
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physicochemical and catalytic properties of polysiloxane network–Pt systems†
Edyta Stochmal *a,
Joanna Strzezikb and
Agnieszka Krowiakb
*a,
Joanna Strzezikb and
Agnieszka Krowiakb
aFaculty of Materials Science and Ceramics, AGH-University of Science and Technology, Al. Mickiewicza 30, 30-059 Kraków, Poland. E-mail: stochmal@agh.edu.pl; Fax: +48 12 6337161; Tel: +48 12 6172845
bDepartment of Chemistry, Silesian Technical University, ul. Strzody 9, 44-100 Gliwice, Poland
First published on 18th May 2017
Abstract
Different polysiloxane networks obtained via a cross-linking process have served as matrices for the incorporation of metallic Pt particles by chemical reduction of metal ions from PtCl4 in THF solution in the presence of active Si–H groups remaining in the networks. Polysiloxane networks have been prepared by hydrosilylation of D4/V4 polysiloxane with branched (Q(MH)4) or cyclic (DH4) hydrosiloxanes, at different molar ratios of reagents. The influence of various topologies of matrices on the amount of introduced metal particles and their distribution in the matrix was investigated. Network–Pt systems, thus formed, have been characterized using FTIR spectroscopy, swelling measurements, X-ray diffraction, SEM and TEM microscopy combined with EDX microanalysis, and thermogravimetric studies. The reduction of platinum ions was monitored using UV-vis spectroscopy. The consumption of Si–H groups, accompanying the reduction, was investigated by IR measurements. Depending on the applied matrix, different amounts of platinum were introduced. XRD studies have confirmed the incorporation of Pt(0) into all obtained systems. It was established that the systems contained metal nanoparticles (size 3–6 nm). Microscopic investigations have shown that the size and the arrangement of Pt crystallites formed depend on the type of matrix applied. Catalytic performance of examined systems investigated using isopropyl alcohol conversion as the test reaction indicated mainly redox type activity. It was found that Pt dispersed in Q–P type supports, i.e. the polysiloxane networks obtained using the branched hydrosiloxane as the cross-linking agent, showed higher catalytic activity than Pt dispersed in C–P type matrices – the networks obtained with the application of the cyclic hydrosiloxane. The comparison of the behavior of synthesized samples with standard Pt/alumina catalyst in isopropyl alcohol conversion revealed higher redox activity of polysiloxane-supported systems in the lower temperature range.
Introduction
Supported noble metal catalysts are widely used in diverse commercial processes namely: automobile exhaust treatment, reforming of hydrocarbons or ethylene oxidation.1,2 The composition of such systems is often very complex.3 Apart from the properties of active agents, the nature and type of the support are the most important features affecting activity and application of these catalysts.4 The supports commonly used for deposition of noble metals are alumina, silica, aluminosilicates, zeolites and activated carbon.4 The main role of these additives is to disable sintering and induce uniform and nanoscale dispersion of metal particles.5 Despite constant development of methods for the synthesis of supported noble metal catalysts, there is still a challenge to obtain metal particles with well-defined shape and size, since these parameters determine the surface structure, electronic and oxidation states of metal and thus strongly impact upon the rate of the catalytic reaction and selectivity of the desired product.6 There are numerous publications concerning the improvement of properties of conventional supports,7 however great efforts have also been made to establish new types of matrices by adopting, among others, polymer networks as hosts for noble metals. For example, metal organic frameworks (MOFs) with palladium were applied for oxidation of cinnamyl alcohol, hydrogenation of 1-octene and cyclododecene,8 hydrogenation of ethyl cinnamate,9 hydrogenation of styrene 1-octene and cis cyclooctene.10 Covalent triazine frameworks (CTFs) with Pd were tested in liquid phase oxidation of glycerol.11 In turn, Au and Pd incorporated into the gel-type polyacrylic resins were active in the rapid oxidation of n-butanal to n-butanoic acid by dioxygen under mild conditions in water, and also in the oxidation of n-butanol to n-butanal under the same conditions.12 Conjugated microporous polymers (CMPs) containing platinum or palladium were active in the oxidation of methane to methanol and Suzuki coupling reactions.13In the literature there are only a few reports describing the deposition of platinum group metals into polysiloxane networks. Motoyama et al. applied polysiloxane gels containing ruthenium or platinum species for isomerization of alkenes14,15 or for reduction of nitro compounds to the corresponding amines.16 In those works metal species were incorporated into insoluble gels synthesized by polymethylhydrosiloxane (PMHS) treatment with amides, diols or dienes in the presence of catalyst containing an appropriate metal. There are some works devoted to the incorporation of platinum group metals, mainly palladium, into polysiloxanes in order to obtain catalytically active systems. In reported studies, metal ions have been introduced into appropriately functionalized polymers17–19 or metal particles nanoclusters have been generated and stabilized with polymethylhydrosiloxane.20–26 Cypryk et al. applied soluble polysiloxane as supports for palladium catalysts. They investigated the influence of different factors, such as various topologies (linear, star-shaped and hyperbranched) of polymers as well as different ligand (vinyl, butylthioetyl or diphenylphosphinoethyl pendant side groups) density on catalysts stability in the Mizoroki–Heck reaction.17,18 PMHS functionalized with cinchonidine was used for the stabilization of palladium particles, which were generated ex situ during thermal decomposition of palladium trifluoroacetate and followed by the incorporation into polymer in order to prevent Pd from agglomeration. These systems were tested in hydrogenation of isophorone.19 In Chauhan et al. works, catalytically active metal nanoparticles incorporated into polysiloxane matrix were obtained by in situ reduction of metal salts in the presence of PMHS. This polymer served as a reducing agent and as a protective agent for the stabilization of metal particles. Obtained systems, mainly Pd–polysiloxane nanocomposites, were applied as catalysts in silaestryfication, chemoselective hydrogenation of polymethylhydrosiloxane or for macromolecular grafting of PMHS via alcoholysis.20–23 Polymethylhydrosiloxane was also used for preparation of polysiloxane-stabilized silver20 or platinum nanoparticles.24–26 Pt/PMHS catalysts were applied in regioselective polyhydrosililation of PMHS, functionalization of polybutadiene or in hydrolitic oxidation of organofunctional hydrogenosilanes to silanols.24–26
As already mentioned, the papers devoted to the preparation of catalytic materials containing platinum deposited on polysiloxane matrix support are very rare in the literature, despite the great catalytic potential of these compounds. Furthermore many of these papers describe materials for homogenous catalysis. Presented work is one of the very few focused on the application of polysiloxane networks–Pt systems in heterogenous catalysis. The use of polysiloxane networks of reducing properties as supports for platinum catalysts is a convenient way of deposition of platinum particles in the matrix by reduction of metal ions. The advantage of synthesized supports is the possibility of their modification in a wide range, namely the application of cross-linking agents of a different structure or functionality can provide final supports of varying topology. Even the starting polymer, undergoing then the cross-linking process can be modified. In this paper the purpose was realized using cross-linking agents of the same functionality but of different structure. There are some points in our experiments which we would like to be highlighted:
- The polysiloxane networks are applied, which can act both as reducing agents for platinum ions and simultaneously as the matrices for the incorporation of metal species.
- The arrangement of catalytic centers is strongly related to the localization and to the number of active Si–H groups participating in the reduction, and can be controlled to some extent.
- There is the possibility of obtaining catalytic systems containing well dispersed metal nanoparticles using described procedure.
- The ability to modify the topology of networks can affect accessibility of the active phase in the matrix and consequently influence catalytic properties of final products.
- The introduction of different amounts of platinum species into polysiloxane networks is possible depending on the applied type of matrix.
This work is a continuation of our earlier research.27 In the previous paper we reported the successful application of the method allowing the incorporation of metallic Pt particles into polysiloxane network by a chemical reduction of metal ions. Process was conducted in the presence of cross-linking products containing active Si–H groups exhibiting reducing properties.28,29 It was concluded, that polysiloxane networks are able to be multipurpose and can play simultaneously the role of reducing agents and the role of matrices hosting metallic Pt particles. Comparing the matrices obtained with the application of the linear or cyclic cross-linking agent, we observed that the topology of network affected significantly the catalytic properties of obtained Pt–network systems. The cross-linking agents with substantially different structure and functionality, significantly influenced the topology of network and a number of the remaining Si–H groups, which in turn affected the dispersion of platinum particles and catalytic properties of obtained Pt-systems – in favor of these obtained using cyclic cross-linking agent. Moreover, networks obtained with the use of cross-linking agent of cyclic type proved to be more promising matrices also for the introduction of a higher – than used in previous experiments – amount of metal species (i.e. higher than 1.0 wt% of Pt).
In the present study, for the preparation of networks, D4/V4 polymer and the selected hydrosiloxane compounds of the same functionality but various structures, i.e. cyclic 2,4,6,8-tetramethylcyclotetrasiloxane (DH4) or branched tetrakis(dimethylsilyloxy)silane (Q(MH)4) were applied (Fig. 1a). D4/V4 polymer was obtained by equilibrium cationic ring-opening polymerization of octamethylcyclotetrasiloxane (D4) and 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane (V4). The preparation of networks involved cross-linking of D4/V4 polysiloxane containing vinyl groups with two different hydrosiloxane compounds used as cross-linking agents. For this purpose, hydrosilylation reaction – widely utilized in organosilicon chemistry – was applied.30 The scheme of a catalytic addition of a hydrosilane group (Si–H) to a double carbon–carbon bond was shown in Fig. 1b.
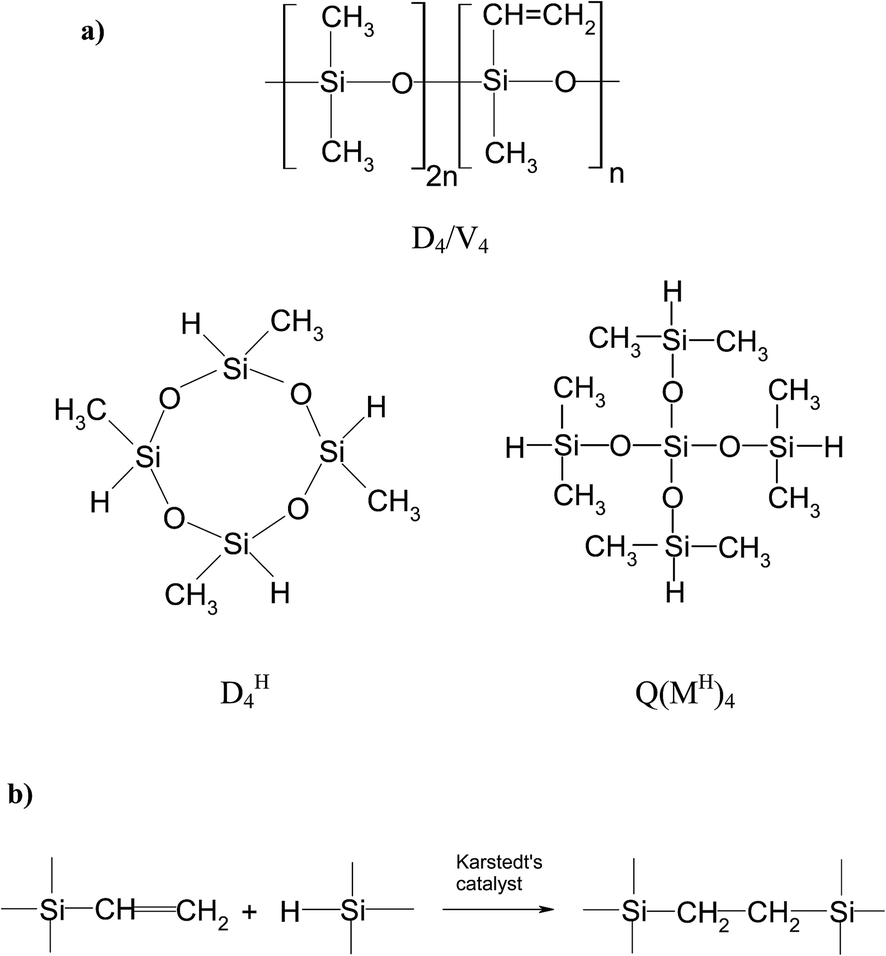 | ||
| Fig. 1 (a) Structures of compounds used during the preparation of polysiloxane networks: polysiloxane D4/V4 and cross-linking agents – DH4, Q(MH)4; (b) scheme of hydrosilylation of vinyl compounds. | ||
It has been demonstrated in our earlier experiments, that it is possible to increase the dispersion of platinum particles in the final product – which can improve its catalytic properties – by the application of better-swelling solvent during the reduction step. Therefore the introduction of platinum species into the structure of Pt–network system via reduction of Pt4+ ions in the presence of cross-linked products was carried out in tetrahydrofuran solution.
