
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
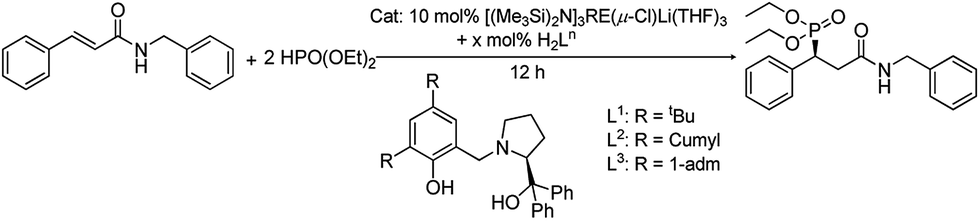
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient asymmetric hydrophosphonylation of unsaturated amides catalyzed by rare-earth metal amides [(Me3Si)2N]3RE(μ-Cl)Li(THF)3 with phenoxy-functionalized chiral prolinols†
Zenghui Fei,
Chao Zeng,
Chengrong Lu,
Bei Zhao * and
Yingming Yao*
* and
Yingming Yao*
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Key Laboratory of Organic Synthesis of Jiangsu Province, Dushu Lake Campus, Suzhou 215123, P. R. China. E-mail: zhaobei@suda.edu.cn; yaoym@suda.edu.cn; Fax: +86 512 65880305; Tel: +86 512 65880305
First published on 31st March 2017
Abstract
An asymmetric hydrophosphonylation reaction of diethyl phosphite with α,β-unsaturated amides catalyzed by [(Me3Si)2N]3RE(μ-Cl)Li(THF)3 (RE = Sc (1), Y (2), La (3), Yb (4) and Lu (5)) with H2Ln ((S)-2,4-R2-6-[[2-(hydroxydiphenylmethyl)pyrrolidin-1-yl]methyl]phenol) (R = tBu (H2L1); R = 1-cumyl (H2L2) and R = 1-adm (H2L3)) was disclosed. The effects of different central metals and proligands on the addition reaction were tested and it was found that the combination of Sc complex 1 and ligand H2L2 gave the best results. An excellent chemical yield (up to 99%) and good to high enantioselectivities (varied from 73 to 89%) were achieved with a relatively broad scope of the unsaturated amides. The active species in the current system was also discussed.
Introduction
In recent years, the catalytic asymmetric synthesis of optically active phosphonates has attracted increasing attention due to their important applications in biology and medical science.1 The asymmetric conjugate addition reactions of phosphite to α,β-unsaturated carbonyl compounds, including aldehydes,2 ketones,3 amides4 and esters,3i,4g,5 promoted by metal-based catalysts are regarded as simple and efficient methods for C–P bond formation. Among the investigations, the phospha-Michael reaction of α,β-unsaturated amides is quite limited, with only a few reports focused on the subject. As early as 2001, Quirion realized the synthesis of amidophosphonates, which was the first case of a phospha-Michael reaction of an α,β-unsaturated amide.4a Thirteen years later, Leung disclosed the first asymmetric phospha-Michael addition of diarylphosphines to N-enoyl phthalimides catalyzed by a chiral palladacycle catalyst.4b Soon after, they achieved the first catalytic asymmetric phospha-Michael addition of Ph2PH to 4-oxo-enamides.6 Almost at the same period, Wang successfully employed a dinuclear zinc catalyst to catalyze the conjugate addition reactions of phosphite to α,β-unsaturated N-acylpyrroles.4c–e,7 Subsequently, his group continued to develop the phospha-Michael reaction of dialkyl phosphine oxide with various α,β-unsaturated substrates, such as N-acylpyrroles, N-acylated oxazolidinones and β,β-disubstituted α,β-unsaturated carbonyl compounds. Most of them gave good yields and high enantioselectivities. In 2011, a palladium-catalyzed asymmetric addition of diarylphosphines to α,β-unsaturated N-acylpyrroles was explored by Duan and his results were as good as those previously reported.4f Ishihara conducted a 1,4-addition reaction to an α,β-unsaturated amide with chiral magnesium(II)binaphtholates in 2013, which gave an 80% yield and 94% enantioselectivity.4g However, the diversity of metal-based catalysts used in the phospha-Michael reaction of α,β-unsaturated amide still needs to be developed.Our group has been devoted to studying lanthanide chemistry for several years.8 Recently, we turned to investigate the enantioselective synthesis of some valuable target molecules using rare-earth metal catalysts. Fortunately, a series of rare-earth metal complexes together with phenoxy-functionalized chiral prolinols were proven to be highly efficient catalysts in the epoxidation of α,β-unsaturated ketones9 and also effective in the Michael addition of malonates to unsaturated ketones.10 All these wonderful results encouraged us to seek for new applications of the current catalytic system. Since the asymmetric phospha-Michael reaction of α,β-unsaturated amide is still challenging, herein, we report this novel transformation catalyzed using rare-earth amides [(Me3Si)2N]3RE(μ-Cl)Li(THF)3 with phenoxy-functionalized chiral prolinols.
