
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromotional effect of Ti doping on the ketonization of acetic acid over a CeO2 catalyst†
Feipeng Lua,
BinBo Jiang *a,
Jingdai Wanga,
Zhengliang Huanga,
Zuwei Liaoa,
Yongrong Yanga and
Jie Zheng*b
*a,
Jingdai Wanga,
Zhengliang Huanga,
Zuwei Liaoa,
Yongrong Yanga and
Jie Zheng*b
aState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: jiangbb@zju.edu.cn
bDepartment of Chemical and Biomolecular Engineering, The University of Akron, Akron, Ohio, USA 44325
First published on 19th April 2017
Abstract
A series of Ce1−xTixO2−δ mixed oxide catalysts were synthesized using a homogeneous precipitation method and characterized, and then these catalysts were applied to convert acetic acid to acetone using a gas-phase ketonization reaction. Ti-doped Ce1−xTixO2−δ catalysts (x = 0.1–0.5) exhibited much better ketonization performance than their parent catalysts of CeO2 and TiO2, and such Ti-doping-induced catalysis improvement is attributed to the formation of a Ce–O–Ti structure depending on the Ti content. Among the different Ce1−xTixO2−δ catalysts, the Ce0.7Ti0.3O2−δ catalyst calcinated at 500 °C showed the best catalysis activity and high stability. A combination of techniques (i.e. TEM, FTIR, H2-TPR, NH3/CO2-TPD and XPS) further revealed that the formation of the Ce–O–Ti structure modified the surface acid–base properties and thus enhanced the redox properties. Moreover, the introduction of Ti into CeO2 also increased the number of oxygen vacancies on the catalysts’ surface that favored the ketonization of acid molecules. This work provides valuable insights into the design of highly efficient CeO2-based catalysts for acid removal in the upgrading process of bio-oil.
1 Introduction
As an alternative energy source, biomass-derived fuels have attracted much attention because of abundant sources, cheap price, and environmentally benign properties. Different synthesis routes have been developed for converting biomass into liquid fuels. For example, the fast pyrolysis method usually converts the whole biomass into a liquid bio-oil typically containing carboxylic acids, alcohols, aldehydes and other oxygenated compounds. However, when further upgrading the derived bio-oil into high-grade fuels (e.g., gasoline, diesel, and jet fuels), it is crucial to reduce the substantial oxygen content and generate C–C bonds, coupling the light oxygenates to increase the molecular weight of the products, thus improving the total energy density of the bio-fuels. Deng et al.1 proposed an efficient catalytic method to upgrade acidic components of bio-oil into non-corrosive compounds using the catalytic ketonization reaction. Ketonization, as a reaction that converts two carboxylic acid molecules into a ketone, carbon dioxide, and water (eqn (1)), was considered as a green, non-polluting, and economical method to obtain ketones. The ketonization reaction can efficiently reduce small-molecule carboxylic acids in bio-oil and yield abundant ketones with different increased lengths of carbons. The obtained ketones can be further upgraded into liquid fuels by the aldol condensation and hydrodeoxygenation process.| 2RCOOH → R2CO + CO2 + H2O | (1) |
Significant efforts have been made to develop different materials as catalysts for ketonization. Among them, metal oxides are considered as the most suitable and efficient ones due to their acid–base and redox properties. Based on different catalysis mechanisms, these metal oxides can be classified into two categories.2 The first category includes alkali and alkaline earth oxides with low lattice energies, which can catalyze the decarboxylation by decomposition of the corresponding carboxylate salts. These oxides are known as bulk ketonization catalysts. The second category includes CeO2, MnO2, TiO2, ZrO2, and others with higher lattice energies. These oxides can form surface intermediates including β-ketoacids, ketene etc., and are thus defined as surface ketonization catalysts. Moreover, compared with pure acidic or basic oxides, the amphoteric oxides, especially ceria-based catalysts, usually possess excellent ketonization activity and selectivity. Different ceria-based catalysts such as CeO2–Fe2O3,3,4 CeO2–Mn2O3,3,5,6 and CeO2–ZrO2 (ref. 5, 7–12) have been developed for ketonization of carboxylic acids, and the activity of these ketonization reactions was largely improved by the modified CeO2 oxides. Since CeZrOx catalysts exhibit outstanding reducibility and oxygen storage capacity, they are often used as preferable catalysts for the ketonization of carbohydrate-derived carboxylic acids and help to improve the catalytic activity of the ketonization reaction compared with other catalysts. It is generally believed that the excellent performance of CeZrOx catalysts is attributed to the Lewis acid–base site pairs and the enhanced reducibility.10 However, the structure–property–activity relationship of CeO2-based catalysts between ketonization activity, surface acid–base and redox characteristics, and structure of active composites still remains unclear, which hinders the development of applicable catalysts for the ketonization reaction.
In this work, we synthesized and characterized a series of Ce–Ti composite oxides using a homogeneous precipitation method, and then used them to catalyze the ketonization of acetic acid in the gas phase. Ce–Ti mixed oxides have been mainly used as catalysts for selective catalytic reduction (SCR) of NOx, while TiO2-anatase is the most commonly used supporting material for SCR catalysts that offers excellent dispersion of active components.13–15 The Ti4+ cations regarded as same-valence dopants to CeO2 could actually affect the properties of the host oxide, especially the redox properties.16 Furthermore, the modification of acid–base sites can also be obtained for Ti-doped CeO2, which is caused by the introduction of both Ti4+ (Lewis acid) and O2− (Lewis base) ions.17 However, to our knowledge, Ce–Ti composite oxides have not been reported as solid acid–base catalysts for acid ketonization to date. Herein, the ketonization process on Ce–Ti mixed oxides was examined to illustrate the relationship between surface structures and properties of Ce–Ti composite oxides and their catalytic activities. In addition, synergistic effects of the catalysts’ acid–base sites and reducibility on the catalytic activity of Ce–Ti mixed oxides were also studied using NH3/CO2-TPD and H2-TPR.
2 Material and methods
2.1 Catalyst preparation
A homogeneous precipitation method was adopted to prepare the Ce1−xTixO2−δ mixed oxide catalysts, where x means the molar ratio of Ti. In a typical procedure, appropriate amounts of Ti (SO4)2 and Ce (NO3)3·6H2O were dissolved in deionized water under vigorous stirring conditions until the formation of a clear solution. Excess urea solution was then added into the mixed solution, with a urea/(Ce + Ti) molar ratio of 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The solution was heated to 90 °C and held for 5 h with continuous stirring. The obtained yellow precipitate was decanted, filtered off, and washed with distilled water multiple times to remove the weakly adhered ions on the sample surface, followed by oven drying at 120 °C for 12 h. Subsequently the oven-dried sample was calcined at a certain temperature (400, 500, 650, 800 °C) for 5 h in air at a heating rate of 5 °C min−1. Pure CeO2 and TiO2 were also prepared for comparison purposes by adopting the same method.
:1. The solution was heated to 90 °C and held for 5 h with continuous stirring. The obtained yellow precipitate was decanted, filtered off, and washed with distilled water multiple times to remove the weakly adhered ions on the sample surface, followed by oven drying at 120 °C for 12 h. Subsequently the oven-dried sample was calcined at a certain temperature (400, 500, 650, 800 °C) for 5 h in air at a heating rate of 5 °C min−1. Pure CeO2 and TiO2 were also prepared for comparison purposes by adopting the same method.
2.2 Catalyst characterization
The powder X-ray diffraction (XRD) patterns of the samples were recorded on a Philips PW3050/60 vertical goniometer using Ni-filtered Cu Kα1 radiation (λ = 1.5406 Å). A proportional counter and a 0.02° step size in the 2θ range from 20 to 80° were used. The assignment of the various crystalline phases is based on the JPDS powder diffraction file cards. The crystal size of the prepared oxide was calculated using the Scherrer equation:
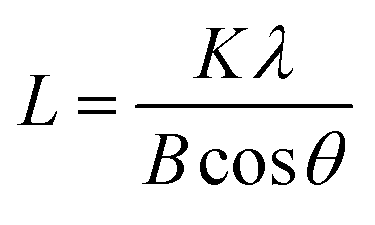 | (2) |
Raman spectra were recorded on a Thermo Scientific DXR Raman microscope with a He–Cd laser of excitation wavelength 532 nm.