In the present work, we aimed to investigate how the type of network obtained with the use of a cyclic or of a branched cross-linking agent, influences the amount of metal introduced into the matrix and its arrangement in the system. We have explored, how the application of cross-linking agents of the same functionality but different structure affects the physicochemical and catalytic properties of the network–Pt systems. We have examined the products taking into account various topology of the matrices and also differences in their cross-linking degree. In this paper, we have presented the synthesis of network–Pt systems and the results of the physicochemical research, such as swelling investigations, UV-vis spectroscopy, Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and thermogravimetric studies. Catalytic activity of obtained materials was determined using isopropyl alcohol conversion as the test reaction. The aim of this investigations was to evaluate the influence of the type of network matrix as well as the amount of platinum introduced on catalytic properties of the network–Pt systems.
Experimental
Chemicals
2,4,6,8-Tetramethylcyclotetrasiloxane (DH4) and tetrakis(dimethylsilyloxy)silane Q(MH)4 (cross-linking agents), received from ABCR Germany, were applied during the syntheses as supplied. Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex (Karstedt's catalyst) solution in xylene (2% Pt) was purchased from Aldrich. Toluene (analytical grade) – solvent used in networks preparation, obtained from POCh (Poland) was initially dried over CaCl2 and subsequently distilled over P2O5 in the flow of argon. Tetrahydrofuran (THF) (Sigma-Aldrich) – solvent used in network–Pt systems preparation and swelling studies, was initially treated with KOH to remove peroxides, then dried by heating over metallic Na and benzophenone mixture and finally distilled in the flow of argon. PtCl4 was purchased from Alfa Aesar (Germany). Al2O3 was received from ERG Bieruń (Poland). H2Cl6Pt × H2O (99.9%) was purchased from Sigma-Aldrich (Germany).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The product of the polymerization – a colorless and transparent, viscous liquid – was subsequently subjected to cross-linking with two selected hydrosiloxanes (DH4 or Q(MH)4). For both types of cross-linking agents, the preparation of polysiloxane networks involved two different molar ratio values of the reagents, i.e. molar ratios of Si–H groups from hydrosiloxane to vinyl groups from the polymer, equaled 1 or 2. Karstedt's catalyst31,32 was used in all hydrosilylation reactions. Its volume applied corresponded to the molar ratio of Pt to Si–H groups (equal to 1 × 10−5). In each experiment, the reagents were introduced into the reaction flask in the flow of argon and the process was conducted with stirring in toluene at the temperature of 60 °C for three days. Finally, the products were dried under vacuum. All cross-linking products were colorless solids and products obtained via cross-linking with the use of a branched hydrosiloxane (Q(MH)4) possessed higher elasticity.
000. The product of the polymerization – a colorless and transparent, viscous liquid – was subsequently subjected to cross-linking with two selected hydrosiloxanes (DH4 or Q(MH)4). For both types of cross-linking agents, the preparation of polysiloxane networks involved two different molar ratio values of the reagents, i.e. molar ratios of Si–H groups from hydrosiloxane to vinyl groups from the polymer, equaled 1 or 2. Karstedt's catalyst31,32 was used in all hydrosilylation reactions. Its volume applied corresponded to the molar ratio of Pt to Si–H groups (equal to 1 × 10−5). In each experiment, the reagents were introduced into the reaction flask in the flow of argon and the process was conducted with stirring in toluene at the temperature of 60 °C for three days. Finally, the products were dried under vacuum. All cross-linking products were colorless solids and products obtained via cross-linking with the use of a branched hydrosiloxane (Q(MH)4) possessed higher elasticity.Network–Pt systems were prepared similarly to the procedure described in our previous work,27 by reduction of Pt4+ ions from PtCl4 solution in THF in the presence of cross-linking products containing un-reacted active Si–H groups possessing reducing properties. The reagents were introduced into the reaction flask under argon. Reactions were carried out by stirring reagents at room temperature for 24 h. In a typical experiment, 1 g of a cross-linked polymer was introduced to the reaction flask and subsequently 9.4 ml of PtCl4 solution was added under vigorous magnetic stirring. In particular experiments, such amount of PtCl4 was used to supply the desired amount of Pt, e.g. 1.5, 3.0 or 6.0 wt%, in the network–Pt system. During syntheses, color change of both the network and the solution was observed, mainly at the initial steps of the process – solids changed from colorless to gray or black-gray and solutions from orange-yellow through gray-brown to almost colorless. These changes occurred faster when solutions contacted with networks containing a branched hydrosiloxane (Q(MH)4). Differences in color change was also observed between similar types of networks but obtained from varying amounts of cross-linking agent. Final solid products of a black-gray color, were filtered, washed with THF, and dried under vacuum. Process of the introduction of metal was monitored using UV-vis spectroscopy. Reference spectra for starting solutions were measured. During syntheses, samples of the reaction mixtures were withdrawn and filtered at selected time intervals. Subsequently, UV-vis spectra of the filtrate solutions were registered. This enabled us to monitor the progress of Pt4+ ions reduction in the presence of a different type of network, especially at its initial stage, when most of the changes was observed. As the reaction proceeded, the absorbtion spectra changed. Finally we could observe very low intensity of absorbtion maxima which allowed us to conclude that practically whole amount of metal from the initial solutions was incorporated into particular matrix, except of C–P_(1:1)–Pt_3 system, for which the final absorbtion spectrum indicated the lower than expected platinum content.
Symbolic descriptions of the prepared polysiloxane networks and the network–Pt systems have been summarized in Table 1.
| Cross-linking agent | Si–H:vinyl ratio |
Symbol of polysiloxane network | Pt content [wt%] | Symbol of network–Pt system |
|---|---|---|---|---|
| DH4 | 1:1 |
C–P_(1:1) |
1.5 | C–P_(1:1)–Pt_1.5 |
| 3.0 | C–P_(1:1)–Pt_3 |
|||
| DH4 | 2:1 |
C–P_(2:1) |
1.5 | C–P_(2:1)–Pt_1.5 |
| 3.0 | C–P_(2:1)–Pt_3 |
|||
| 6.0 | C–P_(2:1)–Pt_6 |
|||
| Q(MH)4 | 1:1 |
Q–P_(1:1) |
1.5 | Q–P_(1:1)–Pt_1.5 |
| Q(MH)4 | 2:1 |
Q–P_(2:1) |
1.5 | Q–P_(2:1)–Pt_1.5 |
| 3.0 | Q–P_(1:1)–Pt_3 |
The catalyst Al2O3–Pt_0.1 – used for comparison – was prepared by dissolving H2Cl6Pt × H2O in deionized water at 80 °C. Aluminum oxide (SBET = 112 m2 g−1) was added to the hot solution of chloroplatinic acid and stirred at 80 °C until a paste was formed. The resulting paste was dried at 120 °C for 24 h. After that the calcination of the sample in static air at 550 °C for 6 h was performed. Final catalyst contained 0.1 wt% of platinum.
| nFR = −[ν2 + ν22χ + ln(1 − ν2)]/V1(ν21/3 − 0.5ν2) |
Measurements of samples' density were conducted on a helium pycnometer (Micromeritics, AccuPyc II 1340). Samples were initially purged with helium (100 times) and subsequently analyzed at room temperature. For each sample 30 single measurements were performed (standard deviation lower than 1%). The term “density” instead of “samples' density” will be applied further in the document.
Fourier transform infrared (FTIR) spectra in the middle infrared range (400–4000 cm−1) were recorded in the transmission mode on a BioRad FTS60v spectrometer using KBr pellet technique. The resolution in the single experiment was equal to 4 cm−1. Quantitative analysis was performed using baseline-corrected spectra. Area ratios of bands due to Si–H bond at 2170 cm−1 or 2135 cm−1 (depending on cross-linking product) and C–H bond from SiCH3 groups at 1261 cm−1 were calculated.
UV-vis spectra were recorded on a Hewlett-Packard HP 8453 spectrophotometer equipped with a diode array detector. All spectra were measured in the range of 190–1100 nm with the resolution equal to 1 nm.
XRD patterns of the powder samples were measured on a Philips X'Pert XRD diffractometer using Cu Kα radiation. Reflections positions were determined using Panalytical X'Pert HighScore software.
Scanning electron microscopic (SEM) analyses were conducted on a Nova Nanosem 200, FEI Co. microscope equipped with the back-scattered electrons (BSE) detector and the EDS system. The samples attached to the SEM stubs by conductive silver paste were vacuum-sputtered with a thin carbon layer.
Transmission electron microscopy (TEM) studies were performed on a Jeol JEM 1011 microscope (with maximum magnification 500000×) at 100 kV equipped with energy dispersive X-ray (EDX) detector. Samples were suspended in ethanol. A drop of suspension poured onto a carbon film equipped copper grid and followed by evaporation of the solvent.
Thermogravimetric analyses were carried out on a NETZSCH STA 449 F3 Jupiter® TA instrument in the temperature range of 20–1100 °C, at a rate of 10 K min−1 under argon. Samples were placed in Al2O3 crucibles.
000 h−1 in all measurements. In each experiment approximately 0.5 g of the sample was used. The conversion of isopropyl alcohol in the applied temperature range was lower than 20% for all experiments. In order to preserve steady state conditions, after each increase of temperature, several portions of the products mixture were analyzed. The main measurements were performed, when the increase of the products concentration stopped. The separation of the reaction products was performed using the capillary column of DB FFAP type with the diameter 0.32 mm and the length 60 m, while the thickness of the film was 0.25 μm. The products were analyzed in situ applying the gas chromatograph Agilent 7890A with a flame ionization detector (FID). Before the reaction all catalysts were standardized in the reactor at 400 K for 1 h in the nitrogen flow in order to remove residual water.Based on the experimental data the rates of the reaction, the selectivities and the apparent activation energies were calculated.
Results and discussion
Characterization of polysiloxane networks
Polysiloxane networks examined in present work were obtained by the hydrosilylation reaction of polyvinylsiloxane D4/V4 with two hydrogenosiloxanes DH4 or Q(MH)4. The formulas of the compounds used in reactions are shown in Fig. 1. Unlike our previous work, the applied cross-linking agents possess different structure but the same functionality – the same number of Si–H groups in each compound. We expected, however, that the application in hydrosilylation reaction of cross-linking compounds containing Si–H groups in the flexible, branched molecule Q(MH)4 or in the rigid cyclic units of DH4 may result in receipt of systems of variable cross-linking density. This in turn may affect different availability of desirable groups during the reduction of platinum ions, at the stage of introduction of metal particles into the polysiloxane network matrix. The degree of cross-linking was an important parameter characterizing the received networks. Cross-linking density of obtained products was calculated on the basis of their swelling in tetrahydrofuran (THF). This solvent was used for two reasons. The first one was a good solubility of polysiloxanes in THF and in turn, easily swelling of cross-linked polysiloxanes in this solvent. The second reason was the planned use of THF as a medium in the reduction process due to a good solubility of PtCl4 in this solvent. The introduction of platinum from the metal salt solution in THF required knowledge of the amount of solvent, which would provide a contact of the solution with the entire volume of the sample, and thereby better distribution of metal in the matrix.The results of swelling research has been shown in Table 2. They indicate that both the type of applied cross-linking agent as well as its amount affect on swelling degree of particular networks. Despite the same functionality of both cross-linking agents, these results clearly reveal the influence of a variety of structure of these compounds on swelling. Swelling degree of networks obtained with the application of Q(MH)4 are higher, thus cross-linking density (nRF) lower than in networks obtained with using DH4. This is particularly evident after application of the excess of cross-linking agent. The value of average molecular weight between cross-links (Mc) calculated for Q–P_(2:1) sample is almost sixfold higher than for C–P_(2:1). The differences between the samples in which the ratio of Si–H to vinyl groups was 1:1, are not so significant. In those cases, Mc values are 822 g mol−1 and 701 g mol−1 for Q–P_(1:1) and C–P_(1:1) respectively. It is worth mention, that the application of the excess of cross-linking agent has a different influence on the cross-linking degree in particular types of networks. Namely, at the excess of DH4 the network of a higher cross-linking density (sample C–P_(2:1)) is formed but in the case of a product obtained with the application of the excess of Q(MH)4, almost threefold decrease of cross-linking density is observed (sample Q–P_(2:1) vs. sample Q–P_(1:1)).
| Network | Swelling in THF: weight increase [%] | nRF [mol cm−3] | Mc [g mol−1] | Density [g cm−3] |
|---|---|---|---|---|
| C–P_(1:1) |
196 | 14.26 × 10−4 | 701 | 1.0517 |
| C–P_(2:1) |
168 | 23.08 × 10−4 | 433 | 1.0806 |
| Q–P_(1:1) |
203 | 12.87 × 10−4 | 822 | 1.0574 |
| Q–P_(2:1) |
287 | 4.36 × 10−4 | 2397 | 1.0461 |
We would also like to pay attention to the density of cross-linked products (Table 2). The decrease of density for particular samples is as follows: C–P_(2:1) > Q–P_(1:1) > C–P_(1:1) > Q–P_(2:1). It should be noted a good correlation between density and cross-linking density in the case of samples with significantly different cross-linking degree and samples of similar type (i.e. C–P or Q–P networks). Thus, the highest density is observed for the highest cross-linked sample, in turn, the lowest cross-linked product shows the lowest density.
