
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The unprecedented iridal lactone and adducts of spiroiridal and isoflavonoid from Belamcanda chinensis†
Gang Ni ,
Guo-Ru Shi,
Jia-Yuan Li and
De-Quan Yu*
,
Guo-Ru Shi,
Jia-Yuan Li and
De-Quan Yu*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: dqyu@imm.ac.cn; Fax: +86-10-63017757; Tel: +86-010-63165224
First published on 6th April 2017
Abstract
Five novel iridal-type triterpenoid derivatives, including three unprecedented adducts of spiroiridal and isoflavonoid, named belamcandanins A–C (1–3), one unprecedented iridal lactone, named belamcanolide A (4), and one new and rare iridal epoxide, named belamcanoxide A (5), have been isolated from the rhizomes of Belamcanda chinensis, along with five known compounds. Their structures were elucidated on the basis of extensive spectroscopic analysis. Compounds 1–3 represent the first meroterpenoids composed of an isoflavonoid linked to a spiroiridal-triterpenoid. To the best of our knowledge, compound 4 is the first example of 5/6-fused bicyclic iridal lactone. Compounds 1–8 were evaluated for their in vitro cytotoxicity against five tumor cell lines. Among them, compounds 5–8 showed moderate cytotoxic activities against five cancer cell lines with IC50 values from 3.26 to 8.63 μM.
Introduction
Belamcanda chinensis (L.) DC belongs to the family of Iridaceae, which comprises 92 genera and 1800 species worldwide.1 B. chinensis is a perennial herb that is mainly distributed in South-East Asia.2 Its rhizome has been used for many centuries as a Chinese traditional medicine for the treatment of throat ailments such as asthma and tonsillitis. Previous phytochemical studies on this plant have led to the isolation of a series of isoflavonoids,3 iridal-type triterpenoids,4 and benzoquinones.5 In the last decade, a number of pharmacological investigations have been conducted on isoflavonoids including tectoridin, irigenin, and tectorigenin, which exhibited anti-inflammatory activity,6 antioxidant activity,7 hepatoprotection activity,8 antidiabetic activity,9 and selective estrogen receptor modulating activity.10Iridals are characteristic constituents of the iridaceous plants. These substances are derived from squalene and may exist in two forms, free iridals and iridal esters. Based on the carbon skeleton, free iridals may be divided into three classes: monocyclic iridals, bicyclic iridals, and spiroiridals.4 Common features include a prenylated side chain and an α,β-unsaturated aldehyde. The intriguing structures of iridal-type triterpenoids have attracted wide interesting from phytochemists and pharmacologists. Up to now, more than seventy iridals have been isolated from various iris and B. chinensis. This group of triterpenes exhibited a variety of biological activities including cytotoxicity,11 PKC activation,12 pesticidal activity,13 neuroprotective activity,14 and ichthyotoxicity.15 Some iridals showed remarkable cytotoxic activities comparable to that of doxorubicine.11 Furthermore, iritectol B exhibited dose-dependent apoptotic effect against COR-L23 cells in vitro.16 In addition, a spiro-bicyclic iridal, 28-deacetylbelamcandal, has been shown to be an activating ligand for protein kinase C (PKC) and Ras activator RasGRP3, and induces production of tumor necrosis factor-α (TNF-α).17 Due to the appealing biological properties and unique structural features, iridals have attracted attention to the total synthesis research. Recently, the enantioselective synthesis of iridal has been reported.18
To further explore the potential medicinal values, the authors have lately examined the ethanolic extracts of B. chinensis and performed an intensive investigation of their triterpenoid constituents. The current program has resulted in the isolation of three unprecedented adducts of spiroiridal and isoflavonoid (Fig. 1), belamcandanins A–C (1–3), one unprecedented iridal lactone (Fig. 2), named belamcanolide A (4), and one new and rare iridal epoxide (Fig. 2), named belamcanoxide A (5), together with five known compounds. Compounds 1–3 represent the first meroterpenoids composed of an isoflavonoid linked to a spiroiridal-triterpenoid. Furthermore, compound 4 is the first example of 5/6-fused bicyclic iridal lactone. Compound 5 is a structurally unique iridal with an isolated aldehyde instead of the α,β-unsaturated aldehyde function typical for this group of triterpenoids. Compounds 1–8 were evaluated for their in vitro cytotoxicity against five tumor cell lines. Among them, compounds 5–8 showed cytotoxic activities against five cancer cell lines with IC50 values from 3.26 to 8.63 μM.
 | ||
| Fig. 1 Structures of compounds 1–3. | ||