The BET surface area, pore volume, and pore size distribution measurements were performed using a Quantachrome Autosorb-iQ instrument. Prior to analysis, the samples were degassed at 300 °C for 3 h to remove any residual moisture. The BET surface areas were calculated from adsorption data in the relative pressure (P/P0) range of 0.04–0.25. Pore size and pore volume were calculated using the BJH method applied to the desorption leg of the isotherms.
Temperature programmed reductions (TPR) were recorded using a thermal conductivity detector (TCD) and a reducing gas of 10% H2 in Ar set at a flow rate of 40 mL min−1. The analysis regime was as follows. Approximately 50 mg samples were pre-treated at 450 °C for 60 min, the sample cell was purged with a H2/Ar mixture for 30 min and the analysis run from 50 °C to 800 °C at a heating rate of 10 °C min−1. Water produced by the reduction was trapped in an ice bath upstream of the TCD. H2 consumption was quantified by comparison to the reduction of CuO.
X-ray photoelectron spectroscopy (XPS) measurements were performed using an Escalab 250Xi with a monochromatized Al K source (1486 eV). Narrow spectra were collected with a pass energy of 30 eV. The charge correction was made considering that the C 1s signal of contaminating carbon (C–C or C–H bonds) was centered at 284.8 eV. The C 1s, Ti 3d, Ce 3d, and O 1s levels were chosen for characterization because their signals are the most intense and do not overlap.
A Fourier transform IR spectrometer (NICOLET 5700, USA) was used to acquire the IR spectra of the samples. For the transmission FTIR, each disc was mixed with 1 wt% sample and 99 wt% KBr.
The nature and concentration of acid–base sites were studied by means of TPD of pre-adsorbed NH3/CO2 and the desorbed NH3/CO2 on a Quantachrome Instrument. Samples of approximately 100 mg were outgassed and dried at 450 °C for 30 min under Ar flow (40 mL min−1, purity >99.999% wt%), cooled to room temperature, and then treated with a flow of 10 vol% NH3/He or 10% vol% CO2/Ar (40 mL min−1) for 120 min. The sample cell was then evacuated for 20 min and a He flow at 40 mL min−1 was started. This continued for 15 min prior to the start of data collection. The sample was heated from room temperature to 500 °C at 10 °C min−1. NH3/CO2 was quantified by comparing the areas under the curve with the sample of known amounts of NH3/CO2 using thermal conductivity.
2.3 Catalyst tests
The gas-phase ketonization of carboxylic acids was carried out in a down-flow quartz fixed-bed reactor (9 mm i.d.) under atmospheric pressure. The temperature of heating furnace externally was measured using a K-type thermocouple attached to the reactor and controlled with a 708P temperature controller (Yudian Automation Technology Co., Ltd). A HPLC pump was used to introduce the liquid feed solution into the reactor. The ketonization reaction was operated in the temperature range of 250 °C to 350 °C. Typically, 0.5 g of the as-prepared catalyst (40–60 mesh) was placed in the middle region of the reactor between two plugs of quartz wool and fused SiO2 granules and the reaction was carried out at a liquid acetic acid flow rate of 3 mL h−1 with an N2 flow rate of 30 mL min−1. For the co-feed of CO2, the total molar flow of the gas feed through the reactor was kept constant by modifying the molar ratio of CO2 and N2. For the H2O co-feeding experiment, the weight hourly space velocity (WHSV) of acetic acid was kept the same by adjusting the pump rate of H2O and the N2 flow rate. The mass transfer limitation was excluded during the tests.The feed and gas-phase reaction mixtures were analyzed with online GC analysis equipped with two columns in parallel and two detectors to obtain reactant conversion and product selectivity. The outlet line was heated to 150 °C along with the injection valve of the gas chromatograph to avoid product condensation. For organic compounds, a FFAP capillary column (30 m × 0.32 mm × 0.5 μm) was connected to a flame ionization detector, while for gas products, a TDX-01 column was connected to a TCD detector. Prior to the catalytic test, each sample was pretreated under a nitrogen flow at the lowest reaction temperature in the stream of reactant for 2 h. The selectivity was defined as the percentage of carboxylic acid converted into a particular product taking into account the reaction stoichiometry; thus, 100% ketone selectivity would mean 1 mol of carboxylic acid converted to form 0.5 mole of the ketone. The mean absolute percentage error in conversion and selectivity was ≤10% and the carbon balance was maintained in the range of 100 ± 5%.
In the absence of catalyst, no conversion of acetic acid was observed. However, when SiO2 granules were used, a background conversion of 2–3% was detected for our typical reaction condition.
3 Results and discussion
3.1 Ce1−xTixO2−δ catalyst performance
