
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow temperature hydrothermal synthesis of battery grade lithium iron phosphate†
Peter Benedek ,
Nils Wenzler,
Maksym Yarema and
Vanessa C. Wood*
,
Nils Wenzler,
Maksym Yarema and
Vanessa C. Wood*
Laboratory of Nanoelectronics, Department of Information Technology and Electrical Engineering, ETH Zurich, CH-8092 Zurich, Switzerland. E-mail: vwood@ethz.ch
First published on 23rd March 2017
Abstract
Lithium ion transport through the cathode material LiFePO4 (LFP) occurs predominately along one-dimensional channels in the [010] direction. This drives interest in hydrothermal syntheses, which enable control over particle size and aspect ratio. However, typical hydrothermal syntheses are performed at high pressures and are energy intensive compared to solid-state reactions, making them less practical for commercial use. Here, we show that the use of high precursor concentrations enables us to achieve highly crystalline material at record low-temperatures via a hydrothermal route. We produce LFP platelets with thin [010] dimensions and low antisite defect concentrations that exhibit specific discharge capacities of 150 mA h g−1, comparable to material produced with higher temperature syntheses. An energy consumption analysis indicates that the energy required for our synthesis is 30% less than for typical hydrothermal syntheses and is comparable to solid-state reactions used today, highlighting the potential for low temperature hydrothermal synthesis routes in commercial battery material production.
Lithium iron(II) phosphate (LFP) is a commercially-used lithium ion battery (LIB) cathode material that offers some advantages over other cathode materials due to the fact that it does not contain cobalt, and that it has a flat voltage profile and a high rate capability.1 It is commercially produced in a solid-state synthesis route; however, this well-established preparation offers less morphology and composition control than wet chemistry approaches.2 A large number of solvothermal syntheses have emerged for LFP,3 especially using water as the solvent (i.e., hydrothermal syntheses).3–12 Typically, these hydrothermal syntheses are performed with a precursor concentration in the range of 0.1–0.3 M, at temperatures between 150 °C and 200 °C, and for a time in the order of ten hours.5,7–9 Rather high process temperatures (150–200 °C) pose two issues towards commercialization: (i) the energy consumption for hydrothermal production of LFP is significantly higher than for the solid-state LFP synthesis and (ii) the vapor pressure of water can reach more than 10 bar, requiring a special reactor design.13 A commercially viable LFP hydrothermal synthesis will therefore require reduction of the reaction temperature.
To find the parameters at which the specific energy consumption of a hydrothermal synthesis process could compete with the solid state one, we develop a model to estimate the energy required for the hydrothermal-based synthesis of LFP from mine to ready-to-use cathode material. Extending upon existing literature,13–18 we consider the impact of the key parameters such as synthesis temperature, precursor concentration, and reaction time on the specific energy consumption. Details of the model are given in the ESI,† and the dependence of the specific energy on reaction temperature and precursor concentration is shown in Fig. 1. The energy consumption of the hydrothermal synthesis is largely related to the large heat capacity of water, which makes the heating of the reactor (not the time held at a specific temperature) energy intensive. Our model shows that the reaction temperature as well as the precursor concentration, Ctot play a role in energy usage. The larger Ctot, the smaller the specific energy consumption because, at lower concentrations, a smaller mass of LFP is created for the same heated volume of water. For temperatures of 150 °C, a hydrothermal synthesis can only compete with the 3 MJ kg−1 energy consumption of a solid-state synthesis if the precursor concentration were at the solubility limit of LiOH in water. With decreasing the reactor temperature, the dependence of energy consumption on concentration weakens, allowing a larger range of possible precursor concentrations.
