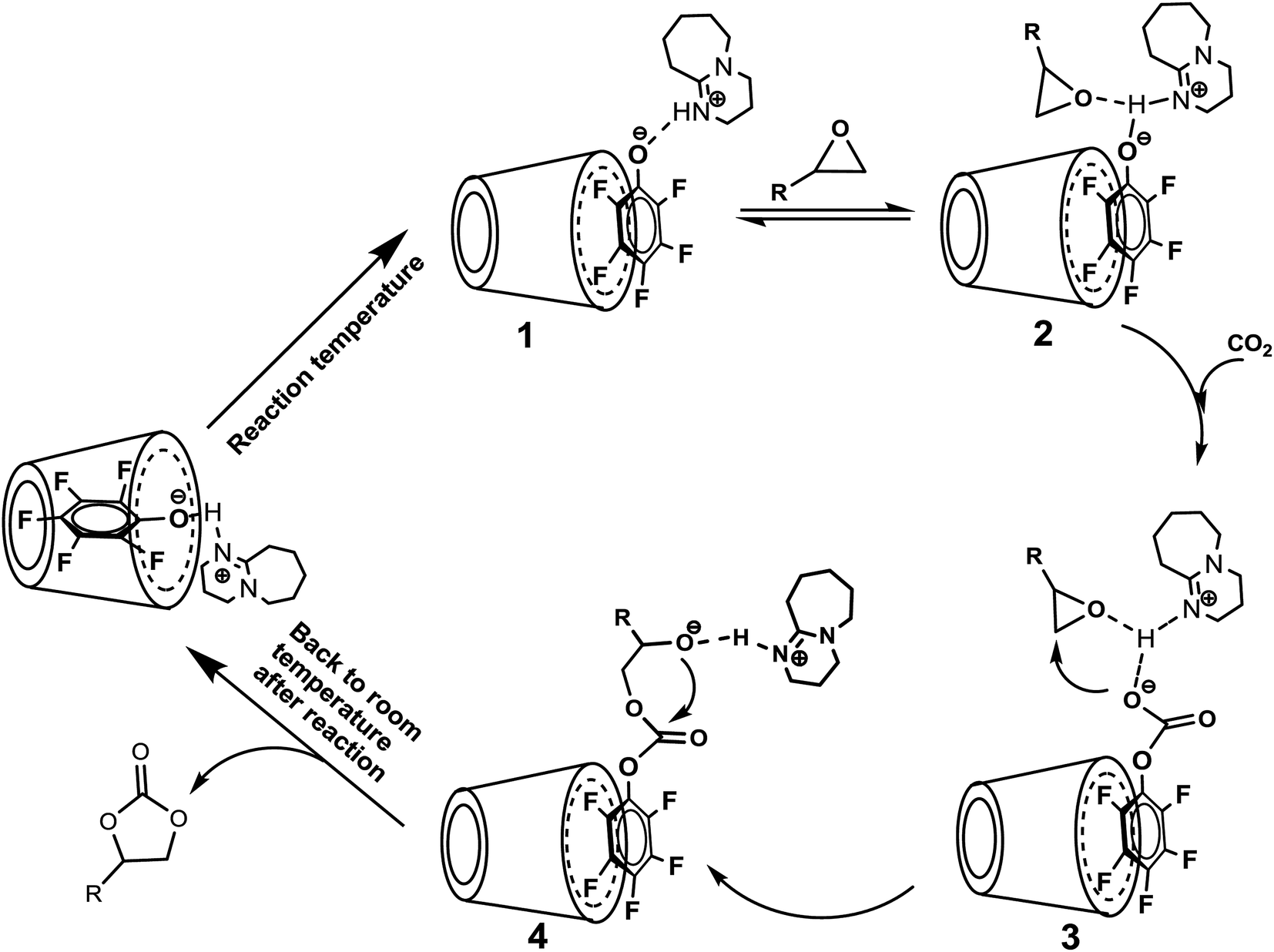 | ||
| Scheme 3 The proposed reaction mechanism for cycloaddition of epoxide and CO2 catalyzed by [DBUH][PFPhO]/β-CD catalyst. | ||
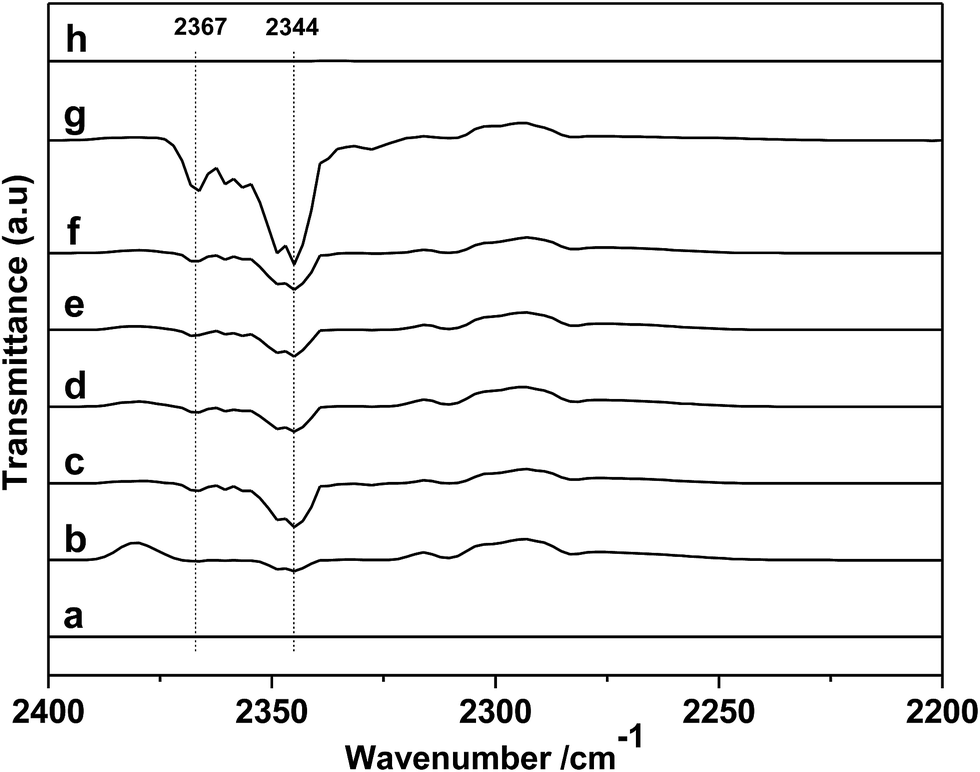 | ||
| Fig. 9 DRIFT spectra of (a) baseline; (b) gas-phase CO2; (c) gas-phase CO2 on [DBUH][PFPhO]/β-CD; (d) in situ CO2 adsorbed on [DBUH][PFPhO]/β-CD samples with purging CO2 for 5 min; (e) for 15 min; (f) gas-phase CO2 and PO on [DBUH][PFPhO]/β-CD; (g) in situ CO2 and PO co-adsorbed on [DBUH][PFPhO]/β-CD samples with purging CO2; (h) all samples purging with Ar (50 mL min−1) for 5 min. | ||
From what has been discussed above, we may draw the conclusion that 2,3,4,5,6-pentafluorophenol had CO2-philic properties, and it may capture the CO2 and forms a critical intermediate species during the reaction. Thus we attempted to characterize this probable critical intermediate species formed with [PFPhO]− anion and CO2. A stainless autoclave was charged with [DBUH][PFPhO] (336 mg, 1.0 mmol) and CO2 (1.0 MPa), and the mixture was stirred at a constant temperature (60 °C) for 24 h. Carefully release of CO2 gave a white powder. The product was characterized by 13C NMR spectra, a new signal at 159.2 ppm produced after the absorption of CO2 (Fig. S9b†), which can be assigned to carbonate carbon.37 Based on the previous reports, and observed product, a plausible intermediate species formed with [PFPhO]− anion and CO2 can be proposed (Scheme S1†), which exhibits the interaction between the electronegative oxygen on [PFPhO]− and CO2 to form the intermediate [PFPhO-CO2]− in CO2 cycloaddition reaction.38 Another new signal at 129.4 ppm could be assigned to the gas CO2 which was dissolved in DMSO.39 The solvent of CO2 adduction with [DBUH][PFPhO] was vacuumed and the 13C NMR spectra was recorded again. As shown in Fig. S9c,† it was found that the peaks at 159.2 and 129.4 ppm disappeared completely, which indicated that the peaks at 129.4 ppm and 159.2 ppm were highly relevant to CO2 adduction.