The catalytic ketonization of acetic acid with the Ce1−xTixO2−δ catalysts was obtained at 280–350 °C. As shown in Fig. 1A, the introduction of Ti atoms into CeO2 can promote the ketonization of acetic acid. The catalytic activity increased as the added amount of TiO2 into CeO2 was increased from x = 0.1 to 0.3, with Ce0.7Ti0.3O2−δ catalyst reaching the highest conversion of 100%. But, as the amount of TiO2 was further increased to x = 0.5, the catalytic activity decreased. Pure TiO2 showed a poor catalytic reaction of acetic acid ketonization compared with CeO2 when the reaction temperature was below 350 °C. As one of the ketonization products, the CO2 formation rates at different ratios of Ce/Ti are shown in Fig. S1.† Ce0.7Ti0.3O2−δ catalyst displayed the highest CO2 formation rate (0.12 mol h−1), consistent with acid conversion data (Fig. 1A). In addition, other side-products (e.g. acetic anhydride, CH4, etc.) produced at 350 °C had very low selectivity of <2%. The performance of the Ce0.7Ti0.3O2−δ catalyst was also compared with the results of other active metal oxides in the representative literature, and the results are shown in Table S1.† It can be seen that Ce0.7Ti0.3O2−δ catalyst is superior to those catalysts reported previously under similar reaction conditions.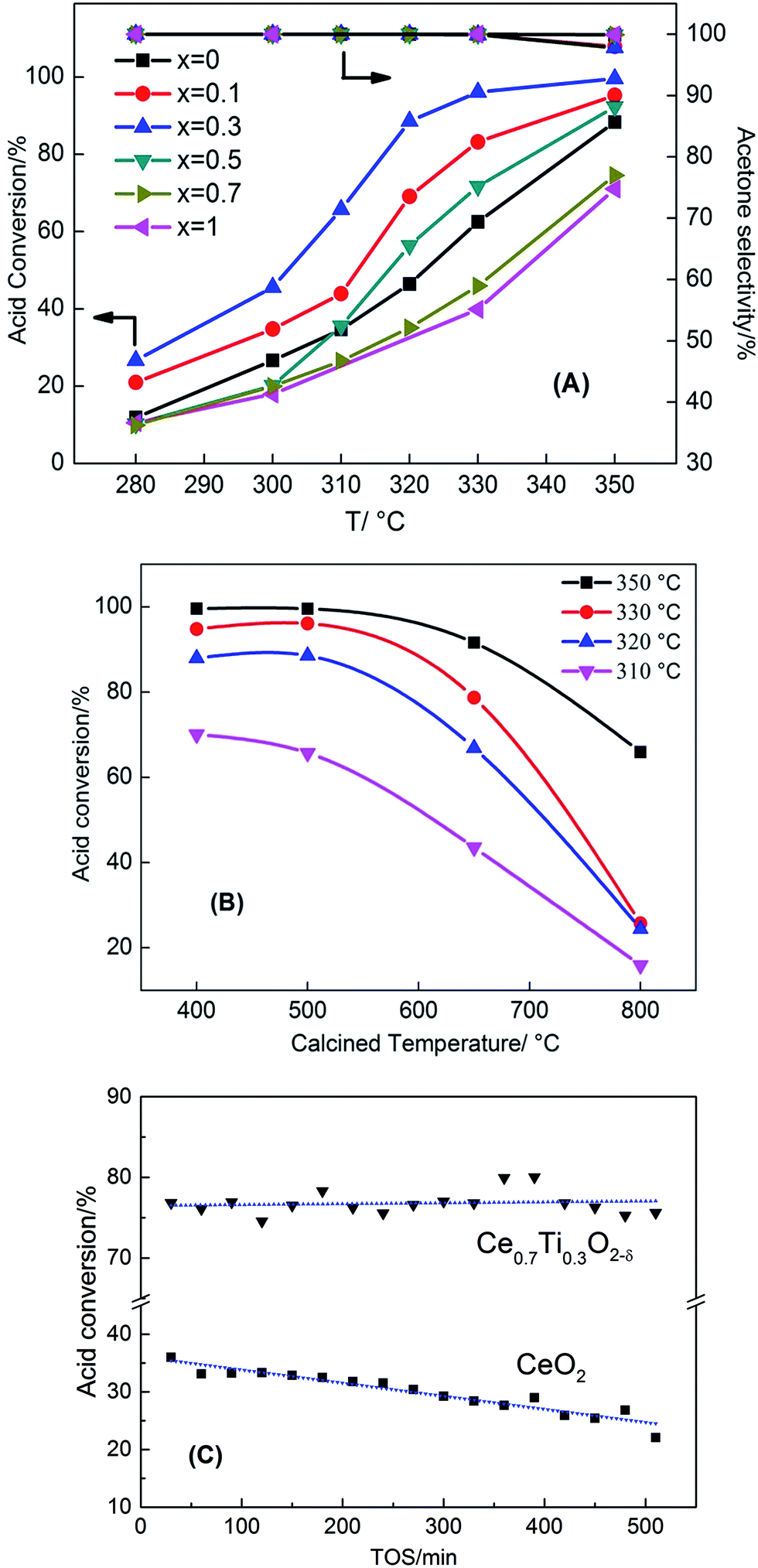 | ||
| Fig. 1 Acetic acid conversion as a function of temperature for (A) Ce1−xTixO2−δ catalysts and (B) Ce0.7Ti0.3O2−δ catalysts calcined at different temperatures, (C) acetic acid conversion of Ce0.7Ti0.3O2−δ and pure CeO2 at 315 °C as a function of time on stream (reaction condition: 0.5 g catalyst, 30 mL min−1 N2 flow rate, 3 mL h−1 liquid flow rate). | ||
Fig. 1B shows the influence of calcination temperature on the catalytic performance of the ketonization reaction. The Ce0.7Ti0.3O2−δ catalyst was selected for the ketonization reaction because of its excellent activity. It can be seen that Ce0.7Ti0.3O2−δ catalysts calcined at 400 °C and 500 °C displayed almost the same ketonization activity. At a calcined temperature of 650 °C, the conversion of acetic acid reacted at 350 °C decreased from 100% to 91%. When the calcined temperature further increased to 800 °C, the reaction activity dropped drastically, leading to 66% conversion at 350 °C. These results indicate that calcination temperature has a significant impact on acid conversion and ketone yield. Furthermore, regarding the stability of Ce0.7Ti0.3O2−δ catalyst, Fig. 1C shows that the Ti-doped catalyst exhibited very high stability to retain a higher acid conversion of 75% for 500 min. In sharp contrast, CeO2 deactivated rapidly from 36% to 22% within 500 min.
3.2 Catalyst characterization
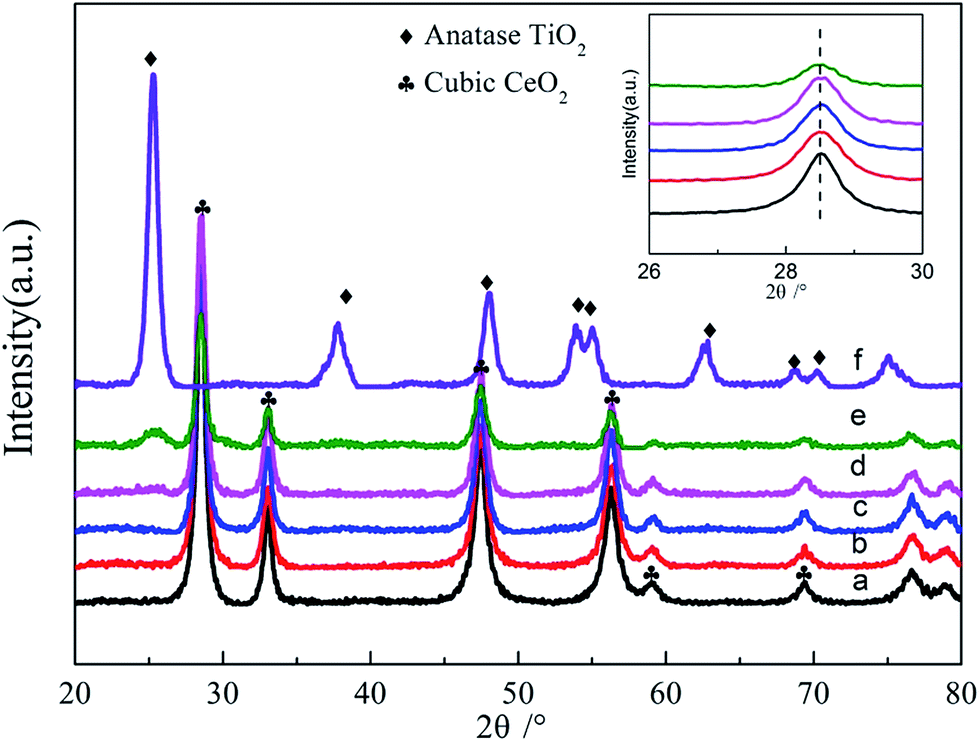 | ||
| Fig. 2 XRD patterns of as-prepared Ce1−xTixO2−δ catalysts: (a) x = 0, (b) x = 0.1, (c) x = 0.3, (d) x = 0.5, (e) x = 0.7, (f) x = 1. The inset shows the shifting of the CeO2 (111) peak to higher energies with increasing amount of doping Ti. | ||
| Samples | SBET (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | Crystallite structure | Lattice parametera (nm) | Mean crystallite size (nm) |
|---|---|---|---|---|---|---|
| a Estimated by the formulate a = (h2 + k2 + l2)1/2 × (λ/2sinθ) using the {111} lattice face.b Determined by the main peak of CeO2 cubic phase.c Determined by the main peak of TiO2 anatase phase. |
||||||
| CeO2 | 117 | 0.129 | 3.71 | Cubic | 0.543 | 12.2b |
| Ce0.9Ti0.1O2−δ | 81 | 0.144 | 3.72 | Cubic | 0.542 | 11.1b |
| Ce0.7Ti0.3O2−δ | 86 | 0.284 | 5.05 | Cubic | 0.541 | 12.4b |
| Ce0.5Ti0.5O2−δ | 94 | 0.204 | 4.74 | Cubic | 0.539 | 13.1b |
| Ce0.3Ti0.7O2−δ | 99 | 0.197 | 5.06 | Anatase + cubic | 0.538 | 5.1b |
| TiO2 | 90 | 0.149 | 4.47 | Anatase | — | 10.3c |
Fig. S2† shows the N2 adsorption–desorption isotherms and corresponding pore size distributions of Ce1−xTixO2−δ mixed oxides, in comparison with those of pure CeO2 and TiO2. All isotherms displayed a type IV shape with a H3 hysteresis loop at high relative pressures of 0.4–1, suggesting the presence of slit-like mesopores, associated with aggregates of plate-like particles. Table 1 summarizes the physical properties, crystalline structure, and crystallite size obtained from N2 adsorption–desorption isotherms and XRD data. With the increasing amount of Ti being doped into CeO2, the surface area increased gradually from 81.5 m2 g−1 to 93.7 m2 g−1 and the crystallite size increased from 11.1 nm to 13.1 nm, respectively. From the calculated crystallite sizes of Ce1−xTixO2 catalysts in Table 1, Ce0.3Ti0.7O2−δ showed a relatively smaller crystallite size than pure CeO2, because of the appearance of the TiO2 anatase phase in Ce0.3Ti0.7O2−δ, which restrained the growth of CeO2 crystal particles. The HRTEM micrographs and SAED images of Ce0.7Ti0.3O2−δ sample are displayed in Fig. S3.† The TEM images show that the Ce0.7Ti0.3O2−δ sample is composed of nanosized particles with an average particle size of ∼11 nm, coinciding with the results obtained from XRD patterns. The continuous rings in the SAED patterns (shown as the inset in Fig. S3†) confirm the high crystallinity and the fluorite structure of Ce0.7Ti0.3O2−δ sample.