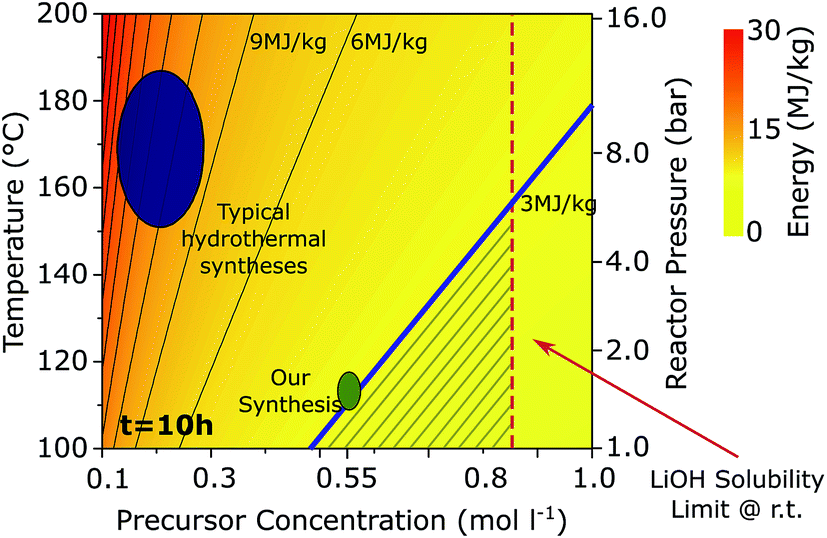 | ||
| Fig. 1 Specific energy consumption of the LFP synthesis step as a function of hydrothermal reaction temperature and total precursor concentration. The blue shaded region indicates the energy consumption of typical hydrothermal syntheses, the synthesis reported here is indicated green. The iso-energy line corresponding to 3 MJ kg−1 is shown in bold, corresponding to the specific energy consumption of a solid-state synthesis. The shaded triangular region below this line represents a temperature and precursor concentration window where a hydrothermal synthesis consumes less energy than a solid-state synthesis. | ||
Based on this analysis, we aim to develop a synthesis at 115 °C that still yields battery grade material. At this temperature, the specific energy consumption can be lowered to 3 MJ kg−1 using a feasible precursor concentration (i.e., one below the solubility limit) and the vapor pressure in the reactor will be about 1.5 bar, presenting convenient operating conditions, similar to a pressure cooker.
The hydrothermal synthesis of high-quality LFP at low temperatures is a challenging chemistry task.4–6 Low temperature syntheses result in slow growth kinetics and produce LFP with poor crystallinity and a large number of antisite defects (i.e., Fe atoms populating the Li positions). These antisite defects block the [010] Li transport channels in the LFP structure and ultimately decrease specific charge capacities.19–22 Moreover, the slow growth kinetics lead to a low number of nuclei, leading to thicker LFP particles and thus longer [010] Li channel lengths such that there is a higher probability of antisite defects blocking them.23
In this letter, we present a study of low-temperature hydrothermal synthesis of LFP platelets. In particular, we optimize the precursor concentration and reaction time in order to achieve battery-grade LFP material. We then perform the carbon coating of LFP platelets and show electrochemical performance on par with that synthesized at higher temperatures with specific discharge capacities of up to 150 mA h g−1 at C/5 and 120 mA h g−1 at 1C rate.
Scheme 1 shows our low temperature synthesis approach. As detailed in the Experimental methods, we use typical precursors for hydrothermal synthesis of LFP: LiOH, H3PO4, and FeSO4·7H2O.4–11 We also use ethylene glycol (EG) as an additive,5,7 which is known to decrease the solubility of the precursors, increase the number of nucleation sites, and thereby achieve smaller particle dimensions.7 Here, to ensure that the role of the EG is primarily in coordination of the LFP surfaces and to be close to a possible commercial application, we work with small additive concentrations and select a fixed molar ratio of [LiOH]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[FeSO4·7H2O]:[H3PO4]:[EG] = 3:1:1:0.1 for all syntheses.
:[FeSO4·7H2O]:[H3PO4]:[EG] = 3:1:1:0.1 for all syntheses.