Based on the results we have obtained, we proposed a reasonable mechanism for this chemical fixation reaction of CO2 (Scheme 3). As the temperature increasing, the guest compound [DBUH][PFPhO] dissociated from the cavity of β-CD under the reaction temperature, and could immobilize on the surface through the hydrogen bond. The proton in DBU-based salts was coordinated with the oxygen of the epoxide through a hydrogen bond, resulting in the activation of an epoxide,2c,4h,12b,28 and this is followed by CO2 insertion to form the intermediate species [PFPhO-CO2]− bounding with the [DBUH]+ and PO through the hydrogen bond. Simultaneously, the nucleophilic attack of [PFPhO-CO2]− on the less sterically hindered β-carbon atom of the epoxide furnished the ring-opened intermediate species, and then the intermediate species made a subsequent intramolecular ring-closure, cyclic carbonate was formed. With the temperature cooled down after reaction, the guest compound [DBUH][PFPhO] could be back to the cavity of β-CD and the catalyst can be regenerated. It should be noticed that the hydroxyl groups of the glucopyranose monomers (such as cellulose and β-CD etc.) act as hydrogen bond donor and thus activate the ring-opening reaction of epoxides through the hydrogen bond on the oxygen atom of the epoxide.20,30,40 In the present catalyst system, we could regard β-CD as an additional activator for the ring-opening of epoxide and thus promoted the cycloaddition of carbon dioxide with epoxide.
Conclusions
In summary, we have developed a series of DBU-based phenolates and their inclusion complexes with β-CD to act as the supramolecular organocatalysts in chemical fixation of CO2 into cyclic carbonates. High epoxides conversion and excellent cyclocarbonates selectivity can be achieved under the optimal reaction conditions. Among these catalysts, the organocatalyst [DBUH][PFPhO]/β-CD afforded the best catalytic performance. Furthermore, it can be easily separated after reaction and reused with high activity and selectivity. Based on all characterization, we proposed a reasonable mechanism for this reaction, where β-CD played a crucial role in immobilizing catalytically active species and thus improving the recyclability of the supramolecular catalyst. This easily-prepared, metal and halogen ion-free, inexpensive and recyclable catalytic system has potential application for catalytically synthesizing cyclic carbonates from CO2 and epoxides.Experimental
Chemical materials
β-Cyclodextrins (β-CDs) was purchased from Sinopharm Chemical Reagent Co., Ltd., Shanghai. Organic bases 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) was also purchased from Sinopharm Chemical Reagent Co., Ltd., and directly used without any further purification. Phenol and its other kinds of substituted derivatives such as 2,3,4,5,6-pentafluorophenol, 4-fluorophenol, 2-fluorophenol and 4-nitrophenol were all purchased from Shanghai Macklin Biochemical Co., Ltd., and were also used without further purification. CO2 was supplied by Shanghai Shangnong Gas Factory with a purity of >99.95%. All kinds of epoxides were analytic grade and were used directly without further purification. All solvents (analytic grade) were dried by using the methods that have been previously reported.Characterizations
1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE 400 MHz instrument (400 MHz 1H NMR), and 19F NMR spectra were recorded on a Bruker AVANCE 500 MHz instrument (500 MHz 19F NMR). The concentration of samples in deuterated solvent was 0.054 mmol mL−1, unless indicated otherwise. The FT-IR spectra were recorded at RT on a Nicolet Magna 550 FT-IR spectrometer. The thermal stability of catalysts was determined by Thermogravimetry analysis (TGA) method (heating rate: 10 °C min−1; N2 flow: 100 mL min−1) using Perkin Elmer Pyris Diamond Analyzer. The scanning electron microscopy (SEM) images were performed on JSM electron microscopes (JEOL JSM-6360LV, Japan). In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) experiments were conducted on a Nicolet 6700 FTIR fitted with a liquid nitrogen cooled mercury-cadmium-telluride detector (MCT). The DRIFTS cell (Harrick, HVC-DRP) fitted with CaF2 windows were used as the reaction chamber that allowed samples to be heated to 150 °C. All the spectra were within the range of 4000–1200 cm−1 at the resolution of 4 cm−1 and 64 scans. Prior to the adsorption experiments, the samples were pretreated in Ar at 150 °C for 4 h. Then the samples were cooled down to 25 °C in order to remove the contaminants and the PO/CO2-adsorbed FT-IR spectra of the samples (40 mg) were recorded at 25 °C.Preparation of DBU-based phenolates