Fig. 3 shows Raman spectra of various Ce1−xTixO2−δ catalysts. Pure CeO2 exhibited a prominent band at ca. 461 cm−1 corresponding to the F2g Raman active mode in metal dioxides with a fluorite-like structure.18 Three Raman bands at 391, 515, and 638 cm−1 were identified for pure anatase TiO2. The former two bands were assigned to the B1g mode, while the latter band was assigned to the E2g mode.19 Addition of Ti led to a gradual attenuation of the F2g band intensity, and this is likely caused by the decrease of Ce amount and different crystallite sizes of CeO2. Ce1−xTixO2−δ samples at x < 0.5 retained their sole F2g mode of CeO2 with the increasing amount of doped Ti, and this further confirms that the Ce–O–Ti linkage bonds are formed for Ce1−xTixO2−δ (x < 0.5). The Ce0.5Ti0.5O2−δ catalyst showed slight Raman peaks of three modes (391, 515, and 638 cm−1) similar to anatase TiO2, while Ce0.3Ti0.7O2−δ showed distinct Raman lines characteristic of anatase TiO2 together with a large drop in F2g band intensity. Besides, the visible Raman spectroscopy employed in this work mainly supplies the bulk information of the catalysts.20,21 Hence, no related peak was observed in around 585 cm−1 (ref. 22) or 601 cm−1 (ref. 23) that were attributed to production of oxygen vacancies, indicating that the bulk oxygen vacancies might not be formed on these catalysts.
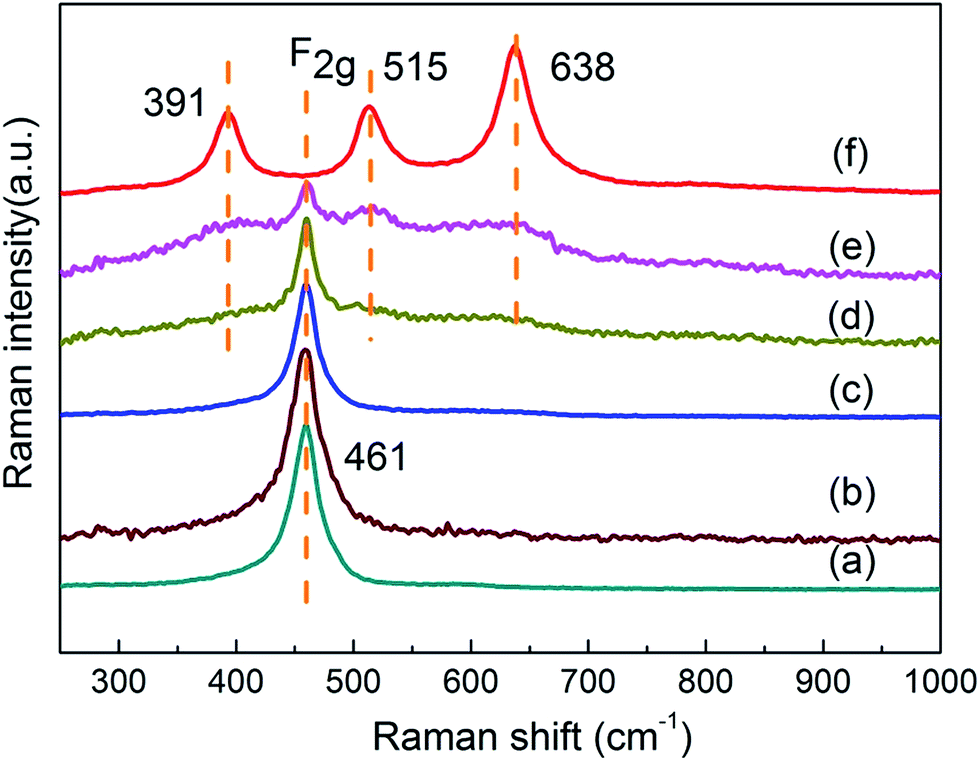 | ||
| Fig. 3 Raman spectra of various Ce1−xTixO2−δ catalysts: (a) x = 0, (b) x = 0.1, (c) x = 0.3, (d) x = 0.5, (e) x = 0.7, (f) x = 1. | ||
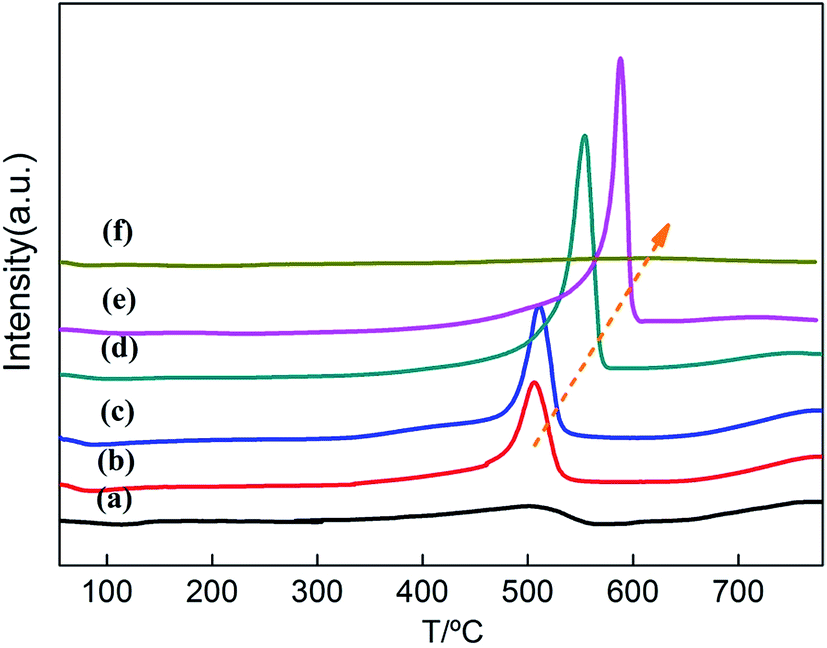 | ||
| Fig. 4 H2-TPR profiles of Ce1−xTixO2−δ catalysts at (a) x = 0, (b) x = 0.1, (c) x = 0.3, (d) x = 0.5, (e) x = 0.7, (f) x = 1. | ||
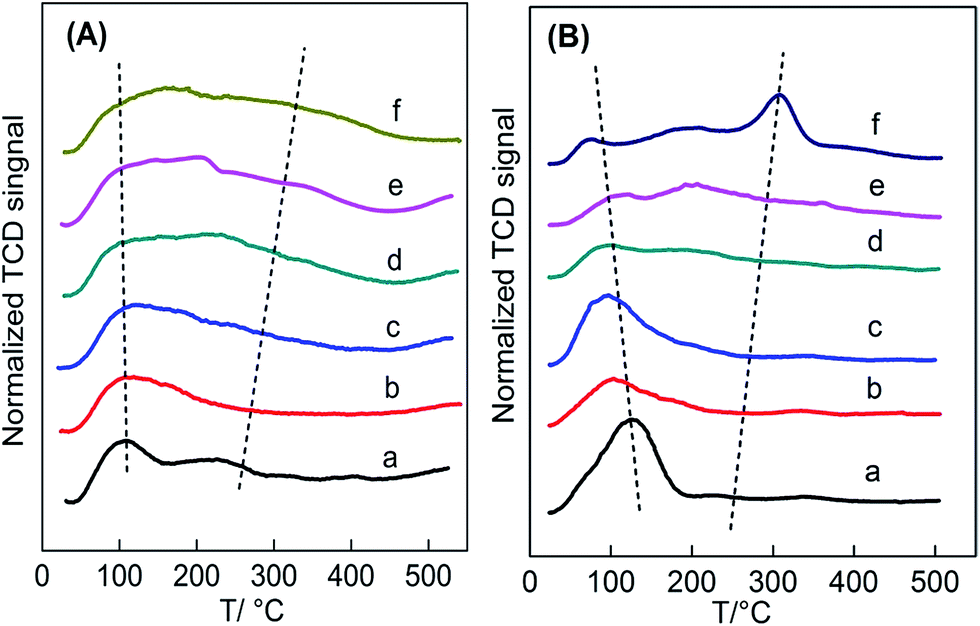 | ||
| Fig. 5 (A) NH3-TPD and (B) CO2-TPD profiles of Ce1−xTixO2−δ catalysts at (a) x = 0, (b) x = 0.1, (c) x = 0.3, (d) x = 0.5, (e) x = 0.7, (f) x = 1. | ||
Fig. 5B presents the CO2-TPD profiles of the Ce1−xTixO2−δ catalysts. Similar to the NH3-TPD profiles, these profiles can be divided into two regions of weak adsorption sites (50–250 °C) and strong adsorption sites (250–500 °C) in a wide temperature range (50–500 °C). Since CeO2 only had weak basic sites,33 desorbed CO2 was detected at a low temperature range. Weak basic sites in CeO2 lead to the formation of surface –OH groups because they act as both acidic and basic sites. Differently, the presence of strong basic sites in TiO2 caused CO2 to be desorbed at a high temperature range. In fact, there are two types of surface –OH groups due to various structures.35 CeO2 hydroxyl groups are mainly nucleophilic groups that always serve as reactive basic (O2−) sites, while TiO2 hydroxyl groups are essentially H-bond donors. Thus, the two oxide catalysts expose variously structured surface-OHs, which may cause the discrepancy of CO2 adsorption on the oxide surface. It can also be seen that the low-temperature desorbed peak moved towards lower temperatures with increasing addition of Ti, which could be attributed to the gradual formation of Ce–O–Ti linkages instead of Ce–O–Ce in CeO2.