Results and discussion
First, to test the reactivity and selectivity of our catalytic systems, we prepared five rare-earth metal amides [(Me3Si)2N]3RE(μ-Cl)Li(THF)3 (RE = Sc (1), Y (2), La (3), Yb (4) and Lu (5)) according to the previous study.11 Then, three phenoxy-functionalized chiral prolinols with bulky groups ((S)-2,4-di-R2-6-[[2-(hydroxydiphenylmethyl)pyrrolidin-1-yl]methyl]phenol) (R = tBu (H2L1); R = cumyl (H2L2) and R = 1-adm (H2L3)) were synthesized as previously reported.9 With the ytterbium amide 4 in hand, the model reaction of N-benzyl unsaturated amide 6 with HPO(OEt)2 was carried out in the presence of the proligand H2L1. To our delight, the yields of the corresponding 1,4-adducts were satisfying and the enantiomeric excess was moderate, from 47% to 59% (Table 1, entries 1–3). The steric effect of the different proligands was investigated and the bulky cumyl groups in proligand H2L2 revealed a slight ascendant in controlling the enantioselectivity (Table 1, entries 2, and 4–5). A relatively low temperature is usually helpful to improve the enantioselectivity of the asymmetric reaction. Attempts to lower the reaction temperature in our case have a positive effect on the enantioselectivity. Although the yields dramatically dropped from 99% to 72% when the temperature was decreased, it was an acceptable yield, considering the good enantiomeric excess achieved (Table 1, entries 4, and 6–7).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | x | Ligand | T (°C) | Solvent | Yieldb (%) | eec (%) |
| a Reactions were performed with unsaturated amide (0.5 mmol), HPO(OEt)2 (1.0 mmol) in 2 mL solvent.b HPLC yield.c Determined by chiral HPLC analysis.d The reaction was conducted with dimethyl phosphite.e The reaction was conducted with diisopropyl phosphite.f The reaction was conducted with diphenyl phosphite. | |||||||
| 1 | Yb | 10 | H2L1 | 25 | THF | 99 | 47 |
| 2 | Yb | 15 | H2L1 | 25 | THF | 96 | 59 |
| 3 | Yb | 20 | H2L1 | 25 | THF | 99 | 58 |
| 4 | Yb | 15 | H2L2 | 25 | THF | 99 | 59 |
| 5 | Yb | 15 | H2L3 | 25 | THF | 99 | 58 |
| 6 | Yb | 15 | H2L2 | 0 | THF | 99 | 61 |
| 7 | Yb | 15 | H2L2 | −20 | THF | 72 | 64 |
| 8 | La | 15 | H2L2 | −20 | THF | 10 | 70 |
| 9 | Y | 15 | H2L2 | −20 | THF | 69 | 64 |
| 10 | Lu | 15 | H2L2 | −20 | THF | 65 | 63 |
| 11 | Sc | 15 | H2L2 | −20 | THF | 80 | 83 |
| 12 | Sc | 15 | H2L2 | −20 | Tol | 22 | 80 |
| 13 | Sc | 15 | H2L2 | −20 | DME | 9 | 82 |
| 14 | Sc | 15 | H2L2 | −20 | Et2O | 10 | 80 |
| 15 | Sc | 15 | H2L2 | −20 | CH3CN | 99 | 57 |
| 16 | Sc | 15 | H2L2 | 20 | 1,4-Dioxane | 99 | 81 |
| 17 | Sc | 10 | H2L2 | 20 | 1,4-Dioxane | 92 | 78 |
| 18 | Sc | 20 | H2L2 | 20 | 1,4-Dioxane | 99 | 85 |
| 19d | Sc | 20 | H2L2 | 20 | 1,4-Dioxane | 69 | 75 |
| 20e | Sc | 20 | H2L2 | 20 | 1,4-Dioxane | 21 | 81 |
| 21f | Sc | 20 | H2L2 | 20 | 1,4-Dioxane | Trace | — |
The effects of the center rare-earth metals of the amides were next to be explored. As expected, the radius of the center metals significantly influenced both the yield and enantioselectivity, that is, the outcome of the transformation improved when the radius of center metals decreased. When scandium amide 1 was employed as the catalyst in the model reaction, the ee value reached 83%, while the yield was maintained at 80% (Table 1, entries 7–11). As we know, solvent often plays an important role in homogenous reactions, and some commonly used solvents were screened subsequently. Fortunately, the reaction gave an excellent yield and high enantioselectivity in 1,4-dioxane at 20 °C (Table 1, entries 11–16). Finally, the ratio of scandium amide [(Me3Si)2N]3Sc(μ-Cl)Li(THF)3 to the proligand was tested again in 1,4-dioxane and the results showed that the model reaction proceeded better with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of the Sc complex to the proligand H2L2 at 20 °C (Table 1, entries 16–18). The effect of the ester moiety in different phosphites was then studied since it may have had an influence on the level of the asymmetric induction.12 Dimethyl phosphite gave the target product in 69% yield and 75% ee, while diisopropyl phosphite gave a 21% yield and 81% ee. As to diphenyl phosphite, only a trace amount of the corresponding adduct was observed. As a result, the aliphatic ester moiety matched better in the enantioselective transformation in the present case, particularly diethyl phosphite (Table 1, entries 18–21).
