
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic lipids with a cyclen headgroup: synthesis and structure–activity relationship studies as non-viral gene vectors†
De-Chun Chang,
Yi-Mei Zhang,
Ji Zhang*,
Yan-Hong Liu and
Xiao-Qi Yu *
*
Key Laboratory of Green Chemistry and Technology (Ministry of Education), College of Chemistry, Sichuan University, Chengdu 610064, PR China. E-mail: xqyu@scu.edu.cn; jzhang@scu.edu.cn; Fax: +86-28-85415886
First published on 28th March 2017
Abstract
The clarification of the structure–activity relationships of non-viral gene delivery vectors is of great importance. A series of novel cyclen-based cationic lipids were synthesized and applied as gene carriers. The structures of these fifteen lipids varied with different linking groups and hydrophobic tails. The liposomes formed by these lipids and helper lipid DOPE could efficiently bind DNA and condense it into nano-sized particles with positive zeta-potentials and protect DNA from enzymatic digestion. Results revealed that the hydroxyl group or aromatic ring in the lipid structure would affect their DNA binding and transfection ability. Lipids with double hydrophobic tails gave much higher transfection efficiencies than those with a single tail, and aromatic rings in the lipid backbone might facilitate the transfection. The transfection efficiency could be further improved by optimizing the hydrophobic tails. The lipid with C14 tails gave a much higher efficiency than its analogs. A cytotoxicity assay showed that the lipids generally gave higher cell viabilities than a commercially available transfection reagent. It was suggested that such cationic lipids may act as promising non-viral gene delivery carriers.
1. Introduction
The success of gene therapy, which is considered as an innovative method to treat some diseases including genetic disorders, chronic and acute diseases and cancer, depends largely on the properties of the gene delivery vectors.1,2 Comparing with highly efficient viral vectors, non-viral vectors such as cationic lipids, cationic polymers and other nanoparticles have attracted more attention and been developed substantially in the past decades due to their low immunogenicity and controlled release.3–5 Among non-viral vectors, cationic lipids have clear morphological structure,6 ease of preparation, modification and characterization,7 and the ability to collaboratively assemble with other materials.8 Therefore, they have been used as an advanced platform for delivering various nucleic acid molecules in a diverse range of sizes to cells. Since the first study of DOTMA as a cationic lipid gene vector by Felgner in 1987,9 various types of lipids and liposomes for gene delivery have been reported.10 However, although the molecular architecture of cationic lipids has a significant impact on their cytotoxicity, intracellular trafficking and gene transfection efficiency (TE), the detailed structure–activity relationship (SAR) of such type of vectors remains unclear. Thus elaborate design of lipid structure is needed to comprehensively understand the SAR of the lipidic vectors.Generally speaking, cationic lipids contain three parts including polar headgroups, linking groups and hydrophobic tails. The change of each part can influence their physical and chemical characteristics along with TE.11 Various cationic lipids with heterocyclic headgroups such as imidazolium,12,13 pyridinium14,15 and macrocyclic polyamines16,17 were introduced in recent years to study their SAR and be used as promising gene vectors. In our previous studies, 1,4,7,10-tetraazacyclododecane (cyclen) with wide range of pKa values and the property of easy modification was used as an appropriate cationic headgroup to interact with negatively charged nucleic acids by electrostatic attraction.18–21 Although the positively charged nitrogen atoms in the headgroup may promote DNA condensation and endocytosis, it also increases the toxicity and reduces the tolerability in serum.22 Some strategies were applied to overcome these disadvantages such as the introduction of PEG,23 hydroxyl group,24 hyaluronic acid,25 or other negatively charged moieties.26 Besides, a few studies also found that lipidic vectors with aromatic groups in the structure might give improved TE and biocompatibility.27–29 It was speculated that aromatic groups may have some interactions with DNA base pairs through π–π interaction,30 also intercalation,31 leading to improved DNA binding ability.32 For examples, Ilies et al. designed a set of cationic lipids based on dopamine backbone which showed high TE.33 Hiremath and co-workers revealed that cationic surfactant lipids containing aromatic units at different locations of hydrophobic segments displayed remarkable thermal stability due to inter-monomer stacking.34 Meanwhile, the structures of hydrophobic tails also largely affect the TE of cationic lipids.20,35,36 The influences of the amounts, length, or saturation degrees of hydrophobic tails on the gene transfection still need in-depth studies for further clarification.
Herein, we designed and synthesized a series of single and double tailed cationic lipids with different liking moieties which may introduce hydroxyl or aromatic groups to the structure. The SAR of these cyclen-based cationic lipids was systematically studied, and the lipid which shows the best TE was identified. Further, the effect of hydrophobic alkyl tails was also investigated based on the optimized structure. We believe that the studies may give us some clues for the design of novel lipidic gene delivery vectors with high efficiency and biocompatibility.