Physicochemical properties of network–platinum systems
FTIR spectra of all starting polysiloxane networks obtained from D4/V4 polymer cross-linked with the hydrogenosiloxanes DH4 or Q(MH)4, and the spectra of systems with incorporated Pt, are shown in Fig. 2 and 3. All recorded spectra are characteristic for polysiloxane network formed by the hydrosilylation reaction.35,36 Characteristic bands attributed to Si–CH2–CH2–Si linkages appear at about 2880 cm−1 and 2910–2917 cm−1 for C–H stretching vibrations, and in the range of 1137–1142 cm−1 for C–H bending vibrations. In the range of 1017–1095 cm−1 double band corresponding to Si–O–Si stretching vibrations is observed. Set of bands characteristic of C–H bond from Si–CH3 groups appears at about 1262 cm−1 (symmetric bending vibrations), 1409–1413 cm−1 (asymmetric bending vibrations), 2904–2908 cm−1 (symmetric stretching vibrations) and 2963–2965 cm−1 (asymmetric stretching vibrations).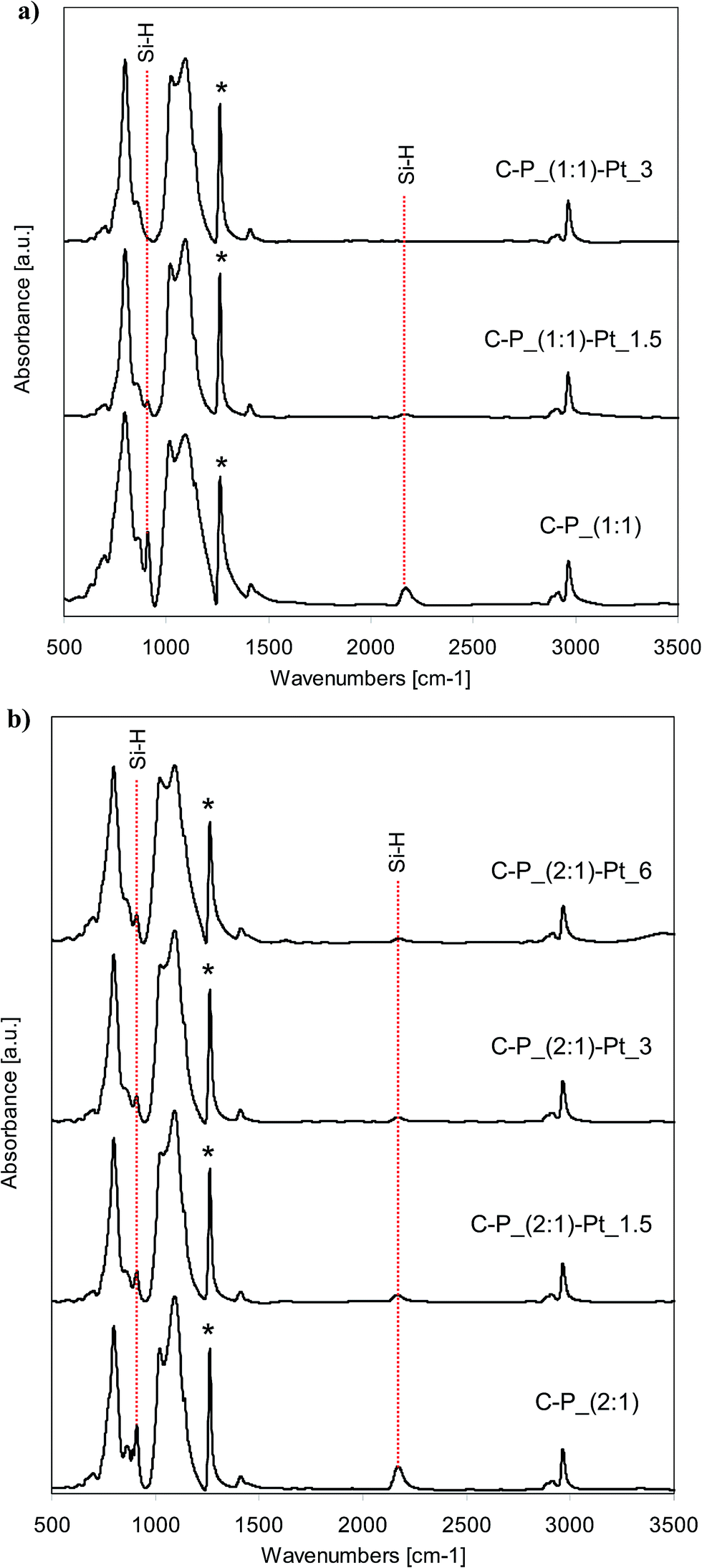 | ||
| Fig. 2 FTIR spectra of selected networks before and after the introduction of platinum: (a) C–P_(1:1) type; (b) C–P_(2:1) type. | ||
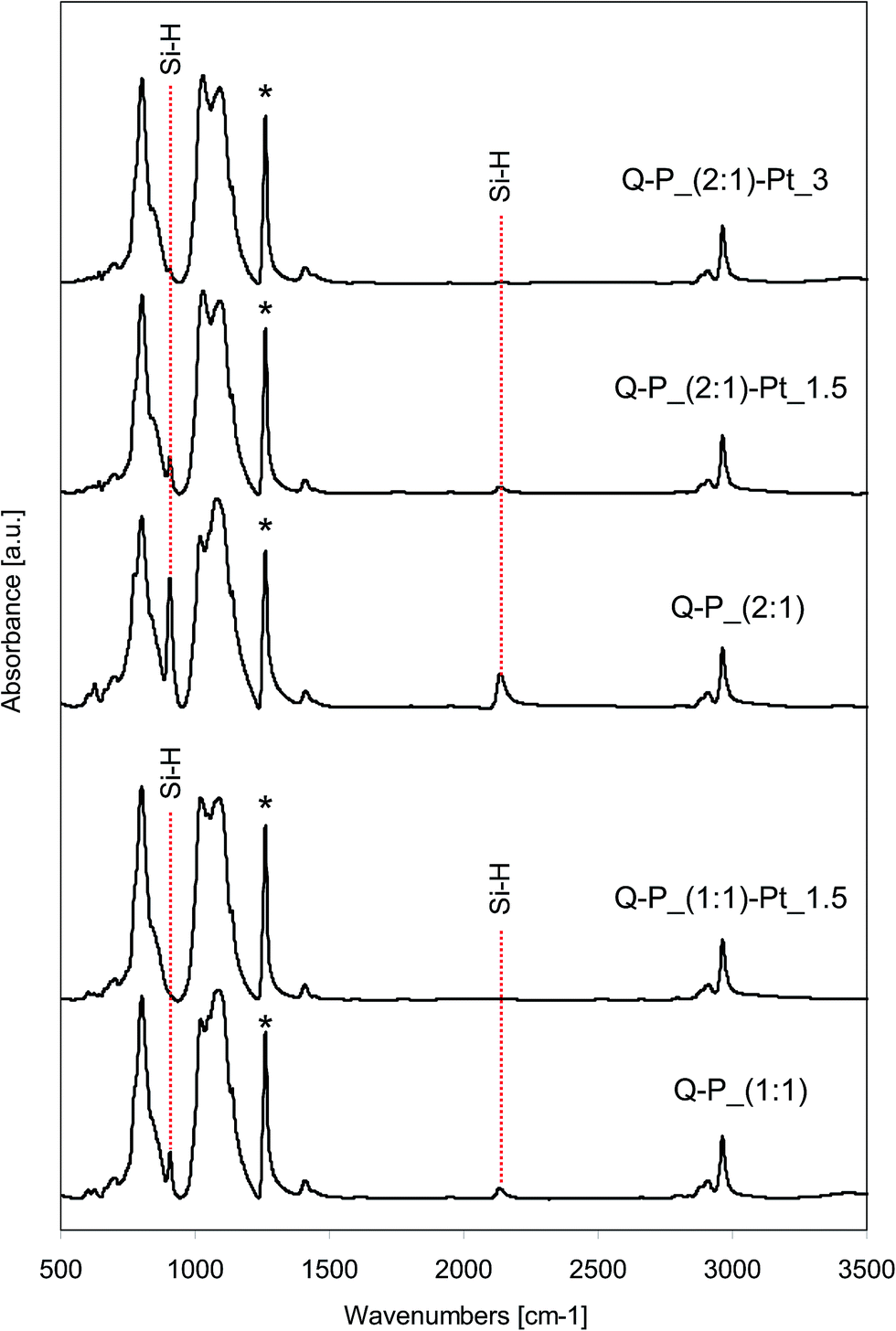 | ||
| Fig. 3 FTIR spectra of selected networks before and after the introduction of platinum: Q–P_(1:1) and Q–P_(2:1) type. | ||
All spectra of starting networks contain the bands corresponding to Si–H groups. Depending on the cross-linking agent applied, they are located at 2170 cm−1 (Si–H stretching vibrations) and 910 cm−1 (Si–H bending vibrations) for DH4, while in the case of Q(MH)4 the bands attributed to the mentioned vibrations are observed at 2136 cm−1 and 905 cm−1 respectively. Remaining Si–H groups are essential for the reducing properties of the networks. In the spectra of systems after the incorporation of platinum, the intensities of bands originating from Si–H groups decrease significantly when compared to the spectra of starting samples (Fig. 2 and 3). Pt4+ ions present in solutions of PtCl4 in THF interact with networks containing active sites causing the reduction of metal ions. The consumption of Si–H groups changes depending on the amount of platinum ions applied for the reduction. Thus, the more Pt ions the lower intensity of characteristic bands is observed. However, the differences in consumption of Si–H groups depend on the applied network. Namely, in the case of C–P_(2:1)–Pt_6, into which 6 wt% of platinum was incorporated, the bands ascribed to Si–H groups are still seen after the reduction, while in the spectrum of Q–P_(2:1)–Pt_3 sample (3 wt% of Pt introduced), these bands are of relatively lower intensity. Comparing the spectra of samples with the same amount of platinum incorporated (C–P_(1:1)–Pt_1.5 and Q–P_(1:1)–Pt_1.5), in the first one the characteristic bands at 2170 cm−1 and 910 cm−1 are seen, whereas in the spectrum of the second sample, the respective bands at 2136 cm−1 and 905 cm−1 are not observed. As FTIR spectroscopy allows the study of changes in the amount of Si–H groups in polysiloxanes,35,37,38 in order to assess the degree of conversion of Si–H groups, quantitative analysis of the spectra of the starting networks and the systems after the incorporation of metal has been carried out. Area ratios of bands due to Si–H bond and C–H bond from SiCH3 (which remain unchanged during the reaction – in the spectra denoted with the asterisk) has been calculated. The results are collected in Table 3. Cross-linking agents, i.e. DH4 or Q(MH)4, used for the preparation of networks contain the same number of Si–H groups in the molecule but a different number of Si–CH3 groups. Therefore, the absolute values of area ratio can not be compared in the systems obtained with the application of different cross-linking agents. In such cases, it is useful to compare the conversion of Si–H groups (values in Table 3, last column), calculated according to the formula given under the table. Decrease of Si–H groups is much more significant in the (1:1) than in (2:1) type of networks, when the excess of a cross-linking agent was applied. This phenomenon may be explained by the different content of free Si–H groups remained in the cross-linked systems. In the case of (1:1) networks obtained with a less amount of cross-linking agent and containing – most probably – lower number of Si–H groups, to introduce a comparable amount of platinum the relatively higher conversion of Si–H groups must take place, than in the case of (2:1) networks containing more Si–H groups.