Preparation of inclusion complexes
The inclusion complexes were prepared by mixing the DBU-based phenolates (1 mmol) and β-CD (1 mmol, 1.135 g) in aqueous solution at 60 °C for 6 h under the N2 atmosphere. After the solution was cooled to room temperature, the water was removed by rotary evaporation to get the solid powder. Then drying the powder at 70 °C for 2 h under the vacuum afforded the inclusion complexes as white powder (Fig. S3†).Reaction of cycloaddition from CO2 and peroxides
The cycloaddition reaction of propylene oxide (PO) and CO2 to propylene carbonate (PC) was used as a model reaction to evaluate the catalytic performance. The catalyst (0.15 mmol) and PO (10 mmol) were added into the glass bush, and then put the bush into the 50 mL stainless autoclave and then sealed it. The loading of supramolecular organocatalyst was based on that of DBU-based phenolates for the reaction. The reactor was purged with CO2 for 3 times to remove the air into the reactor, and finally charged with CO2 to the desired pressure. After the reaction, the reactor was cooled in ice-water bath immediately, and the CO2 was slowly vented while anhydrous ether was as absorption liquid to absorb the PO and products entrained by CO2. Then, the anhydrous ether in the trap was added to the reactor and washed the catalyst to extract products and substrate. The catalyst was separated simply by standing and filtration, and the filtrate was subjected to GC-MS and GC analysis. The catalyst was washed with anhydrous ether 3 times and dried in a vacuum at 60 °C to be used directly in next reaction.Acknowledgements
The authors are grateful for support from the National Natural Science Foundation of China (21373082) and innovation Program of Shanghai Municipal Education Commission (15ZZ031), and the Fundamental Research Funds for the Central Universities.Notes and references
- (a) K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266 CrossRef CAS PubMed; (b) M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2 CrossRef CAS; (c) S. Wang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 2308 CrossRef CAS PubMed; (d) J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981 RSC; (e) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375 RSC; (f) P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562 RSC; (g) G. Ji, Z. Yang, H. Zhang, Y. Zhao, B. Yu, Z. Ma and Z. Liu, Angew. Chem., Int. Ed., 2016, 55, 9685 CrossRef CAS PubMed; (h) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (i) M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 RSC; (j) M. Peters, B. Kohler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Muller, ChemSusChem, 2011, 4, 1216 CrossRef CAS PubMed.
- (a) P. Z. Li, X. J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142 CrossRef CAS PubMed; (b) T. T. Liu, J. Liang, Y. B. Huang and R. Cao, Chem. Commun., 2016, 52, 13288 RSC; (c) M. Ding and H. L. Jiang, Chem. Commun., 2016, 52, 12294 RSC; (d) Y. Toda, Y. Komiyama, A. Kikuchi and H. Suga, ACS Catal., 2016, 6, 6906 CrossRef CAS; (e) S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2016, 18, 4855 RSC; (f) X. Liu, S. Zhang, Q. W. Song, X. F. Liu, R. Ma and L. N. He, Green Chem., 2016, 18, 2871 RSC; (g) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC; (h) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC; (i) J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588 CrossRef CAS; (j) D. H. Lan, F. M. Yang, S. L. Luo, C. T. Au and S. F. Yin, Carbon, 2014, 73, 351 CrossRef CAS.
- (a) M. Liu, B. Liu, L. Liang, F. Wang, L. Shi and J. Sun, J. Mol. Catal. A: Chem., 2016, 418–419, 78 CrossRef CAS; (b) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC; (c) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (d) M. Yoshida and M. Ihara, Chem.–Eur. J., 2004, 10, 2886 CrossRef CAS PubMed.
- (a) F. D. Bobbink and P. J. Dyson, J. Catal., 2016, 343, 52 CrossRef CAS; (b) V. Besse, N. Illy, G. David, S. Caillol and B. Boutevin, ChemSusChem, 2016, 9, 2167 CrossRef CAS PubMed; (c) V. Butera, N. Russo, U. Cosentino, C. Greco, G. Moro, D. Pitea and E. Sicilia, ChemCatChem, 2016, 8, 1167 CrossRef CAS; (d) H. Yang, X. Wang, Y. Ma, L. Wang and J. Zhang, Catal. Sci. Technol., 2016, 6, 7773 RSC; (e) S. Liu, N. Suematsu, K. Maruoka and S. Shirakawa, Green Chem., 2016, 18, 4611 RSC; (f) V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561 CrossRef; (g) Y. Zhou, S. Hu, X. Ma, S. Liang, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2008, 284, 52 CrossRef CAS; (h) N. Aoyagi, Y. Furusho and T. Endo, Tetrahedron Lett., 2013, 54, 7031 CrossRef CAS.