The changes in the distribution and strength of surface acid–base sites versus Ti content were summarized in Table S3† and shown in Fig. 6a–d. The total surface density of Ce–Ti composite oxides gradually increased with amount of Ti, while the density of strong acid sites on the surface increased almost linearly with Ti (Fig. 6c), further confirming that the addition of Ti can greatly enhance the strength of strong acids. In addition, the Ce0.7Ti0.3O2−δ sample achieved a maximal value for the density of weak acid sites, while the Ce0.5Ti0.5O2−δ sample enabled the transformation of weak acid sites to strong acid sites. This could be attributed to NH3 adsorption on the strong Lewis acids provided by sufficiently doped Ti4+.32,35 Due to the co-existence of both acid–base pairs in TiO2, the density of strong basic sites increased linearly with the added amount of Ti (Fig. 6d). On the other hand, this kind of strong basic site was not greatly enhanced by adding TiO2 until x ≥ 0.5. In other words, CeO2-rich samples, which showed a typical CeO2 cubic phase in the XRD patterns, contained more weak acid–base sites compared with others. In the case of Ce0.5Ti0.5O2−δ, desorbed CO2 was detected at high temperature at the expense of the loss of weak basic density, and the number of strong basic sites continued to increase with increasing x.
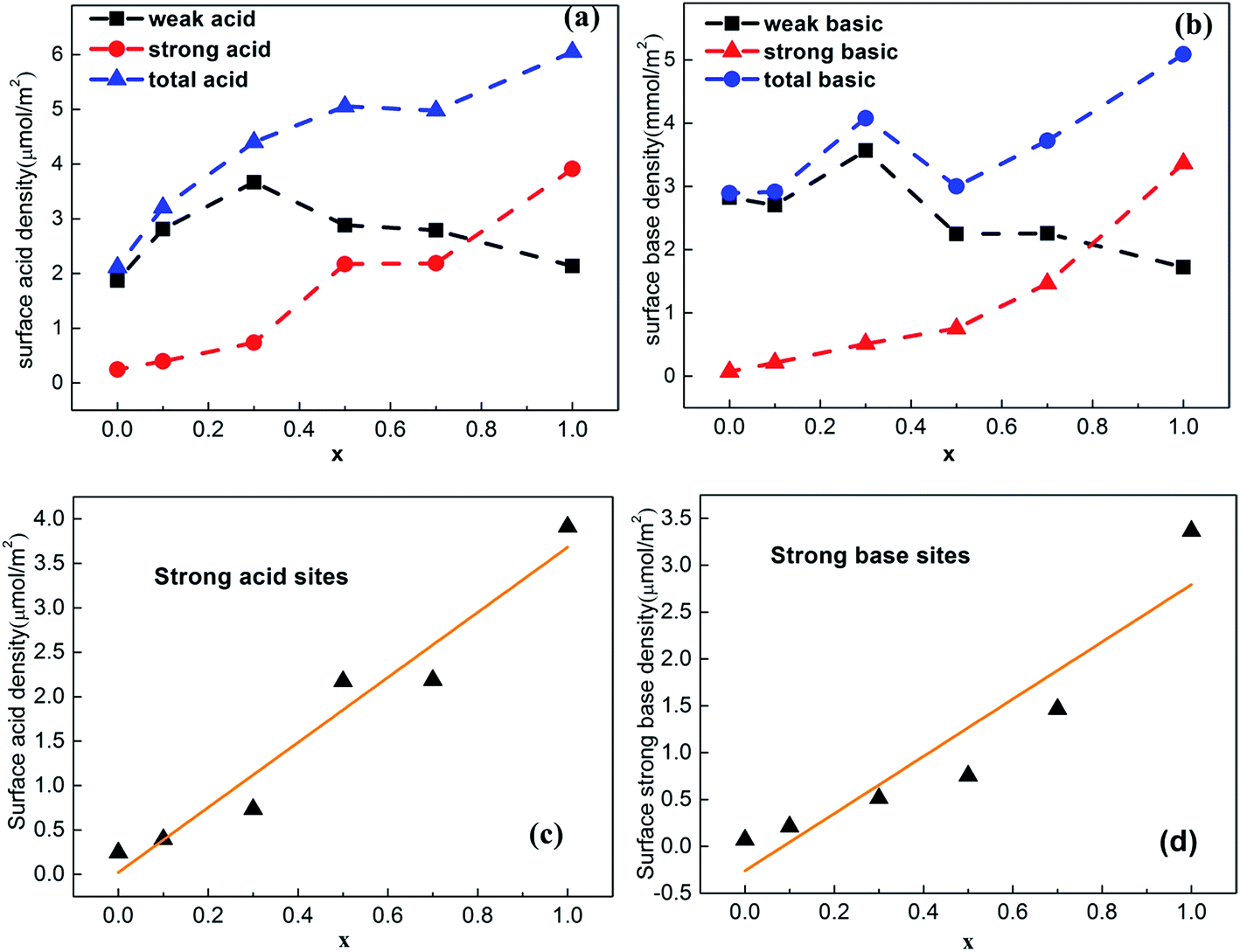 | ||
| Fig. 6 Change of (a) surface acidity and (b) surface basicity of Ce–Ti with different Ce/Ti ratio, (c) and (d) linear plots of calculated surface strong acidity and basicity, respectively. | ||
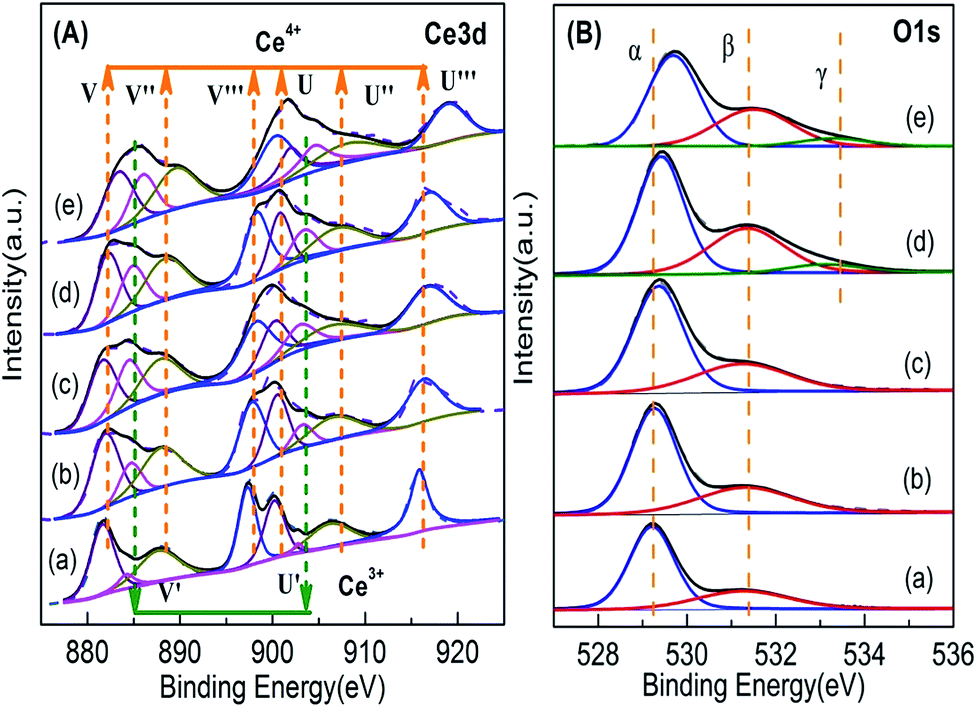 | ||
| Fig. 7 XPS results of Ce1−xTixO2−δ catalysts at (a) x = 0, (b) x = 0.1, (c) x = 0.3, (d) x = 0.5, (e) x = 0.7. | ||
| x | Surface Ce3+ ratio (%) | Surface molar fraction of O 1s | |
|---|---|---|---|
| Ce3+/(Ce4+ + Ce3+) | Oα/Ototal | Oβ/Ototal | |
| 0 | 6.5 | 0.70 | 0.30 |
| 0.1 | 11.9 | 0.69 | 0.31 |
| 0.3 | 21.1 | 0.66 | 0.34 |
| 0.5 | 17.9 | 0.57 | 0.36 |
| 0.7 | 17.3 | 0.58 | 0.32 |
Fig. 7B shows the O 1s spectra of the Ce1−xTixO2−δ samples. The peaks at 527–536 eV were decomposed into three components: (1) binding energy (BE) peak at ∼529.1 eV corresponding to lattice oxygen (Oα), (2) BE peak at ∼531.0–532.5 eV corresponding to surface absorbed oxygen and surface hydroxyl groups (Oβ), and (3) BE peak at ∼533.5 eV corresponding to –OH species (Oγ) in the Ti–O–Ti bridge bonds.15,36–38 The molar ratio of Oβ/Ototal was calculated by the area ratio of the areas under the corresponding peaks. In Table 2, the molar ratio of Oβ/Ototal of the Ce1−xTixO2−δ catalysts increased obviously with increasing doping ratio of Ti, suggesting the formation of oxygen vacancies which promotes the adsorption and activation of oxygen species. In the case of the Ce0.3Ti0.7O2−δ catalyst, it showed a significant decrease in Oβ/Ototal molar ratio. This is likely due to the partial deposition of TiO2 on the surface. It was also noted that the binding energy of O 1s for the lattice oxygen of Ce–Ti composite oxides increased with Ti concentration, suggesting that Ti and Ce indeed react with each other to form chemical bonds in composite oxides, instead of physically mixing together.
3.3 Correlation between the reducible acid–base property and catalytic performance of Ce/Ti mixed oxides