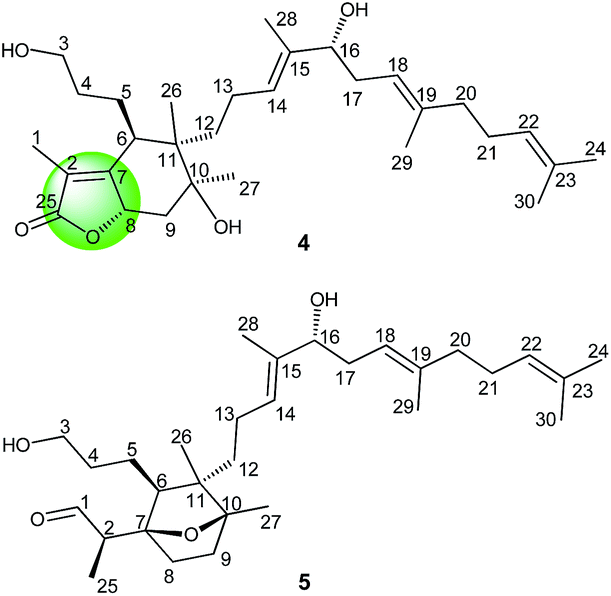 | ||
| Fig. 2 Structures of compounds 4 and 5. | ||
Results and discussion
Belamcandanin A (1) was obtained as yellow solid. Its molecular formula C48H62O13 was established from HRESIMS (m/z 847.42285, [M + H]+). The IR spectrum showed the absorption bands of hydroxyl, benzene ring, and α,β-unsaturated aldehyde groups at 3389, 1582, 1511, 1701, and 1651 cm−1, respectively. The 13C and DEPT NMR spectra of 1 showed 48 carbon signals comprising eighteen quaternary carbons (sixteen olefinic), eleven methines (two oxygenated and six olefinic), eight methylenes (one oxygenated), six methyls, three OMe groups, one carbonyl, and one aldehyde group. Further analysis of its 1H (Table 1), 13C (Table 2), and HMQC NMR data revealed characteristic signals for one aromatic proton [δH 8.01 (1H, s, H-2′); δC 153.0 (C-2′)] and a hydrogen-bonded hydroxyl proton [δH 13.09 (1H, s)], suggesting the presence of an isoflavonoid unit. Besides, six methyl singlets [δH 1.46 (3H, s, H-27), 1.55 (3H, s, H-30), 1.63 (3H, s, H-24), 1.69 (3H, s, H-28), 1.72 (3H, s, H-29), 1.77 (3H, s, H-1); δC 28.0 (C-27), 17.7 (C-30), 25.7 (C-24), 12.0 (C-28), 16.4 (C-29), 11.6 (C-1)] and an α,β-unsaturated aldehyde [δH 10.21 (1H, s, H-25); δC 132.7 (C-2), 162.7 (C-7), and 190.8 (C-25)] suggested the presence of an iridal-type tritepenoid unit. By inspection of the HMBC and 1H–1H COSY spectra, it revealed 1 was an unprecedented adduct of isoflavonoid and iridal-type tritepenoid.| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| a Overlapped signals. | |||||
| 1 | 1.77 s | 10.12 s | 1.79 s | 1.83 d (1.2) | 1.02 d (6.6) |
| 2 | 2.84 q (6.6) | ||||
| 3 | 3.53 m | 3.55 m | 3.57 m; 3.61 m | 3.63 | 3.65 t (6.0) |
| 4 | 1.29 m; 1.39 m | 1.24 m; 1.41 m | 1.31 m; 1.43 m | 1.37 m; 1.43 m | 1.47 m; 1.71 m |
| 5 | 1.94 m; 2.21 m | 1.97 m; 2.12 m | 1.96 m; 2.23 m | 1.83a; 1.94 m | 1.42 m |
| 6 | 2.42 br d (11.4) | 2.99 d (10.8) | 2.43 d (10.8) | 2.80 (3.0, 12.0) | 1.46a |
| 8 | 2.66 m; 3.16 m | 2.46 m; 2.69 dt (4.8, 13.8) | 2.68 m; 3.17 br d (13.8) | 5.11 (6.6, 12.0) | 1.54 m |
| 9 | 1.50 m; 1.69a | 1.48a; 1.67a | 1.51 m; 1.69 m | 1.60a; 2.32 (6.6, 13.2) | 1.38 m; 2.07 m |
| 12 | 1.52 m; 1.71a | 1.51 m; 1.85a | 1.54 m; 1.73 m | 1.13a | 1.44 m |
| 13 | 3.94 m | 3.92a | 3.94a | 1.97 m | 2.00 m |
| 14 | 2.60 t (10.8) | 2.62 t (10.8) | 2.60 t (10.8) | 5.25 br t (6.6) | 5.37 br t (6.6) |
| 16 | 5.94 d (8.4) | 5.94 d (8.4) | 5.95 d (9.0) | 3.95 dd (4.8, 7.8) | 3.98 dd (5.4, 7.8) |
| 17 | 5.14 t (8.4) | 5.14 t (8.4) | 5.15 t (8.4) | 2.19 m; 2.27 dd (7.8, 14.4) | 2.23 m; 2.30 m |
| 18 | 5.64 d (8.4) | 5.64 d (8.4) | 5.64 d (8.4) | 5.07 br t (7.2) | 5.10 br t (7.2) |
| 20 | 1.98 m | 1.98 m | 1.98 m | 2.03 m | 2.04 m |
| 21 | 2.05 m | 2.05 m | 2.05 m | 2.08 m | 2.08 m |
| 22 | 5.03 br t (7.2) | 5.03 br t (6.6) | 5.03 br t (6.6) | 5.05 br t (6.6) | 5.07 br t (7.2) |
| 24 | 1.63 s | 1.63 s | 1.63 s | 1.68 s | 1.69 s |
| 25 | 10.21 s | 1.83 s | 10.23 s | 9.91 d (0.6) | |
| 26 | 3.69 d (10.8) | 3.67 d (10.8) | 3.69 d (10.8) | 1.13 s | 1.00 s |
| 27 | 1.46 s | 1.47 s | 1.47 s | 1.21 s | 1.30 s |
| 28 | 1.69 s | 1.67 s | 1.69 s | 1.60 s | 1.64 s |
| 29 | 1.72 s | 1.72 s | 1.73 s | 1.62 s | 1.64 s |
| 30 | 1.55 s | 1.55 s | 1.56 s | 1.58 s | 1.60 s |
| 2′ | 8.01 s | 8.01 s | 8.01 s | ||
| 2′′ | 6.72 s | 6.72 s | 7.15 br s | ||
| 5′′ | 7.00 d (8.4) | ||||
| 6′′ | 6.72 s | 6.72 s | 6.98 br d (8.4) | ||
| OMe-6′ | 4.02 s | 4.03 s | 4.06 s | ||
| OMe-3′′ | 3.90 s | 3.91 s | 3.95 s | ||
| OMe-4′′ | 3.93 s | 3.93 s | |||
| OH-5′ | 13.09 s | 13.10 s | 13.14 s | ||
| OH-7′ | 7.10 br s | 7.03 br s | |||
| OH-4′′ | 5.73 br s | ||||
| OH-5′′ | 6.00 br s | 5.96 br s | |||
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 11.6 | 190.3 | 11.6 | 8.80 | 9.8 |
| 2 | 132.7 | 133.3 | 132.7 | 122.5 | 46.9 |
| 3 | 62.8 | 62.7 | 62.9 | 62.7 | 63.0 |
| 4 | 32.3 | 32.5 | 32.4 | 32.3 | 33.3 |
| 5 | 27.4 | 27.1 | 27.4 | 27.2 | 23.3 |
| 6 | 54.0 | 49.7 | 54.0 | 43.5 | 57.8 |
| 7 | 162.7 | 161.9 | 162.5 | 164.9 | 88.2 |
| 8 | 19.8 | 23.7 | 19.8 | 76.4 | 34.4 |
| 9 | 38.8 | 37.6 | 38.9 | 43.1 | 32.9 |
| 10 | 75.4 | 75.2 | 75.3 | 76.9 | 88.5 |
| 11 | 52.1 | 51.7 | 52.2 | 44.7 | 48.4 |
| 12 | 43.2 | 43.3 | 43.3 | 36.7 | 41.3 |
| 13 | 68.1 | 67.8 | 68.2 | 22.7 | 24.1 |
| 14 | 64.3 | 64.3 | 64.3 | 125.1 | 125.8 |
| 15 | 130.8 | 130.8 | 130.7 | 137.2 | 136.8 |
| 16 | 133.3 | 133.4 | 133.5 | 76.6 | 76.7 |
| 17 | 33.2 | 33.2 | 33.2 | 34.3 | 34.3 |
| 18 | 125.4 | 125.3 | 125.4 | 119.8 | 119.9 |
| 19 | 135.8 | 135.8 | 135.8 | 139.0 | 138.8 |
| 20 | 39.6 | 39.6 | 39.6 | 39.8 | 39.8 |
| 21 | 26.5 | 26.5 | 26.5 | 26.5 | 26.5 |
| 22 | 124.0 | 124.0 | 124.0 | 124.0 | 124.1 |
| 23 | 131.5 | 131.5 | 131.5 | 131.7 | 131.7 |