| Network | SiH/SiCH3 area ratio | Network–Pt system | SiH/SiCH3 area ratio | Conversion of Si–H groupsa [%] |
|---|---|---|---|---|
| a The conversion of Si–H groups (%) is calculated as follows: 100% − (the molar ratio of Si–H/SiCH3 (from spectra of network–Pt systems) to Si–H/SiCH3 (from spectra of starting networks) × 100%). | ||||
| C–P_(1:1) |
0.373 | C–P_(1:1)–Pt_1.5 |
0.059 | 84.18 |
| C–P_(1:1)–Pt_3 |
— | 100.00 | ||
| C–P_(2:1) |
0.747 | C–P_(2:1)–Pt_1.5 |
0.130 | 82.60 |
| C–P_(2:1)–Pt_3 |
0.073 | 90.23 | ||
| C–P_(2:1)–Pt_6 |
0.045 | 93.98 | ||
| Q–P_(1:1) |
0.086 | Q–P_(1:1)–Pt_1.5 |
— | 100.00 |
| Q–P_(2:1) |
0.372 | Q–P_(2:1)–Pt_1.5 |
0.048 | 87.10 |
| Q–P_(1:1)–Pt_3 |
0.007 | 98.12 | ||
Analyzing the results of IR studies and swelling investigations, it can be concluded that the cross-linking degree of the applied networks affects significantly the conversion of Si–H groups during the introduction of platinum. In the systems obtained with the application of Q(MH)4 the loss of the greater number of these groups is observed. Q–P_(1:1) and Q–P_(2:1) networks exhibited a lower degree of cross-linking (Table 2). The lower conversion of Si–H groups has been found in systems prepared using DH4, which matrices show a higher cross-linking degree (Table 2). These results can be explained taking into account the availability of Si–H groups for metal ions in the reaction with the polysiloxane networks. The reduction process was conducted in solution of PtCl4 in THF in which polysiloxane networks were swollen (see section Experimental). In systems with a lower cross-linking degree, exhibiting higher swelling, Si–H groups were available not only on the surfaces of the matrix but also in its bulk, which led to higher degrees of conversion of Si–H groups. In the case of systems with a higher cross-linking degree, thereby indicating lower swelling, the metal ions could react mainly with Si–H groups located on the surface of matrix, hence lower conversion of these groups is observed.
Both compounds applied as cross-linking agents are of the same functionality but Si–H groups present in the flexible, branched molecule Q(MH)4 are more accessible to Pt4+ ions than those at the rigid cyclic units derived from DH4. Thus, the extent of the loss of Si–H groups shows their availability for the metal ions during the reaction with the polysiloxane networks.
Moreover, the analysis of Si–H groups consumption in these systems allows to evaluate simultaneously the amount of metal which can be incorporated into the particular networks.
The consumption of Si–H groups, accompanying the reduction of Pt ions which was controlled using UV-vis spectroscopy (see section Experimental) can lead to the conclusion, that depending on the matrix applied different amount of platinum can be incorporated into investigated networks.
Analysis of the UV-vis spectra clearly shows the progress of the reduction of platinum ions in the presence of polysiloxane networks (Fig. 4). In all cases, the UV-vis spectra change rapidly after contacting the networks with PtCl4 solutions. Characteristic absorption for Pt4+ in UV-vis spectral range vanishes which proves gradual disappearance of Pt4+ ions from the solutions due to their reduction. It should be noted, that changes between investigated systems was observed mostly at first minutes or hours of reduction process. Based on the selected set of spectra we would like to pay attention to the essential differences between particular samples. Comparing systems Q–P_(1:1)–Pt_1.5 and Q–P_(2:1)–Pt_1.5 (Fig. 4a and b), much quicker loss of Pt4+ ions in the latter system can be seen. This steams undoubtedly from the amount of Si–H groups in matrices. Moreover, so pronounced decrease of the absorption – after 15 min in the case of Q–P_(2:1)–Pt_1.5 system – may also indicate the much easier migration of Pt ions in the matrix of lower cross-linking density (Table 2).
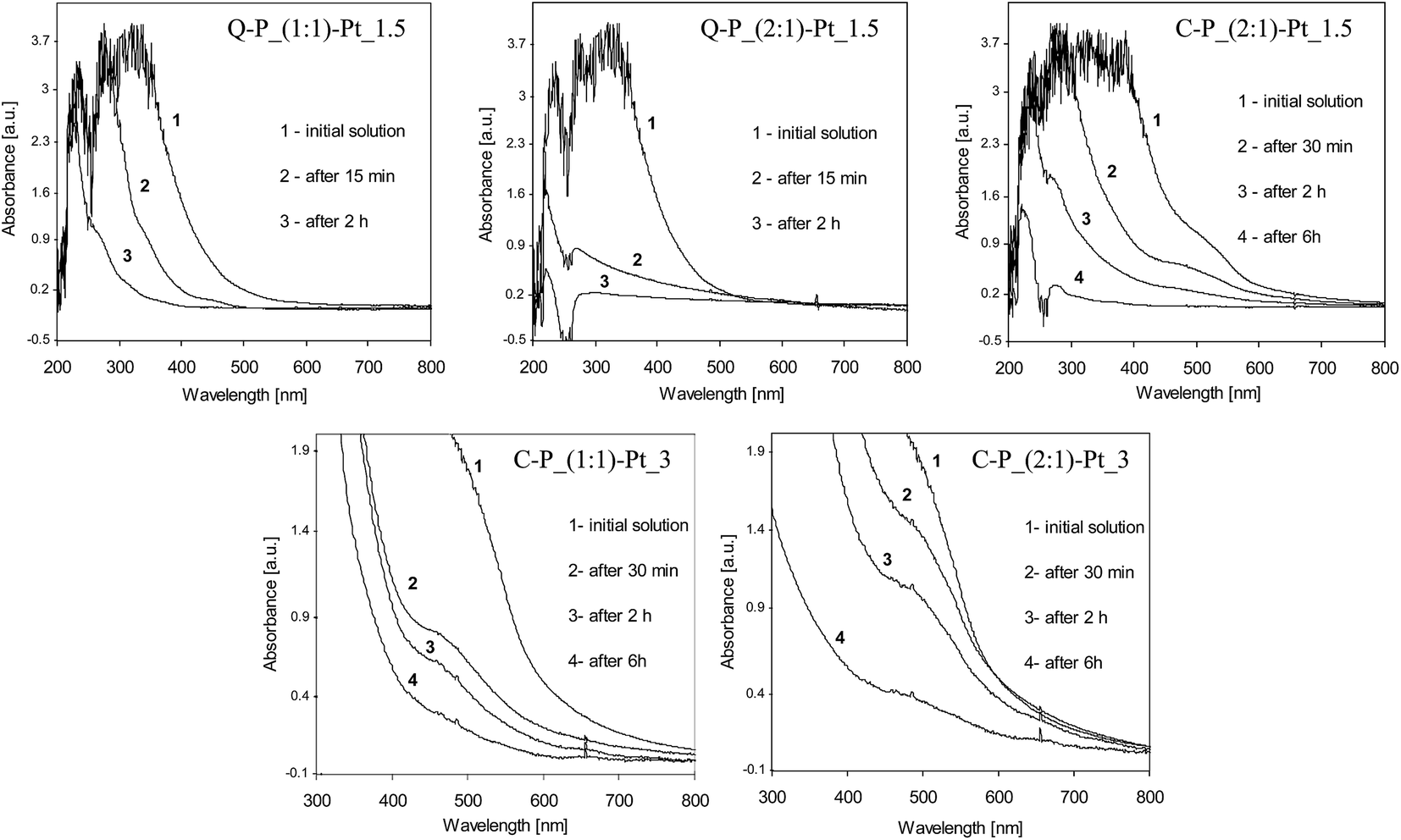 | ||
| Fig. 4 UV-vis spectra of the PtCl4 solutions contacting with different polysiloxane networks recorded during syntheses of the selected network–Pt systems. | ||
As one can see, the structure of cross-linking agents is the important factor influencing the rate of the reduction. Significant differences between systems Q–P_(2:1)–Pt_1.5 and C–P_(2:1)–Pt_1.5 are observed (Fig. 4b and c). A similar effects of reduction are achieved after 15 min in Q–P_(2:1)–Pt_1.5 system or after more than 2 hours in the case of C–P_(2:1)–Pt_1.5. In spite of potentially similar amount of Si–H groups, these matrices exhibit significant differences of cross-linking degree. The active sites are not so easily available and the migration of Pt ions is more difficult in C–P_(2:1) matrix. Hence, the reduction process occurs much slower in this case.
Differences between matrices C–P_(1:1) and C–P_(2:1) were more evident during the introduction of the higher amount of platinum (C–P_(1:1)–Pt_3 and C–P_(2:1)–Pt_3 systems – Fig. 4d and e; for better clarity, the closer inspection of these spectra have been presented). The behaviour of above mentioned matrices differs from the one observed for matrices Q–P_(1:1) and Q–P_(2:1). Namely, in the case of C–P_(1:1) matrix the reduction occurs much quicker at the beginning (after 30 min, as registered) of the process and then it becomes slower. On the contrary, in C–P_(2:1) matrix, the reduction at the beginning of the process occurs not so rapidly. Decrease of the number of Pt ions is more gradual, but after about 6 hours the greater loss of Pt ions than in the C–P_(1:1) matrix can be observed. Hence, the reduction occurs at first in the areas where the migration of metal ions is easier, e.g. the less cross-linked ones. The metal ions migrate then into the higher cross-linked areas containing less accessible active sites, and the process becomes slower.
Therefore it can be concluded, that the cross-linking density is the essential factor influencing the rate of the reduction, and – most probably – is prior to the amount of the potential reactive Si–H groups.
Reduction of Pt4+ ions from the solutions resulting in the formation of metallic platinum particles which are introduced into polysiloxane networks has been confirmed by XRD studies. X-ray diffraction patterns of network–Pt systems as well as of the starting networks are shown in Fig. 5 and 6. It can be seen, that all patterns contain a broad reflections located at the 2θ range of 3–30° corresponding to the amorphous polymer phase39,40 and indicating the short-range ordering only. After incorporation of Pt, these reflections are preserved. However, additional new reflections at 2θ values equal to 39.8°, 46.2° and also 67.5° (more pronounced in diffractograms of the samples with higher Pt contents) appear. These additional reflections can be unambiguously ascribed to crystalline metallic Pt phase, e.g. Pt (111), Pt (200) and Pt (220) planes, respectively.41–43 It is worth mention that relative intensities of the reflections ascribed to metallic Pt and the network depend on the amount of metal in the sample. They are higher for the samples containing more Pt. It can be also seen that the XRD patterns of Pt-systems with Q–P_(2:1) matrix show reflections more sharp than the corresponding systems with C–P_(2:1) matrix. This can indicate various size of Pt crystallites in appropriate matrices. Based on X-ray diffraction patterns, the average size values of platinum crystallites have been calculated (basing on the width of [111] reflection) using Scherer's equation.44 Obtained values are in the range from 2.7 to 4.8 nm in the case of Pt dispersed in C–P_(1:1) or C–P_(2:1) matrices, and 5.4–6.3 nm for Pt dispersed in Q–P_(1:1) or Q–P_(2:1) ones. Hence, on the basis of XRD results it can be concluded that metal nanoparticles have been formed in all Pt-containing systems. The greater mean size of Pt crystallites has been appointed for systems based on Q–P type matrices with the lower cross-linking degree (the higher distances between cross-links (Mc) – Table 2). For the selected Pt-systems containing 1.5 wt% of platinum, the mean size of crystallites grows as follows: C–P_(2:1)–Pt_1.5 < C–P_(1:1)–Pt_1.5 < Q–P_(1:1)–Pt_1.5 < Q–P_(2:1)–Pt_1.5 which corresponds to the order of the cross-linking density of starting networks.
