
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CASSCF and CASMP2 study on the photoisomerization mechanisms of [tris(trialkylsilyl)silyl]cyclotetrasilene and related cyclobutene molecules†
Ming-Der Su *ab
*ab
aDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan. E-mail: midesu@mail.ncyu.edu.tw
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 3rd February 2017
Abstract
The mechanisms for photochemical isomerization reactions are studied theoretically using the model systems, cyclotetrasilene and cyclobutene, both of which feature one four-membered-ring and one double bond, using the CAS(6,6)/6-311G(d) and CAS-MP2-(6,6)/6-311++G(3df,3pd) methods. The mechanisms, which are governed by the conical intersection concept, play a crucial role in these photorearrangement reactions, and explain the reaction pathways. The model computations demonstrate that the preferred reaction path for the photoisomerization reactions for both cyclotetrasilene and cyclobutene is as follows: four-membered-ring reactant → Franck–Condon region → conical intersection → butterfly-like photoproduct. These results allow a number of predictions to be made.
I. Introduction
For at least three decades, the chemistry of disilene, which features a silicon–silicon double bond, has been the subject of extensive study because of its novel structure and bonding properties.1,2 However, only very few photochemical reactions that involve the disilene systems have been studied experimentally.3About 15 years ago, Kira and co-workers reported a distinctive photochemical reaction that rearranges hexakis(tert-butyldimethylsilyl)cyclotetrasilene (I) to its corresponding isomer, hexakis(tert-butyldimethylsilyl)-bicyclo[1,1,0]tetrasilane (II), as shown in Scheme 1.4 After heating, (II) is isomerized back to the initial reactant (I). However, to the best of the author's knowledge, there has been no theoretical study of the mechanism for this photo-isomerization reaction for cyclotetrasilene (I).4c This may be because the computational methods for studying photochemistry were not so mature at that time. The author uses a simplified cyclotetrasilene (Si-Rea-S0) as a model to study its photochemical mechanism for the isomerization reaction to bicyclo[1,1,0]tetrasilane (Si-Pro-S0) using modern theoretical methods, i.e., CASSCF (the complete-active-space SCF)5 and the multi-reference Møller–Plesset (CAS-MP2) algorithm.6 For the purposes of comparison, a similar mechanistic study is also made of a four-carbon system, C-Rea-S0, as represented in Scheme 2.7
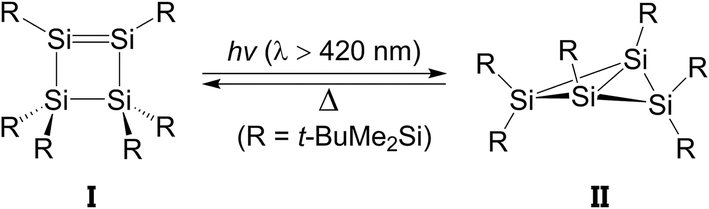 | ||
| Scheme 1 | ||
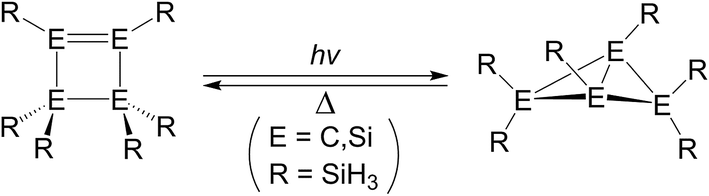 | ||
| Scheme 2 | ||
Conical intersection (CI) points8 can occur on the potential surfaces, from which the excited state decays to the ground state surface at the same multiplicity (usually from singlet to singlet). Therefore, the CI mechanism is used in this work to interpret the reaction trajectories for the photochemistry of both Si-Pro-S0 and C-Pro-S0 molecules. This paper shows that the CIs8 play a decisive role in the photo-rearrangement reactions for cyclotetrasilene and cyclobutene molecules, which have a four-membered-ring.