| 24 | 25.7 | 25.7 | 25.7 | 25.7 | 25.7 |
| 25 | 190.8 | 11.1 | 190.7 | 174.9 | 205.3 |
| 26 | 80.3 | 80.5 | 80.4 | 17.3 | 17.2 |
| 27 | 28.0 | 28.2 | 28.1 | 26.4 | 19.0 |
| 28 | 12.0 | 12.0 | 12.1 | 11.9 | 12.0 |
| 29 | 16.4 | 16.4 | 16.4 | 16.3 | 16.3 |
| 30 | 17.7 | 17.7 | 17.7 | 17.7 | 17.7 |
| 2′ | 153.0 | 153.0 | 152.6 | ||
| 3′ | 123.1 | 123.1 | 123.3 | ||
| 4′ | 181.4 | 181.4 | 181.7 | ||
| 4′a | 106.3 | 106.3 | 106.4 | ||
| 5′ | 150.6 | 150.6 | 150.6 | ||
| 6′ | 130.5 | 130.5 | 130.4 | ||
| 7′ | 152.4 | 152.3 | 152.2 | ||
| 8′ | 109.7 | 109.6 | 109.5 | ||
| 8′a | 150.2 | 150.2 | 150.1 | ||
| 1′′ | 126.5 | 126.5 | 122.5 | ||
| 2′′ | 105.3 | 105.3 | 111.7 | ||
| 3′′ | 152.4 | 149.4 | 146.5 | ||
| 4′′ | 135.9 | 135.8 | 146.1 | ||
| 5′′ | 149.5 | 152.4 | 114.6 | ||
| 6′′ | 108.7 | 108.7 | 121.8 | ||
| OMe-6′ | 61.0 | 60.9 | 60.9 | ||
| OMe-3′′ | 56.0 | 56.0 | 56.1 | ||
| OMe-4′′ | 61.0 | 61.0 |
Interpretation of the 1H–1H COSY data allowed the assignment of five subunits (Fig. 3): one –CH2–CH2–CH2–CH– system (C-3/C-4/C-5/C-6), one –CH2–CH2– system (C-8/C-9), one –CH2–CH–CH–CH– system (C-12/C-13/C-14/C-26), one ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH–CH system (C-16/C-17/C-18), and one –CH2–CH2–CH system (C-20/C-21/C-22). In the HMBC spectrum, the C-1 methyl group showed the long-range correlations with the aldehyde, C-2, and C-7, indicating the presence of an α-methyl-acryl aldehyde group. The C-27 methyl singlet exhibited the HMBC correlations with C-9, C-10, and C-11, indicating the linkages from C-9 to C-11. The HMBC correlations from H-6 to C-7, C-8, C-10, C-11, C-12 and C-26, from H-13 and H-14 to C-11 and C-12, from H-26 to C-6, C-10, C-11, and C-12 allowed us to construct a spiro[4,5]decane moiety containing a hydroxypropane side chain at C-6 and an α-methyl-acryl aldehyde group at C-7. Additionally, the HMBC correlations from CH3-28 to C-14, C-15, and C-16 supported the linkages from C-14 to C-16 and from C-28 to C-15. Similarly, the linkages from C-18 to C-20 and from C-29 to C-19 were determined on the basis of correlations between CH3-29 with C-18, C-19, and C-20. Finally, correlations from CH3-24 and CH3-30 to C-22 and C-23 permitted the assignment of one dimethylallyl residue at the terminal position of the side chain. The 1D NMR spectroscopic data of the isoflavonoid unit were found to be similar to those reported for irigenin,19 one of the main isoflavonoid aglycones isolated from B. chinensis. The only difference between the isoflavonoid unit and irigenin was replacement of H-8 in irigenin by an iridal-type triterpenoid unit in 1. The strong deshielding of H-17 (δH 5.14) implied the linkage position of two units. Finally, the HMBC correlations from H-16 and H-18 to C-8′ and from H-17 to C-7′, C-8′, and C-8′a proved the isoflavonoid moiety was attached to C-17 of the iridal-type triterpenoid unit via C-17/C-8′, thus establishing the planar structure of 1.
CH–CH–CH system (C-16/C-17/C-18), and one –CH2–CH2–CH system (C-20/C-21/C-22). In the HMBC spectrum, the C-1 methyl group showed the long-range correlations with the aldehyde, C-2, and C-7, indicating the presence of an α-methyl-acryl aldehyde group. The C-27 methyl singlet exhibited the HMBC correlations with C-9, C-10, and C-11, indicating the linkages from C-9 to C-11. The HMBC correlations from H-6 to C-7, C-8, C-10, C-11, C-12 and C-26, from H-13 and H-14 to C-11 and C-12, from H-26 to C-6, C-10, C-11, and C-12 allowed us to construct a spiro[4,5]decane moiety containing a hydroxypropane side chain at C-6 and an α-methyl-acryl aldehyde group at C-7. Additionally, the HMBC correlations from CH3-28 to C-14, C-15, and C-16 supported the linkages from C-14 to C-16 and from C-28 to C-15. Similarly, the linkages from C-18 to C-20 and from C-29 to C-19 were determined on the basis of correlations between CH3-29 with C-18, C-19, and C-20. Finally, correlations from CH3-24 and CH3-30 to C-22 and C-23 permitted the assignment of one dimethylallyl residue at the terminal position of the side chain. The 1D NMR spectroscopic data of the isoflavonoid unit were found to be similar to those reported for irigenin,19 one of the main isoflavonoid aglycones isolated from B. chinensis. The only difference between the isoflavonoid unit and irigenin was replacement of H-8 in irigenin by an iridal-type triterpenoid unit in 1. The strong deshielding of H-17 (δH 5.14) implied the linkage position of two units. Finally, the HMBC correlations from H-16 and H-18 to C-8′ and from H-17 to C-7′, C-8′, and C-8′a proved the isoflavonoid moiety was attached to C-17 of the iridal-type triterpenoid unit via C-17/C-8′, thus establishing the planar structure of 1.