:2 molar ratio of the Sc complex to the proligand H2L2 at 20 °C (Table 1, entries 16–18). The effect of the ester moiety in different phosphites was then studied since it may have had an influence on the level of the asymmetric induction.12 Dimethyl phosphite gave the target product in 69% yield and 75% ee, while diisopropyl phosphite gave a 21% yield and 81% ee. As to diphenyl phosphite, only a trace amount of the corresponding adduct was observed. As a result, the aliphatic ester moiety matched better in the enantioselective transformation in the present case, particularly diethyl phosphite (Table 1, entries 18–21).
Thus, various substrates were investigated using 10 mol% of Sc catalyst 1 in 1,4-dioxane at 20 °C in the presence of 20 mol% of proligand H2L2, and the results are summarized in Scheme 1. As we can see, almost all of the substrates participated in the reaction and gave excellent yields and good to high enantioselectivities under the optimal conditions. Whatever the substrate was, α,β-unsaturated secondary amides (7a–7p) or tertiary amides (7q–7t) bearing either aliphatic or aromatic N-substituents, the yields were excellent, from 90% to 99%, except in two cases (7b and 7c). Perhaps the flexible n-butyl and i-propyl groups attached to the nitrogen atom in the molecule are relatively crowded and blocked the interaction between the catalyst and the substrate, which also affected the enantioselectivity (only 74% ee for 7b and 64% ee for 7c). In addition, there was no evidence that the electronic effect of the groups on the phenyl rings had an significant influence on the results. Moreover, we successfully realized the asymmetric hydrophosphonylation of unsaturated amide with an aliphatic acyl group in our system (7l).
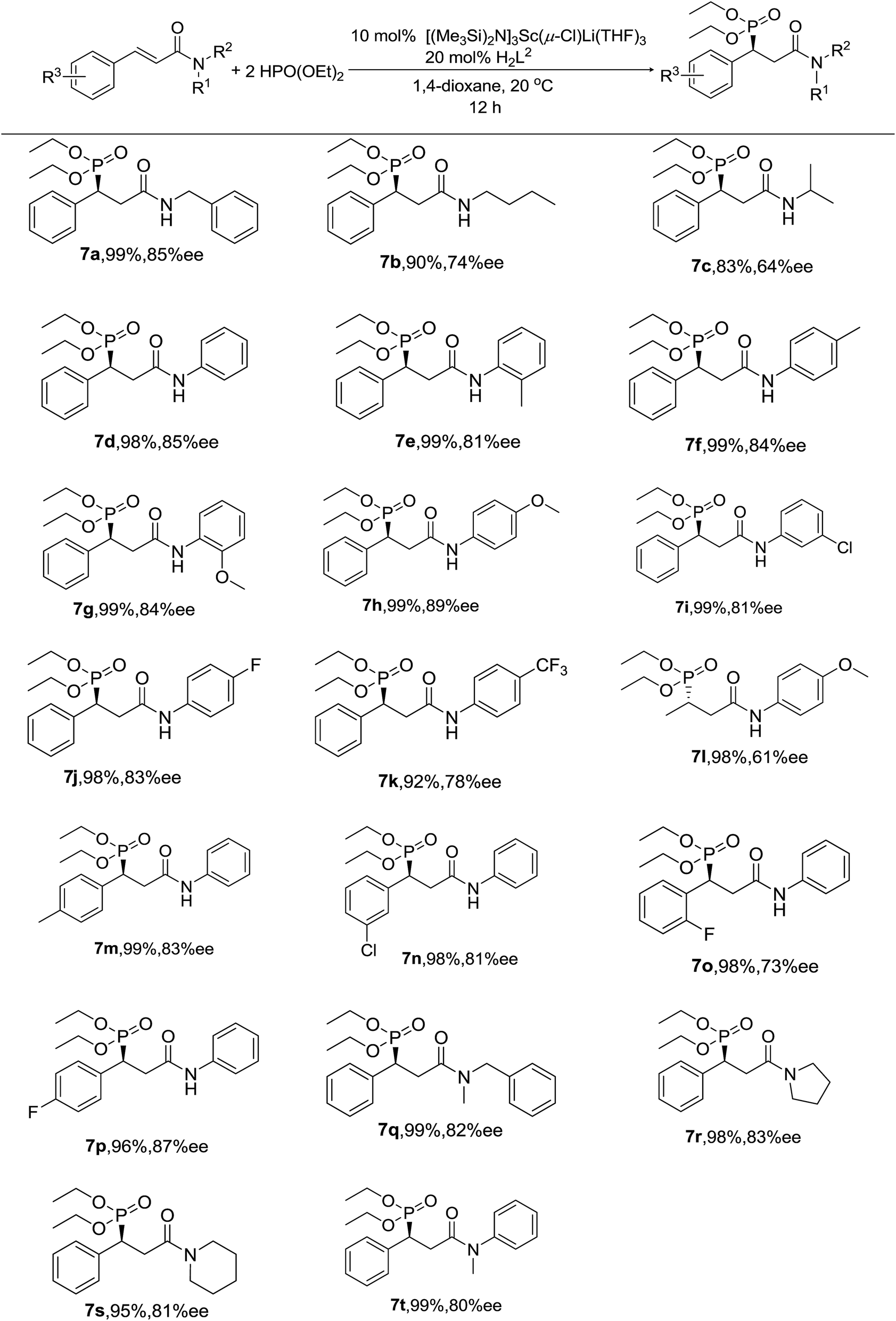 | ||