II. Theoretical methods
The CASSCF computations are executed using the MCSCF9 and CAS-MP2 (ref. 9) programs in GAUSSIAN 09.10 The active space that is used to describe the photo-rearrangements for the four-membered-ring molecules (Scheme 2) that are studied in this work include six electrons in six orbitals, i.e., 1π(E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E) + 1π*(EE) + 2σ(E–E) + 2σ*(E–E) orbitals. The state-averaged CASSCF(6,6) method was adopted to verify geometry on the intersection space (i.e., root = 2). Moreover, the excited state gradient was obtained by using the CSASSCF method. The harmonic frequencies are computed analytically at the CAS(6,6)/6-311G(d) level of theory and are used to characterize either minima or transition states. Unless otherwise stated, the relative energies that are shown in this study are those that are ascertained at the CAS-MP2(6,6)/6-311++G(3df,3pd) level, using the CAS(6,6)/6-311G(d) geometry. These are designated CAS-MP2 and CASSCF, respectively. The Cartesian coordinates and the energies that are computed for the various points are available as ESI.†
E) + 1π*(EE) + 2σ(E–E) + 2σ*(E–E) orbitals. The state-averaged CASSCF(6,6) method was adopted to verify geometry on the intersection space (i.e., root = 2). Moreover, the excited state gradient was obtained by using the CSASSCF method. The harmonic frequencies are computed analytically at the CAS(6,6)/6-311G(d) level of theory and are used to characterize either minima or transition states. Unless otherwise stated, the relative energies that are shown in this study are those that are ascertained at the CAS-MP2(6,6)/6-311++G(3df,3pd) level, using the CAS(6,6)/6-311G(d) geometry. These are designated CAS-MP2 and CASSCF, respectively. The Cartesian coordinates and the energies that are computed for the various points are available as ESI.†
III. General considerations
According to the available experimental results,4 there is no indication that a triplet state participates in these photo-isomerization reactions. The CAS-MP2 computational results also show that the relative energies (kcal mol−1) for the Franck–Condon (FC) regions are as follows: Si-Rea-S0 (0.0) < Si-FC-S1 (68.8) < Si-FC-T1 (93.6) and C-Rea-S0 (0.0) < C-FC-S1 (116.8) < C-FC-T1 (121.8).11 From these theoretical results, it is concluded that triplet states do not play any role in the photochemical reactions for these cyclic disilene molecular systems, so this study concentrates on singlet surfaces for both Si-Rea-S0 and C-Rea-S0 reactants.12In order to determine possible excited-state reaction paths that lead to CIs, a molecular orbital representation of the relationship between the likely reaction pathway and intersection is schematically illustrated in Scheme 3 (E = C and Si),13 which is a foundation for these theoretical studies. Upon irradiation with light, a singlet four-membered-ring molecule (1) is moved up to a singlet excited species (2), succeeding an intramolecular double bond (EE) rotation. Then, as shown in (3), this excited molecule 1 can be considered as a composition of four kinds of carbenes (>E), existing four spin-up σ orbitals and four spin-down σ orbitals.13 Consequently, through the conical intersection, a two-three-membered-ring photoproduct (4) can be obtained.
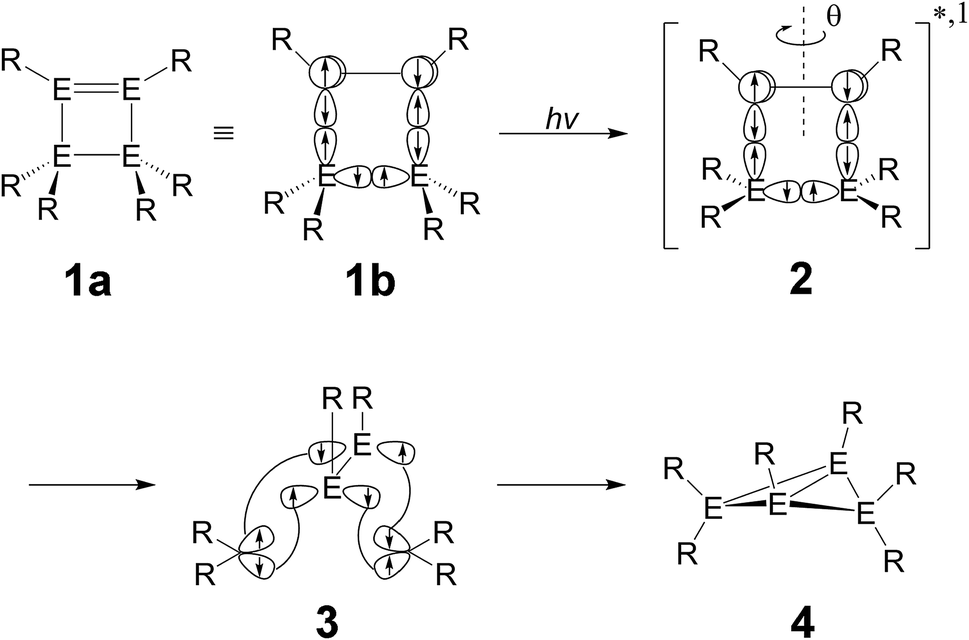 | ||
| Scheme 3 | ||
Using the molecular orbital analysis that is given in Scheme 3, since the narrow energy gap between S0 and S1 energy surfaces strongly implies the existence of a CI point, Si-Rea-S0 and C-Rea-S0 are used as examples to determine whether there is a low-lying S1/S0 surface crossing. Fig. 1 shows the potential energy profiles for the rotational angle, θ, as shown in Scheme 3.14 Fig. 1(A) and (B) show that the reaction pathways all terminate at a very similar S1–S0 energy gap at around 75° and 60°, respectively. These energy profile results suggest that a CI point occurs in the intramolecular rotation for the four-membered-ring geometries. In the next section, these findings are used to explain the mechanisms for the photochemical isomerization reactions of Si-Rea-S0 and C-Rea-S0.