As discussed above, the macro-structure of Ce1−xTixO2−δ catalysts shows a strong dependence on the doped Ti content. When the additional amount of Ti reaches a certain degree, composite oxides possess modified acid–base sites different from either pure CeO2 or TiO2, which likely results from the surface heterogeneity by substituting Ce4+ with Ti4+ ions. The macro-structure of the Ce1−xTixO2−δ catalysts was found to correlate with both catalytic properties (redox properties and surface acid–base density) and catalytic performance (acid conversion or ketone yield) during the ketonization reaction.It can be seen that CeO2-rich Ce–Ti composite oxides (x = 0.1–0.5) show better ketonization reaction activity than the pure parent oxides. It was reported that carboxylic acids utilize both dehydration and dehydrogenation sites, indicating the involvement of both acid and base sites for ketonization.39 Therefore, pure CeO2 that has abundant weak basic sites but lacks acid sites, showed a lower ketonization activity compared with the Ce0.9Ti0.1O2, Ce0.7Ti0.3O2−δ and Ce0.5Ti0.5O2−δ samples. Furthermore, based on the ketonization mechanism with β-ketoacid as an intermediate,3,40 the formation of surface carboxylates was the first step. The α-H atom was then abstracted from one carboxylate and a corresponding anionic radical was formed, while the surface Ce4+ cations were partly reduced by picking up the redundant electrons from the abstracted α-H atoms. Thereafter, the anionic radical subsequently attacked another carboxylate to form the β-ketoacid intermediate. At the same time, the reduced Ce3+ cations were reoxidized by the hydroxyl radical desorbed from the intermediate. Finally, the β-ketoacid intermediate decarboxylated to form the ketone product. It should be noted that this surface mechanism involves consecutive reduction–oxidation cycles of the redox oxide catalyst.35,41 Therefore, the redox properties should play a critical role during the ketonization process. As a result, pure CeO2, which is less reducible than Ti-doped CeO2, will be restricted in taking part in the ketonization reaction.
In the case of Ce0.7Ti0.3O2−δ, it displays the highest ketonization activity and the highest weak acid–base density. This modified surface has proper acid–base properties due to the formation of Ce–O–Ti linkage bonds. These Ce–O–Ti species are considered to be obtained via the incorporation of Ti4+ into the lattice of CeO2 to form a CeO2 solid solution, which has been confirmed by the results of TEM, XRD, Raman and FTIR. The redox properties of Ce–O–Ti bonds are superior to those of Ce–O–Ce and Ti–O–Ti bonds, which was manifested by the fact that the H2 consumed for Ce–Ti mixed-oxides is much higher than for pure oxides. Additionally, when the Ti content is lowered to x < 0.5, the surface concentration of oxygen vacancies increases due to the formation Ce–O–Ti bonds. It is believed that the increasing amount of surface oxygen vacancies will facilitate the absorption and activation of acid molecules on the catalyst surface.42–44 The acid consumption rate is notably increased as a function of the ratio of surface Ce3+ and the Ce0.7Ti0.3O2−δ catalyst with the highest surface concentration of oxygen vacancies showed the best ketonization activity. So, it gives a direct experimental proof that the presence of oxygen vacancies or coordinatively unsaturated surface metal cations could contribute to the improvement of ketonization performance at low Ti content (x ≤ 0.3).