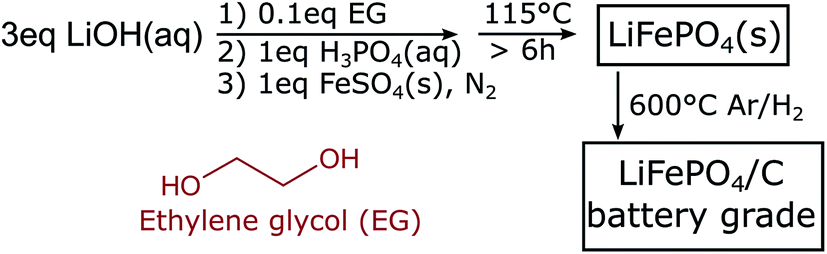 | ||
| Scheme 1 Preparation procedure of battery grade LiFePO4. | ||
To assess our ability to control material quality as well as particle shape and size with our low temperature hydrothermal synthesis, we systematically look at the effect of (1) precursor concentration and (2) reaction time. Since density function theory (DFT) calculations suggest that the symmetric stretching band of the PO4 group is redshifted when the antisite defect concentration is increased,5 we use Fourier transform infrared (FTIR) spectroscopy to assess material quality. Scanning electron microscopy (SEM) enables us to obtain approximate the particle morphology.
Even though it is generally accepted that the total precursor concentration influences the supersaturation24 and therefore the reaction kinetics, crystallinity, and particle size, it has not been extensively studied in hydrothermal syntheses of LFP.11 We investigate measurements performed on LFP samples after 16 hours of reaction. The FTIR spectra of the PO4 stretching modes of each sample at different concentrations are shown in Fig. 2a. Increasing the precursor concentration, Ctot, results in LFP particles that show sharper spectral features and a shift of the PO4 stretching band towards lower wavenumbers, both indicating smaller defect concentration (Fig. 2b). These FTIR results are consistent with Rietveld refinements on X-Ray diffractograms (Fig. 2S†), from which we find a decrease of antisite defects with increased Ctot (Fig. 3S†). The decrease in antisite defect with increasing precursor concentration can be explained by faster reaction kinetics at higher concentrations, whereby the LFP crystals form faster and have more time within the set reaction time of 16 hours to recrystallize. This is supported by SEM images (Fig. 2c) showing that, at low Ctot, the particles are not all evolved, while uniform platelets are formed for increasing Ctot.
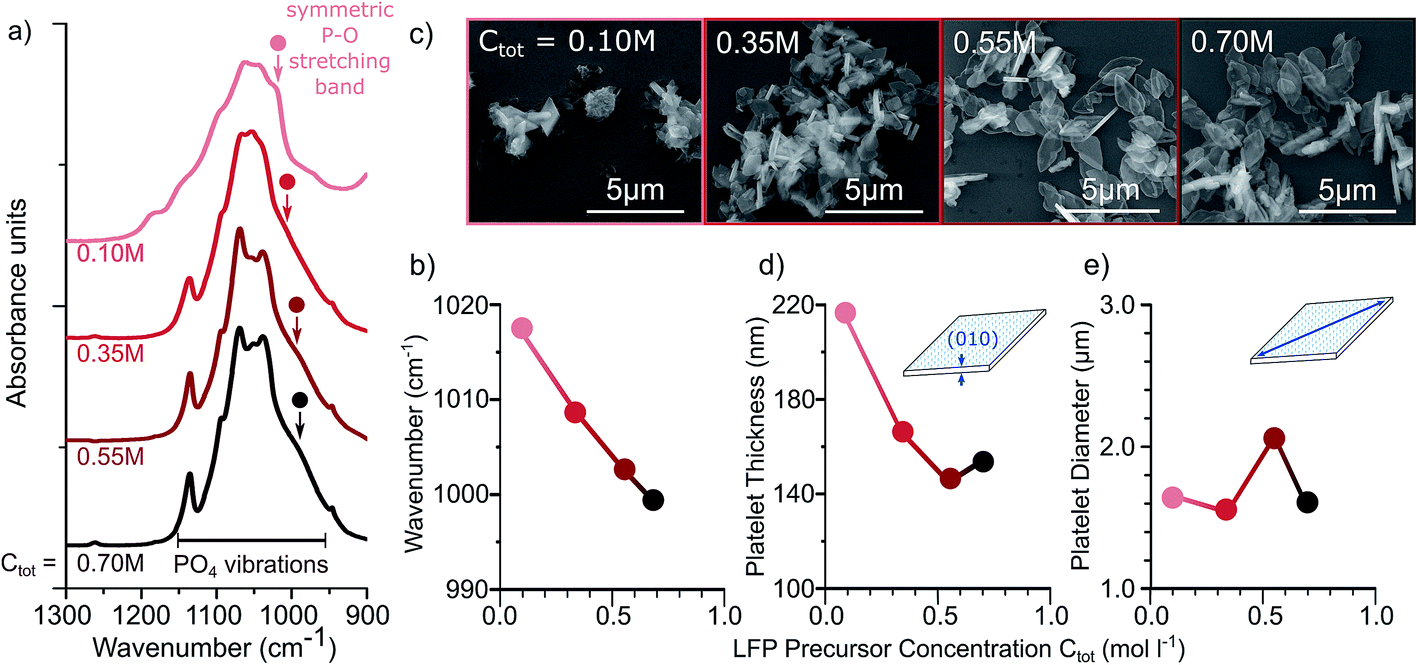 | ||