| Scheme 1 The asymmetric hydrophosphonylation of unsaturated amides. Reactions were performed with the substrate (0.5 mmol), HPO(OEt)2 (1.0 mmol) in 2 mL of 1,4-dioxane at 20 °C. The ee values were determined by chiral HPLC. The isolated yields are shown. | ||
To further investigate the possible active species in the current catalytic system based on the optimal molar ratio of the Sc amide 1 to proligand H2L2, crystals of 8 were obtained from the reaction of the Sc-based precatalyst 1 with 2 equiv. of proligand H2L2 in THF. The NMR spectra and elemental analysis results of crystal 8 were informative. One peak at 8.86 was assigned to the hydrogen of the hydroxyl group and was observed in the proton NMR spectrum; the chemical shifts at 1.64 and 1.36 were ascribed to the hydrogens in the C(CH3)2 group the peaks at 3.88 and 3.73 were ascribed to hydrogens of the methine in group NCH2Ar. These observations prove the existence of the ligand in the crystal. Furthermore, the elemental analysis shows that the ratio of Sc:L2 was 1:2, and the LiCl component was not observed. However, the crystals of 8 obtained in the abovementioned reaction were not good enough for the X-ray diffraction analysis and only the skeleton of the molecular structure could be presumed. Fortunately, a colourless crystal of 8 suitable for the X-ray diffraction analysis could also be obtained by treating tris((trimethylsilyl)methyl)scandium with 2 equiv. of proligand H2L2 in hexane (Scheme 2). Finally, the definite solid structure of 8 [L2Sc(L2H)] was determined as expected and is depicted in Fig. 1. The scandium complex has a mononuclear structure with two chiral ligands in the unit cell. One hydrogen atom of the hydroxyl group in one of the chiral prolinolates remains to meet the requirement of the trivalence of the central metal.