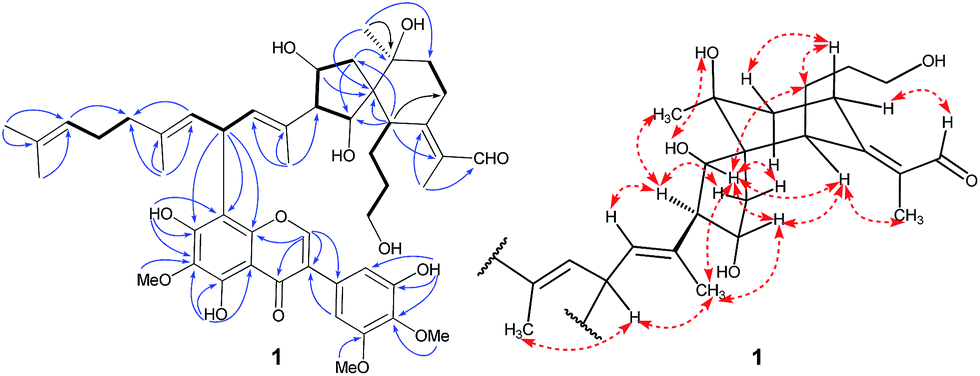 | ||
| Fig. 3 Selected COSY (bold bonds), HMBC (H→C), and ROESY (↔) correlations of compound 1. | ||
The relative configuration of 1 was assigned by analysis of its 1H–1H coupling constants and ROESY data. The 10.8 Hz vicinal coupling constant for H-13/H-14 and H-14/H-26 revealed their anti-relationship. The NOE correlations of H-13/H-26, H-13/H-28, and H-26/H-28 indicated that these protons are on the same face of the ring system (Fig. 3). The NOE correlations of H-6/H-13 and CH3-1/H-6 indicated that H-6 should be in an equatorial position. An NOE correlation between H-5 and H-8β was further observed, indicating that the hydroxypropane side chain at C-6 and H-8β were in an axial. A NOESY correlation between H-14 and CH3-27 supported the assignment of CH3-27 as equatorial. The geometries of four double-bonds were assigned by the correlations of H-28/H-17, H-29/H-17, H-22/H-24, and CH3-1/H-6.
The molecular formula of belamcandanin B (2) was determined to be C48H62O13 by HRESIMS at m/z 847.42346 (calcd for C48H63O13, 847.42632), suggesting that 1 and 2 might be isomer. Comparison of its NMR data with those of compounds 1 revealed that they share the same carbon skeleton except for the geometry of Δ,2,7 which was confirmed by the NOE correlations of CH3-25 with H-8 and CHO-1 with H-6. Extensive analysis of the 2D NMR spectra including HMQC and HMBC supported the structure of 2.
The relative configuration of 2 was also assigned by analysis of its 1H–1H coupling constants and ROESY data. The 10.8 Hz vicinal coupling constant for H-13/H-14 and H-14/H-26 revealed their anti-relationship. ROESY correlations of H-13/H-26, H-13/H-28, H-26/H-28, H-6/H-26, H-6/H-13, H-5/H-8β, and H-14/CH3-27 reflected the same stereostructure in the spirocyclic unit as that in 1.
The molecular formula of belamcandanin C (3) was determined to be C47H60O12 by HRESIMS at m/z 839.39386 [M + Na]+ (calcd for C47H60O12Na, 839.39770), suggesting that 3 might be also an adduct of isoflavonoid and iridal-type triterpenoid. The 1 D NMR spectroscopic data of 3 (Tables 1 and 2) were found to be similar to those of 1, but the constitution of C-ring of isoflavonoid unit was different. The 1H NMR spectrum of 3 showed signals for three aromatic protons in an ABX system at δH 6.98 (1H, br d, J = 8.4 Hz, H-6′′), 7.00 (1H, d, J = 8.4 Hz, H-5′′), and 7.15 (1H, br s, H-2′′), suggesting the presence of a 1,3,4-trisubstituted benzene ring in 3 instead of a 1,3,4,5-tetrasubstituted benzene ring in 1. This was confirmed by the HMBC correlations from H-2′ to C-1′′, from H-2′′ to C-1′′, C-3′′, C-4′′, and C-6′′, from H-5′′ to C-1′′ and C-3′′, from H-6′′ to C-2′′ and C-4′′, and from OH-4′′ to C-3′′, C-4′′, and C-5′′. The methoxyl group at the C-3′′ position was assigned by the NOESY spectrum which exhibited NOE an interaction between the methoxyl group and the aromatic proton at C-2′′. Further assignment of the methoxyl group was obtained by the HMBC spectrum, revealing correlation to the C-3′′ carbon (δC 146.5). The relative configuration of 3 was also established by analysis of its 1H–1H coupling constants and ROESY data. The 10.8 Hz vicinal coupling constant for H-13/H-14 and H-14/H-26 revealed their anti-relationship. Similarly, ROESY correlations of H-13/H-26, H-13/H-28, H-26/H-28, H-6/H-26, H-14/H-12β, H-6/H-13, CH3-1/H-6, H-5/H-8β, and H-14/CH3-27 revealed the same stereostructure in the molecule as that in 1. The biosynthetic pathway for compounds 1–3 was proposed as shown in Scheme 1. Belamcandal or its derivatives were considered as the biogenetic precursors. In brief, belamcandal might undergo deacetylation, hydrogenation, and oxidation to produce intermediate i, which was attacked by nucleophilic isoflavonoids to form intermediate ii. Subsequent dehydration and hydrogenation would lead to compounds 1–3.
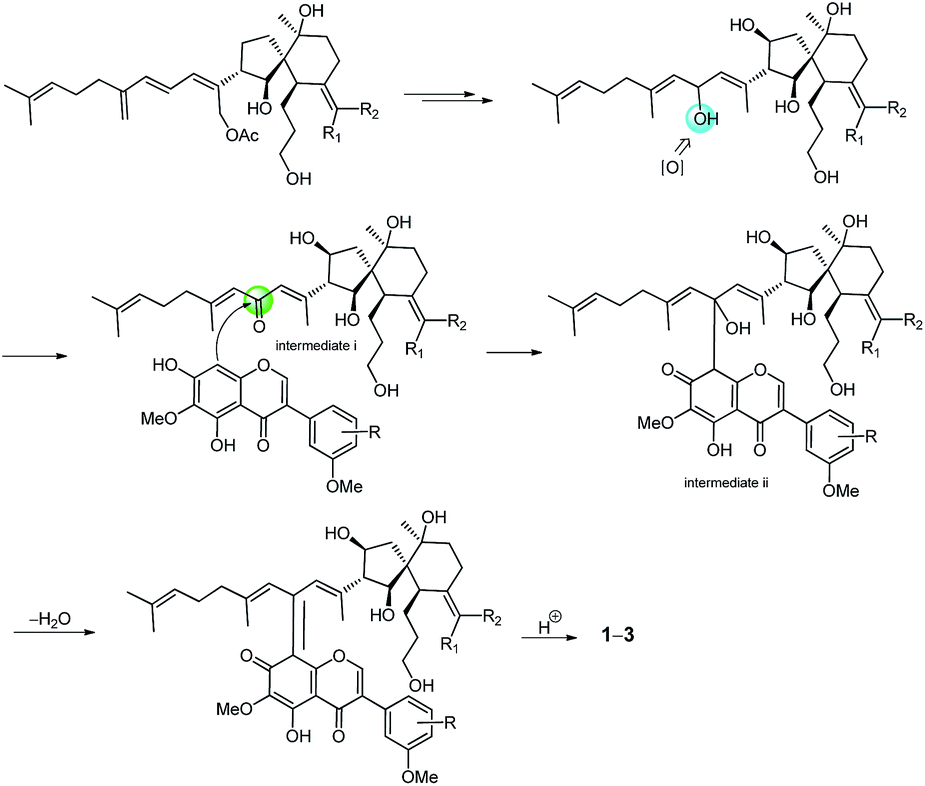 | ||
| Scheme 1 Proposed biosynthesis of compounds 1–3. | ||
Belamcanolide A (4) exhibited a molecular formula of C30H48O5, as established by HRESIMS at m/z 511.33795 [M + Na]+ (calcd for C30H48O5Na, 511.33940), indicating seven degrees of unsaturation in its structure. The IR spectrum showed a characteristic absorption band at 1734 cm−1, suggesting the presence of a lactone group. The 1H NMR spectrum (Table 1) showed three distinctive olefinic protons [δH 5.05 (1H, br t, J = 6.6 Hz); 5.07 (1H, br t, J = 7.2 Hz); 5.25 (1H, br t, J = 6.6 Hz)], suggested the presence of a homofarnesyl side chain. In addition, seven methyl singlets and two oxygenated methine protons [δH 5.11 (6.6, 12.0) and 3.95 (4.8, 7.8)] were also observed in the 1H NMR spectrum. The 13C NMR (Table 2) and DEPT spectra of 4 displayed resonances for 30 carbons, ascribed to seven methyls, nine methylenes (one oxygenated), six methines (two oxygenated and three olefinic), seven quaternary carbons (one oxygenated and five olefinic), and one ester carbonyl. Because four double bonds and one carbonyl accounted for five degrees of unsaturation, 4 should possess a bicyclic framework.