| Fig. 2 Dependence of defect concentration and particle morphology as a function of molar concentration of precursors, Ctot, in water. Precursors are fixed in a ratio of [LiOH]:[FeSO4·7H2O]:[H3PO4]:[EG] = 3:1:1:0.1. (a) FTIR spectra and (b) position of the symmetric P–O stretching band peak for samples synthesized at different precursor concentrations. (c) SEM images, (d) mean particle thickness, and (e) mean particle diameter are also shown for different precursor concentrations. | ||
Increasing the precursor concentration also affects the particle morphology. With increasing Ctot, the average particle thickness decreases from 220 nm to 150 nm (Fig. 2d) while the platelet diameter remains approximately constant around 1.6 μm (Fig. 2e). These results can be explained by DFT calculations21 and previous experimental results,5,7 which suggest that both water and EG cap the (010) facet, inhibiting growth in the [010] direction.
To investigate the optimal reaction time for a low temperature hydrothermal synthesis, we construct a sampling reactor that enables us to remove material at different times during the reaction (Fig. 4S†). We consider a reaction with a precursor concentration Ctot = 0.55 M. As shown in Fig. 3a, with increasing reaction time, the average particle thickness tends to increase slightly and the distribution of thicknesses broadens (i.e., 130 nm ± 50 nm at 3 hours to 150 nm ± 70 nm at 72 hours), reflecting the Ostwald ripening process.25 At the same time, defect concentration (as quantified by the peak position of the symmetric PO4 stretching band) decreases with longer reaction times (Fig. 3b) due to recrystallization of LFP. This poses a trade-off, because for the highest charge capacity both thin platelet morphology LFP particles (i.e., short [010] Li channels) and low antisite defect concentrations are required. We thus expect a reaction time in the middle of those tested (e.g., 48 hours) to exhibit the best electrochemical performance.