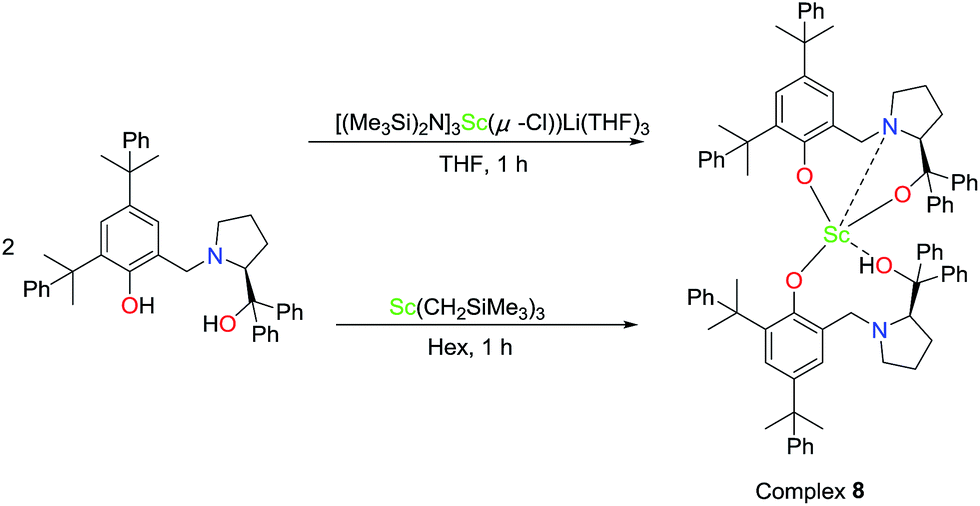 | ||
| Scheme 2 Synthesis of complex 8. | ||
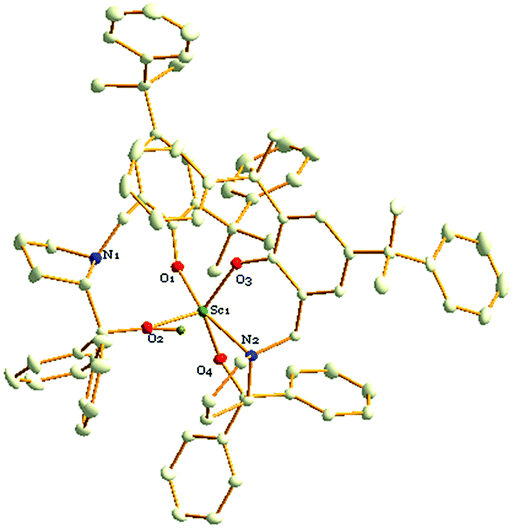 | ||
| Fig. 1 The molecular structure of 8 showing 20% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Sc(1)–O(1) 2.0350(16), Sc(1)–O(2) 1.9518(17), Sc(1)–O(3) 2.0141(16), Sc(1)–O(4) 1.9833(16), Sc(1)–N(2) 2.3539(19), O(2)–Sc(1)–O(4) 108.05(7), O(2)–Sc(1)–O(3) 115.27(7), O(4)–Sc(1)–O(3) 132.87(7), O(2)–Sc(1)–O(1) 97.92(7), O(4)–Sc(1)–O(1) 99.48(7), O(2)–Sc(1)–O(1) 92.29(6), O(2)–Sc(1)–N(2) 99.29(7), O(4)–Sc(1)–N(2) 74.83(7), O(3)–Sc(1)–N(2) 80.64(7), O(1)–Sc(1)–N(2) 162.78(7). | ||
Subsequently, complex 8 was added to the model reaction of the asymmetric hydrophosphonylation of N-benzyl unsaturated amide and the target molecule was obtained in 74% yield and 79% ee (Table 2, entry 1). Taking into consideration the difference between the catalyst system generated in situ and the abovementioned structure of complex 8, 10 mol% of lithium chloride and 20 mol% of HNSiMe3 were deliberately added to the reaction. The outcome was satisfactory with a 98% yield and 85% ee, almost the same as the in situ catalyst system (Table 2, entry 2). The following test without the addition of lithium chloride indicates that lithium chloride was essential. Numerous additives such as DBU, Et3N, TMEDA, Cy2NMe, and DABCO were added to make clear whether the aminosilane HNSiMe3 was essential in the reaction. The results revealed that the base is helpful to the transformation; however, all the results are not as good as those achieved using aminosilane (Table 2, entries 4–8). Therefore, we came to the conclusion that the bases are beneficial for the asymmetric hydrophosphonylation of unsaturated amides and aminosilane is optimal. Because complex 8 was recrystallized in a mixed solvent of toluene and hexane, the lithium chloride molecule was removed after work up due to its poor solubility in toluene; it was almost impossible for us to gain the expected crystals bearing LiCl in the crystal cell with the current situation. A detailed study of the mechanism is underway in our laboratory.