2. Experimental section
2.1 Materials and methods
All chemicals and reagents were obtained commercially and were used as received. Anhydrous acetonitrile, dichloromethane (DCM), chloroform (CHCl3) and acetonitrile were dried and purified under nitrogen by using standard methods and were distilled immediately before use. [4,7,10-Tris(tert-butoxycarbonyl)-1,4,7,10-tetraazacyclododecan-1-yl]acetic acid (tri-boc-cyclen-acetic acid, compound 6, Scheme S2†), tri-tert-butyl 10-(4-(bromomethyl)benzyl)-1,4,7,10-tetraazacyclododecane-1,4,7-tricarboxylate (compound 7, Scheme S2†) were synthesized according to the literature.37,38 1,4,7,10-Tetraazacyclododecanes (cyclen) was purchased from Quzhou Synpartner Pharmaceutical Technology Co., Ltd. 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was purchased from Avanti Polar Lipids, Inc. The supercoiled plasmid DNA (pUC-19) used in agarose-gel assay was purchased from Takara (Dalian, China). The plasmids used in the study were pEGFP-N1 (Clontech, Palo Alto, CA, USA). MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfo-phenyl)-2H tetrazolium, inner salt) was obtained from Promega (Madison, WI, USA). The Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum and Lipofectamine® 2000 were purchased from Invitrogen Corp. MicroBCA protein assay kit was obtained from Pierce (Rockford, IL, USA). Luciferase assay kit was purchased from Promega (Madison, WI, USA). Endotoxin free plasmid purification kit was purchased from TIANGEN (Beijing, China). HEK 293 human embryonic kidney cell lines and HeLa cervical carcinoma cell lines were purchased from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. The NMR spectra were measured on a Agilent 400-MR DD2 spectrometer and the d scale in parts per million was referenced to residual solvent peaks or internal tetramethylsilane (TMS). MS-ESI spectra data were recorded on a Bruker Daltonics BioTOF mass spectrometer.2.2 Preparation of cationic liposome
The cationic lipid and the neutral lipid DOPE in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio were dissolved in 2.5 mL anhydrous chloroform in a glass vial. The solvent was removed under reduced pressure with a thin flow of moisture-free nitrogen gas. And the dried lipid film was then kept under high vacuum overnight. The lipid film was hydrated with 2.5 mL of Tris–HCl buffer (10 mM, pH 7.4) to the final lipid concentration of 1 mM. The samples were sonicated in a bath sonicator to generate small unilamellar vesicles according to previously described procedures.21
:1 mole ratio were dissolved in 2.5 mL anhydrous chloroform in a glass vial. The solvent was removed under reduced pressure with a thin flow of moisture-free nitrogen gas. And the dried lipid film was then kept under high vacuum overnight. The lipid film was hydrated with 2.5 mL of Tris–HCl buffer (10 mM, pH 7.4) to the final lipid concentration of 1 mM. The samples were sonicated in a bath sonicator to generate small unilamellar vesicles according to previously described procedures.21
2.3 Amplification and purification of plasmid DNA
pGL-3 and pEGFP-N1 plasmids were used. The former one was seed as the luciferase reporter gene, which was transformed in M109 Escherichia coli, and the latter one was used as the enhanced green fluorescent protein reporter gene, which was transformed in E. coli DH5α. Both plasmids were amplified in E. coli grown in LB medium at 37 °C and 220 rpm overnight. The plasmids were purified by an EndoFree Tiangen™ Plasmid Kit. Then, the purified plasmids were dissolved in TE (Tris + EDTA) buffer solution and stored at −80 °C. The integrity of plasmids was confirmed by agarose gel electrophoresis. The purity and concentration of plasmids were determined by the ratio of ultraviolet (UV) absorbances at 260 nm/280 nm (∼1.9). The concentrations of plasmids were determined by ultraviolet (UV) absorbance at 260 nm.2.4 Preparation of lipid/DOPE/DNA complexes (lipoplexes)
To prepare the lipid/DOPE/pDNA complexes (lipoplexes), various amounts of cationic lipids were mixed with a constant amount of DNA by pipetting thoroughly at various N/P ratios, and the mixture was incubated for 30 min at room temperature. The theoretical N/P ratio represents the charge ratio of cationic lipid to nucleotide base (mole ratios) and was calculated by considering the average nucleotide mass of 309.2.5 Agarose-gel retardation assay
Lipid/DOPE/pDNA complexes at different N/P ratios (the amino groups of lipids to phosphate groups of DNA) ranging from 0 to 8 were prepared by adding an appropriate volume of lipids to 0.125 μg (0.150 μg for the lipids D2 with five protonated N atoms) pUC-19 DNA. The complexes were incubated at 37 °C for 30 min. Then the complexes were electrophoresed on the 1.0% (w/v) agarose gel containing gel-red and with Tris-acetate (TAE) running buffer at 140 mV for 30 min. DNA was visualized with a UV lamp at wavelength of 312 nm by using BioRad Universal Hood II. SDS 1% solution and DNase I enzyme (5 μL DNase (2 unit per μL) and 5 μL 1% SDS per well) were added to the samples to evaluate the release and protection, respectively in similar method.2.6 Ethidium bromide replacement assay
The ability of lipids to condense DNA was studied using ethidium bromide (EB) exclusion assays. Fluorescence spectra were measured at room temperature in air by a Horiba Jobin Yvon Fluoromax-4 spectrofluorometer and corrected for the system response. EB (5 μL, 1.0 mg mL−1) was put into quartz cuvette containing 2.5 mL of 10 mM 4-(2-hydroxyethyl)-1-piperazinee-thanesulfonic acid (HEPES) solution (pH 7.4). After shaking, the fluorescence intensity of EB was measured. Then CT-DNA (10 μL, 1.0 mg mL−1) was added to the solution and mixed symmetrically, and the measured fluorescence intensity is the result of the interaction between DNA and EB. Subsequently, the solutions of lipid S1–S4 and D1–D6 (1 mmol L−1, 4 μL for each addition except for D2: 3.25 μL for each addition) were added to the above solution for further measurement. All the samples were excited at 520 nm and the emission was measured at 600 nm. The pure EB solution and DNA/EB solution without cationic liposome were used as negative and positive controls, respectively. The percent relative fluorescence (% F) was determined using the equation % F = (F − FEB)/(F0 − FEB), wherein FEB and F0 denote the fluorescence intensities of pure EB solution and DNA/EB solution, respectively.2.7 Lipoplex particle sizes and zeta potentials
Sizes and zeta potentials of the lipoplexes at various N/P ratios were analyzed at room temperature on a dynamic light scattering system (Zetasizer Nano ZS, Malvern instruments Led) at 25 °C. The lipoplexes particle solutions were first prepared by mixing S1/DOPE lipids and pUC-19 (2 μg mL−1) under diverse N/P ratios in 1 mL deionized water.2.8 Transmission electron microscopy (TEM)
TEM images were obtained on a JEM-100CX (JEOL) transmission electron microscope at an acceleration voltage of 100 kV. The TEM samples were prepared by dipping a copper grid with Formvar film into the freshly prepared nanoparticles solution (10 μL). A few minutes after the deposition, the aqueous solution was blotted away with a strip of filter paper and then the samples were dried for 2 min at room temperature. The samples were stained with phosphotungstic acid (ATP) aqueous solution and dried in air.2.9 Cell culture
Human embryonic kidney transformed 293 (HEK 293) cells and HeLa cervical carcinoma cells were incubated in DMEM containing 10% fetal bovine serum (FBS) and 1% antibiotics (penicillin–streptomycin, 10000 U mL−1) at 37 °C in a humidified atmosphere containing 5% CO2.
2.10 In vitro transfection procedure
Gene transfection of a series of complexes was investigated in HEK 293 and HeLa cells. In order to obtain about 80% confluent cultures at the time of transfection, 24-well plates were seeded with 85000 cell per well in 500 μL of serum-free media 24 h before transfection. For the preparation of lipoplexes applied to cells, various amounts of liposomes and DNA were serially diluted separately in antibiotic-free DMEM culture medium; then, the DNA solutions were added into liposome solutions and mixed briefly by pipetting up and down several times, after which the mixtures were incubated at room temperature for about 30 min to obtain lipoplexes of desired N/P ratios, the final lipoplexes volume was 100 μL, and the DNA was used at a concentration of 1 μg per well. After 30 min of complexation, old cell culture medium was removed from the wells, cells were washed once with serum-free DMEM, and the above 100 μL lipoplexes was added to each well. The plates were then incubated for 4 h at 37 °C in a humidified atmosphere containing 5% CO2. At the end of incubation period, medium was removed, and 500 μL of fresh DMEM medium containing 10% FBS was added to each well. Plates were further incubated for a period of 24 h before checking the reporter gene expression.