- J. A. Kozak, J. Wu, X. Su, F. Simeon, T. A. Hatton and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 18497 CrossRef CAS PubMed.
- (a) Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513 RSC; (b) H. Yang, Y. Gu, Y. Deng and F. Shi, Chem. Commun., 2002, 274 RSC; (c) J. Sun, L. Han, W. Cheng, J. Wang, X. Zhang and S. Zhang, ChemSusChem, 2011, 4, 502 CrossRef CAS PubMed; (d) A.-L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Gonçalves and J. Dupont, Green Chem., 2014, 16, 2815 RSC; (e) L. Xiao, D. Su, C. Yue and W. Wu, J. CO2 Util., 2014, 6, 1 CrossRef CAS; (f) J. Palgunadi, O. S. Kwon, H. Lee, J. Y. Bae, B. S. Ahn, N. Y. Min and H. S. Kim, Catal. Today, 2004, 98, 511 CrossRef CAS.
- Y. M. Shen, W. L. Duan and M. Shi, J. Org. Chem., 2003, 68, 6705 CrossRef PubMed.
- (a) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC; (b) T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. Y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270 CrossRef CAS PubMed; (c) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134 CrossRef CAS PubMed; (d) Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, Green Chem., 2015, 17, 2853 RSC.
- H. Yasuda, L. He, T. Sakakura and C. Hu, J. Catal., 2005, 233, 119 CrossRef CAS.
- (a) H. Zhou, G. X. Wang, W. Z. Zhang and X. B. Lu, ACS Catal., 2015, 5, 6773 CrossRef CAS; (b) L. Martinez-Rodriguez, J. Otalora Garmilla and A. W. Kleij, ChemSusChem, 2016, 9, 749 CrossRef CAS PubMed; (c) C. Kohrt and T. Werner, ChemSusChem, 2015, 8, 2031 CrossRef CAS PubMed; (d) D. H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C. T. Au and S. F. Yin, Chin. J. Catal., 2016, 37, 826 CrossRef CAS.
- (a) Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 Search PubMed; (b) L. F. Xiao, F. W. Li, J. J. Peng and C. G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265 CrossRef CAS; (c) D. H. Lan, L. Chen, C. T. Au and S. F. Yin, Carbon, 2015, 93, 22 CrossRef CAS; (d) D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81 CrossRef CAS.
- (a) L. Han, S. W. Park and D. W. Park, Energy Environ. Sci., 2009, 2, 1286 RSC; (b) W. Cheng, X. Chen, J. Sun, J. Wang and S. Zhang, Catal. Today, 2013, 200, 117 CrossRef CAS; (c) A. S. Aquino, F. L. Bernard, J. V. Borges, L. Mafra, F. D. Vecchia, M. O. Vieira, R. Ligabue, M. Seferin, V. V. Chaban, E. J. Cabrita and S. Einloft, RSC Adv., 2015, 5, 64220 RSC.
- (a) Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066 CrossRef CAS PubMed; (b) W. Wang, C. Li, L. Yan, Y. Wang, M. Jiang and Y. Ding, ACS Catal., 2016, 6, 6091 CrossRef CAS; (c) Z. Dai, Q. Sun, X. Liu, C. Bian, Q. Wu, S. Pan, L. Wang, X. Meng, F. Deng and F.-S. Xiao, J. Catal., 2016, 338, 202 CrossRef CAS; (d) A. H. Jadhav, G. M. Thorat, K. Lee, A. C. Lim, H. Kang and J. G. Seo, Catal. Today, 2016, 265, 56 CrossRef CAS; (e) W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767 RSC.
- W. H. Zhang, P. P. He, S. Wu, J. Xu, Y. Li, G. Zhang and X. Y. Wei, Appl. Catal., A, 2016, 509, 111 CrossRef CAS.
- L. Han, H. Li, S. J. Choi, M. S. Park, S. M. Lee, Y. J. Kim and D. W. Park, Appl. Catal., A, 2012, 429–430, 67–72 CrossRef CAS.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660 RSC.
- (a) J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS PubMed; (b) M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875 CrossRef CAS PubMed.
- (a) F. Hapiot, H. Bricout, S. Menuel, S. Tilloy and E. Monflier, Catal. Sci. Technol., 2014, 4, 1899 RSC; (b) A. H. Karoyo, P. S. Sidhu, L. D. Wilson, P. Hazendonk and A. Borisov, J. Phys. Chem. C, 2015, 119, 22225 CrossRef CAS.
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071 RSC.