|
|||
|---|---|---|---|
| Entry | Additive | Yieldb (%) | eec (%) |
| a Reactions were performed with unsaturated amide (0.5 mmol), HPO(OEt)2 (1.0 mmol), complex 8 (10 mol%), LiCl (10 mol%), additive (20 mol%) in 2 mL of 1,4-dioxane at 20 °C.b HPLC yield.c Determined by chiral HPLC analysis.d Reaction was performed without LiCl and HNSiMe3.e Reaction was performed without LiCl. | |||
| 1d | — | 74 | 79 |
| 2 | HNSiMe3 | 98 | 85 |
| 3e | HNSiMe3 | 98 | 78 |
| 4 | DBU | 98 | 79 |
| 5 | Et3N | 96 | 77 |
| 6 | TMEDA | 81 | 76 |
| 7 | Cy2NMe | 73 | 77 |
| 8 | DABCO | 98 | 75 |

Furthermore, the outcome of the model reaction on a gram scale was satisfactory with an appropriate prolonging of the reaction time (Scheme 3). The absolute configuration of the target phosphonate 7a was determined by a combination of single crystal diffraction, chiral HPLC and optical rotation analysis (Schemes 3 and 4).
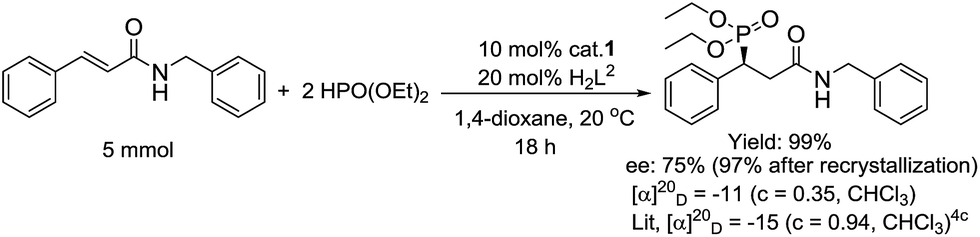 | ||
| Scheme 3 The amplification reaction of the preparation of compound 7a and its optical rotation analysis. | ||
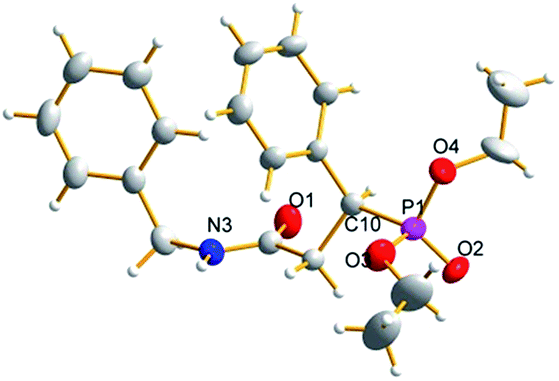 | ||
| Scheme 4 The molecular structure of compound 7a. | ||
Conclusion
In summary, the asymmetric hydrophosphonylation of α,β-unsaturated amides was successfully catalyzed by rare-earth metal amides with phenoxy-functionalized chiral prolinols. After careful screening, 10 mol% of the Sc complex [(Me3Si)2N]3Sc(μ-Cl)Li(THF)3 together with the proligand H2L2 ((S)-2,4-dicumyl-6-[[2-(hydroxydiphenylmethyl)pyrrolidin-1-yl]methyl]phenol) in a 1:2 molar ratio were found to be the optimal catalytic conditions in 1,4-dioxane at 20 °C. Substrates bearing various groups were examined in this catalytic system, including secondary amides and tertiary amides, which gave excellent yields (up to 99%) and good to high enantioselectivities (73–89%). The absolute configuration of the target phosphonate was determined. During the exploration of the definite structure of the actual catalyst, complex 8 was obtained. It was proven to be an efficient catalyst in the presence of 1 equiv. LiCl and 2 equiv. base in the hydrophosphonylation reaction. We are still on the way to deduce the clear catalytic cycle occurring in the reaction.