For fluorescence microscopy assays, cells were transfected by complexes containing pEGFP-N1. After 24 h incubation, GFP-expressed cells were observed using an inverted fluorescence microscope (Nikon Eclipse TE 2000E) equipped with a cold Nikon camera. Control transfection was performed in each case using a commercially available transfection reagent Lipofectamine 2000® based on the standard conditions specified by the manufacturer.
For fluorescent microscopy assays, cells were transfected by complexes containing pEGFP-N1. After 24 h incubation, the microscopy images were obtained at the magnification of 100 and recorded using Viewfinder Lite (1.0) software. Control transfection was performed in each case using a commercially available transfection reagent Lipofectamine 2000® based on the standard conditions specified by the manufacture.
For luciferase assays, cells were transfected by complexes containing pGL-3. For a typical assay in a 24-well plate, 24 h post transfection as described above, the old medium was removed from the wells, and the cells were washed twice with 500 μL of prechilled PBS. According to luciferase assay kit (Promega) manufacture, 100 μL of 1 × cell lysis buffer diluted with PBS was then added to each well, and the cells were lysed for 30 min in a horizontal rocker at room temperature. The cell lysate was transferred completely to Eppendorf tubes and centrifuged (4000 rpm, RT) for 2 min; the supernatant was transferred to Eppendorf tubes and stored in ice. For the assay, 20 μL of this supernatant and 100 μL of luciferase assay substrate (Promega) were used. The lysate and the substrate were both thawed to RT before performing the assay. The substrate was added to the lysate, and the luciferase activity was measured in a luminometer (Turner designs, 20/20, Promega, USA) in standard single-luminescence mode. The integration time of measurement was 10000 ms. A delay of 2 s was given before each measurement. The protein concentration in the cell lysate supernatant was estimated in each case with BCA protein t assay kit (PIERCE, Rockford, IL, USA) at a wavelength of 620 nm. Comparison of the TEs of the individual lipids was made based on the data for luciferase expressed as relative light units (RLU) per mg of protein. All the experiments were done in triplicates, and results presented are the average of at least two such independent experiments done on the same days.
2.11 Cytotoxicity of the lipoplexes
The cytotoxicity assay of the lipoplexes was carried out by using MTS with HEK 293 and HeLa cells. About 1 × 104 cells per well HEK 293 and HeLa cells were respectively seeded in 96-well plates. After 24 h culture, the medium was replaced with 50 μL of fresh medium with 10% FBS, to which 50 μL of lipoplexes at various N/P ratios of the liposome relative to DNA was added to achieve a final volume of 100 μL. After 24 h of incubation, lipoplexes solutions were removed, and 20 μL of MTS in 80 μL PBS was added to each well for additional 1 h incubation. The absorbance was measured using an ELISA plate reader (model 550, BioRad, USA) at a wavelength of 490 nm. The relative cell viability was calculated by using the following equation: viability (%) = (optical density of sample/optical density of control) × 100. The cell viability of Lipofectamine 2000® was also determined by MTS.3. Results and discussion
3.1 Synthesis of target cationic lipids