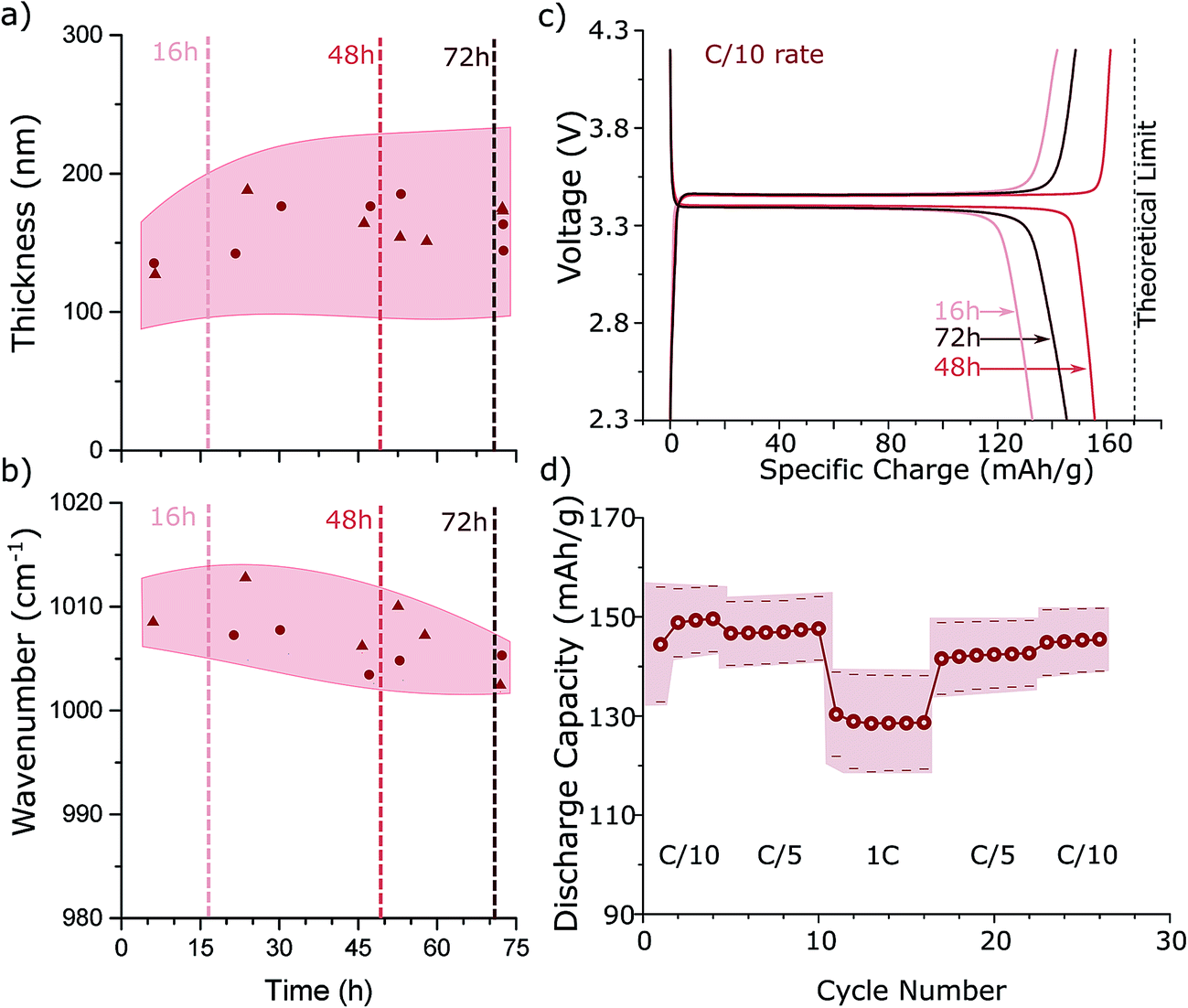 | ||
| Fig. 3 Two different batches (circles, triangles) of LFP platelets synthesized under the same conditions are sampled as a function of reaction time and their thickness (a) and the peak position of the symmetric P–O stretching mode (b) is plotted. Shading indicates the standard deviation of the measurements. (c) Polarization curves at C/10 rate of LFP samples prepared with different reaction times. (d) Specific discharge capacity at different C rates (1C = 0.17A g−1) of LFP synthesized at 115 °C at a precursor concentration of Ctot = 0.55 M and a reaction time of 48 h. Shading indicates the error bars on the measurements, which are determined by measuring 5 cells containing different electrodes prepared from two different batches of material. | ||
To test the electrochemical performance of our newly obtained material, we prepare the LFP particles for electrochemical cycling by carbon coating them. The LFP particles are mixed with D-glucose, pressed into pellets, and annealed at 600 °C resulting in LFP particles with a coating of 3 wt% of carbon. XRD, FTIR, SEM, and transmission electron microscope (TEM) measurements suggest that the carbon coating step removes the solvent incorporated in the crystal and provides a conductive coating but does not affect the particle crystal structure or the antisite defect concentration. As described in the methods, electrodes are made from different particle batches and cycled vs. lithium in a half-cell configuration.