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 21572151, 21372172), PAPD and the Qing Lan Project.Notes and references
- (a) H. Sasai, S. Arai, Y. Tahara and M. Shibasaki, J. Org. Chem., 1995, 60(21), 6656–6657 CrossRef CAS; (b) I. Schlemminger, Y. Saida, H. Gröger, W. Maison, N. Durot, H. Sasai, M. Shibasaki and J. Martens, J. Org. Chem., 2000, 65(16), 4818–4825 CrossRef CAS PubMed; (c) X. Chen, A. J. Wiemer, R. J. Hohl and D. F. Wiemer, J. Org. Chem., 2002, 67(26), 9331–9339 CrossRef CAS PubMed; (d) G. D. Joly and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126(13), 4102–4103 CrossRef CAS PubMed; (e) B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2005, 44(29), 4600–4602 CrossRef CAS PubMed; (f) J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2008, 130(32), 10521–10523 CrossRef CAS PubMed; (g) D. Uraguchi, T. Ito and T. Ooi, J. Am. Chem. Soc., 2009, 131(11), 3836–3837 CrossRef CAS PubMed; (h) F. Yang, D. Zhao, J. Lan, P. Xi, L. Yang, S. Xiang and J. You, Angew. Chem., Int. Ed., 2008, 47(30), 5646–5649 CrossRef CAS PubMed; (i) S. Nakamura, M. Hayashi, Y. Hiramatsu, N. Shibata, Y. Funahashi and T. Toru, J. Am. Chem. Soc., 2009, 131(51), 18240–18241 CrossRef CAS PubMed; (j) X. Zhou, D. Shang, Q. Zhang, L. Lin, X. Liu and X. Feng, Org. Lett., 2009, 11(6), 1401–1404 CrossRef CAS PubMed; (k) W. Chen, Y. Hui, X. Zhou, J. Jiang, Y. Cai, X. Liu, L. Lin and X. Feng, Tetrahedron Lett., 2010, 51(32), 4175–4178 CrossRef CAS; (l) Y. Zhao, X. Li, F. Mo, L. Li and X. Lin, RSC Adv., 2013, 3(29), 11895–11901 RSC; (m) J. Guin, Q. Wang, M. van Gemmeren and B. List, Angew. Chem., Int. Ed., 2015, 54(1), 355–358 CrossRef CAS PubMed; (n) Y. Gao, X. Li, W. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2015, 80(22), 11398–11406 CrossRef CAS PubMed; (o) M.-X. Cheng, R.-S. Ma, Q. Yang and S.-D. Yang, Org. Lett., 2016, 18(13), 3262–3265 CrossRef CAS PubMed.
- (a) I. Ibrahem, R. Rios, J. Vesely, P. Hammar, L. Eriksson, F. Himo and A. Córdova, Angew. Chem., Int. Ed., 2007, 46(24), 4507–4510 CrossRef CAS PubMed; (b) A. Carlone, G. Bartoli, M. Bosco, L. Sambri and P. Melchiorre, Angew. Chem., Int. Ed., 2007, 46(24), 4504–4506 CrossRef CAS PubMed; (c) E. Maerten, S. Cabrera and K. A. Jørgensen, J. Org. Chem., 2007, 72(23), 8893–8903 CrossRef CAS PubMed; (d) Y.-R. Chen and W.-L. Duan, Org. Lett., 2011, 13(21), 5824–5826 CrossRef CAS PubMed; (e) X. Luo, Z. Zhou, X. Li, X. Liang and J. Ye, RSC Adv., 2011, 1(4), 698–705 RSC.
- (a) D. Zhao, Y. Yuan, A. S. C. Chan and R. Wang, Chem.–Eur. J., 2009, 15(12), 2738–2741 CrossRef CAS PubMed; (b) Y. Huang, S. A. Pullarkat, Y. Li and P.-H. Leung, Chem. Commun., 2010, 46(37), 6950–6952 RSC; (c) J.-J. Feng, X.-F. Chen, M. Shi and W.-L. Duan, J. Am. Chem. Soc., 2010, 132(16), 5562–5563 CrossRef CAS PubMed; (d) D. Zhao, L. Mao, D. Yang and R. Wang, J. Org. Chem., 2010, 75(20), 6756–6763 CrossRef CAS PubMed; (e) S. Wen, P. Li, H. Wu, F. Yu, X. Liang and J. Ye, Chem. Commun., 2010, 46(26), 4806–4808 RSC; (f) A. Russo and A. Lattanzi, Eur. J. Org. Chem., 2010, 2010(35), 6736–6739 CrossRef; (g) M.-J. Yang, Y.-J. Liu, J.-F. Gong and M.-P. Song, Organometallics, 2011, 30(14), 3793–3803 CrossRef CAS; (h) Y.-R. Chen, J.-J. Feng and W.-L. Duan, Tetrahedron Lett., 2014, 55(3), 595–597 CrossRef CAS; (i) X.-Q. Hao, J.-J. Huang, T. Wang, J. Lv, J.-F. Gong and M.-P. Song, J. Org. Chem., 2014, 79(20), 9512–9530 CrossRef CAS PubMed; (j) C. Li, Q.-L. Bian, S. Xu and W.-L. Duan, Org. Chem. Front., 2014, 1(5), 541–545 RSC; (k) Y.-X. Jia, B.-B. Li, Y. Li, S. A. Pullarkat, K. Xu, H. Hirao and P.-H. Leung, Organometallics, 2014, 33(21), 6053–6058 CrossRef CAS; (l) G. Li, L. Wang, Z. Yao and F. Xu, Tetrahedron: Asymmetry, 2014, 25(13–14), 989–996 CrossRef CAS.