The chemical structures of cyclen-based cationic single-tailed (S1–S4) and double-tailed (D1–D6) lipids are shown in Fig. 1. To introduce hydroxyl or aromatic group to the structure, we chose some natural amino acids, such as glycine, serine, phenylalanine, tyrosine and their derivatives, to act as linking moiety. The single-tailed lipids S1–S4 were prepared by using above-mentioned four amino acids respectively via 4–5 steps (Scheme S1†). For the double-tailed lipids D1–D6, diethylenetriamine (DETA) and serine were used as tri-functional building block for the preparation, and some additional steps were needed (Scheme S2†). The preparation of the lipids has a common starting material cyclen-acetic acid except D2, which has a phenyl group on the backbone but not side chain. All of the hydrophobic parts in the lipids are oleyl chains, and the target compounds could be smoothly obtained with good yields and were characterized by NMR and HRMS. Thus we obtained ten cationic lipids with novel structures. These lipids have different amount of hydrophobic tails, various types of hydroxyl groups (alcohol or phenol), and different locations of aromatic group (backbone or side chain). The structure diversity would help us to study the SAR of such type of lipidic gene vectors.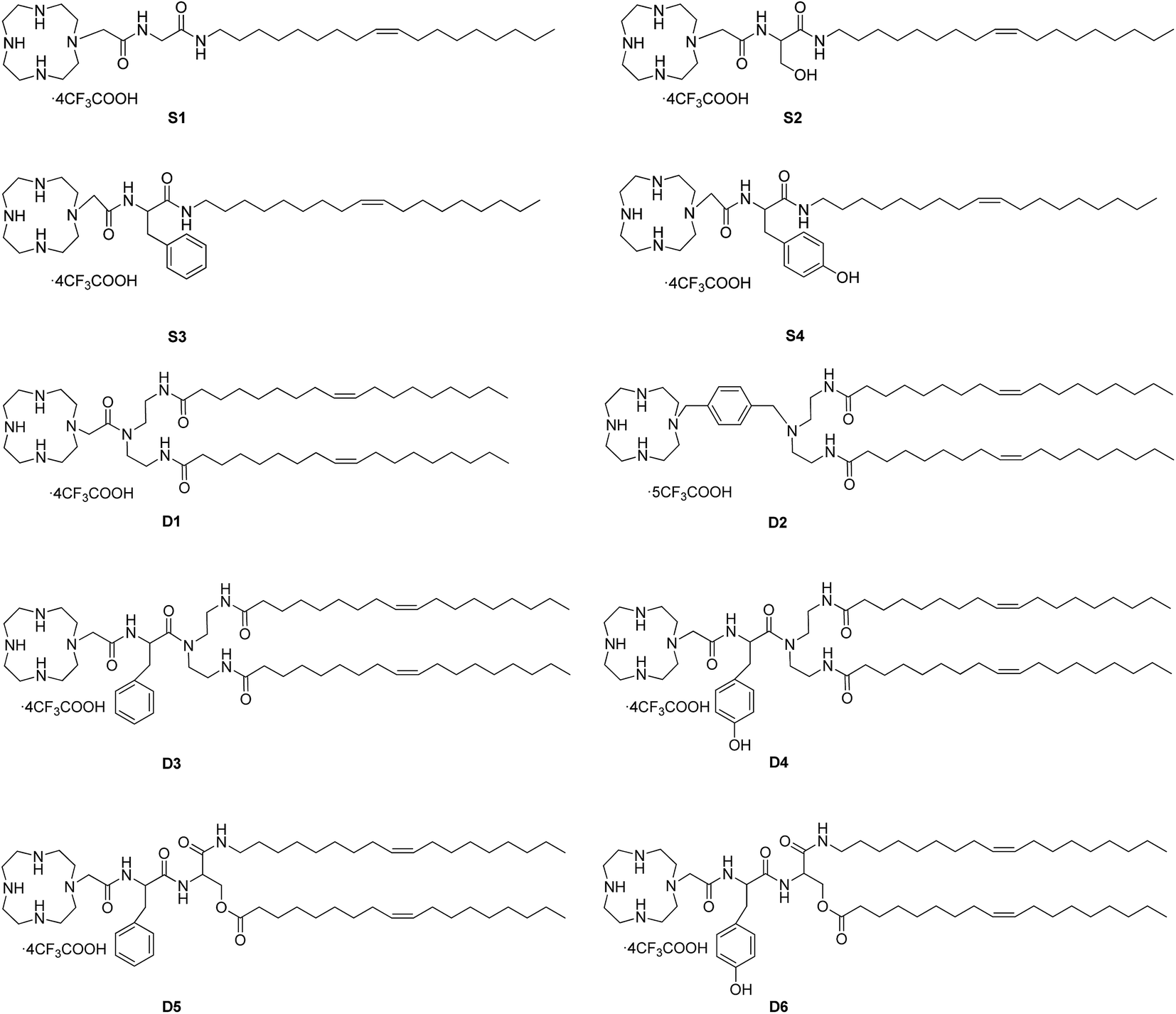 | ||
| Fig. 1 Chemical structures of prepared cationic lipids for SAR study. | ||
3.2 Interactions between the liposomes and plasmid DNA
Cationic liposomes may be formed from either individual cationic lipids or in combination with co-lipids. In this study, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was used as a co-lipid to facilitate hexagonal phase formation in order to enable endosomal escape and release of the nucleic acid cargo.39 Liposome was prepared by lipids mixed with DOPE in a molar ratio of 1:1. The DNA condensation capability of lipids was first studied by gel retardation assay. The lipoplexes were prepared at different N/P ratios which were calculated by the N atoms of cationic lipids and P atoms of plasmid DNA. As shown in Fig. 2, completely DNA retardation for lipid S1 and S2 without aromatic ring was achieved at N/P ratio of 8 which was higher than that needed for phenyl-contained S3 and S4 (N/P of 6 for full retardation). Such results reveal that the aromatic group may facilitate the DNA binding.40 For D1–D6, aromatic ring seemed not to have remarkable effect on the binding, this might be due to the enhanced hydrophobic effect of the liposomes formed from the double-chain tail, leading to improved DNA condensation capability. Besides, the binding abilities of the liposomes were also studied by the ethidium bromide (EB) dye replacement assay. Results shown in Fig. S1† exhibit continuously decreased fluorescent intensities along with the increase of liposome dosage (depicted as N/P ratios), indicating that EB was expelled by the cationic species. For single-tailed lipids, liposome S4 with benzene on the side chain showed stronger DNA binding ability, while D2 with benzene on the backbone has the strongest DNA affinity among the double-tailed lipids. Furthermore, the capacity of liposome to protect DNA from enzymatic digestion was examined via the same method (Fig. S2†). Results show that DNA could be well released from the D1–D6/DNA complexes after the sequential treatment of DNase and SDS. However, for the cases involving single-tailed lipids, the original DNA bands were vague, indicating the enzyme degradation of DNA. This also reveals that liposomes formed from double-tailed lipids have much better DNA binding and protection ability than those comprising single-tailed ones.
 | ||