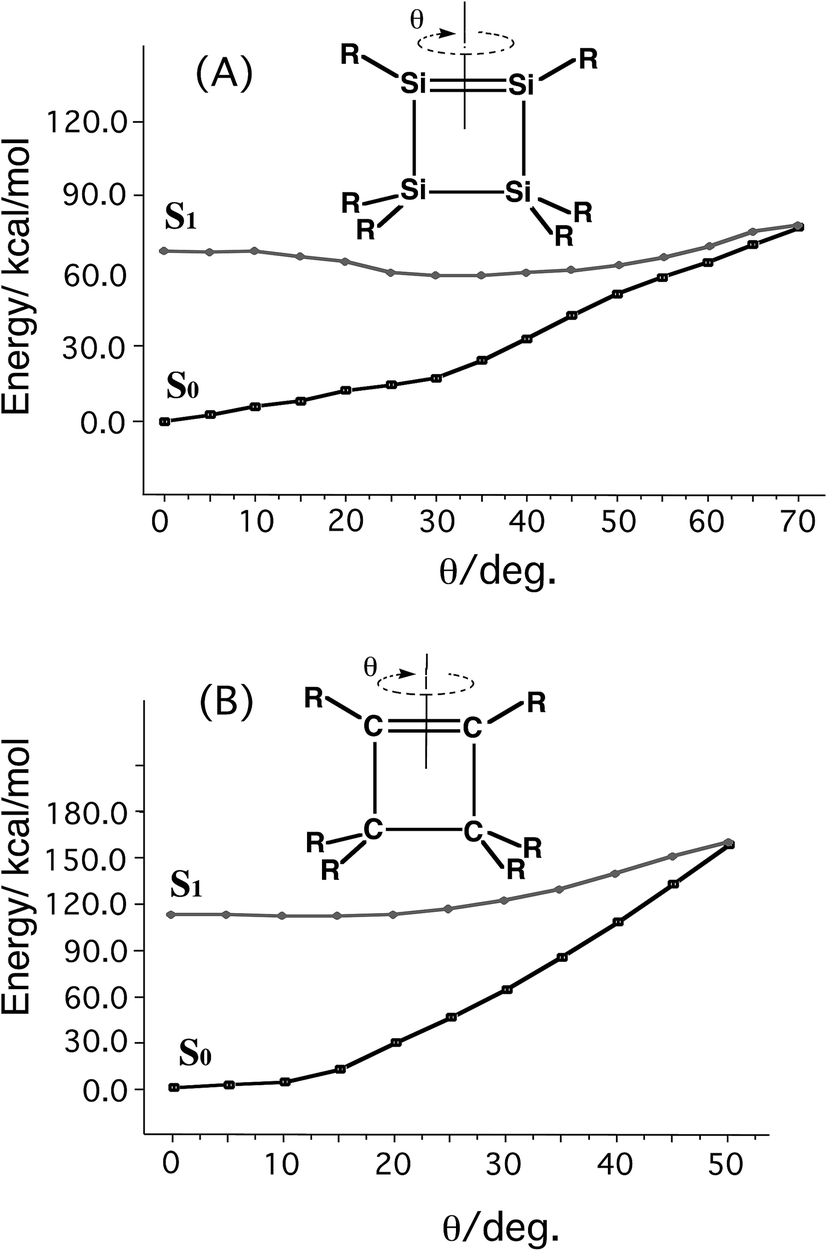 | ||
| Fig. 1 The scans for finding a CI pathway using a EE (E = C and Si) rotation in the reactants ((A) Si-Rea-S0 and (B) C-Rea-S0; R = SiH3) for the angle, θ, optimized for the S0 and S1 states at the CAS(6,6)/6-311G(d) level of theory. See the text and ESI.† | ||
IV. Results and discussion
The discussion in Section III shows that there should exist a reaction route for photochemical isomerization reactions on the singlet excited energy surfaces of Si-Rea-S0 that contains a conical intersection point. Fig. 2 shows the energies for the key points relative to the energy of the reactant, Si-Rea-S0. The optimized geometries for these important points on the mechanistic pathways in Fig. 2 are also shown in the same figure.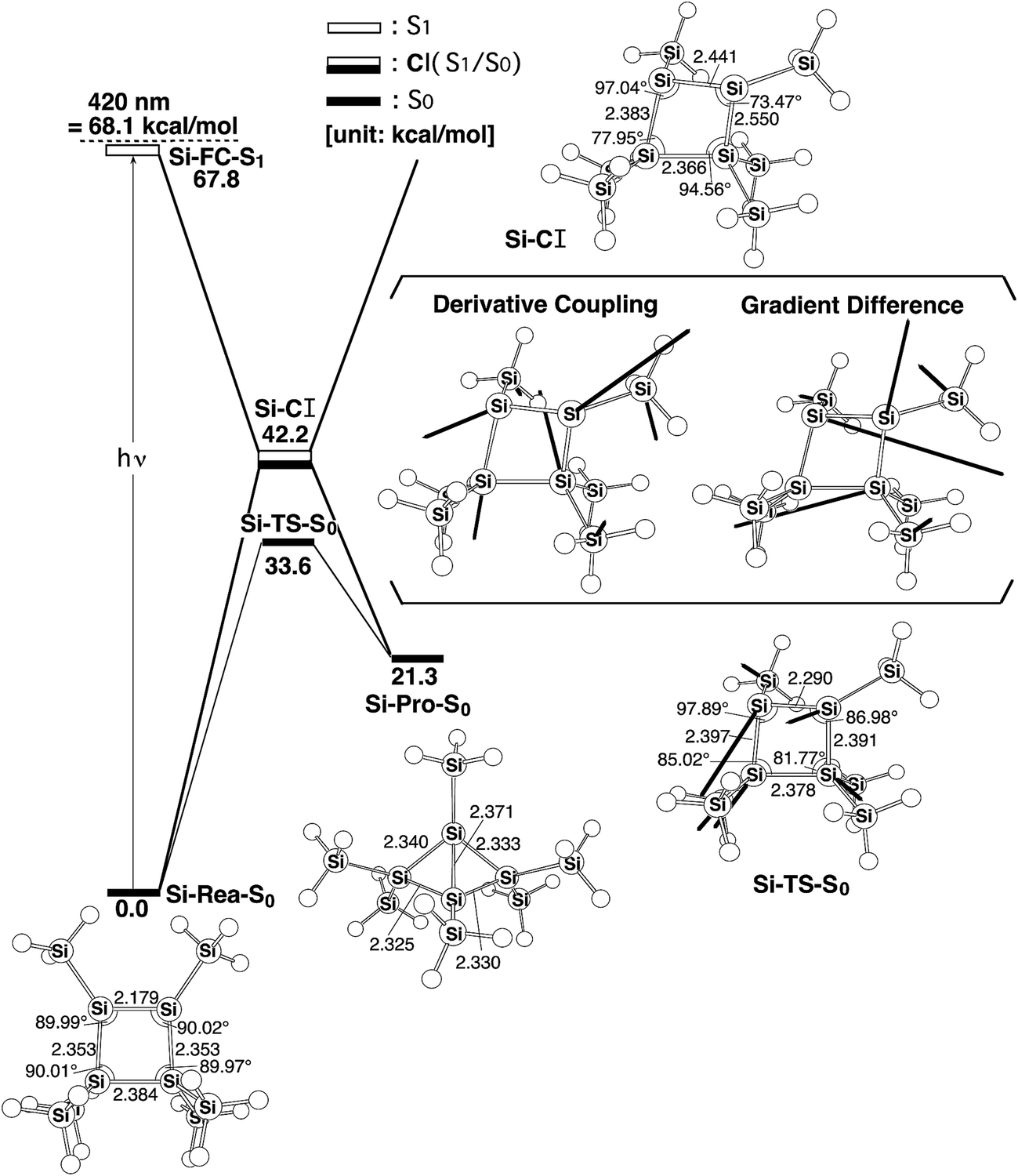 | ||
| Fig. 2 The energy surfaces for the photochemical isomerization reaction for Si-Rea-S0. The abbreviations, FC, TS and CI, denote Franck–Condon, transition state and conical intersection, respectively. The relative energies are obtained at the CAS-MP2-(6,6)/6-311++G(df,pd)//CAS(6,6)/6-311G(d) level of theory. The geometrical parameters for the CASSCF-optimized structures for the stationary points are also shown. Hydrogen atoms are omitted for clarity. The bold arrows show the main atomic motions in the Si-TS-S0 eigenvector. The derivative coupling and the gradient difference vectors – those which increase the degeneracy – are computed using CASSCF at the conical intersection, Si-CI. For more information see the text. | ||
A comparison with the CASSCF-optimized geometry for the silicon reactant (Si-Rea-S0), as shown in Fig. 2, shows that the theoretical values for the bond lengths and the bond angles in these four-membered-ring species are in good agreement with the experimental values.4 The bond lengths and the bond angles are within 0.07 Å and 5.9°, respectively. Therefore, CASSCF and CAS-MP2 computations should be satisfactory for a theoretical study of the photochemical and thermal reactions.