 | ||
| Fig. 5 XRD patterns of selected networks and products after incorporation of Pt species: (a) C–P_(1:1) type; (b) C–P_(2:1) type. | ||
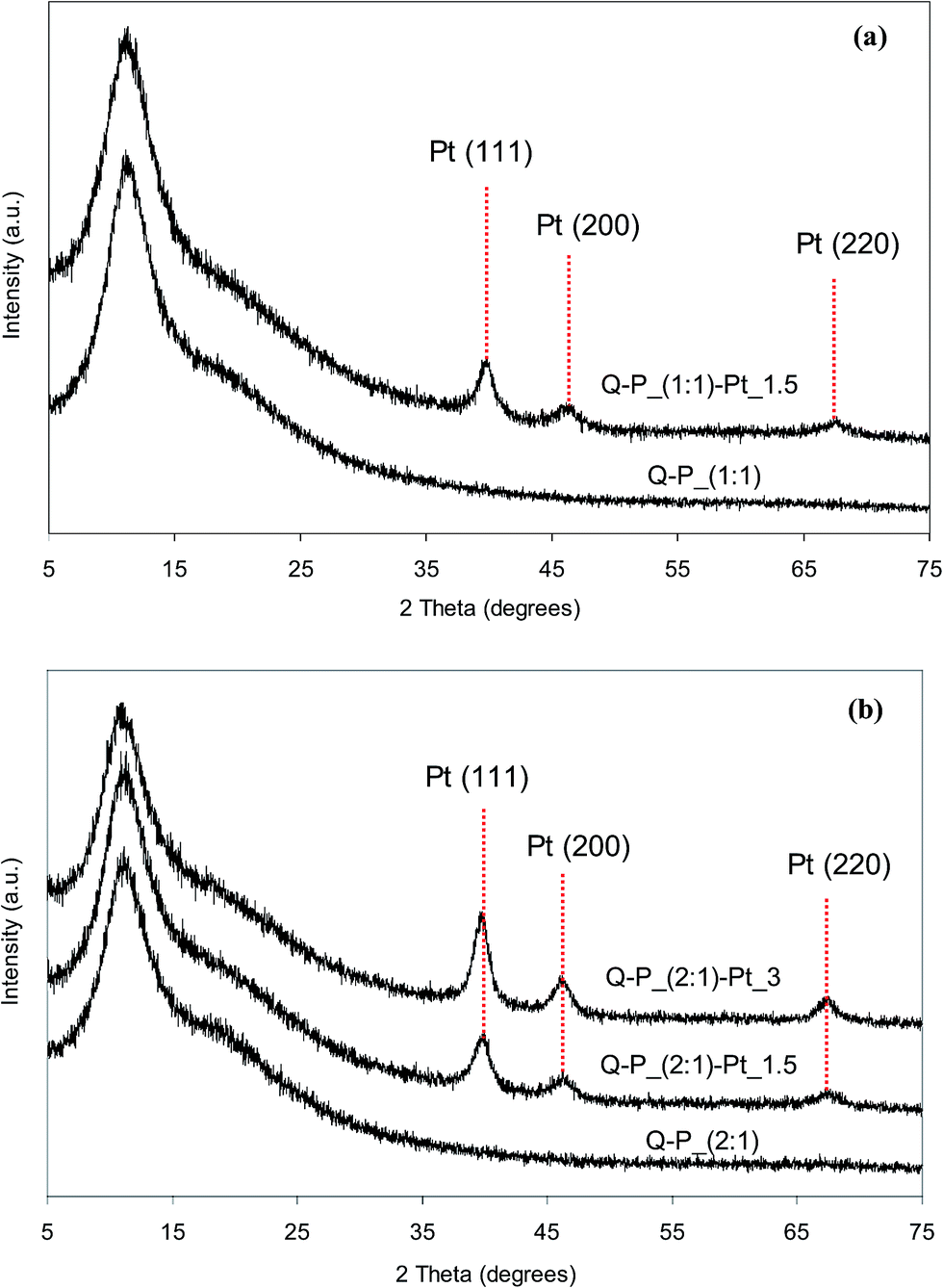 | ||
| Fig. 6 XRD patterns of selected networks and products after incorporation of Pt species: (a) Q–P_(1:1) type; (b) Q–P_(2:1) type. | ||
Thus, XRD results prove that reduction of Pt4+ ions with Si–H groups in networks investigated, yields two-component systems consisting of the polysiloxane network phase into which various amounts of metallic Pt nanoparticles are dispersed. The size of crystallites is connected with a cross-linking density of matrices.
For further characterization of materials after incorporation of platinum particles, swelling investigations and density tests were carried out. The results of these experiments compared with data obtained for starting networks are shown in diagrams (Fig. 7 and 8). It can be seen, that the incorporation of platinum into polysiloxane networks causes the increase of density in all obtained products (Fig. 7) – density of Pt-containing systems increases with the amount of platinum introduced. The most distinct change of this property is observed after the incorporation of 1.5% of platinum, while the introduction of larger amount of metal has only minor influence, probably due to the agglomeration of metal particles. Relatively higher increase is observed for samples containing C–P_(1:1) or C–P_(2:1) than Q–P_(1:1) or Q–P_(2:1) networks. Such behavior is undoubtedly connected with the structure of a cross-linking agent. The application of rigid molecule of DH4 creates networks with a higher cross-linking density and a lower Mc of starting networks (Table 2). In these Pt-systems “gaps” created between polysiloxane chains can be filled with metal particles (smaller ones, accordingly to XRD results), giving a more significant increase of density as compared with samples containing Q–P_(1:1) or Q–P_(2:1) matrices with a lower cross-linking density.
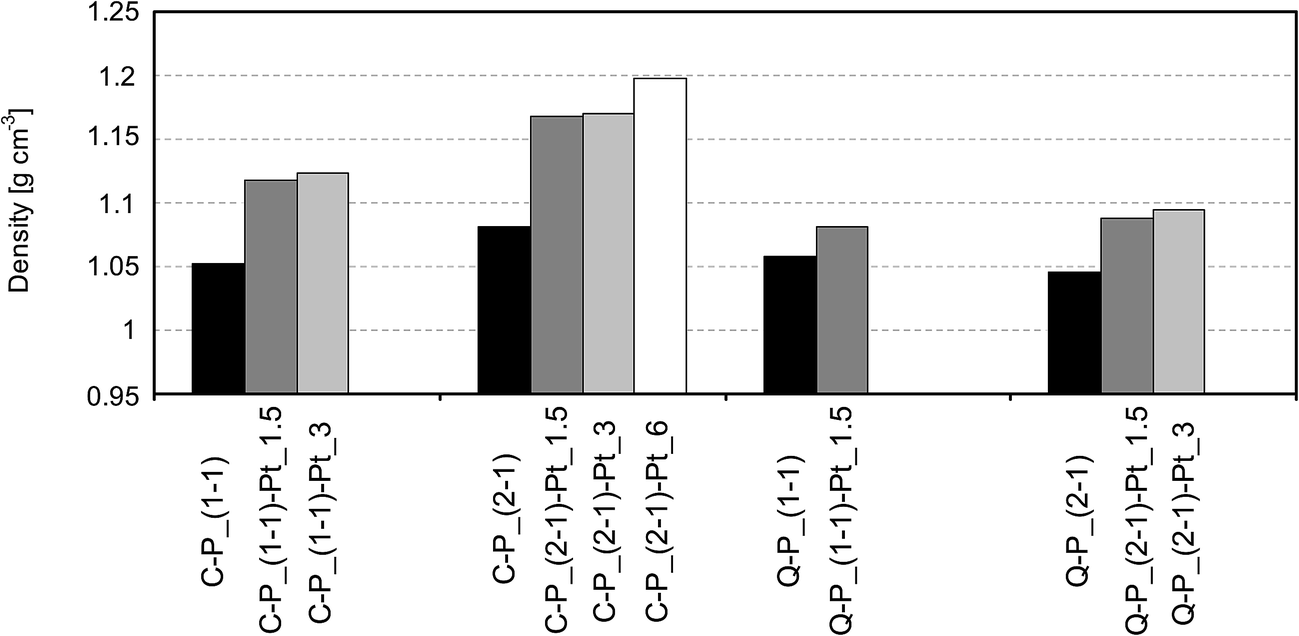 | ||
| Fig. 7 Density comparison for networks and network–Pt systems. | ||
 | ||
| Fig. 8 The results of swelling measurements for networks and network–Pt systems. | ||
The comparison of swelling of Pt-systems with the starting networks shows the increase of swelling degree in each case and furthermore its higher growth with the amount of metal introduced (Fig. 8). This may be caused by a disintegration of polysiloxane networks that can occur as a result of the reaction with platinum ions during the reduction process. Significantly higher increase of swelling is observed in the case of samples with matrices Q–P_(1:1) and Q–P_(2:1). These results can indicate the higher level of disintegration in systems of lower cross-linking degree. Although, comparing Pt-systems prepared on the basis of C–P_(1:1) and Q–P_(1:1) networks, for which nRF do not differ significantly (Table 2), it can be concluded, that the crucial factor of such behavior is the structure of cross-linking agents used for the preparation of these networks. The use of Q(MH)4 causes linking of the polysiloxane chains by branched and more flexible bridges which enable better migration of Pt ions and better accessibility of Si–H groups during the reduction, resulting in a higher consumption of these groups, as demonstrated by IR studies (Table 3). This in turn, may cause more evident changes in interactions between polysiloxane chains in a cross-linked structure of polymer in these systems.
Electron microscopic studies of the Pt-containing systems have been undertaken in order to investigate how the type of network as well as the amount of platinum introduced into the matrix influence the metal particles size and their arrangement in particular systems. Two complementary methods, namely, SEM – a surface probe and TEM – the transmission method have been applied. These techniques can provide information about the surface of the analyzed sample or about its bulk. Moreover, due to different magnifications attainable, they allow to examine objects of different size.
SEM micrographs of selected samples, obtained with the application of BSE detector have been presented in Fig. 9–11 and in ESI (Fig. S1 and S2).† It should be noted, that in the case of each examined system, distinct white inclusions are present. It is characteristic of each sample, that these inclusions form mainly areas covering matrice's surface. These areas, as established by EDX, contain mainly Pt atoms (Fig. S1 and S2 in ESI†). There are also other regions visible with lower coverage of platinum or those quite without Pt particles. In SEM images presented in Fig. 9a–c the Pt-enriched regions as well as those with lower coverage of platinum are clearly visible. The micrographs obtained at magnification 20000× show the presence of well separated Pt-particles of very small size and of regular shape, which diameter in Pt-enriched region (Fig. 9b) is about 25–50 nm and smaller. In areas with lower coverage of platinum (Fig. 9c) the larger regular particles are also present with sizes in the range of 0.1–0.3 μm. However, some white inclusions of different shape and size are also seen and indicate, that the small particles may agglomerate (Fig. 9a). Their sizes are in the range of 1–7 μm.
 | ||
| Fig. 9 BSE images of C–P_(1:1)–Pt_3 at magnification: 2000 – overall look (a), and magnification 20000 – selected regions (b) and (c). | ||
It has been found, that the Pt-enriched regions possess different Pt-layer thickness and the occupancy by metal particles in various samples (Fig. 10 and 11). On the basis of the results of average EDX analysis of a typical Pt-enriched area, the weight% ratios of Pt to Si for the selected samples have been calculated (Table 4). These results show the higher metal content in Pt-enriched regions in the case of samples with the higher amount of platinum introduced. The differences are also observed between systems containing (2:1) or (1:1) type of matrices. In (2:1) systems the higher Pt-content has been found (for both C–P and Q–P type matrices). These results indicate higher coverage of platinum in the case of networks with higher amount of Si–H groups involved in the reduction of Pt ions. These differences are clearly visible in Fig. 10, where the images of the edge of the Pt-enriched layers are compared. Their thicknesses were estimated at about 0.7–1.0 μm for C–P_(1:1)–Pt_1.5 and about 1.8 μm for C–P_(2:1)–Pt_1.5. The higher occupation of this layer by Pt particles is observed in the case of C–P_(2:1)–Pt_1.5 sample. Comparing values of Pt-layer thickness recorded for the samples C–P_(2:1)–Pt_1.5 and C–P_(2:1)–Pt_3 it is seen, that this parameter is larger for the latter one (Fig. 10 and 11), what, as mentioned previously, results from different amount of platinum incorporated. The observed increase of Pt-layer thickness with the amount of platinum introduced indicates the presence of regions where the formation of metal particles is favoured. It leads to the conclusion, that these areas were enriched in Si–H groups at the stage of reduction process, so they are less cross-linked as well.
 | ||
| Fig. 10 BSE images of Pt-enriched regions and the corresponding edges at magnifications: 50000 or 10000 respectively for C–P_(1:1)–Pt_1.5 and C–P_(2:1)–Pt_1.5 samples. | ||
 | ||
| Fig. 11 BSE images of Pt-enriched regions and the corresponding edges at magnifications: 50000 or 10000 respectively for C–P_(2:1)–Pt_3 and Q–P_(2:1)–Pt_3 samples. | ||
| Network–Pt system | Pt/Si content ratioa |
|---|---|
| a The weight% ratio of Pt to Si, calculated on the basis of the average EDX analysis at the magnification 10000×. |
|
| C–P_(1:1)–Pt_1.5 |
0.89 |
| C–P_(1:1)–Pt_3 |
1.26 |
| C–P_(2:1)–Pt_1.5 |
1.23 |
| C–P_(2:1)–Pt_3 |
1.43 |
| Q–P_(1:1)–Pt_1.5 |
0.59 |
| Q–P_(2:1)–Pt_1.5 |
0.95 |
| Q–P_(2:1)–Pt_3 |
1.96 |
Comparing systems based on Q–P matrices and the corresponding materials containing C–P ones, the lower platinum content in Pt-enriched coverages is observed in the samples Q–P_(1:1)–Pt_1.5 and Q–P_(2:1)–Pt_1.5 in comparison with C–P_(1:1)–Pt_1.5 and C–P_(2:1)–Pt_1.5 respectively. Simultaneously the same factor is higher for Q–P_(2:1)–Pt_3 sample than for C–P_(2:1)–Pt_3 one (Table 4). It can be suggested, that the cross-linking density of networks may affect the occupation with platinum particles in those cases where the lower amount of metal has been incorporated. Easier migration of Pt ions into Q–P matrices with a lower cross-linking density causes the formation of Pt-particles on the surface of matrix as well as in its bulk, while in the case of C–P matrix, platinum is deposited mainly on the surface. The incorporation of the higher amount of platinum induces the increase of metal content in the Pt-enriched layers in both cases. However, there are regions indicating the much higher coverage by platinum in the Q–P_(2:1) matrix than in the C–P_(2:1) one (Fig. 11).