| Fig. 2 Electrophoretic gel retardation assays of lipid/DOPE/pDNA complexes at different N/P ratios. The molar ratio of lipid/DOPE was 1:1. | ||
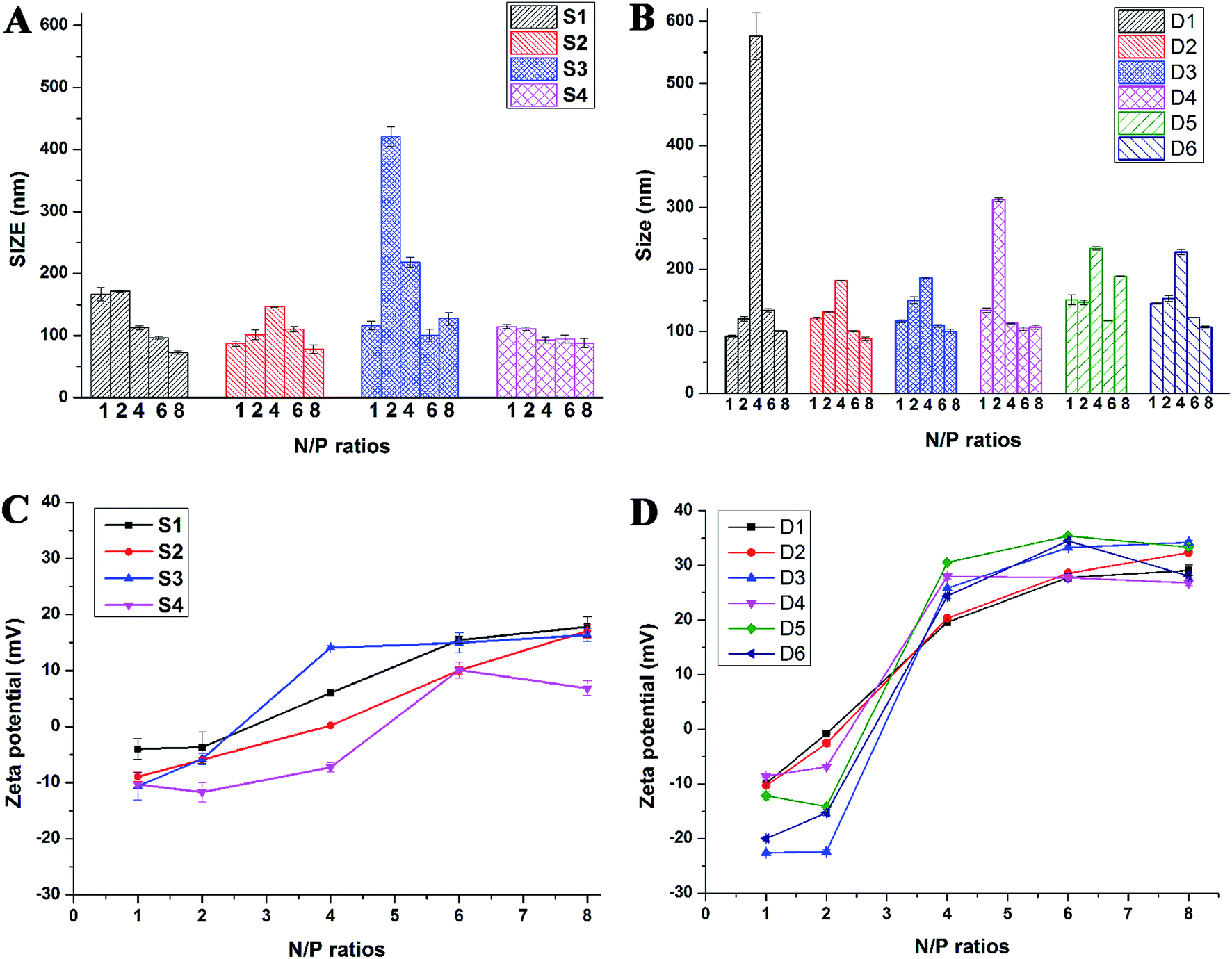
Proper sizes and zeta potentials of liposome are important factors for long circulation in the blood41 and efficient uptake by endocytosis.42 Dynamic light scattering assay (DLS) was utilized to examine these characteristics of liposomes and liposome/DNA complexes (lipoplexes). The sizes of liposome formed from S1 and S3 were smaller than those from S2 and S4 (Fig. S3A†). This might be attributed to the hydroxyl group which may influence the self-assembly of the lipids. However, such effect was not observed in the double-tailed lipids D1–D6, which gave similar particle sizes (Fig. S3B†). All liposomes gave positive zeta potentials (25–55 mV) needed for DNA condensation, and double-tailed ones have slightly higher potentials. After complexation with DNA (Fig. 3), the lipoplexes formed from double-tailed lipids gave obviously higher zeta-potentials than those from single-tailed lipids. The charge conversion was observed at N/P of 2–4, this was also lower than the latter. Such results demonstrate the higher DNA condensation ability of double-tailed liposomes, which is consistent with the gel retardation results in Fig. 2. The size of lipoplexes at low N/P ratios (1–4) were unstable, and very large aggregates might appear due to the lack of electrostatic repulsion at neutral surface potential. With the rise of N/P ratio, the particle sizes became stable at ∼100 nm, which is appropriate for cell endocytosis. The shape and morphology of the lipoplex was also confirmed by transmission electron microscope (TEM) directly. Spherical and homogeneous particles with the diameter of 50–100 nm were observed. As shown in Fig. 4, after complexation with DNA, the lipoplexes gave similar particle size to the empty liposomes (especially for D2), suggesting good DNA condensation. The particle size determined by TEM was much smaller than that obtained by DLS (100–150 nm). This might be attributed to the different experimental conditions: DLS examines the particles in the hydrated state in solution, while TEM observes the particles under dehydrated state.43
 | ||
| Fig. 3 Mean particle sizes (A and B) and zeta-potentials (C and D) of the lipoplexes formed from S1–S4 (A and C) and D1–D6 (B and D) under N/P ratios of 1, 2, 4, 6, 8. Data represent mean ± SD (n = 3). | ||
 | ||
| Fig. 4 TEM images of liposome D1 (A), liposome D2 (B), D1/DNA lipoplex (C, N/P = 6), and D2/DNA lipoplex (D, N/P = 6). | ||
3.3 In vitro gene transfection
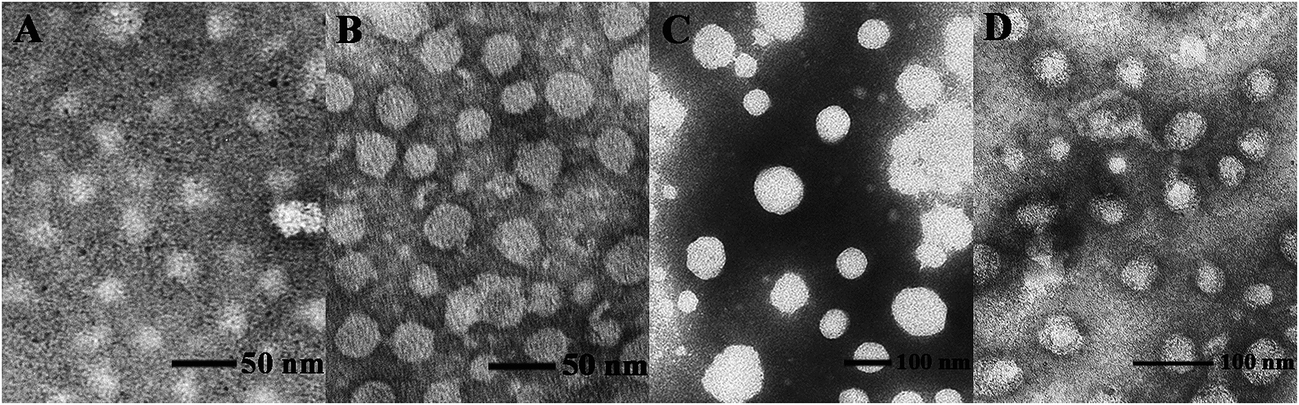
To investigate the gene transfection efficiency, pEGFP-N1 expression assays were carried out with lipoplexes prepared by target lipids in different cell lines (HEK 293 and HeLa). The N/P ratios of the lipoplexes were selected according to the complete retardation N/P obtained from gel retardation assay (Fig. 2). For the single-tailed lipids, as shown in Fig. 5, aromatic group is necessary for the transfection, and almost no green fluorescence of expressed GFP was found for the transfection by S1 and S2. Tyrosine-derived lipid S4 gave slightly higher TE than phenylalanine-derived lipid S3, suggesting that the hydroxyl group may benefit the transfection. However, serine-derived lipid S2 gave poor TE, indicating that for such type of lipidic vector, aromatic ring is more necessary than hydroxyl group for better TE. For double tailed-lipids, the transfection was much better (Fig. 6). Similar to single-tailed ones, aromatic ring is essential for the transfection, and D1 