Initially, cyclotetrasilene (Si-Rea-S0) with a four-silicon-membered-ring is irradiated by visible light and is promoted to the Franck–Condon region at the first excited singlet state (Si-FC-S1), which is shown on the left side of Fig. 2. Subsequently, this species relaxes to the conical intersection point (Si-CI), from which it can go to either the final photoproduct (Si-Pro-S0) or the initial reactant (Si-Rea-S0). These CAS-MP2 computations show that the relative energies for Si-FC-S1, Si-CI, and Si-Pro-S0 with respect to Si-Rea-S0 are 68, 42, and 21 kcal mol−1, respectively. The theoretical evidence strongly suggests that when cyclotetrasilene (Si-Rea-S0) is irradiated by visible light, it easily undergoes a SiSi double bond rotation to produce either the reactant (Si-Rea-S0) or the corresponding bicyclo[1,1,0]tetrasilane (Si-Pro-S0). The photochemical isomerization mechanism for Si-Rea-S0 that is shown in Fig. 2 is represented as follows:
| Cyclotetrasilene (photo): Si-Rea-S0 + hν → Si-FC-S1 → Si-CI → Si-Pro-S0 |
The thermal (dark) reaction on the ground singlet state energy surface that is shown in Fig. 2 is also studied. Fig. 2 shows that the Si-TS-S0 point combines Si-Rea-S0 and Si-Pro-S0 on the ground singlet surface and that its energy lies 15 kcal mol−1 below the energy of the Si-CI point. It is worthy of note that the CAS-MP2 calculations give barrier heights for Si-Rea-S0 → Si-TS-S0 and Si-Pro-S0 → Si-TS-S0 that are approximately 34 and 12 kcal mol−1, respectively. This theoretical evidence points to the fact that cyclotetrasilene (Si-Rea-S0) is thermodynamically more stable than bicyclo[1,1,0]tetrasilane (Si-Pro-S0) and producing the latter from the former using the thermal reaction would be difficult, which is in good agreement with the experimental results.4 Fig. 2 also predicts that the thermal reversion from Si-Pro-S0 to Si-Rea-S0 is relatively easily accomplished, since the energy that is required to surmount the reverse barrier (12 kcal mol−1) is less than that which is required to surmount forward barrier (34 kcal mol−1). This theoretical prediction is again in good agreement with the available experimental results.4