- Y. M. Shen, W. L. Duan and M. Shi, Adv. Synth. Catal., 2003, 345, 337–340 CrossRef CAS.
- A. Chen, C. Chen, Y. Xiu, X. Liu, J. Chen, L. Guo, R. Zhang and Z. Hou, Green Chem., 2015, 17, 1842 RSC.
- (a) L. Branco, G. Carrera, A. Costa and M. da Ponte, Synlett, 2013, 24, 2525 CrossRef; (b) B. Wang, Z. Luo, E. H. M. Elageed, S. Wu, Y. Zhang, X. Wu, F. Xia, G. Zhang and G. Gao, ChemCatChem, 2016, 8, 830 CrossRef CAS; (c) I. Cota, R. Chimentao, J. Sueiras and F. Medina, Catal. Commun., 2008, 9, 2090 CrossRef CAS.
- (a) Y. Xie, R. Parnas, B. Liang, Y. Liu, C. Tao and H. Lu, Chin. J. Chem. Eng., 2015, 23, 1728 CrossRef CAS; (b) L. Mino, A. Zecchina, G. Martra, A. M. Rossi and G. Spoto, Appl. Catal., B, 2016, 196, 135 CrossRef CAS.
- J. W. Wiench, L. Stefaniak, E. Grech and E. Bednarek, J. Chem. Soc., Perkin Trans., 1999, 2, 885 RSC.
- (a) H. J. Schneider, F. Hacket and V. Rudiger, Chem. Rev., 1998, 98, 1755 CrossRef CAS PubMed; (b) K. Srinivasan, K. Sivakumar and T. Stalin, Carbohydr. Polym., 2014, 113, 577 CrossRef CAS PubMed.
- (a) H. S. Kim, J. J. Kim, B. G. Lee, O. S. Jung, H. G. Jang and S. O. Kang, Angew. Chem., Int. Ed., 2000, 39, 4096 CrossRef CAS; (b) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS.
- Y. M. Shen, W. L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080 CrossRef CAS.
- (a) Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Energy Environ. Sci., 2013, 6, 3684 RSC; (b) Z. Z. Yang, Y. Zhao, G. Ji, H. Zhang, B. Yu, X. Gao and Z. Liu, Green Chem., 2014, 16, 3724 RSC.
- J. Song, Z. Zhang, B. Han, S. Hu, W. Li and Y. Xie, Green Chem., 2008, 10, 1337 RSC.
- (a) K. Srinivasan, T. Stalin and K. Sivakumar, Spectrochim. Acta, Part A, 2012, 94, 89 CrossRef CAS PubMed; (b) R. Rajamohan, S. Kothai Nayaki and M. Swaminathan, J. Solution Chem., 2011, 40, 803 CrossRef CAS.
- B. Song, L. Guo, R. Zhang, X. Zhao, H. Gan, C. Chen, J. Chen, W. Zhu and Z. Hou, J. CO2 Util., 2014, 6, 62 CrossRef CAS.
- X. Wu, M. Wang, Y. Xie, C. Chen, K. Li, M. Yuan, X. Zhao and Z. Hou, Appl. Catal., A, 2016, 519, 146 CrossRef CAS.
- S. Supasitmongkol and P. Styring, Catal. Sci. Technol., 2014, 4, 1622 CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 91, 133 CrossRef CAS.
- (a) S. Chen, T. Cao, Y. Gao, D. Li, F. Xiong and W. Huang, J. Phys. Chem. C, 2016, 120, 21472 CrossRef CAS; (b) J. Yu and S. S. C. Chuang, Energy Fuels, 2016, 30, 7579 CrossRef CAS.
- (a) X. Luo, Y. Guo, F. Ding, H. Zhao, G. Cui, H. Li and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 7053 CrossRef CAS PubMed; (b) C. Wang, H. Luo, H. Li, X. Zhu, B. Yu and S. Dai, Chem.–Eur. J., 2012, 18, 2153 CrossRef CAS PubMed; (c) C. Wang, H. Luo, D. E. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978 CrossRef CAS PubMed.
- Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728 CrossRef CAS PubMed.
- S. Morooka, C. Wakai, A. Nobuyuki Matubayasi and M. Nakahara, J. Phys. Chem. A, 2005, 109, 6610 CrossRef CAS PubMed.
- (a) K. R. Roshan, G. Mathai, J. Kim, J. Tharun, G. A. Park and D. W. Park, Green Chem., 2012, 14, 2933 RSC; (b) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00416h |
| This journal is © The Royal Society of Chemistry 2017 |