According to micrographs presented in Fig. 10 and 11 obtained at 50000× magnification, Pt-enriched large areas covering surfaces of selected matrices are very tightly occupied with very small metal particles, and no agglomeration is observed. Although some differences in the occupation of metal particles in particular systems can be seen, such as between C–P_(1:1)–Pt_1.5 and C–P_(2:1)–Pt_1.5 or between Q–P_(2:1)–Pt_3 and C–P_(2:1)–Pt_3 samples. The size of platinum particles in these coverages has been estimated at about 20–50 nm and smaller. However, the exact determination of the size of such small particles using SEM method is rather difficult. For this reason TEM, the complementary microscopic method, has been used. This method enables the analysis of the size of objects at higher magnifications. TEM studies have revealed the presence of Pt nanoparticles in obtained Pt-systems. In micrograph presented in Fig. 12 there are regions where well separated very small particles of spherical shape are clearly visible. The size of these particles was estimated at 2–3 nm. As one can see, these small nanoparticles can form agglomerates, with sizes in the range of 5–30 nm. Similarly to SEM analysis, TEM studies have shown some differences between particular systems. The selected micrographs are presented in Fig. 13. TEM images exhibit more exactly the differences between Q–P_(2:1)–Pt_3 and C–P_(2:1)–Pt_3 samples, already observed in SEM analysis. In the micrographs presented in Fig. 13a and b much more platinum particles in the system with Q–P matrix can be seen. More detailed analysis reveals Pt-particles of different sizes and shapes (Fig. 13c and d). In micrograph of C–P_(2:1)–Pt_3 sample, very small (of 2–3 nm size), spherical particles are visible, which have a tendency to agglomerate. The larger, 4–8 nm spherical particles dominate in Q–P_(2:1)–Pt_3 sample. It can be assumed, that differences in metal particles size in mentioned cases are a consequence of the removal of solvent after the introduction of platinum into the matrix. During the reduction of Pt ions, matrices were swollen and the removal of a solvent caused the shrinkage of samples. The shrinkage of more swollen matrix Q–P_(2:1) led to the formation of larger particles, although of regular shape. The sample containing C–P_(2:1) matrix was less swollen and the smaller Pt particles are present. However, the formation of agglomerates due to the removal of a solvent can not be excluded in that case.
 | ||
| Fig. 12 TEM images of C–P_(2:1)–Pt_6 sample: overall look and selected regions. | ||
 | ||
| Fig. 13 TEM images of selected Pt-containing systems: at magnifications 100000: (a) and (b); 300000: (c)–(f). | ||
Similarly to SEM results, more platinum particles are visible in TEM image corresponding to the sample with higher metal content (Fig. 13c and e). Wherein weaker tendency to the agglomeration in the sample C–P_(2:1)–Pt_1.5 (e.g. the sample containing the lower amount of platinum) can be noted. TEM analysis has revealed also differences between systems containing C–P_(2:1) or C–P_(1:1) matrices (Fig. 13c, e and f). Apart from the presence of small particles (of 2–5 nm size) in the system with C–P_(1:1) matrix, much bigger spherical agglomerates are visible. Their size has been estimated at about 25–30 nm. This indicates better dispersion of metal particles in the matrix containing more active sites (Si–H groups) during the reduction of Pt ions.
Thus, TEM results relating to the particle size remain in a good agreement with results of XRD studies presented earlier. The mean sizes of Pt crystallites determined for systems with Q–P matrices were higher than for those with C–P ones, and higher for systems containing C–P_(1:1) matrix than for those containing C–P_(2:1) one.
Analyzing the results of microscopic studies we can conclude, that the type of matrix applied as well as the amount of platinum introduced affect significantly the size of Pt crystallites, their tendency to the agglomeration and also the manner of metal deposition on the matrix surface. However, it should be emphasized that well dispersed spherical platinum nanoparticles dominate in the investigated Pt-systems.
Thermogravimetric analysis was carried out primarily to determine the stability of cross-linking products and the Pt-containing systems due to their application in catalytic tests. The typical measurement conducted from the room temperature up to 1100 °C enabled also the comparison of the cross-linking degree between particular networks, basing on a ceramic residue. The selected TG curves of obtained products in the full measuring range have been presented in ESI (Fig. S3).† The knowledge of temperatures when the mass loss begins was the prior aim of these measurements. According to the results obtained, TG curves indicate straight course up to about 300 °C and no significant changes in weight loss for both the networks and the network–Pt systems. A strong weight loss is observed in a wide range of 400–800 °C for all samples, and the difference in the rate of weight loss between particular samples was recorded. Different ceramic residue have been obtained for particular networks, indicating various cross-linking degree of these samples.45,46 These data are in a good agreement with the results of the swelling investigations (Table 2). As mentioned previously, the knowledge of the temperature range of the thermal stability of the systems is very important for the application of Pt-systems as heterogeneous catalysts. Detailed analysis of TG curves at about 300 °C has been carried out. Temperature values at which about 1 wt% of mass loss is reached were determined for samples studied and collected in Table 5. In the case of networks such a loss was recorded in temperatures between 341 and 424 °C. The introduction of platinum into matrices causes a drop in temperature of the mass loss beginning in all cases but one, namely Q–P_(2:1) matrix, for which the increase of that temperature has been detected. Furthermore, the Pt-containing samples of Q–P series found to be more stable than the corresponding C–P ones. The lowest temperature was determined for C–P_(2:1)–Pt_3 sample (313 °C) and the highest for Q–P_(2:1)–Pt_3 one (375 °C). Some differences were also observed in the case of samples containing (2:1) or (1:1) type of matrices. A little better stability have shown Pt-systems with (1:1) type of matrix.
| Sample | Temperature [°C] | Sample | Temperature [°C] |
|---|---|---|---|
| C–P_(1:1) |
402 | Q–P_(1:1) |
424 |
| C–P_(1:1)–Pt_1.5 |
357 | Q–P_(1:1)–Pt_1.5 |
373 |
| C–P_(1:1)–Pt_3 |
328 | ||
| C–P_(2:1) |
395 | Q–P_(2:1) |
341 |
| C–P_(2:1)–Pt_1.5 |
337 | Q–P_(2:1)–Pt_1.5 |
361 |
| C–P_(2:1)–Pt_3 |
313 | Q–P_(2:1)–Pt_3 |
375 |
Catalytic properties of Pt-systems
Catalytic tests of platinum supported on the different types of polymer network were performed to elaborate the influence of the structure of polymer matrices and the amount of catalytically active agent on the activity of the samples. The conversion of isopropyl alcohol, often applied as test reaction in the examination of metal oxides47,48 and supported metals,49,50 seemed to be suitable for studies of the catalytic behavior of investigated systems. This process consists of three parallel reactions, namely: dehydrogenation to acetone, dehydration to propene and intermolecular dehydration to diisopropyl ether. First reaction runs over redox or base sites of the catalyst, whereas the latter two use acid active centers. Thus in a single experiment it is possible to get information about the activity of catalytic sites of different nature.The results of activity measurements of examined samples are presented in a form of Arrhenius plots and collected in Fig. 14a and b for conversion to acetone, propene and diisopropyl ether. The selectivities and activation energies are gathered in Table 6.
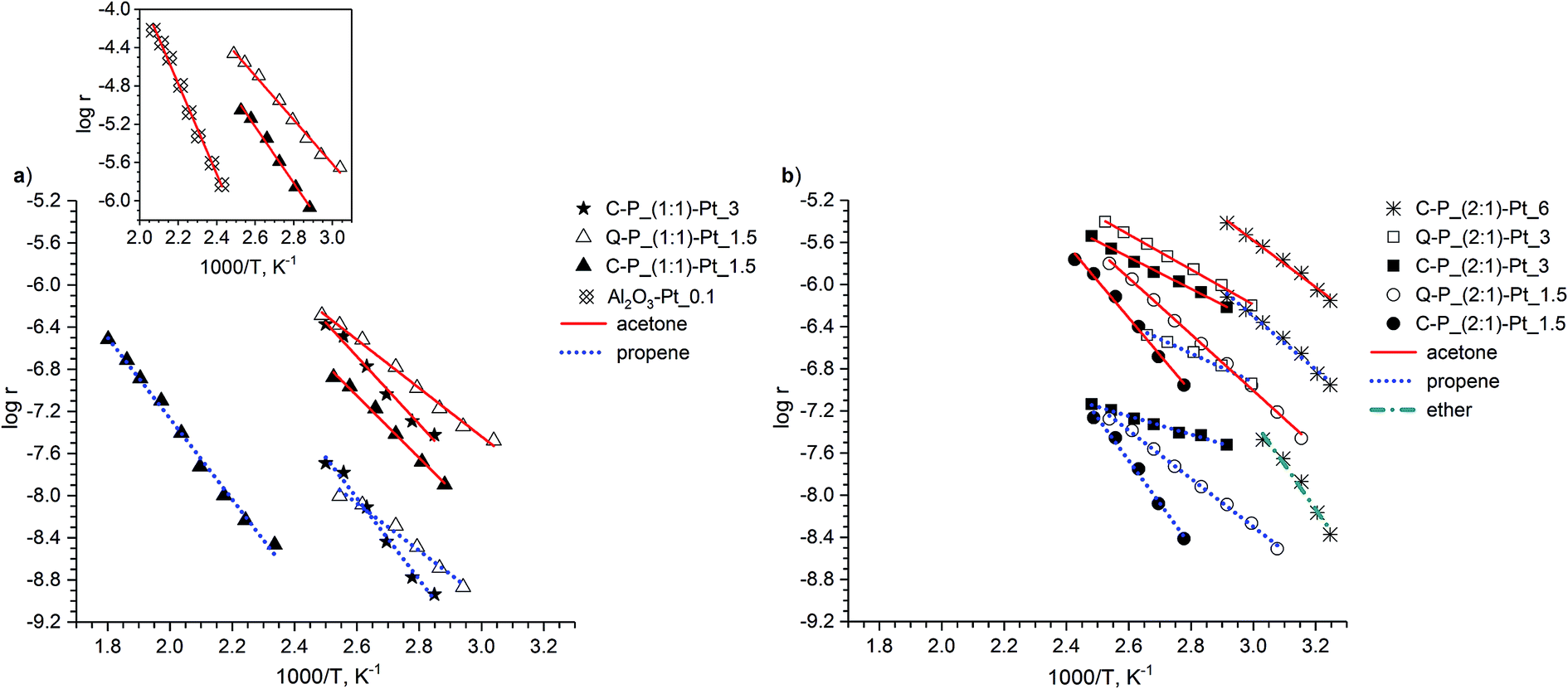 | ||
| Fig. 14 Arrhenius plots of isopropyl alcohol conversion to acetone, propene and diisopropyl ether conducted over examined samples containing: (a) C–P_(1:1) or Q–P_(1:1) matrices, (b) C–P_(2:1) or Q–P_(2:1) matrices. The reaction rate (r) has been expressed (per 1 g of catalyst) in mol gcat−1 s−1. Inset in (a): over selected Pt–network samples and Pt/Al2O3 – conversion to acetone only; for better clarity the reaction rate (r) has been expressed (per 1 g of platinum) in mol gPt−1 s−1. | ||
| Catalyst | Selectivity [%] | Activation energya [kJ mol−1] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Propene | Ether | Acetone | Propene | Ether | Acetone | Propene | Ether | Acetone | Ea1 | Ea2 | Ea3 | |
| T = 360 K | T = 380 K | T = 400 K | ||||||||||
| a Ea – activation energy of isopropyl alcohol conversion: to propene (Ea1); to ether (Ea2); to acetone (Ea3). | ||||||||||||
| C–P_(1:1)–Pt_1.5 |
2.6 | — | 97.4 | 2.7 | — | 97.3 | 2.8 | — | 97.2 | 73.6 ± 2.3 | 56.5 ± 2.4 | |
| C–P_(1:1)–Pt_3 |
3.3 | — | 96.7 | 4.1 | — | 95.9 | 5.0 | — | 95.0 | 74.1 ± 4.5 | 61.5 ± 3.4 | |
| C–P_(2:1)–Pt_1.5 |
3.4 | — | 96.6 | 4.1 | — | 95.9 | 4.8 | — | 95.2 | 77.8 ± 4.3 | 67.3 ± 3.0 | |
| C–P_(2:1)–Pt_3 |
3.9 | — | 96.1 | 3.1 | — | 96.9 | 2.6 | — | 97.4 | 16.8 ± 0.7 | 28.7 ± 1.0 | |
| T = 330 K | T = 340 K | T = 350 K | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C–P_(2:1)–Pt_6 |
13.8 | 0.8 | 5.4 | 14.4 | 1.3 | 84.3 | 5.1 | 2.0 | 82.9 | 35.6 ± 5.6 | 73.0 ± 11.8 | 29.8 ± 3.2 |
| T = 340 K | T = 360 K | T = 380 K | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q–P_(1:1)–Pt_1.5 |
2.8 | — | 97.2 | 2.8 | — | 97.2 | 2.7 | — | 97.3 | 43.1 ± 2.9 | 44.0 ± 1.5 | |
| Q–P_(2:1)–Pt_1.5 |
4.6 | — | 95.4 | 4.0 | — | 96.0 | 3.5 | — | 96.5 | 45.7 ± 0.9 | 51.2 ± 0.8 | |
| Q–P_(2:1)–Pt_3 |
15.0 | — | 85.0 | 13.6 | — | 86.3 | 12.5 | — | 87.5 | 26.2 ± 1.7 | 31.9 ± 0.6 | |
| T = 440 K | T = 460 K | T = 480 K | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3–Pt_0.1 | 2.4 | 0.4 | 97.2 | 5.4 | 0.8 | 97.8 | 11.2 | 1.7 | 87.1 | 163.5 ± 3.3 | 160.9 ± 2.7 | 90.4 ± 2.6 |
Closer inspection to the data reveals that examined catalysts exhibit activity mainly in the dehydrogenation reaction, thus it is suitable to start the discussion of catalytic properties with the matter concerning activity of redox sites. In the formation of acetone the most active is the sample with the highest loading of platinum i.e. C–P_(2:1)–Pt_6 (Fig. 14b). It is also clearly visible that for each type of matrices C–P_(1:1), C–P_(2:1) or Q–P_(2:1) the increase of the amount of platinum entails the rise of activity (Fig. 14a and b). For example, the activity of C–P_(2:1)–Pt series grows as follows: C–P_(2:1)–Pt_1.5 < C–P_(2:1)–Pt_3 < C–P_(2:1)–Pt_6, and this tendency remains the same for other systems. Thus the activity of the samples with the same type of support depends mostly of the concentration of active sites. This assumption is also confirmed relating the values of the density of Pt-containing catalysts (Fig. 7) to their activity. As it was mentioned in the previous part of the discussion, higher loading of platinum implicates higher value of density for both C–P–Pt and Q–P–Pt samples series. It is seen that the enhancement of activity is achieved for samples with higher density, which, in turn, correlates with the rising content of platinum particles. Additional evidence of positive impact of the Pt-doping level on the activity of examined samples gives SEM-EDX analysis (Fig. 9–11). According to those results the most exposed area of catalysts surface, susceptible for penetration of alcohol molecules are those of Pt-enriched regions. It is observed that higher content of platinum in Pt-enriched regions are characteristic of samples with higher amount of platinum introduced.