The photochemical conversion of C-Rea-S0 to C-Pro-S0 and its thermal reversion are studied. The computational results are all listed in Fig. 3. Similarly to the model for cyclotetrasilene (Si-Rea-S0), the singlet photo-rearrangement reaction for C-Rea-S0 that is studied in this work follows similar reaction paths to those that are shown in Fig. 2. The CAS-MP2 computations suggest that the photo-isomerization reaction for C-Rea-S0 proceeds as follows:
| Cyclobutene (photo): C-Rea-S0 + hν → C-FC-S1 → C-CI → C-Pro-S0 |
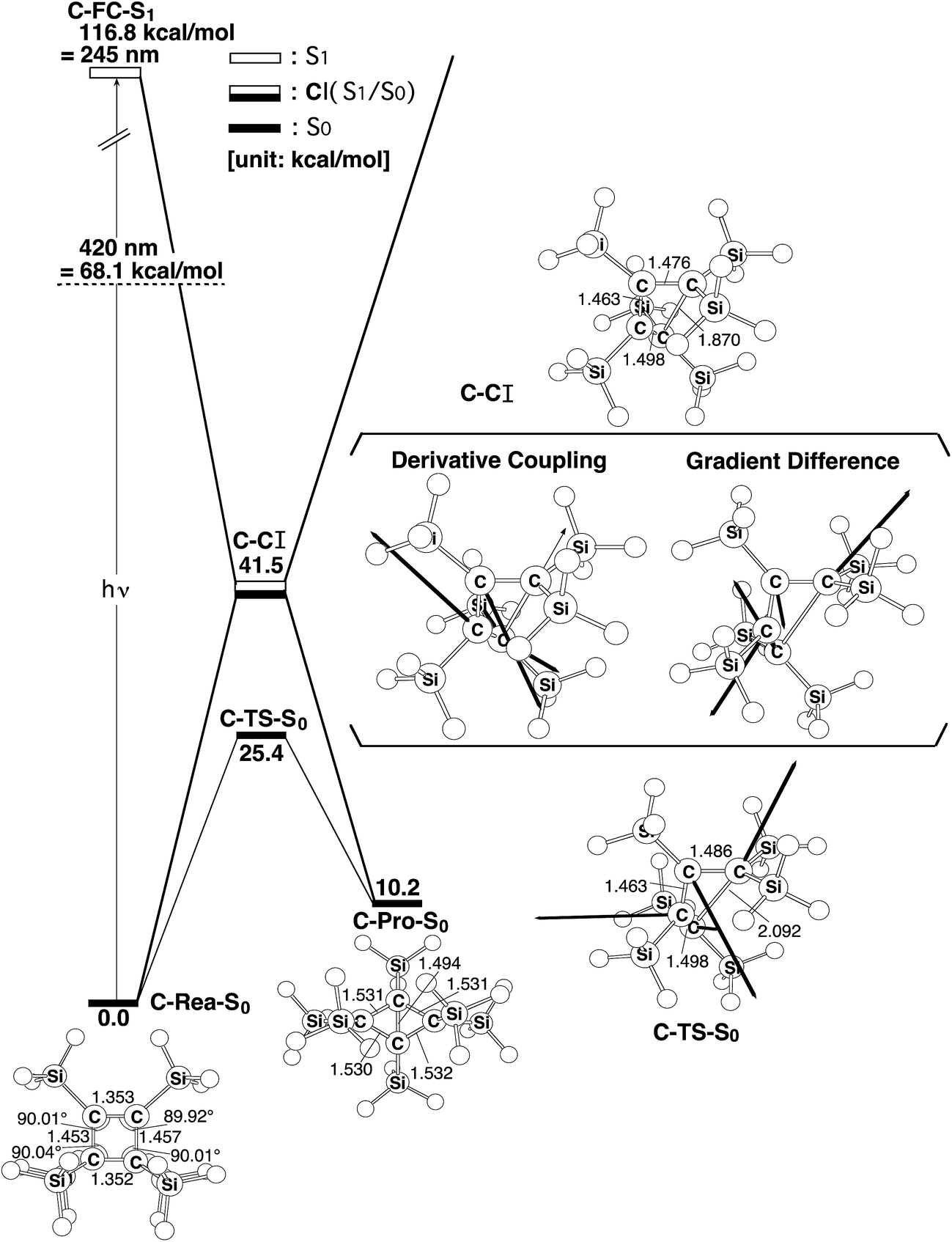 | ||
| Fig. 3 The energy surfaces for the photochemical isomerization reaction for C-Rea-S0. The abbreviations, FC, TS and CI, denote Franck–Condon, transition state and conical intersection, respectively. The relative energies are obtained at the CAS-MP2-(6,6)/6-311++G(df,pd)//CAS(6,6)/6-311G(d) level of theory. The geometrical parameters for the CASSCF-optimized structures for the stationary points are also shown. Hydrogen atoms are omitted for clarity. The bold arrows show the main atomic motions in the C-TS-S0 eigenvector. The derivative coupling and the gradient difference vectors – those that increase the degeneracy – are computed using CASSCF at the conical intersection, C-CI. For more information, see the text. | ||
However, the CAS-MP2 results demonstrate that much higher energy (117 kcal mol−1) of C-FC-S1 than that of the irradiating light (420 nm = 68 kcal mol−1) makes this photo-rearrangement reaction energetically unfeasible,15 so the energy of photo-irradiation is insufficient to generate the butterfly-like product, C-Pro-S0 via radiation-less decay (i.e., the CI channel). Consequently, no