Fig. 3c shows the electrochemical voltage vs. capacity curves for C/10 (dis)charge for three LFP batches prepared with the same precursor concentration of Ctot = 0.55 M and reaction times of 16, 48, and 72 hours. All three samples exhibit a flat voltage plateau and low polarization at a C/10 rate. Our parameter sweep indicated that the best electrochemical performance should result from particles synthesized for ∼48 hours, and indeed this sample performs best with a specific discharge capacity >150 mA h g−1. Importantly, such high discharge capacity of LFP material is already comparable to values obtained by high temperature hydrothermal syntheses and improves upon previous reports for low temperature hydrothermal LFP synthesis.4,5 Furthermore, our LFP samples show only very small capacity fading after 50 charge/discharge cycles (Fig. 6S†). Using galvanostatic cycling, the specific discharge capacity at different C rates (1C = 0.17 A g−1) is determined from the LFP particles synthesized for 48 hours. Fig. 3d shows the average discharge capacity and standard deviation of sample electrodes prepared in different batches. The specific charge capacity drops to 130 mA h g−1, when the cycling rate is increased to 1C, which is comparable to high temperature hydrothermal LFP syntheses.4–11
Table 1 summarizes the specific energy consumption of our proposed hydrothermal synthesis, a high temperature hydrothermal approach proposed by Chen et al.,6 and a solid state synthesis. This comparison includes the carbon-coating step in our hydrothermal synthesis for which we assume an extra energy consumption of 2 MJ kg−1. By increasing the concentration and decreasing reaction time, our synthesis saves more than 30% compared to high temperature hydrothermal syntheses, but still consumes more energy than solid state syntheses. However, an excess of lithium is used in the hydrothermal process, which leads to the formation of Li2SO4. If this co-product was recycled, the precursor energy consumption would decrease by up to 50% eventually making the synthesis beneficial from an energy perspective. Of course, energy consumption during synthesis is only a small part of the costs of the overall material production. For instance, the aforementioned temperature dependent pressure in the reactor has a high impact on investment cost due to increased safety requirements. However, while increased reaction time has only a small effect on energy consumption (heating up to and not holding the temperature is the energy intensive step as highlighted in Fig. 7S†), it does impact throughput. Therefore, a multi-parameter optimization of temperature, reaction time, and the process (e.g. using flow reactor set-up) would be necessary for commercial introduction of a hydrothermal process.
In conclusion, we synthesized LFP platelet particles in a hydrothermal synthesis with a low reaction temperature of 115 °C. Increased precursor concentration together with small amounts of EG as an additive lead to highly crystalline particles with a platelet-shaped morphology having small dimensions along [010]. Using a sampling setup, we found the counteracting effects of recrystallization and Ostwald ripening lead to an optimal reaction time in the range of 48 hours. Our analysis shows that reduction of reaction temperature and increase of precursor concentration can bring the energy consumption of a hydrothermal synthesis in line with that of a solid state approach, while enabling particle size and shape control not available with solid state approaches. This work highlights the open potential for optimization of hydrothermal processes at lower temperatures and higher concentrations.
Experimental methods
In a typical synthesis of LFP, a 20 mL solution of LiOH (Sigma) and the ethylene glycol (Sigma) is transferred into a 50 mL stainless steel reactor. While stirring, 20 mL of H3PO4 solution is added dropwise. A white precipitate of Li3PO4 forms. Ground FeSO4·7H2O (Sigma) powder is quickly added to the dispersion and the reactor is purged with N2 for 10 min. The final molar ratio [LiOH]:[FeSO4·7H2O]:[H3PO4]:[EG] in the solution is kept 1:1:3:0.1. After purging, the reactor is heated with a heating mantle. When the synthesis is finished, the resulting off-white powder is washed in H2O and ethanol and dried at 80 °C. To coat the LFP particles with 3 wt% of carbon, glucose is mixed thoroughly with the LFP particles. The mixture is pressed in a pellet and then heated at 600 °C for 6 h in Ar/3 wt% H2 stream. The resulting powder is black.