gave almost no fluorescence dots (data not shown). On the other hand, lipids D2–D4 may induce more fluorescent protein than D5 and D6, indicating that as linking group, DETA might be more suitable than amino acid serine, and this is consistent with our previous results.19 Such transfection study was also performed in HeLa cells by using D1–D4 as vectors (Fig. S4†). It was shown that as the only lipid with aromatic ring on the backbone, D2 gave the closest TE to lipofectamine 2000. The difference of TE between single-tailed and double-tailed lipids might be attributed to the shape of the liposomes. Considering the cross-sectional area of hydrophilic cyclen headgroup and the hydrophobic domain, the single-tailed lipids are liable to form aggregates like of spherical micelles, while the double-tailed lipids are tend to adopt a bilayer supramolecular structure, which is more liable to compact DNA into small nanoparticles.44 | ||
| Fig. 5 Fluorescent microscope images of HEK 293 cells transfected by lipoplexes S1–S4 (A, C, E, G: N/P = 6; B, D, F, H: N/P = 8). Lipofectamine 2000 was used as control (I). Lipid/DOPE ratio was 1:1. The cells were observed by inversion fluorescence microscope after 24 h transfection. | ||
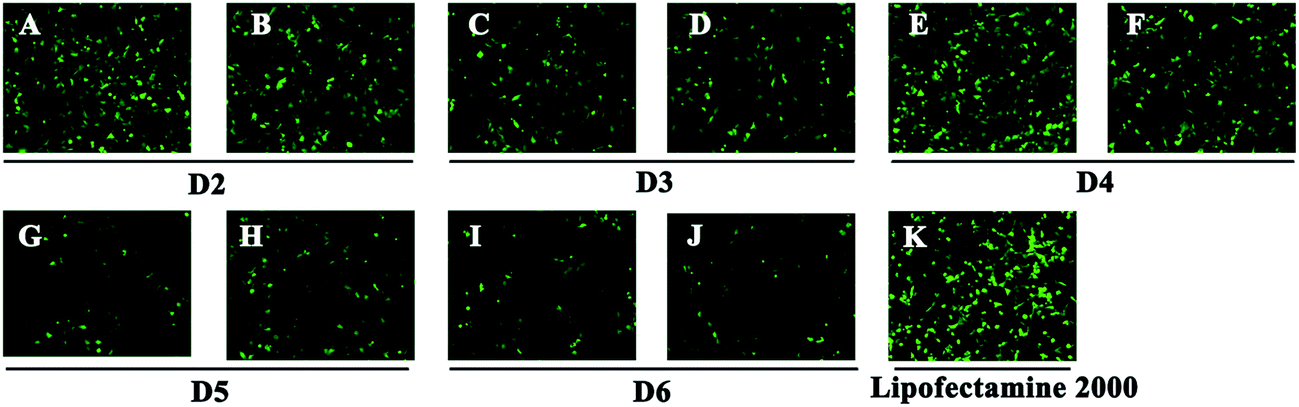 | ||
| Fig. 6 Fluorescent microscope images of HEK 293 cells transfected by lipoplexes D2–D6 (A, C, E, G, I: N/P = 6; B, D, F, H, J: N/P = 8). Lipofectamine 2000 was used as control (K). Lipid/DOPE ratio was 1:1. The cells were observed by inversion fluorescence microscope after 24 h transfection. | ||
Subsequently, luciferase reporter gene expression assay was carried out to quantitatively study the transfection mediated by the liposomes. As shown in Fig. 7A, D2 exhibited the best TE among the six materials, which is consistent with the GFP results. This also suggests that aromatic ring on the backbone but not side chain may better facilitate the gene transfection.45 Since the TE of D2 was still lower than the comparison reagent lipofectamine 2000, we adjusted the lipid structure by varying the length of hydrophobic chains of D2 in order to further optimize the TE. Lipids D2–10–D2–18, which contain different saturated alkyl chains with 10–18 carbon atoms respectively, were prepared according to the synthesis method of D2 (Scheme S3†). Gel retardation assay exhibit that the DNA binding ability increased with the extension of chain length, and became constant from the length of 14 carbon atoms (Fig. S5†). To our delight, luciferase assay showed that the TE might be largely improved by such optimization. Lipid with C14 chains (D2–14) gave 6.4 times higher TE than D2 (Fig. 7B), and the TE was close to lipofectamine 2000. Besides, transfection study performed in HeLa cells also exhibited the advantage of C14-containing hydrophobic chains. However, its TE was similar to D2 (Fig. S6†). Recently Shi et al. proved that the length of alkyl chain could influence the fluidity of the bilayer.35 Short or unsaturated alkyl chain both favor a higher rate of intermembrane transfer of lipid monomers and lipid membrane mixing, leading to better endocytosis.
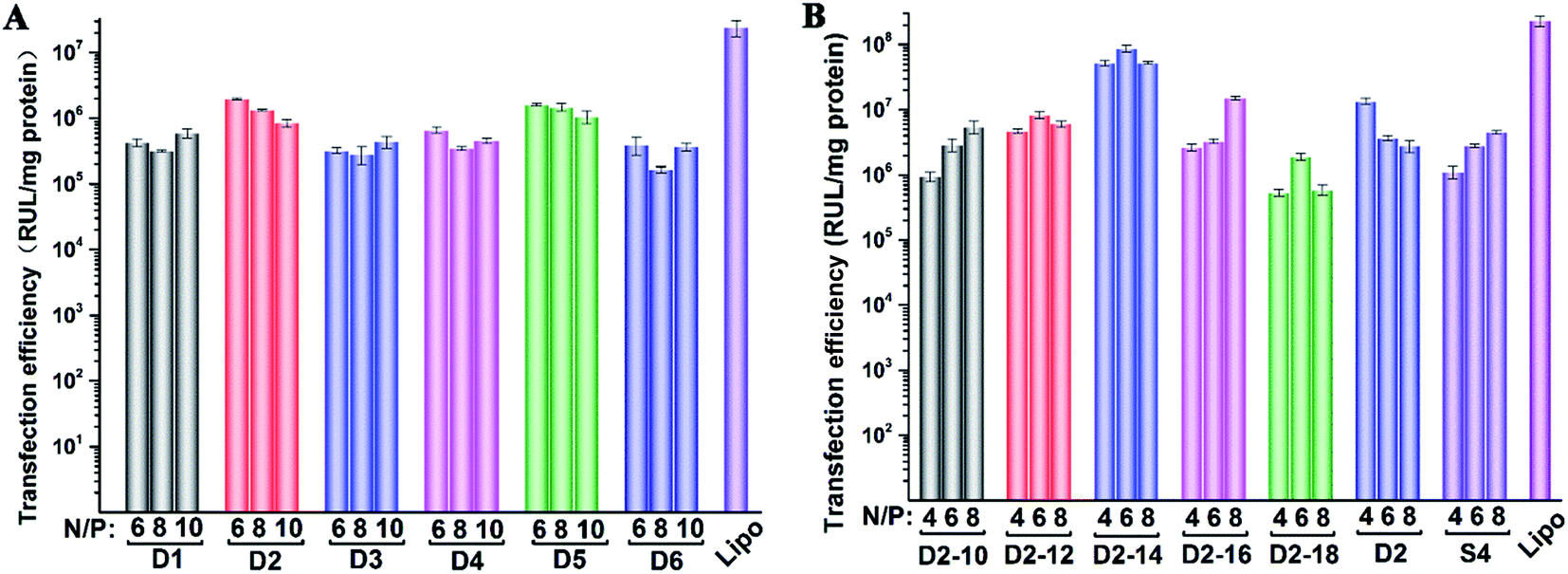 | ||
| Fig. 7 Luciferase expression in HEK 293 cells transfected by lipid/DOPE/DNA lipoplexes D1–D6 (A) and D2–10–D2–18, D2, S4 (B) at various N/P ratios under lipid/DOPE ratio of 1:1. Data represent mean ± SD (n = 3). | ||
3.4 Cytotoxicity