To visualize the influence of the properties of polymer matrices it is convenient to compare activities of corresponding type of the supports with the same loading of platinum. For these reasons the catalytic behavior of three sets of catalysts i.e.: Q–P_(1:1)–Pt_1.5 and C–P_(1:1)–Pt_1.5, Q–P_(2:1)–Pt_1.5 and C–P_(2:1)–Pt_1.5, Q–P_(2:1)–Pt_3 and C–P_(2:1)–Pt_3 are taken into account (Fig. 14a and b). Closer inspection at the results of the conversion to acetone reveals that in each case Q–P supported systems are more active than these with corresponding C–P network. This behavior is evidently connected with the structure of the cross-linking agent. The use of more flexible bridge of Q(MH)4 induces better accessibility of active centers and allows for easier penetration of the reacting molecules in the bulk of corresponding matrix. It is even more clear, regarding the values of density of the examined samples (Fig. 7). The Q–P–Pt samples, with lower density and higher degree of swelling (Fig. 8) than corresponding C–P–Pt ones, reveal higher level of the disintegration of the system, which in turn induces better accessibility of the platinum sites. The accessibility seems to be the most important feature predominating the crystallite size factor. The average size of platinum crystallites is higher for more active Q–P–Pt systems comparing to C–P–Pt. Most likely the shielding effect of the network is stronger for polymer with more rigid cross-linking agent DH4, which induces narrow gaps for incorporated platinum particles, higher density (Fig. 7) and as a result worse accessibility of active sites. TEM analysis (Fig. 13) reveals that for Q–P_(2:1)–Pt_3 sample Pt particles number is higher and the size of crystallites is about two times greater than for C–P_(2:1)–Pt_3. It can suggest, that larger platinum particles in Q–P matrix are sticking out of the polymer network and in such position are more exposed to the adsorption of reacting molecules, which ameliorates catalytic activity. Additionally SEM results show much greater content of platinum in the Pt-enriched regions in Q–P_(2:1)–Pt_3 matrix than in C–P_(2:1)–Pt_3 one (Table 4), which can probably enhance concentration and attainability of platinum sites incorporated into Q–P network in this case. Unlike to systems with higher platinum content, the drop of platinum amount in Pt-enriched regions is observed for Q–P_(1:1)–Pt_1.5 and Q–P_(2:1)–Pt_1.5 in comparison with C–P_(1:1)–Pt_1.5, and C–P_(2:1)–Pt_1.5. However Q–P–Pt samples are still more active than corresponding C–P–Pt ones. It seems that in Q–P network active sites are localised both on the surface and in the bulk, while in C–P matrix platinum is deposited mainly on the surface. It leads to the conclusions that less dense structure of Q–P network facilitates access to active centers more efficiently regardless their localization. The amount of cross-linking agent seems to have great influence on the activity of examined samples. The comparison of the catalytic performance of Q–P_(1:1)–Pt_1.5 and Q–P_(2:1)–Pt_1.5, C–P_(1:1)–Pt_1.5 and C–P_(2:1)–Pt_1.5, C–P_(1:1)–Pt_3 and C–P_(2:1)–Pt_3 materials in the conversion of isopropyl alcohol to acetone reveals, in each case, higher activity of samples with (2:1) type of matrix. According to SEM-EDX analysis (Table 4), content of the platinum in Pt-enriched regions is always higher for (2:1) systems. It implies higher coverage of platinum in the networks with higher amount of Si–H groups participating in the reduction. Higher coverage gives higher concentration of active sites, which in turn improves catalytic activity of these samples.
As it was shown in the previous part of discussion, all examined samples reveal mainly redox activity. Therefore it is necessary to compare these properties with commonly used standard platinum catalytic system. For this reason the study of redox activity of selected samples referred to the Pt/Al2O3 catalyst was performed in the isopropyl alcohol conversion. To avoid ambiguities concerning the influence of different amount of platinum, rates of the conversion to acetone for chosen samples and for Pt/Al2O3 system were related to the 1 g of Pt. According to the results of conversion to acetone (Fig. 14a – inset) in the lower range of temperature even less active samples such as C–P_(1:1)–Pt_1.5 and Q–P_(1:1)–Pt_1.5 reveal stronger redox sites than Al2O3–Pt_0.1 catalyst. It suggests better redox activity in lower temperature of all examined platinum polymer supported catalysts than the standard Pt/Al2O3 system used for comparison.
Regarding the results of dehydration of isopropyl alcohol to propene (Fig. 14a and b) it is evident that the activity of samples rises with the increase of the amount of incorporated platinum. The most active catalyst is C–P_(2:1)–Pt_6 whereas the lowest activity reveals C–P_(1:1)–Pt_1.5 sample. Similarly, for Q–P–Pt systems, the most active is Q–P_(2:1)–Pt_3 with highest loading of platinum and the least active is Q–P_(1:1)–Pt_1.5 with lowest content of Pt. Such behavior is probably due to the generation of acid sites during the reaction of the reduction of Pt4+ ions by the hydrogen from Si–H groups. Higher amount of platinum ions demands higher number of reactive groups of polymer network and induces higher concentration of available acid centers. Comparing catalytic properties of Q–P–Pt samples and corresponding C–P–Pt ones in the conversion to propene it is well seen that Q–P supported samples are more active in each case. It might be explained by the fact that the reduction of platinum ions proceeds mainly on the surface of C–P support whereas in Q–P matrix Si–H groups are available both on the surface and in the bulk (see SEM discussion). Higher concentration of active sites together with better accessibility explains higher activity of Q–P supported catalyst in comparison with the corresponding C–P supported ones. Such behavior can also be related to the consumption of the Si–H groups during the reduction of Pt4+ ions (Table 3). SiH/SiCH3 area ratio is always lower for Q–P–Pt catalyst compared to C–P–Pt ones. It indicates that, for Q–P supported systems, higher amount of Si–H groups is involved in the reduction, which probably results in higher concentration of new sites active in conversion to propene. Additional evidence of such correlation supplies the sequence of the activity in conversion to propene which for all examined samples is the same as for dehydrogenation to acetone. Since one of the most important features of catalysts studied, influencing redox activity is the amount of incorporated platinum, and the Pt-doping level is strongly connected with the consumption of Si–H groups. More efficient generation of acid sites is achieved for samples with higher platinum content as well.
The reaction of isopropyl alcohol conversion to diisopropyl ether is an intermolecular dehydration of two alcohol molecules involving two adjacent acid sites. Diisopropyl ether formation was registered for one sample C–P_(2:1)–Pt_6 only (Fig. 14b), where the concentration of acid sites is high enough to ensure the appropriate distance of active centers.
The selectivity values to acetone exceed 80% for all samples including Al2O3–Pt_0.1 catalyst (Table 6), therefore the activity of redox sites for these systems predominates the activity of acid–base ones. Higher selectivities to propene (13.8 and 15%) are characteristic of C–P_(2:1)–Pt_6 and Q–P_(2:1)–Pt_3. These results are consistent with the assumption that more effective reduction of Pt4+ ions in the samples with the highest loading of platinum is responsible for creation of more efficient acid sites. The changes in selectivity to acetone and propene within the temperature for each sample depend on the relation between values of activation energy of conversion to propene and acetone (Table 6). For Q–P supported catalysts and C–P_(2:1)–Pt_3 sample, the activation energies are higher in conversion to acetone than to propene, therefore the selectivities to propene decrease with the increase of temperature. Unlike to C–P–Pt samples for which the activation energies of conversion to acetone are lower than to propene and as a consequence increase of selectivity to propene within the rise of temperature is observed. Nevertheless, the selectivities to acetone for all catalysts in the examined temperature range are always much higher than selectivities to propene. The activation energy values of dehydration to propene (Table 6) for C–P_(1:1)–Pt systems and C–P_(2:1)–Pt_1.5 sample differ slightly by about 1–4 kJ mol−1, whereas the values of dehydrogenation to acetone reveal more pronounced drop (5–11 kJ mol−1). Activation energies of conversion to acetone and propene for Q–P_(1:1)–Pt_1.5 and Q–P_(2:1)–Pt_1.5 are much lower than for corresponding C–P–Pt samples, which may explain higher activity of former samples. The catalysts with the highest loading of platinum of both series: Q–P_(2:1)–Pt_3 and C–P_(2:1)–Pt_6 reveal lower values of activation energy of conversion to propene and acetone correlating with higher activity in both reactions in comparison with samples with lower platinum content. It is worth mention that lower values of activation energy of both reactions are observed for samples with 2:1 type matrices and higher platinum content (3% or 6%). It is also well seen that values of activation energy for Al2O3–Pt_0.1 are the highest comparing to these of C–P–Pt or Q–P–Pt systems, which in fact favors the latter samples.