To determine the particle dimensions, scanning electron microscopy (SEM) images in secondary electron mode are taken using a Hitachi SU-8200. The dimensions of 100–200 particles are measured using ImageJ, see Fig. 8S† for details. FTIR spectra are obtained on an attenuated total reflection (ATR) setup with Ge single crystal on a Bruker Vertex 70 Fourier transformed infrared spectrometer. The observed symmetric PO4 stretching band is fit with a Gaussian function. X-ray powder diffraction spectra are measured in a 2θ range between 15° and 85° using a Rigaku Smartlab diffractometer. Rietveld refinements are performed with the Maud software.26 To analyze the Carbon coated samples, transmission electron micrographs are taken with a FEI F30 microscope.
To test LFP electrodes, 70 wt% of active material, 20 wt% of Super C64 carbon black (Timcal), and 10 wt% of Kynar HSV900 polyvinylidene fluoride binder are dispersed in N-methyl-2-pyrrolidone (NMP, Sigma). Resulting slurries are blade coated on an aluminum sheet and dried at 120 °C under vacuum for 8 h. Half cells were prepared under argon atmosphere, using glass fiber separator soaked with 500 μl of the electrolyte, a 1 M solution of LiPF6 in 1:1 ethylene carbonate and dimethyl carbonate (BASF), between the LFP cathode and lithium metal reference. The cycling measurements are performed at room temperature on a Biologic VMP3 potentiostat. The reported values (data points) and errors (shaded regions) in Fig. 3 come from the average and standard deviation of measurements on different electrodes prepared with materials from different synthesis batches, but with the same reaction conditions.
Acknowledgements
The authors acknowledge funding from the European Research Council (ERC), the Swiss National Science Foundation (SNSF) precoR program. TEM measurements were performed at the Scientific Center for Optical and Electron Microscopy of the Swiss Federal Institute of Technology.References
- B. Kang and G. Ceder, Battery materials for ultrafast charging and discharging, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- M. Yoshimura and K. Byrappa, Hydrothermal processing of materials: past, present and future, J. Mater. Sci., 2008, 43, 2085–2103 CrossRef CAS.
- C. Masquelier and L. Croguennec, Polyanionic (phosphates, silicates, sulfates) frameworks as electrode materials for rechargeable Li (or Na) batteries, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- O. Xiuqin, P. Lin, G. Haichen, W. Yichen and L. Jianwei, Temperature-dependent crystallinity and morphology of LiFePO4 prepared by hydrothermal synthesis, J. Mater. Chem., 2012, 22, 9064–9068 RSC.
- X. Qin, J. Wang, J. Xie, F. Li, L. Wen and X. Wang, Hydrothermally synthesized LiFePO4 crystals with enhanced electrochemical properties: simultaneous suppression of crystal growth along [010] and antisite defect formation, Phys. Chem. Chem. Phys., 2012, 14, 2669–2677 RSC.
- J. Chen, M. J. Vacchio, S. Wang, N. Chernova, P. Y. Zavalij and M. S. Whittingham, The hydrothermal synthesis and characterization of olivines and related compounds for electrochemical applications, Solid State Ionics, 2008, 178, 1676–1693 CrossRef CAS.
- S. Yang, X. Zhou, J. Zhang and Z. Liu, Morphology-controlled solvothermal synthesis of LiFePO4 as a cathode material for lithium-ion batteries, J. Mater. Chem., 2010, 20, 8086–8091 RSC.
- Y. Li, J. N. Weker, W. E. Gent, D. N. Mueller, J. Lim, D. A. Cogswell, T. Tyliszczak and W. C. Chueh, Dichotomy in the Lithiation Pathway of Ellipsoidal and Platelet LiFePO4 Particles Revealed through Nanoscale Operando State-of-Charge Imaging, Adv. Funct. Mater., 2015, 25, 3677–3687 CrossRef CAS.
- Z. Cabán-Huertas, O. Ayyad, D. P. Dubal and P. Gómez-Romero, Aqueous synthesis of LiFePO4 with Fractal Granularity, Sci. Rep., 2016, 6, 27024 CrossRef PubMed.