Ce0.5Ti0.5O2−δ shows a relatively lower ketonization activity with an obvious transformation of weak acid–base sites to strong acid–base sites and inferior redox properties, compared with CeO2-like catalysts doped with Ti (x < 0.5). This phenomenon is closely related to the change of structural properties caused by excess doped Ti. When abundant Ti is doped into CeO2 (x ≥ 0.5), Ti4+ cations will occupy interstitial sites in the fluorite lattice and/or concentrate on the oxide surface45 as confirmed by XRD, Raman, and FTIR results. The redox properties also become worse with the increased amount of doping Ti (x > 0.5), which indicates that the interaction of Ti and Ce species becomes weaker. Moreover, the Ce0.3Ti0.7O2−δ sample shows a lower ketonization activity compared with the Ce0.5Ti0.5O2−δ catalyst. This is because the worse redox ability with numerous strong acid sites for a TiO2-rich composite oxide, such as the Ce0.3Ti0.7O2−δ sample, will be unfavorable for the ketone formation.46,47
3.4 Effect of co-feeding CO2 and H2O
Since both CO2 and H2O are products from the ketonization reaction, we further examined the effect of co-feeding CO2 and H2O on the ketonization activity of the Ce0.7Ti0.3O2−δ catalyst. It can be seen in Fig. 8 that co-feeding of 50 mol% CO2 reduced the ketonization activity by 13% from that found without co-feeding. When terminating the co-feeding of CO2, the Ce0.7Ti0.3O2−δ catalyst regained its ketonization activity, reaching 78%. This result gives evidence that the Ce0.7Ti0.3O2−δ catalyst has a weak binding ability to CO2 molecules due to the lack of strong Lewis basic sites on the surface, consistent with CO2-TPD data. In parallel, co-feeding of 10 mol% H2O led to a 19% reduction in acetic acid conversion, indicating the inhibition effect of H2O on the ketonization activity. Different from the co-feeding effect of CO2, suspension of co-feeding of H2O did not recover the ketonization activity back to its initial value of 79%. It should be mentioned that the calculated thermodynamic conversions of acetic acid under our conditions are >99.8% (ref. 48) and it is much higher than the ones in our co-feeding test. Hence, the thermodynamic limitation impacts can be neglected here. So, the inhibition effect of water is mainly attributed to the hydrolyzation of surface intermediates, such as β-ketoacids. Besides, the adsorption of strong –OH groups onto the surface, which is competitive with acetic acid conversion, may also cause the decline in activity. The observation that the ketonization activity did not recover with the absence of water may be due to the modification of the surface acid–base sites, which is caused by the hydrothermal treatment to the catalysts during the reaction.5,40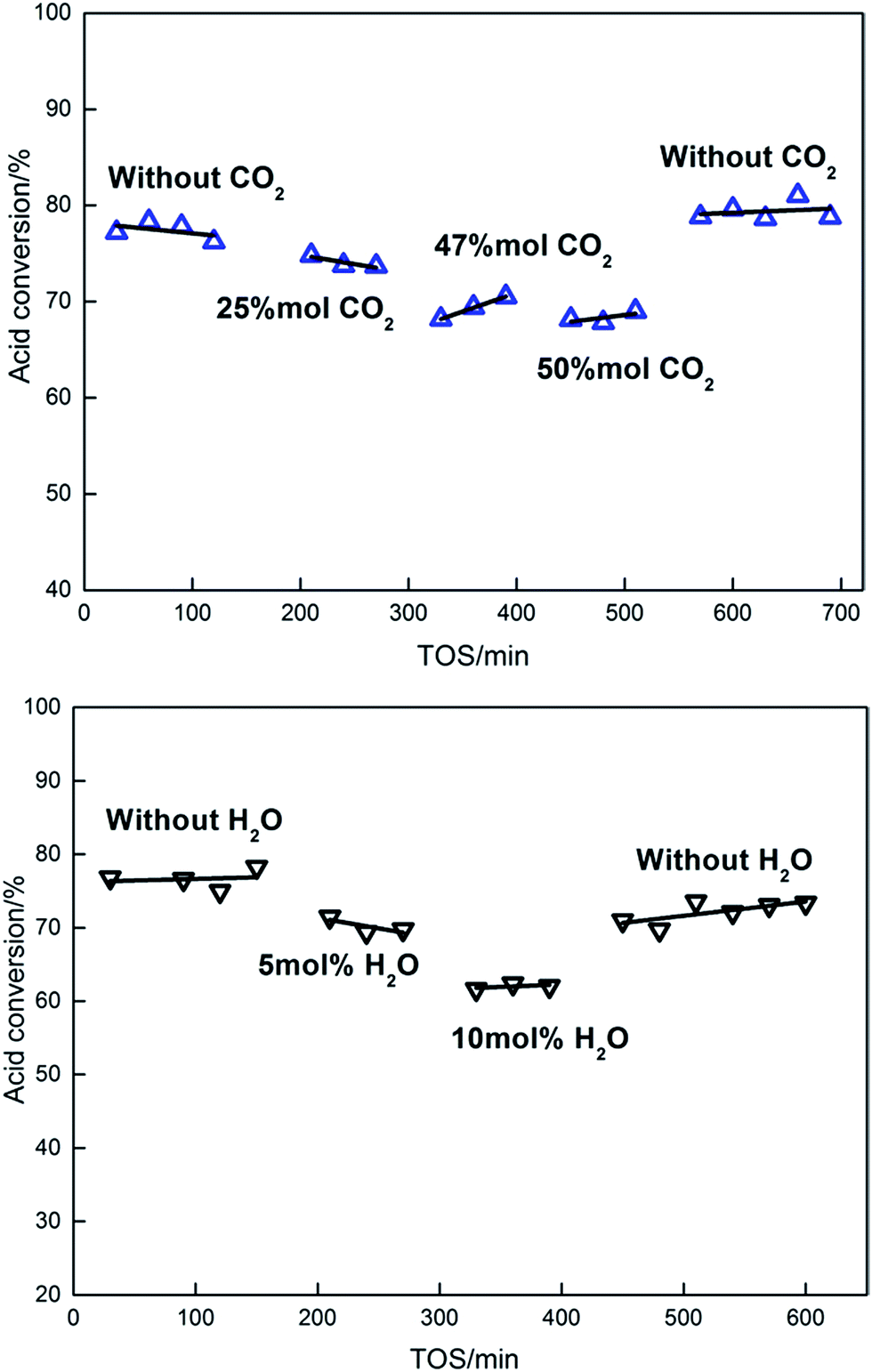 | ||
| Fig. 8 Effects of H2O and CO2 in feeding (reaction conditions: T = 315 °C, 0.5 g catalyst, 30 mL min−1 N2 flow rate, 3 mL h−1 liquid flow rate). | ||
3.5 Post-reaction catalyst characterization
Both the CeO2 and Ce0.7Ti0.3O2−δ catalyst were characterized after 500 min reaction time. The XRD patterns are shown in Fig. S5.† It can be obviously seen that, after 500 min of ketonization reaction, both the CeO2 and Ce–Ti mixed oxide catalysts could maintain the fluorite structure with a little modification of the crystallinity. Some new diffraction peaks also appeared, which can be attributed to the formation of carboxylate. The FTIR spectra of the spent catalysts are shown in Fig. S6.† It is evident from the figure that the Δasy–sym was equal to 119 and 139 cm−1 for CeO2 and Ce–Ti mixed oxides, respectively. The values imply that the surface carboxylate corresponds to a bridging or chelating coordination.49 Ce 3d XPS results (Fig. S7†) of the spent Ce–Ti catalysts also showed significant reduction of the surface cerium, which could be attributed to the presence of Ce(III) in the as-formed metal acetate. Moreover, C 1s spectra are also depicted in Fig. S7.† The C 1s peak at 284.6 eV is attributed to the carbon in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C while the C 1s peak at 288.4 eV is assigned to a carboxylate carbon.50 The O 1s peak at 531.5 eV for the spent catalyst is ascribed based on the presence of a carboxyl group (O–CO).51 Both of the changes in C 1s and O 1s spectra manifested due to the formation of surface carboxylate species. No variation of binding energies for Ti 2p was observed, suggesting that Ti species maintained its chemical state during the ketonization. It should also be noted that the as-formed metal carboxylates on the Ce–Ti catalysts can be completely decomposed thermally and restored to their original status when the spent Ce0.7Ti0.3O2−δ catalyst was then calcined at 450 °C in air for 1 h (shown in Fig. S5†).
C while the C 1s peak at 288.4 eV is assigned to a carboxylate carbon.50 The O 1s peak at 531.5 eV for the spent catalyst is ascribed based on the presence of a carboxyl group (O–CO).51 Both of the changes in C 1s and O 1s spectra manifested due to the formation of surface carboxylate species. No variation of binding energies for Ti 2p was observed, suggesting that Ti species maintained its chemical state during the ketonization. It should also be noted that the as-formed metal carboxylates on the Ce–Ti catalysts can be completely decomposed thermally and restored to their original status when the spent Ce0.7Ti0.3O2−δ catalyst was then calcined at 450 °C in air for 1 h (shown in Fig. S5†).
It has been reported that the performance of the catalytic ketonization was correlated to the efficacy of forming metal acetates versus their decomposition to release a ketone.52 The as-formed surface carboxylate could facilitate the ketonization as reaction intermediates, especially at higher temperatures. As shown in the FTIR spectra, it can be seen that more surface carboxylates were formed over the Ce–Ti mixed oxides catalyst compared with pure CeO2 under the same reaction conditions. Therefore, this phenomenon indicates that the Ti-doped CeO2 with modified acid–base sites and enhanced redox properties could facilitate the formation of surface carboxylates and show better ketonization activity. Although the surface carboxylate could play the role of surface intermediates, the worse redox ability of pure CeO2 will inhibit these surface intermediates from further decomposing to products. In this case, the carboxylate may cause the deactivation.