- (a) G. Castelot-Deliencourt, X. Pannecoucke and J.-C. Quirion, Tetrahedron Lett., 2001, 42(6), 1025–1028 CrossRef CAS; (b) R. J. Chew, Y. Lu, Y.-X. Jia, B.-B. Li, E. H. Y. Wong, R. Goh, Y. Li, Y. Huang, S. A. Pullarkat and P.-H. Leung, Chem.–Eur. J., 2014, 20(44), 14514–14517 CrossRef CAS PubMed; (c) D. Zhao, Y. Wang, L. Mao and R. Wang, Chem.–Eur. J., 2009, 15(41), 10983–10987 CrossRef CAS PubMed; (d) D. Zhao, L. Mao, Y. Wang, D. Yang, Q. Zhang and R. Wang, Org. Lett., 2010, 12(8), 1880–1882 CrossRef CAS PubMed; (e) D. Zhao, L. Mao, L. Wang, D. Yang and R. Wang, Chem. Commun., 2012, 48(6), 889–891 RSC; (f) D. Du and W.-L. Duan, Chem. Commun., 2011, 47(39), 11101–11103 RSC; (g) M. Hatano, T. Horibe and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52(17), 4549–4553 CrossRef CAS PubMed.
- (a) L. Tedeschi and D. Enders, Org. Lett., 2001, 3(22), 3515–3517 CrossRef CAS PubMed; (b) C. Xu, G. Jun Hao Kennard, F. Hennersdorf, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2012, 31(8), 3022–3026 CrossRef CAS.
- R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem.–Eur. J., 2015, 21(12), 4800–4804 CrossRef CAS PubMed.
- D. Zhao, L. Wang, D. Yang, Y. Zhang and R. Wang, Chem.–Asian J., 2012, 7(5), 881–883 CrossRef CAS PubMed.
- (a) X. Hu, C. Lu, B. Wu, H. Ding, B. Zhao, Y. Yao and Q. Shen, J. Organomet. Chem., 2013, 732, 92–101 CrossRef CAS; (b) S. Sun, K. Nie, Y. Tan, B. Zhao, Y. Zhang, Q. Shen and Y. Yao, Dalton Trans., 2013, 42(8), 2870–2878 RSC; (c) S. Sun, Q. Sun, B. Zhao, Y. Zhang, Q. Shen and Y. Yao, Organometallics, 2013, 32(6), 1876–1881 CrossRef CAS; (d) H. Cheng, B. Zhao, Y. Yao and C. Lu, Green Chem., 2015, 17(3), 1675–1682 RSC; (e) B. Zhao, Y. Xiao, D. Yuan, C. Lu and Y. Yao, Dalton Trans., 2016, 45(9), 3880–3887 RSC.
- C. Zeng, D. Yuan, B. Zhao and Y. Yao, Org. Lett., 2015, 17(9), 2242–2245 CrossRef CAS PubMed.
- Q. Qian, W. Zhu, C. Lu, B. Zhao and Y. Yao, Tetrahedron: Asymmetry, 2016, 27(19), 911–917 CrossRef CAS.
- (a) E. Sheng, S. Wang, G. Yang, S. Zhou, L. Cheng, K. Zhang and Z. Huang, Organometallics, 2003, 22(4), 684–692 CrossRef CAS; (b) M. Lu, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2010, 39(40), 9530–9537 RSC; (c) Z. Feng, Y. Wei, S. Zhou, G. Zhang, X. Zhu, L. Guo, S. Wang and X. Mu, Dalton Trans., 2015, 44(47), 20502–20513 RSC.
- (a) J. V. Alegre-Requena, E. Marques-Lopez, P. J. Sanz Miguel and R. P. Herrera, Org. Biomol. Chem., 2014, 12(8), 1258–1264 RSC; (b) M. Nazish, A. Jakhar, N.-U. H. Khan, S. Verma, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, Appl. Catal., A, 2016, 515, 116–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures for the preparation of the complexes and the catalytic reaction; characterization data, including 1H and 13C NMR spectra, HPLC chromatograms, HRMS, crystallographic data for compounds 8 and 7a and figures depicting solid state structures. CCDC 1526584 and 1536158. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00468k |
| This journal is © The Royal Society of Chemistry 2017 |