- Z. Lu, H. Chen, R. Robert, B. Y. X. Zhu, J. Deng, L. Wu, C. Y. Chung and C. P. Grey, Citric Acid- and Ammonium-Mediated Morphological Transformations of Olivine LiFePO4 Particles, Chem. Mater., 2011, 23, 2848–2859 CrossRef CAS.
- B. Ellis, W. H. Kan, W. R. M. Makahnouk and L. F. Nazar, Synthesis of nanocrystals and morphology control of hydrothermally prepared LiFePO4, J. Mater. Chem., 2007, 17, 3248–3254 RSC.
- K. Dokko, S. Koizumi, H. Nakano and K. Kanamura, Particle morphology, crystal orientation, and electrochemical reactivity of LiFePO4 synthesized by the hydrothermal method at 443 K, J. Mater. Chem., 2007, 17, 4803–4810 RSC.
- J. B. Dunn, L. Gaines, J. C. Kelly, C. James and K. G. Gallagher, The significance of Li-ion batteries in electric vehicle life-cycle energy and emissions and recycling's role in its reduction, Energy Environ. Sci., 2015, 8, 158–168 CAS.
- G. Majeau-Bettez, T. R. Hawkins and A. H. Strømman, Life Cycle Environmental Assessment of Lithium-Ion and Nickel Metal Hydride Batteries for Plug-In Hybrid and Battery Electric Vehicles, Environ. Sci. Technol., 2011, 45, 4548–4554 CrossRef CAS PubMed.
- M. C. Johnson, I. Palou-Rivera and E. D. Frank, Energy consumption during the manufacture of nutrients for algae cultivation, Algal Res., 2013, 2, 426–436 CrossRef.
- C.-T. Liu and T. L. William Jr, Vapor pressure of deuterated water from 106 to 300 deg, J. Chem. Eng. Data, 1970, 15, 510–513 CrossRef CAS.
- D. R. Stull, Vapor Pressure of Pure Substances. Organic and Inorganic Compounds, Ind. Eng. Chem., 1947, 39, 517–540 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, W. M. Haynes, ed., CRC Press/Taylor & Francis, Boca Raton, FL, 97th edn (Internet Version), 2017 Search PubMed.
- S.-i. Nishimura, G. Kobayashi, K. Ohoyama, R. Kanno, M. Yashima and A. Yamada, Experimental visualization of lithium diffusion in LixFePO4, Nat. Mater., 2008, 7, 707–711 CrossRef CAS PubMed.
- L. Wang, F. Zhou, Y. Meng and G. Ceder, First-principles study of surface properties of LiFePO4: surface energy, structure, Wulff shape, and surface redox potential, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 16543 Search PubMed.
- C. A. J. Fisher and M. S. Islam, Surface structures and crystal morphologies of LiFePO4: relevance to electrochemical behaviour, J. Mater. Chem., 2008, 18, 1209–1215 RSC.
- S. Chung, S. Choi, T. Yamamoto and Y. Ikuhara, Atomic-Scale Visualization of Antisite Defects in LiFePO4, Phys. Rev. Lett., 2008, 100, 125502 CrossRef PubMed.
- R. Malik, D. Burch, M. Bazant and G. Ceder, Particle size dependence of the ionic diffusivity, Nano Lett., 2010, 10, 4123–4127 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Formation mechanisms of uniform nanocrystals via hot-injection and heat-up methods, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- W. Ostwald, Studien über die Bildung und Umwandlung fester Körper, Z. Phys. Chem., 1897, 22, 289–330 CAS.
- L. Lutterotti, D. Chateigner, S. Ferrari and J. Ricote, Texture, residual stress and structural analysis of thin films using a combined X-ray analysis, Thin Solid Films, 2004, 450, 34–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Energy consumption analysis, description of the sampling tool, X-ray diffraction data and Rietveld refinements, characterization of carbon coatings, additional electrochemical data, and particle morphology characterization. See DOI: 10.1039/c7ra00463j |
| This journal is © The Royal Society of Chemistry 2017 |