Summarizing the results of examination of activity of polymer supported platinum catalysts it is evident that the crucial parameters determining the catalytic behavior of such catalytic systems are accessibility and concentration of platinum particles. The rise of activity is achieved by the increase of the amount of Pt particles in both type of network-polymer support. The most efficient catalysts are those with more flexible Q(MH)4 cross-linking agent, which induces more space for reacting alcohol molecules and better access to the platinum active sites located both on the surface and in the bulk of the polymer matrix.
After catalytic experiments, IR investigations were performed in order to verify if any changes in network structures took place. Some changes have been observed in spectra obtained for samples after catalytic tests compared with the starting ones (Fig. S4 in ESI†). Mainly, the decrease of the intensity of characteristic bands attributed to Si–H groups is clearly visible (marked in the spectra). Some changes have been also noticed in bands corresponding to Si–O–Si stretching vibrations (1017–1095 cm−1). The catalytic tests were carried out up to 300 °C. According to the literature related to cross-linked polysiloxanes, the first step of thermal decomposition of such compounds can occur at the temperature range of 300–600 °C, and is associated with redistribution of Si bonds: Si–O/Si–C, Si–H/Si–O, Si–O/Si–O.51,52 Only slight weight changes of examined samples were recorded by TG analysis at about or above 300 °C. It can be assumed, however, that any redistribution of bonds in the studied networks can occur at higher temperatures of catalytic experiments.
Conclusions
Polysiloxane networks obtained via cross-linking of D4/V4 polysiloxane with branched (Q(MH)4) or cyclic (DH4) hydrosiloxane have been used as supports for platinum catalysts. The influence of various topology of matrices as well as differences in their cross-linking degree on physicochemical and catalytic properties of network–Pt systems has been investigated.It has been found, that both the different structure of the cross-linking agent and the cross-linking density of networks affect significantly the rate of the platinum ions reduction as well as the consumption of Si–H groups, accompanying the reduction process. Different behavior of investigated matrices is related to different availability of reducing groups present in networks.
XRD studies have confirmed the incorporation of Pt(0) into all obtained systems and which contain metal nanoparticles with the average size in the range of 3–6 nm. The size of Pt crystallites depends on the type of matrix applied. Significant differences have been observed between systems containing C–P and Q–P type of matrices, i.e. obtained with the application of a cyclic or a branched hydrosiloxane respectively. Despite the observed tendency to agglomerate, very small, spherical nanoparticles dominate in samples (2–3 nm for C–P–Pt or 4–6 nm for Q–P–Pt) as shown by TEM.
The obtained Pt-systems show high redox activity in catalytic isopropyl alcohol conversion and the essential factor determining the higher activity is the amount of platinum introduced. Taking different supports into consideration, the important factor resulting in varied catalytic activity of studied systems is the different network topology and the availability of active centers associated with this property. The application more rigid cross-linking agent induces formation of narrower gaps for incorporation of platinum particles. In the case of networks containing more flexible bridges platinum particles are sticking out of the network and are more exposed to the adsorption of reacting molecules. Moreover, less dense structure of Q–P networks facilitates access to active centers regardless their localization. The amount of cross-linking agent is also crucial parameter influencing the catalytic activity. Comparison of catalysts supported on different networks shows that (i) higher coverage of platinum in samples with (2:1) type of matrix (more cross-linking agent used) entails higher catalytic activity; (ii) promoting effect on the catalytic activity is observed in the case of Q–P type of matrix, where the accessibility of platinum active centers turns out to be more important feature predominating Pt crystallites size factor.
Comparing to Pt/Al2O3 standard catalyst all examined samples revealed higher redox activity in the lower range of temperature, which is a favorable prognostic for the future applications of such systems.
The results may lead to the conclusion that the application of polysiloxane networks of reducing properties as supports for platinum catalysts is a convenient way of depositing platinum particles into the matrix by reduction of metal ions. The arrangement of catalytic centers is strongly related to the localization and a number of active Si–H groups participating in the reduction and the localization of these latter ones, in turn, associated with the structure of the applied matrix. Pt-containing materials obtained via procedure described in this paper possess good redox activity. Due to the possibility to modify the parameters of their structure and in consequence to optimize their catalytic properties these materials seem to be promising heterogenous catalysts for various applications in redox catalysis.
Acknowledgements
This work was funded under Project No. 11.11.160.767 at the AGH University of Science and Technology in Kraków. Authors would like to thank the staff of Laboratory of Scanning Microscopy and Microanalyses, Dr Agnieszka Różycka, Dr Magdalena Szumera and Mr Łukasz Klita (Faculty of Materials Science and Ceramics, AGH-University of Science and Technology) for SEM, TEM, thermogravimetric and XRD measurements, respectively.References
- J. Ross, in Heterogeneous Catalysis, Elsevier B.V., London, 2012, ch. 3.4, p. 57 Search PubMed.
- Nanoparticle Technology Handbook, ed. M. Hosokawa, K. Nogi, M. Naito and T. Yokoyama, Elsevier B.V., Amsterdam, 2007, ch. A31, p. 550 Search PubMed.
- Catalysis from A to Z.: A Concise Encyclopedia, ed. B. Cornils, W. A. Herrmann, M. Muhler and C. H. Wong, Wiley-VCH, Weinheim, 2006, vol. 1, p. 253 Search PubMed.
- J. Hagen, in Industrial catalysis. A practical Approach, Wiley-VCH, Weinheim, 2nd edn, 2006, ch. 5, p. 181 Search PubMed.
- Synthesis of solid catalysts, ed. K. P. de Jong, Wiley-VCH, Weinheim, 2009, ch. 2, p. 13 Search PubMed.
- Catalytic reactors, ed. B. Saha, Walter de Gruyter GmbH, Berlin, 2016, ch. 6, p. 240 Search PubMed.
- Synthesis of solid catalysts, ed. K. P. de Jong, Wiley-VCH, Weinheim, 2009, ch. 2, pp. 277–297 Search PubMed.
- F. X. Llabrés i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef.
- S. Opelt, S. Turk, E. Dietzsch, A. Henschel, S. Kaskel and E. Klemm, Catal. Commun., 2008, 9, 1286–1290 CrossRef CAS.
- M. Sabo, A. Henschel, H. Froede, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827 RSC.
- C. E. Chan-Thaw, A. Villa, P. Katekomol, D. Su, A. Thomas and L. Prati, Nano Lett., 2010, 10, 537–541 CrossRef CAS PubMed.
- C. Burato, P. Centomo, G. Pace, M. Favaro, L. Prati and B. Corain, J. Mol. Catal. A: Chem., 2005, 238, 26–34 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- Y. Motoyama, K. Mitsui, T. Ishida and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150–13151 CrossRef CAS PubMed.
- Y. Motoyama, M. Abe, K. Kamo, Y. Kosako and H. Nagashima, Chem. Commun., 2008, 5321–5323 RSC.
- Y. Motoyama, K. Kamo and H. Nagashima, Org. Lett., 2009, 11, 1345–1348 CrossRef CAS PubMed.
- M. Cypryk, P. Pośpiech, K. Strzelec and J. W. Sobczak, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 1586–1598 CrossRef CAS.
- M. Cypryk, P. Pośpiech, K. Strzelec, K. Wąsikowska and J. W. Sobczak, J. Mol. Catal. A: Chem., 2010, 319, 30–38 CrossRef CAS.
- M. F. Dumont, S. Moisan, C. Aymonier, J. D. Marty and C. Mingotaud, Macromolecules, 2009, 42, 4937–4940 CrossRef CAS.
- B. P. S. Chauhan, J. Rathore, R. Sardar, P. Tewari and U. Latif, J. Organomet. Chem., 2003, 686, 24–31 CrossRef CAS.
- B. P. S. Chauhan, J. S. Rathore, M. Chauhan and A. Krawicz, J. Am. Chem. Soc., 2003, 125, 2876–2877 CrossRef CAS PubMed.
- B. P. S. Chauhan, J. S. Rathore and T. Bandoo, J. Am. Chem. Soc., 2004, 126, 8493–8500 CrossRef CAS PubMed.
- B. P. S. Chauhan, J. S. Rathore and N. Glloxhani, Appl. Organomet. Chem., 2005, 19, 542–550 CrossRef CAS.
- B. P. S. Chauhan and J. S. Rathore, J. Am. Chem. Soc., 2005, 127, 5790–5791 CrossRef CAS PubMed.
- B. P. S. Chauhan and B. Balagam, Macromolecules, 2006, 39, 2010–2012 CrossRef CAS.
- B. P. S. Chauhan, A. Sarkar, M. Chauhan and A. Roka, Appl. Organomet. Chem., 2009, 23, 385–390 CrossRef CAS.
- E. Stochmal, J. Strzezik and A. Krowiak, J. Appl. Polym. Sci., 2016, 133, 1–14 CrossRef.
- C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS.
- M. A. Brook, in Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000, ch. 7 Search PubMed.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, in Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec and J. Matisons, in Advances in Silicon Science Series, Springer Science+Business Media B.V., Dordrecht, 2009, vol. 1, p. 3 Search PubMed.
- L. N. Lewis, N. Lewis, and R. J. Uriarte, in Homogeneous Transition Metal Catalyzed Reactions, ed. W. R. Moser, and D. W. Slocum, Adv. Chem. Ser. 230, American Chemical Society, Washington DC, 1992, ch. 37, p. 541 Search PubMed.
- B. D. Karstedt, US Pat., 3 775 452, General Electric Co., 1973.
- A. R. Shultz, in Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, N. G. Gayload and N. M. Bikales, Wiley, London, 1966, vol. 4, p. 331 Search PubMed.
- F. Samadi, J. Eckelt, B. A. Wolf, H. Schüle and H. Frey, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1309–1318 CrossRef CAS.
- A. Nyczyk, C. Paluszkiewicz, M. Hasik, M. Cypryk and P. Pośpiech, Vib. Spectrosc., 2012, 59, 1–8 CrossRef CAS.
- A. Lee Smith, Spectrochim. Acta, 1960, 16, 87–105 CrossRef.
- A. C. C. Esteves, J. Brokken-Zijp, J. Laven, H. P. Huinink, N. J. W. Reuvers, M. P. Van and G. de With, Polymer, 2009, 50, 3955–3966 CrossRef CAS.
- J. L. Braun, J. E. Mark and B. E. Eichinger, Macromolecules, 2002, 35, 5273–5282 CrossRef CAS.
- X. Zhang, P. Xie, Y. Shen, J. Jiang, C. Zhu, H. Li, T. Zhang, C. C. Han, L. Wan, S. Yan and R. Zhang, Angew. Chem., Int. Ed., 2006, 45, 3112–3116 CrossRef CAS PubMed.
- L. N. Pankratova, M. G. Zhizhin and L. T. Bugaenko, High Energy Chem., 2005, 39, 382–385 CrossRef CAS.
- JCPDS PDF Card No. 04–0802, ICDD-PDF-4+ 2015 database, www.icdd.com/products/pdf4.htm.
- T. Maiyalagan and F. Nawaz Khan, Catal. Commun., 2009, 10, 433–436 CrossRef CAS.
- T. Maiyalagan, C. Mahendiran, K. Chaitanya, R. Tyagi and F. Nawaz Khan, Res. Chem. Intermed., 2012, 38, 383–391 CrossRef CAS.
- J. A. Anderson, M. Fernandez-Garcia and A. Martines-Arias, in Supported Metals in Catalysis, ed. J. A. Anderson and M. Fernandez-Garcia, Imperial College Press, London, Singapore, 2nd edn, 2012, ch. 2, Determination of Dispersion and Crystallite Sizes for Supported Metal Catalysts Search PubMed.
- M. J. Michalczyk, W. E. Farneth and A. J. Vega, Chem. Mater., 1993, 5, 1687–1689 CrossRef CAS.
- A. Nyczyk-Malinowska, M. Wójcik-Bania, T. Gumuła, M. Hasik, M. Cypryk and Z. Olejniczak, J. Eur. Ceram. Soc., 2014, 34, 889–902 CrossRef CAS.
- M. Radlik, J. Strzezik, A. Krowiak, K. Kozieł, A. Krztoń and W. Turek, React. Kinet., Mech. Catal., 2015, 115, 741–758 CrossRef CAS.
- S. A. El-Molla, Appl. Catal., A, 2005, 280, 189–197 CrossRef CAS.
- Y. Han, J. Shen and Y. Chen, Appl. Catal., A, 2001, 205, 79–84 CrossRef CAS.
- R. M. Rioux and M. A. Vannice, J. Catal., 2005, 233(1), 147–165 CrossRef CAS.
- V. Belot, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 613–623 CrossRef CAS.
- V. Gualandris, D. Bahloul-Houlier and F. Babonneau, J. Sol-Gel Sci. Technol., 1999, 14, 39–48 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00641a |
| This journal is © The Royal Society of Chemistry 2017 |