Cell toxicity is another important issue that has to be addressed for the development of novel cationic lipids. The cytotoxicity of the lipoplexes prepared from D1–D6, S4 and D2–10–D2–18 at various N/P ratios was examined in HEK 293 cells via MTS reduction assay (Fig. 8). It was shown that the lipoplexes gave comparable cytotoxicity to lipofectamine 2000 control. Under low N/P ratios, the percentages of survival cells were higher than those treated with lipofectamine 2000. D2 with benzene on the backbone exhibited higher viability than its analogs, while lipids with shorter hydrophobic tails have higher toxicity, especially at high N/P ratios. Same assay was also carried out in HeLa cells (Fig. S7†). Unlike the results observed in HEK 293 cells, materials with shorter chain led to higher cell viability. It was also reported that toxicity effect is, generally, a cell dependent process where every cell type shows different toleration levels.46 In all, the cytotoxicity of the cyclen-based lipidic materials is lower than commercially available transfection reagent.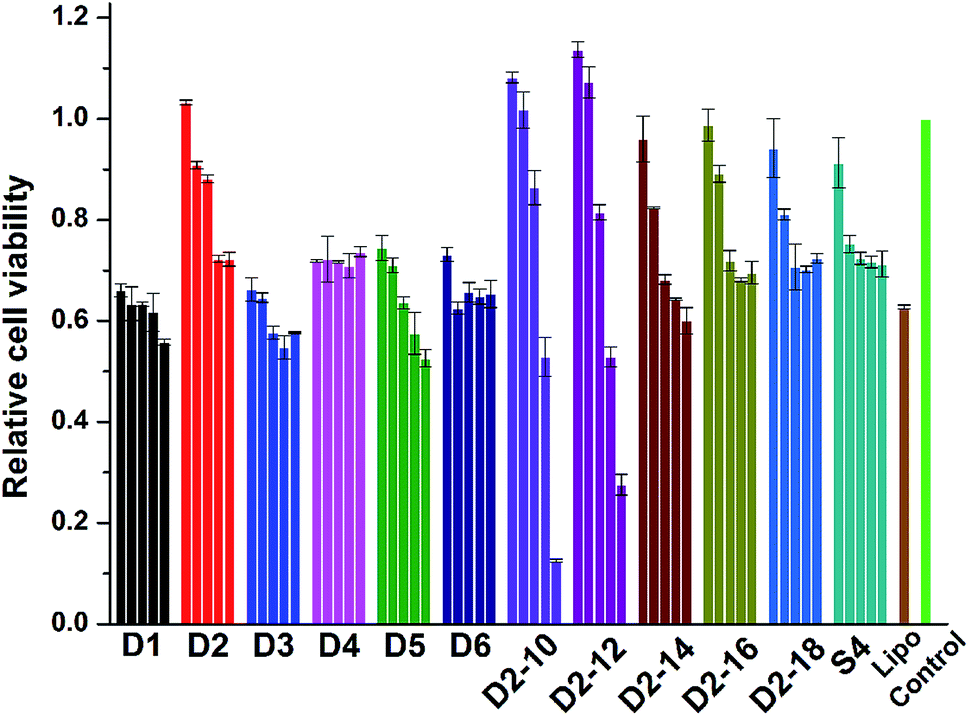 | ||
| Fig. 8 Cytotoxicity of the lipoplexes prepared at various N/P ratios (4, 6, 8, 10 and 12 for D1–D6; 2, 4, 6, 8 and 12 for others) in HEK 293 cells. Data represent mean ± SD (n = 3). | ||
4. Conclusion
In summary, fifteen novel cyclen-based amphiphilic cationic lipids with different linking groups and hydrophobic tails were prepared and characterized. The effect of the structure variety on their activity as non-viral gene vectors was investigated. The liposomes formed by these lipids and helper lipid DOPE could efficiently bind DNA and condense it into nano-sized particles with positive zeta-potentials and protect DNA from enzymatic digestion. Structure–activity relationship studies revealed that aromatic group in the lipid may improve the DNA binding ability of the liposome. Lipids with double hydrophobic tails gave much higher transfection efficiency than those with single tail, and aromatic ring in the lipid backbone might facilitate the transfection. Besides, the transfection efficiency could be further improved by optimizing the hydrophobic tails. The lipid with C14 tails gave much higher efficiency than its analogs. Results show that such type of cationic lipids may act as promising non-viral gene delivery carriers, and further modification towards higher efficiency and biocompatibility is now in progress.Acknowledgements
This work was financially supported by the National Science Foundation of China (No. 21472131, 21232005). We also thank the Comprehensive Training Platform of Specialized Laboratory, College of Chemistry, Sichuan University for sample analysis.References
- M. A. Kay, Nat. Rev. Genet., 2011, 12, 316–328 CrossRef CAS PubMed.
- J. K. B. Deanna Cross, Clin. Med. Res., 2006, 4, 218–227 CrossRef.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS PubMed.
- C. H. Jones, C.-K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharmaceutics, 2013, 10, 4082–4098 CrossRef CAS PubMed.
- B. K. M. Priya Prakash Karmali, B. Sreedhar and A. Chaudhuri, Bioconjugate Chem., 2006, 17, 159–171 CrossRef PubMed.
- C. A. H. Prata, Y. Zhao, P. Barthelemy, Y. Li, D. Luo, T. J. McIntosh, S. J. Lee and M. W. Grinstaff, J. Am. Chem. Soc., 2004, 126, 12196–12197 CrossRef CAS PubMed.
- X. Liu, J. Xiang, D. Zhu, L. Jiang, Z. Zhou, J. Tang, X. Liu, Y. Huang and Y. Shen, Adv. Mater., 2016, 28, 1743–1752 CrossRef CAS PubMed.
- P. L. Felgner, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS.
- S. Bhattacharya and A. Bajaj, Chem. Commun., 2009, 4632–4656 RSC.
- D. Zhi, S. Zhang, B. Wang, Y. Zhao, B. Yang and S. Yu, Bioconjugate Chem., 2010, 21, 563–577 CrossRef CAS PubMed.