photo-isomerization reaction should be observed in these cyclobutene systems for irradiation with visible light. However, the thermal reversion (C-Pro-S0 → C-Rea-S0) is predicted to be easily achieved, since the energy that is required to surmount the reverse barrier (C-Pro-S0 → C-TS-S0) is calculated to be 10 kcal mol−1 less than that which is required to surmount the forward barrier (C-Rea-S0 → C-TS-S0), using the CAS-MP2 calculations. To the best of the author's knowledge, no experimental reports of the photochemical reactions of cyclobutenes have been published so the CAS-MP2 computational results that are shown in Fig. 3 remain a prediction.
V. Conclusion
The photochemical mechanisms for the isomerization reactions for Si-Rea-S0 and C-Rea-S0, which have a skeleton that has one four-membered-ring and one double bond, are studied using the CASSCF and CAS-MP2 levels of theory. This is the first theoretical study of the reaction trajectory and the first theoretical estimation of the activation energy and the reaction enthalpy for both the singlet excited state and the singlet ground state. This study interprets the photochemical behavior of these four-membered-ring species that contain group 14 elements using a simple conical intersection mechanism. The available experimental findings for cyclotetrasilene are verified4 and the photochemical isomerization reactions for analogous cyclobutene systems are predicted.Acknowledgements
The author is grateful to the National Center for High-Performance Computing of Taiwan for generous amounts of computing time, and the Ministry of Science and Technology of Taiwan for the financial support. In particular, the author also wishes to thank Professor Michael A. Robb, Dr S. Wilsey, Dr Michael J. Bearpark, (University of London, UK) and Professor Massimo Olivucci (Universita degli Studi di Siena, Italy), for their encouragement and support during his stay in London. Special thanks are also due to reviewers 1 and 2 for very help suggestions and comments.References