In the HMBC spectrum, the correlations of CH3-28 with C-14, C-15, and C-16, CH3-29 with C-18, C-19, and C-20, and CH3-24 and CH3-30 with C-22 and C-23 were observed. This evidence, along with three proton spin systems deduced from 1H–1H COSY correlations, H2-12/H2-13/H-14, H-16/H2-17/H-18, and H2-20/H2-21/H-22, led to the establishment of the homofarnesyl side chain (Fig. 2). In addition, key HMBC correlations from CH3-26 to C-6, C-10, C-11, and C-12, from CH3-27 to C-9, C-10, and C-11, and from H-6 to C-7, C-8, and C-10, along with the 1H–1H COSY correlations of H-3/H-4/H-5/H-6 and H-8/H2-9, indicated that C-6, C-7, C-8, C-9, C-10, and C-11 constructed a six-membered carbon ring A, which was substituted with a hydroxypropane side chain at C-6, a methyl group (CH3-27) and a hydroxyl group at C-10, and a methyl group (CH3-26) and a homofarnesyl side chain at C-11. Finally, the HMBC correlations from CH3-1 to C-25, C-2, and C-7 and from H-8 to C-25, C-2, and C-7, established a five-membered lactone ring B, which was fused to ring A via C-7 and C-8 (Fig. 4).
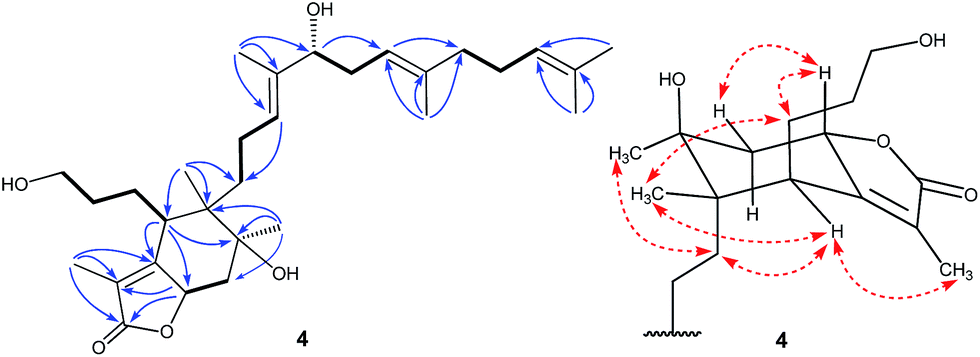 | ||
| Fig. 4 Selected COSY (bold bonds), HMBC (H→C), and ROESY (↔) correlations of compound 4. | ||
The relative configuration of 4 was assigned by analysis of its 1H–1H coupling constants and ROESY data. The large vicinal coupling constant (12.0 Hz) between H-8 and H-9α revealed their trans-diaxial relationship. The NOE correlations of H-8/H-5, H-8/H-9β, and H-5/H-26 indicated that these protons are on the same face of the ring system. The NOE correlations of H-6/H-12 and H-12/H-27 indicated that these protons are on the other face of the ring system. The geometries of three double-bonds of the homofarnesyl side chain were assigned by the correlations of H-14/H-16, H-29/H-17, and H-22/H-24. Biogenetically, compound 4 might be derived from iridobelamal A. The C-25 aldehyde was oxidized to carboxyl group, and C-8 was hydroxylated. Subsequent lactonization might lead to 4 (Scheme 2).