- A. Bhadani, H. Kataria and S. Singh, J. Colloid Interface Sci., 2011, 361, 33–41 CrossRef CAS PubMed.
- M. Mevel, C. Neveu, C. Goncalves, J. J. Yaouanc, C. Pichon, P. A. Jaffres and P. Midoux, Chem. Commun., 2008, 3124–3126 RSC.
- M. A. Ilies, I. Ghiviriga, B. H. Johnson, A. Miller, E. B. Thompson and A. T. Balaban, J. Med. Chem., 2004, 47, 3744–3754 CrossRef CAS PubMed.
- M. A. Ilies, W. A. Seitz, M. T. Caproiu, M. Wentz, R. E. Garfield and A. T. Balaban, Eur. J. Org. Chem., 2003, 2003, 2645–2655 CrossRef.
- Q. D. Huang, W. J. Ou, H. Chen, Z. H. Feng, J. Y. Wang, J. Zhang, W. Zhu and X. Q. Yu, Eur. J. Pharm. Biopharm., 2011, 78, 326–335 CrossRef CAS PubMed.
- Q. F. Zhang, W. H. Yang, W. J. Yi, J. Zhang, J. Ren, T. Y. Luo, W. Zhu and X. Q. Yu, Bioorg. Med. Chem. Lett., 2011, 21, 7045–7049 CrossRef CAS PubMed.
- H.-J. Wang, X. He, Y. Zhang, J. Zhang, Y.-H. Liu and X.-Q. Yu, RSC Adv., 2015, 5, 59417–59427 RSC.
- Y. M. Zhang, D. C. Chang and J. Zhang, Bioorg. Med. Chem., 2015, 23, 5756–5763 CrossRef CAS PubMed.
- Y.-M. Zhang, Y.-H. Liu, J. Zhang, Q. Liu, Z. Huang and X.-Q. Yu, RSC Adv., 2014, 4, 44261–44268 RSC.
- Q. Liu, W. J. Yi, Y. M. Zhang, J. Zhang, L. Guo and X.-Q. Yu, Chem. Biol. Drug Des., 2013, 82, 376–383 CAS.
- D. Zhi, S. Zhang, S. Cui, Y. Zhao, Y. Wang and D. Zhao, Bioconjugate Chem., 2013, 24, 487–519 CrossRef CAS PubMed.
- Y. Li, S. Zheng, X. Liang, Y. Jin, Y. Wu, H. Bai, R. Liu, Z. Dai, Z. Liang and T. Shi, Bioconjugate Chem., 2014, 25, 2055–2066 CrossRef CAS PubMed.
- P. K. D. Rajkumar Banerjee, G. Venkata Srilakshmi and A. Chaudhuri, J. Med. Chem., 1999, 42, 4292–4299 CrossRef.
- Q. Sun, Z. Kang, L. Xue, Y. Shang, Z. Su, H. Sun, Q. Ping, R. Mo and C. Zhang, J. Am. Chem. Soc., 2015, 137, 6000–6010 CrossRef CAS PubMed.
- E. Ojeda, G. Puras and M. Agirre, Biomaterials, 2016, 77, 267–279 CrossRef CAS PubMed.
- H. Yang, S. Y. Fung and M. Liu, Angew. Chem., 2011, 50, 9643–9646 CrossRef CAS PubMed.
- X. X. Zhang, C. A. Prata, J. A. Berlin, T. J. McIntosh, P. Barthelemy and M. W. Grinstaff, Bioconjugate Chem., 2011, 22, 690–699 CrossRef CAS PubMed.
- H. Zeng, H. C. Little, T. N. Tiambeng, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2013, 135, 4962–4965 CrossRef CAS PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef CAS PubMed.
- P. T. Wong, K. Tang, A. Coulter, S. Tang, J. R. Baker Jr and S. K. Choi, Biomacromolecules, 2014, 15, 4134–4145 CrossRef CAS PubMed.
- C.-R. Luan, Y.-H. Liu, J. Zhang, Q.-Y. Yu, Z. Huang, B. Wang and X.-Q. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 10743–10751 CAS.
- S. Savarala, E. Brailoiu, S. L. Wunder and M. A. Ilies, Biomacromolecules, 2013, 14, 2750–2764 CrossRef CAS PubMed.
- M. S. Santanu Bhattacharya and U. S. Hiremath, Chem. Phys. Lipids, 1995, 78, 177–188 CrossRef.
- J. Shi, S. Yu, J. Zhu, D. Zhi, Y. Zhao, S. Cui and S. Zhang, Colloids Surf., B, 2016, 141, 417–422 CrossRef CAS PubMed.
- Z. Huang, Y.-H. Liu, Y.-M. Zhang, J. Zhang, Q. Liu and X.-Q. Yu, Org. Biomol. Chem., 2015, 13, 620–630 CAS.
- J. W. Jeon, C. E. Yoo, I. S. Hong, J. B. Song and J. Suh, Org. Lett., 2002, 4, 4155–4158 CrossRef CAS PubMed.
- C. Q. Xia, N. Jiang, J. Zhang, S. Y. Chen, H. H. Lin, X. Y. Tan, Y. Yue and X. Q. Yu, Bioorg. Med. Chem., 2006, 14, 5756–5764 CrossRef CAS PubMed.
- L. H. Xia Guo, Acc. Chem. Res., 2011, 45, 971–979 Search PubMed.
- W. J. Yi, X. C. Yu, B. Wang, J. Zhang, Q. Y. Yu, X. D. Zhou and X. Q. Yu, Chem. Commun., 2014, 50, 6454–6457 RSC.
- O. C. Boerman, L. v. Bloois, E. B. Koenders, V. d. Meer, F. H. M. Corstens and G. Storm, J. Nucl. Med., 1997, 38, 489–493 CAS.
- J. Rejman, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- W.-J. Yi, Q.-F. Zhang, J. Zhang, Q. Liu, L. Ren, Q.-M. Chen, L. Guo and X.-Q. Yu, Acta Biomater., 2014, 10, 1412–1422 CrossRef CAS PubMed.
- H. J. Wang, Y. H. Liu, J. Zhang, Y. Zhang, Y. Xia and X.-Q. Yu, Biomater. Sci., 2014, 2, 1460–1470 RSC.
- Q. D. Huang, Y. Zhang, J. Ren, Y. Fu, J. Zhang, W. Zhu and X. Q. Yu, PLoS One, 2011, 6, 1–11 Search PubMed.
- S. K. Sohaebuddin, P. T. Thevenot, D. Baker, J. W. Eaton and L. Tang, Part. Fibre Toxicol., 2010, 7, 22 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00422b |
| This journal is © The Royal Society of Chemistry 2017 |