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343 CAS.
- For reviews on disilenes, see: (a) R. West, Pure Appl. Chem., 1984, 56, 163 CrossRef CAS; (b) G. Raabe and J. Michl, Chem. Rev., 1985, 85, 419 CrossRef CAS; (c) A. H. Cowley and N. C. Norman, Prog. Inorg. Chem., 1986, 34, 1 CrossRef CAS; (d) R. West, Angew. Chem., Int. Ed. Engl., 1987, 26, 1201 CrossRef; (e) G. Raabe and J. Michl, in The Chemistry of Organic Silicon Compound, ed. S. Patai and Z. Rappoport, Wiley, New York, 1989, part 2, ch. 17 Search PubMed; (f) T. Tsumuraya, S. A. Batcheller and S. Masamune, Angew. Chem., Int. Ed. Engl., 1991, 30, 902 CrossRef; (g) R. S. Grev, Adv. Organomet. Chem., 1991, 33, 125 CrossRef CAS; (h) M. Weidenbruch, Coord. Chem. Rev., 1994, 130, 275 CrossRef CAS; (i) R. Okazaki and R. West, Adv. Organomet. Chem., 1996, 39, 231 CrossRef CAS; (j) T. Iwamoto, H. Sakurai and M. Kira, Bull. Chem. Soc. Jpn., 1998, 71, 2741 CrossRef CAS; (k) P. P. Power, J. Chem. Soc., Dalton Trans., 1998, 2939 RSC; (l) P. P. Power, Chem. Rev., 1999, 99, 3463 CrossRef CAS PubMed; (m) M. Kira and T. Iwamoto, J. Organomet. Chem., 2000, 610, 236 CrossRef; (n) P. P. Gaspar and R. West, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiely, New York, 2nd edn, 1999, part 3, pp. 2463–2568 Search PubMed; (o) M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704 CrossRef CAS PubMed; (p) B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 607–608, 209 CrossRef; (q) N. J. Hill and R. West, J. Organomet. Chem., 2004, 689, 4165 CrossRef CAS; (r) M. Kira, J. Organomet. Chem., 2004, 689, 4475 CrossRef CAS; (s) O. Kuhl, Coord. Chem. Rev., 2004, 248, 411 CrossRef CAS; (t) R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schutz, Angew. Chem., Int. Ed., 2004, 43, 5896 CrossRef CAS PubMed; (u) B. Gehrhus, P. B. Hitchcock, R. Pongtavornpinyo and L. Zhang, J. Chem. Soc., Dalton Trans., 2006, 15, 1847 RSC.