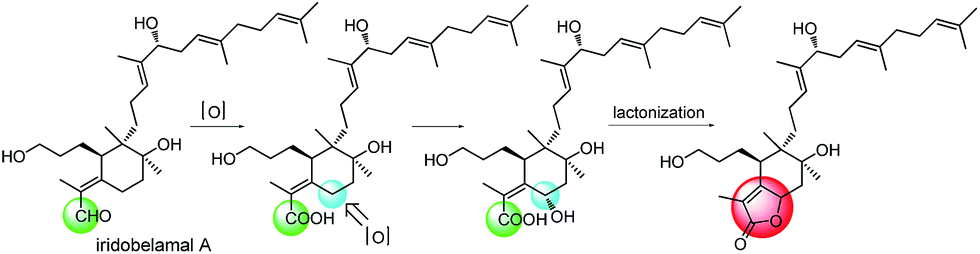 | ||
| Scheme 2 Proposed biosynthesis of compound 4. | ||
Belamcanoxide A (5) had a molecular formula of C30H50O4 determined by HRESIMS at m/z 497.35873 [M + Na]+ (calcd for C30H50O4Na, 497.36013), indicating six degrees of unsaturation in its structure. The IR spectrum showed the absorption bands of hydroxyl and carbonyl groups at 3390 and 1714 cm−1, respectively. The 13C NMR spectrum (Table 2) of 5 displayed 30 carbon resonances, which were further classified by DEPT and HSQC spectra as seven methyls (one secondary and six tertiary), ten methylenes, six methines (one oxygenated and three olefinic), six quaternary (two oxygenated and three olefinic) carbons, and one aldehyde group. The distinctive proton resonances [δH 5.07 (1H, br t, J = 7.2 Hz); 5.10 (1H, br t, J = 7.2 Hz); 5.37 (1H, br t, J = 6.6 Hz)] in the 1H NMR spectrum (Table 1) indicated the presence of a homofarnesyl side chain. The aforementioned data suggested that compound 5 is an iridal-type triterpenoid bearing an aldehyde and a homofarnesyl side chain, similar to iridobelamal A.20 Comparison of the 13C NMR data of the two compounds revealed that the resonances for Δ2,7 in iridobelamal A were replaced by the resonances for an oxygenated quaternary carbon [δC 88.2 (C-7)] and a methine (δC 46.9) in 5. This was confirmed by the HMBC correlations from CH3-1 to C-25, C-2, and C-7, from H-25 to C-1 and C-2, and from H-2 to C-6, C-7, and C-8. Additionally, the resonance C-10 of 5 was significantly shifted by ΔδH +13.2 ppm, when compared with that of iridobelamal A. Accordingly, due to the strong deshielding of C-7 and C-10, it might be proposed that an epoxy bridge was present at C-7 and C-10, which was reinforced by one remaining degree of unsaturation. Interpretation of the HMBC data (Fig. 5) defined the planar structure of compound 5 as shown. The relative configuration of 5 was assigned by ROESY correlations (Fig. 5). The ROESY correlations of H-5/H-26, H-9β/H-27, and H-26/H-27 indicated that H-5, H-26, and H-9β were co-facial, and were randomly assigned to be β-oriented. The ROESY correlations of H-12/H-6, and H-9α/H-12 fixed these protons in an α-oriented. As shown in Fig. 3, the bicyclic system imposes an equatorial orientation for the C-2/C-7 bond. In consequence, the ROESY correlations of CH3-1/H-5, CH3-1/H-8, H-2/H-5, and H-25/H-8 suggested the configuration of C-2 as shown, which was reinforced by the absence of the ROESY correlations of H-25/H-5 and H-2/H-8. Based on above analysis, it might be assumed that rotation about the C-2–C-7 was restricted due to the hydroxypropane side chain at C-6, which resulted in a preferred conformer as shown. Biogenetically, compound 5 might be derived from isoiridogermanal A, which might undergo Michael addition and hydrogenation to generate 5 (Scheme 3).
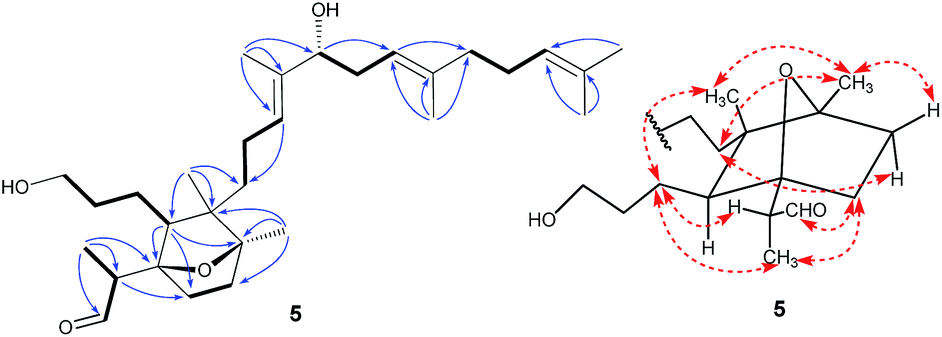 | ||
| Fig. 5 Selected COSY (bold bonds), HMBC (H→C), and ROESY (↔) correlations of compound 5. | ||
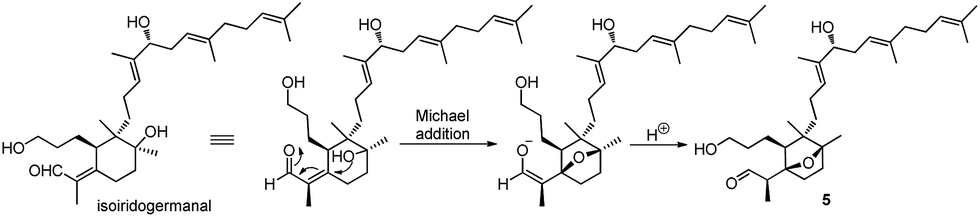 | ||
| Scheme 3 Proposed biosynthesis of compound 5. | ||
Three known iridal-type triterpenoids, iridobelamal A (6),20 isoiridogermanal (7),21 and iridal (8),22 and two known isoflavonoids, irigenin (9),19 and tectorigenin (10),23 were also isolated and identified on the basis of their 1H NMR, 13C NMR, and ESIMS data.
Compounds 1–8 were tested for their in vitro cytotoxicity against NCI-H1650 (non-small-cell lung carcinoma), HepG2 (human hapetocelluar carcinoma), BGC 823 (human stomach carcinoma), HCT-116 (human colon carcinoma), and MCF-7 (human breast carcinoma) cells with paclitaxel as a positive control by means of the MTT assay (Table 3). In consequence, compounds 5–8 showed moderate cytotoxic activities against five cancer cell lines with IC50 values from 3.26 to 8.63 μM.