4 Conclusions
In this work, we prepared a series of Ce1−xTixO2−δ mixed oxide catalysts with different doped Ti contents (x = 0.1–0.9) using the homogeneous precipitation method. Then, we applied these catalysts to the gas-phase ketonization reaction to produce acetone from acetic acids, with special attention to the effects of doped Ti contents on the ketonization activity. The CeO2-rich catalysts (x = 0.1–0.5) with the structure of CeO2-like solution displayed the better redox property and ketonization activity than pure metal oxides and TiO2-rich catalysts. Among the catalysts, the Ce0.7Ti0.3O2−δ catalyst calcinated at 500 °C exhibited the highest activity and super-stability for the ketonization reaction. This superior activity is attributed to the enhanced redox properties together with the modification of surface acid–base sites, providing by the proper formation of Ce–O–Ti linkage bonds. The excess doped Ti catalysts (x > 0.5) with worse redox properties and strong acid sites are unfavorable for ketone formation. Additionally, the doping of Ti promoted the formation of surface oxygen vacancies, which is beneficial for the ketonization reaction. This work demonstrates the promising potential of Ce–Ti mixed oxides for practical bio-oil applications.Acknowledgements
The authors gratefully acknowledge the support and encouragement of the National Natural Science Foundation of China (U1663222) and the National Basic Research Program of China (2012CB720500).References
- L. Deng, Y. Fu and Q. Guo, Energy Fuels, 2009, 23, 564–568 CrossRef CAS.
- R. Pestman, R. M. Koster, A. V. Duijne, J. Pieterse and V. Ponec, J. Catal., 1997, 168, 265–272 CrossRef CAS.
- O. Nagashima, S. Sato, R. Takahashi and T. Sodesawa, J. Mol. Catal. A: Chem., 2005, 227, 231–239 CrossRef CAS.
- Y. Kamimura, S. Sato, R. Takahashi, T. Sodesawa and T. Akashi, Appl. Catal., A, 2003, 252, 399–410 CrossRef CAS.
- C. Liu, A. M. Karim, V. M. Lebarbier, D. Mei and Y. Wang, Top. Catal., 2013, 56, 1782–1789 CrossRef CAS.
- A. D. Murkute, J. E. Jackson and D. J. Miller, J. Catal., 2011, 278, 189–199 CrossRef CAS.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- C. A. Gaertner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, J. Catal., 2009, 266, 71–78 CrossRef CAS.
- S. H. Hakim, B. H. Shanks and J. A. Dumesic, Appl. Catal., B, 2013, 142, 368–376 CrossRef.
- A. Gangadharan, M. Shen, T. Sooknoi, D. E. Resasco and R. G. Mallinson, Appl. Catal., A, 2010, 385, 80–91 CrossRef CAS.
- A. A. Shutilov, M. N. Simonov, Y. A. Zaytseva, G. A. Zenkovets and I. L. Simakova, Kinet. Catal., 2013, 54, 184–192 CrossRef CAS.
- Y. A. Zaytseva, V. N. Panchenko, M. N. Simonov, A. A. Shutilov, G. A. Zenkovets, M. Renz, I. L. Simakova and V. N. Parmon, Top. Catal., 2013, 56, 846–855 CrossRef CAS.
- W. Xu, Y. Yu, C. Zhang and H. He, Catal. Commun., 2008, 9, 1453–1457 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, ChemCatChem, 2011, 3, 1286–1289 CrossRef CAS.
- J. Ding, Q. Zhong and S. Zhang, Ind. Eng. Chem. Res., 2015, 54, 2012–2022 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- G. Martra, Appl. Catal., A, 2000, 200, 275–285 CrossRef CAS.
- M. Luo, J. Chen, L. Chen, J. Lu, Z. Feng and C. Li, Chem. Mater., 2001, 13, 197–202 CrossRef CAS.
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912 CrossRef CAS.
- C. Li and M. Li, J. Raman Spectrosc., 2002, 33, 301–308 CrossRef CAS.
- C. Liu, H. Xian, Z. Jiang, L. Wang, J. Zhang, L. Zheng, Y. Tan and X. Li, Appl. Catal., B, 2015, 176–177, 542–552 CrossRef CAS.
- X. Lu, X. Li, J. Qian, N. Miao, C. Yao and Z. Chen, J. Alloys Compd., 2016, 661, 363–371 CrossRef CAS.
- T. Tsoncheva, A. Mileva, G. Issa, M. Dimitrov, D. Kovacheva, J. Henych, N. Scotti, M. Kormunda, G. Atanasova and V. Štengl, Appl. Surf. Sci., 2017, 396, 1289–1302 CrossRef CAS.
- L. Chen, B. He, S. He, T. Wang, C. Su and Y. Jin, Powder Technol., 2012, 227, 3–8 CrossRef CAS.
- C. X. Gao, Q. F. Liu and D. S. Xue, J. Mater. Sci. Lett., 2002, 21, 1781–1783 CrossRef CAS.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley Online Library, 1986 Search PubMed.
- Y. Zhang, M. Yang, X. Dou, H. He and D. Wang, Environ. Sci. Technol., 2005, 39, 7246–7253 CrossRef CAS PubMed.
- E. Aneggi, M. Boaro, C. D. Leitenburg, G. Dolcetti and A. Trovarelli, J. Alloys Compd., 2006, 408–412, 1096–1102 CrossRef CAS.
- G. Dutta, U. V. Waghmare, T. Baidya, M. S. Hegde, K. R. Priolkar and P. R. Sarode, Chem. Mater., 2006, 18, 3249–3256 CrossRef CAS.
- J. Shen, M. Tu and C. Hu, J. Solid State Chem., 1998, 137, 295–301 CrossRef CAS.
- J. I. Di Cosimo, C. R. Apesteguı, A. M. J. L. Ginés and E. Iglesia, J. Catal., 2000, 190, 261–275 CrossRef CAS.
- S. Watanabe, X. Ma and C. Song, J. Phys. Chem. C, 2009, 113, 14249–14257 CAS.
- R. W. Snell, S. H. Hakim, J. A. Dumesic and B. H. Shanks, Appl. Catal., A, 2013, 464, 288–295 CrossRef.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261–274 CrossRef CAS.
- M. A. Hasan, M. I. Zaki and L. Pasupulety, Appl. Catal., A, 2003, 243, 81–92 CrossRef CAS.
- M. Alifanti, B. Baps, N. Blangenois, J. Naud, P. Grange and B. Delmon, Chem. Mater., 2003, 15, 395–403 CrossRef CAS.
- S. M. Lee, H. H. Lee and S. C. Hong, Appl. Catal., A, 2014, 470, 189–198 CrossRef CAS.
- N. Yan, Z. Zhu, J. Zhang, Z. Zhao and Q. Liu, Mater. Res. Bull., 2012, 47, 1869–1873 CrossRef CAS.
- E. Iglesia, D. G. Barton, J. A. Biscardi, M. J. Gines and S. L. Soled, Catal. Today, 1997, 38, 339–360 CrossRef.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- T. N. Pham, D. Shi, T. Sooknoi and D. E. Resasco, J. Catal., 2012, 295, 169–178 CrossRef CAS.
- K. S. Kim and M. A. Barteau, J. Catal., 1990, 125, 353–375 CrossRef CAS.
- K. M. Dooley, A. K. Bhat, C. P. Plaisance and A. D. Roy, Appl. Catal., A, 2007, 320, 122–133 CrossRef CAS.
- S. D. Randery, J. S. Warren and K. M. Dooley, Appl. Catal., A, 2002, 226, 265–280 CrossRef CAS.
- P. L. Chen and I. W. Chen, J. Am. Ceram. Soc., 1996, 79, 1793–1800 CrossRef CAS.
- M. Renz, Eur. J. Org. Chem., 2005, 2005, 979–988 CrossRef.
- G. Mekhemer, S. A. Halawy, M. A. Mohamed and M. I. Zaki, J. Catal., 2005, 230, 109–122 CrossRef CAS.
- T. N. Pham, D. Shi and D. E. Resasco, Top. Catal., 2014, 57, 706–714 CrossRef CAS.
- R. W. Snell and B. H. Shanks, ACS Catal., 2013, 3, 783–789 CrossRef CAS.
- J. Zhang, Y. S. Choi and B. H. Shanks, ChemSusChem, 2015, 8, 4256–4265 CrossRef CAS PubMed.
- C. H. Kim, C. Joo, H. J. Chun, B. R. Yoo, D. I. Noh and Y. B. Shim, Appl. Surf. Sci., 2012, 262, 146–152 CrossRef CAS.
- R. W. Snell and B. H. Shanks, ACS Catal., 2014, 4, 512–518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00521k |
| This journal is © The Royal Society of Chemistry 2017 |