- (a) M. J. Michalczyk, R. West and J. Michl, Organometallics, 1985, 4, 826 CrossRef CAS; (b) B. D. Shephered, D. R. Powell and R. West, Organometallics, 1989, 8, 2664 CrossRef.
- (a) M. Kira, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 1996, 118, 10303 CrossRef CAS; (b) T. Iwamoto, M. Tamura, C. Kabuto and M. Kira, Organometallics, 2003, 22, 2342 CrossRef CAS; (c) T. Iwamoto and M. Kira, Chem. Lett., 1998, 277 CrossRef CAS.
- For instance, see: F. Bernardi, M. Olivucci and M. A. Robb, J. Photochem. Photobiol., A, 1997, 105, 365 CrossRef CAS.
- J. J. W. McDouall, K. Peasley and M. A. Robb, Chem. Phys. Lett., 1988, 148, 183 CrossRef CAS.
- For the sake of simplicity, the SiH3 substituent groups, instead of t-Bu2MeSi groups, have been used in the present study. Of course, if one uses the above bulky group in the present theoretical study, the relative energies of CI and TS may change. Nevertheless, the basic concepts in these studies are still the same.
- For reviews, see: (a) F. Bernardi, M. Olivucci and M. A. Robb, Isr. J. Chem., 1993, 265 CrossRef CAS; (b) M. Klessinger, Angew. Chem., Int. Ed. Engl., 1995, 34, 549 CrossRef CAS; (c) F. Bernardi, M. Olivucci and M. A. Robb, Chem. Soc. Rev., 1996, 321 RSC; (d) M. Klessinger, Pure Appl. Chem., 1997, 69, 773 CrossRef CAS; (e) M. Klessinger and J. Michl, in Excited States and Photochemistry of Organic Molecules, VCH Publishers, New York, 1995 Search PubMed.
- M. J. Bearpark, M. A. Robb and H. B. Schlegel, Chem. Phys. Lett., 1994, 223, 269 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian, Inc., Wallingford CT, 2013.
- Recently, Matsuo, Tamao, and co-workers experimentally reported an interestingly substituted disilene molecule, (Z)-1,2-di(1-prenyl)disilene, whose first excited state is theoretically estimated to be singlet. See: M. Kobayashi, N. Hayakawa, T. Matsuo, B. Li, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2016, 138, 758 CrossRef CAS PubMed.
- For discussion of excited singlet and triplet states of disilene, for instance, see a comprehensive study: G. A. Oyedepo and A. K. Wilson, J. Phys. Chem. A, 2010, 114, 8806 CrossRef CAS PubMed.
- The MO representation of silylene at the singlet excited state can be found in a recent work. See: M.-D. Su, Chem.–Eur. J., 2014, 20, 9419 CrossRef CAS PubMed.
- The fixed geometrical parameters in (A) Si-Rea and (B) C-Rea are given in ESI.†.
- The reason that the Franck–Condon energy for Si-Rea-S0 (67.8 kcal mol−1) is much less than that for C-Rea-S0 (116.8 kcal mol−1), as already shown in Fig. 2 and 3 is easily understood with reference to the simple molecular systems, i.e., ethylene (H2CCH2) and silene (H2SiSiH2). The energy gap for the HOMO (π) and the LUMO (π*) for ethylene is much larger than that for silene. Silene also has a higher HOMO (π) and a lower LUMO (π*) than ethylene. For details, see ref. 2.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00506g |
| This journal is © The Royal Society of Chemistry 2017 |