Conclusions
In summary, we discovered a novel family of meroterpenoids composed of an isoflavonoid linked to a spiroiridal-triterpenoid. Compound 4 is the first example of 5/6-fused bicyclic iridal lactone. Compound 5 is a structurally unique iridal with an isolated aldehyde and an epoxy bridge. Our bioactivity assay provide further evidence that iridals have significant cytotoxic activities and are potentially promising leads for developing new anticancer drugs.Experimental
General experimental procedures
Optical rotations were recorded on a Perkin-Elmer 341 polarimeter. UV spectra were measured with a Shimadzu UV-2550 spectrophotometer. CD spectra were measured on a JASCO J-810 instrument. IR spectra were recorded on a Perkin-Elmer 577 spectrometer. NMR spectra were obtained on a Varian Mercury-600 spectrometer with TMS as internal standard. HRESIMS data were recorded on a Waters Q-TOF Ultima mass spectrometer. Silica gel (300–400 mesh, Qingdao Marine Chemical Factory), Sephadex LH-20 (Pharmacia Biotech, Sweden), MCI gel (CHP20P, 75–150 μm, Mitsubishi Chemical Industries Ltd), and ODS (40–60 μm, Merck) were used for column chromatography. TLC was performed on GF254 plates (Qingdao Marine Chemical Factory). Preparative HPLC was conducted using LumtechK-501pumps with a UV detector K-2501 and a YMC-Pack ODS-A column (250 × 20 mm, 5 μm). Semi-preparative HPLC was performed on a Shimadzu LC-6AD pump with a Shimadzu SPD-M20A detector and a YMC-Pack ODS-A column (250 × 10 mm, 5 μm). Five cancer cell lines (NCI-H1650, HepG2, BGC 823, HCT-116, and MCF-7) were purchased from ATCC.Plant material
The plant material of Belamcanda chinensis was collected from Anguo, Hebei Province, P. R. China. A voucher specimen has been deposited at Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College accession number: S-2619.Extraction and isolation
The powdered dried twigs and leaves of Belamcanda chinensis (10 kg) were extracted with 95% EtOH at room temperature for three times. After removal of the solvent under reduced pressure, the crude extract (1200 g) was suspended in water and then partitioned with petroleum ether, EtOAc, and BuOH successively. The EtOAc-soluble fraction (370 g) was subjected to macroporous adsorbent resin column, eluting with a gradient of increasing EtOH (0–100%) in H2O to afford five fractions (A–E). Fraction F (90 g) was further applied to a silica gel column and eluted with CHCl3–MeOH (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1, v/v) to give twenty fractions, F1–F20. Fraction F5 (3.2 g) was subjected to an MCI gel column eluted with MeOH–H2O (50% to 100%) to give five fractions, F5a–F5e. F5c (0.5 g) was separated by silica gel column, and further purified by preparative TLC to afford 9 (20 mg) and 10 (30 mg). Fraction F5d (0.7 g) was subjected to Sephadex LH-20 column eluted with MeOH to give three fractions, F5d1–F5d3. Fraction F5d1 (0.2 g) was further purified by preparative HPLC to obtain 8 (30 mg, tR 51 min). Fraction F7 (3.0 g) was chromatographed on a reversed-phase C18 silica gel column (MeOH/H2O, 40% to 100%) to give five fractions, F7a–F7e. F7c (0.4 mg) was separated by a Sephadex LH-20 column eluted with EtOH, and then purified by semi-preparative HPLC to obtain 4 (5.0 mg, tR 18 min). F7d (0.5 mg) was further chromatographed on a Sephadex LH-20 column eluted with EtOH, followed by the purification of semi-preparative HPLC to obtain 5 (6.0 mg, tR 21 min). Fraction F9 (4.3 g) was separated by a reversed-phase C18 silica gel column (MeOH–H2O, 40% to 100%) to generate six fractions F9a–F9f. Fraction F9d was subjected to Sephadex LH-20 column, and was further purified by preparative HPLC to obtain 6 (120 mg, tR 55 min) and 7 (150 mg, tR 58 min). Fraction F11 (2.8 g) was applied to a reversed-phase C18 silica gel column (MeOH–H2O) and eluted with MeOH–H2O (40% to 100%) to give five fractions F11a–F11e. F11c was separated by Sephadex LH-20 (MeOH) and further by preparative HPLC to obtain 1 (15 mg, tR 37 min) and 2 (20 mg, tR 41 min). Fraction F15 (2.5 g) was chromatographed on a reversed-phase C18 silica gel column (MeOH–H2O, 40% to 100%) to give five fractions, F15a–F15e. F15c (0.6) was further chromatographed on a Sephadex LH-20 column eluted with MeOH, followed by the purification on semi-preparative HPLC to obtain 3 (5.0 mg, tR 16 min).
ε) 203.5 (4.66), 260 (4.45) nm; IR νmax 3389, 2971, 2937, 1701, 1651, 1582, 1511, 1454, 1204, 1105 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 847.42285 [M + H]+ (calcd for C48H63O13, 847.42632).ε) 205 (4.65), 265.8 (4.46) nm; CD (MeOH, Δε) λmax 274.5 (+1.12), 232 (−1.19), 210.5 (+3.67) nm; IR νmax 3378, 2921, 2851, 1651, 1582, 1512, 1454, 1205, 1105 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 847.42346 [M + H]+ (calcd for C48H63O13, 847.42632).ε) 205.2 (4.61), 264.2 (4.37) nm; CD (MeOH, Δε) λmax 331 (+0.98), 202.5 (−2.96) nm; IR νmax 3360, 2966, 2924, 1711, 1651, 1616, 1580, 1517, 1454, 1377, 1205, 1045 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 839.39386 [M + Na]+ (calcd for C47H60O12Na, 839.39770).ε) 204 (4.06) and 217 (4.01) nm; CD (MeOH, Δε) λmax 224.5 (+6.17); IR νmax 3413, 2968, 2928, 1734, 1671, 1450, 1377, 1038 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 511.33795 [M + Na]+ (calcd for C30H48O5Na, 511.33940).ε) 205 (3.35) and 263.4 (3.15) nm; CD (MeOH, Δε) λmax 294.5 (−0.29), 200 (−1.51) nm; IR νmax 3390, 2927, 1714, 1453, 1382, 1055 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 497.35873 [M + Na]+ (calcd for C30H50O4Na, 497.36013).
:1 to 1:1, v/v) to give twenty fractions, F1–F20. Fraction F5 (3.2 g) was subjected to an MCI gel column eluted with MeOH–H2O (50% to 100%) to give five fractions, F5a–F5e. F5c (0.5 g) was separated by silica gel column, and further purified by preparative TLC to afford 9 (20 mg) and 10 (30 mg). Fraction F5d (0.7 g) was subjected to Sephadex LH-20 column eluted with MeOH to give three fractions, F5d1–F5d3. Fraction F5d1 (0.2 g) was further purified by preparative HPLC to obtain 8 (30 mg, tR 51 min). Fraction F7 (3.0 g) was chromatographed on a reversed-phase C18 silica gel column (MeOH/H2O, 40% to 100%) to give five fractions, F7a–F7e. F7c (0.4 mg) was separated by a Sephadex LH-20 column eluted with EtOH, and then purified by semi-preparative HPLC to obtain 4 (5.0 mg, tR 18 min). F7d (0.5 mg) was further chromatographed on a Sephadex LH-20 column eluted with EtOH, followed by the purification of semi-preparative HPLC to obtain 5 (6.0 mg, tR 21 min). Fraction F9 (4.3 g) was separated by a reversed-phase C18 silica gel column (MeOH–H2O, 40% to 100%) to generate six fractions F9a–F9f. Fraction F9d was subjected to Sephadex LH-20 column, and was further purified by preparative HPLC to obtain 6 (120 mg, tR 55 min) and 7 (150 mg, tR 58 min). Fraction F11 (2.8 g) was applied to a reversed-phase C18 silica gel column (MeOH–H2O) and eluted with MeOH–H2O (40% to 100%) to give five fractions F11a–F11e. F11c was separated by Sephadex LH-20 (MeOH) and further by preparative HPLC to obtain 1 (15 mg, tR 37 min) and 2 (20 mg, tR 41 min). Fraction F15 (2.5 g) was chromatographed on a reversed-phase C18 silica gel column (MeOH–H2O, 40% to 100%) to give five fractions, F15a–F15e. F15c (0.6) was further chromatographed on a Sephadex LH-20 column eluted with MeOH, followed by the purification on semi-preparative HPLC to obtain 3 (5.0 mg, tR 16 min).
ε) 203.5 (4.66), 260 (4.45) nm; IR νmax 3389, 2971, 2937, 1701, 1651, 1582, 1511, 1454, 1204, 1105 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 847.42285 [M + H]+ (calcd for C48H63O13, 847.42632).ε) 205 (4.65), 265.8 (4.46) nm; CD (MeOH, Δε) λmax 274.5 (+1.12), 232 (−1.19), 210.5 (+3.67) nm; IR νmax 3378, 2921, 2851, 1651, 1582, 1512, 1454, 1205, 1105 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 847.42346 [M + H]+ (calcd for C48H63O13, 847.42632).ε) 205.2 (4.61), 264.2 (4.37) nm; CD (MeOH, Δε) λmax 331 (+0.98), 202.5 (−2.96) nm; IR νmax 3360, 2966, 2924, 1711, 1651, 1616, 1580, 1517, 1454, 1377, 1205, 1045 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 839.39386 [M + Na]+ (calcd for C47H60O12Na, 839.39770).ε) 204 (4.06) and 217 (4.01) nm; CD (MeOH, Δε) λmax 224.5 (+6.17); IR νmax 3413, 2968, 2928, 1734, 1671, 1450, 1377, 1038 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 511.33795 [M + Na]+ (calcd for C30H48O5Na, 511.33940).ε) 205 (3.35) and 263.4 (3.15) nm; CD (MeOH, Δε) λmax 294.5 (−0.29), 200 (−1.51) nm; IR νmax 3390, 2927, 1714, 1453, 1382, 1055 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; HRESIMS m/z 497.35873 [M + Na]+ (calcd for C30H50O4Na, 497.36013).Cytotoxic assay
Compounds 1–5 were tested for cytotoxicity against NCI-H1650 (non-small-cell lung carcinoma), HepG2 (human hepatocelluar carcinoma), BGC 823 (human stomach carcinoma), HCT-116 (human colon carcinoma), and A2780 (human ovarian carcinoma) cells using the MTT assay as described in the literature.24Acknowledgements
This work was financially supported by the National Natural Science Foundation (No. 21472245), CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-1-010), and the State Key Laboratory of Bioactive Substance and Function of Natural Medicines (No. GTZC201101), Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, China.Notes and references
- M. I. Choudhary, S. Hareem, H. Siddiqui, S. Anjum, S. Ali, A. Rahman and M. I. Zaidi, Phytochemistry, 2008, 69, 1880–1885 CrossRef CAS PubMed
.
- D. Woźniak and A. Matkowski, Fitoterapia, 2015, 107, 1–14 CrossRef PubMed
-
(a) S. W. Kang, M. C. Kim, C. Y. Kim, S. H. Jung and B. H. Um, Chem. Pharm. Bull., 2008, 56, 1452–1454 CrossRef CAS PubMed
-
(a) H. Ito, S. Onoue, Y. Miyake and T. Yoshida, J. Nat. Prod., 1999, 62, 89–93 CrossRef CAS PubMed
- K. Seki, K. Haga and R. Kaneko, Phytochemistry, 1995, 38, 703–709 CrossRef CAS
-
(a) Y. P. Kim, M. Yamada, S. S. Lim, S. H. Lee, N. Ryu and K. H. Shin, Biochim. Biophys. Acta, 1999, 1438, 399–407 CrossRef CAS PubMed
- K. A. Kang, K. H. Lee, S. Chae, R. Zhang, M. S. Jung, S. Y. Kim, H. S. Kim, D. H. Kim and J. W. Hyun, Eur. J. Pharmacol., 2005, 519, 16–23 CrossRef CAS PubMed
- S. H. Jung, Y. S. Lee, S. S. Lim, S. H. Lee, K. H. Shin and Y. S. Kim, Arch. Pharmacal Res., 2004, 27, 184–188 CrossRef CAS
- S. H. Jung, Y. S. Lee, S. H. Lee, S. S. Lim, Y. S. Kim and K. H. Shin, Arch. Pharmacal Res., 2002, 25, 306–312 CrossRef CAS
-
(a) C. Morrissey, J. Bektic, B. Spengler, D. Galvin, V. Christoffel, H. Klocker, J. M. Fitzpatrick and R. W. G. Watson, J. Urol., 2004, 172, 2426–2433 CrossRef CAS PubMed
- J. P. Bonfils, F. Pinguet, S. Culine and Y. Sauvaire, Planta Med., 2001, 67, 79–81 CrossRef CAS PubMed
- L. Shao, N. E. Lewin, P. S. Lorenzo, Z. J. Hu, I. J. Enyedy, S. H. Garfield, J. C. Stone, F. J. Marner, P. M. Blumberg and S. M. Wang, J. Med. Chem., 2001, 44, 3872–3880 CrossRef CAS PubMed
-
(a) A. L. Pathiranage, J. M. Stubblefield, X. L. Zhou, J. H. Miao, A. L. Newsome and N. Dunlap, Phytochem. Lett., 2016, 18, 44–50 CrossRef CAS
- C. L. Zhang, Y. Wang, Y. F. Liu, G. Ni, D. Liang, H. Luo, X. Y. Song, W. Q. Zhang, R. Y. Chen, N. H. Chen and D. Q. Yu, J. Nat. Prod., 2014, 77, 411–415 CrossRef CAS PubMed
- H. Ito, S. Onoue, Y. Miyake and T. Yoshida, J. Nat. Prod., 1999, 62, 89–93 CrossRef CAS PubMed
- R. Fang, P. J. Houghton and P. J. Hylands, J. Ethnopharmacol., 2008, 118, 257–263 CrossRef CAS PubMed
-
(a) K. Takahashi, Y. Hano, M. Suganuma, S. Okabe and T. Nomura, J. Nat. Prod., 1999, 62, 291–293 CrossRef CAS PubMed
- A. Corbu, M. Aquino, T. V. Pratap, P. Retaileau and S. Arseniyadis, Org. Lett., 2008, 10, 1787–1790 CrossRef CAS PubMed
- M. Morita, M. Arisawa, Y. Kondo and T. Tokemoto, Chem. Pharm. Bull., 1973, 21, 600 CrossRef
- R. Fang, P. J. Houghton, C. Luo and P. J. Hylands, Phytochemistry, 2007, 68, 1242 CrossRef CAS PubMed
- S. M. Wong, Y. Oshima, J. M. Pezzuto, H. H. S. Fong and N. R. Farnsworth, J. Pharm. Sci., 1986, 75, 317 CrossRef CAS PubMed
- Y. Miyake, H. Ito and T. Yoshida, Can. J. Chem., 1997, 75, 734 CrossRef CAS
- S. W. Kang, M. C. Kim, C. Y. Kim, S. H. Jung and B. H. Um, Chem. Pharm. Bull., 2008, 56, 1452–1454 CrossRef CAS PubMed
- G. Ni, Q. J. Zhang, Z. F. Zheng, R. Y. Chen and D. Q. Yu, J. Nat. Prod., 2009, 72, 966–968 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: The UV, IR, HR-ESIMS, 1D and 2D NMR spectra of compounds 1–5. See DOI: 10.1039/c7ra00614d |
| This journal is © The Royal Society of Chemistry 2017 |