
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Three-component 1D and 2D metal phosphonates: structural variability, topological analysis and catalytic hydrocarboxylation of alkanes†
Konstantinos D. Demadis *a,
Zafeiria Anagnostoua,
Aggeliki Paneraa,
Gellert Mezei*b,
Marina V. Kirillovac and
Alexander M. Kirillov*c
*a,
Zafeiria Anagnostoua,
Aggeliki Paneraa,
Gellert Mezei*b,
Marina V. Kirillovac and
Alexander M. Kirillov*c
aCrystal Engineering, Growth and Design Laboratory, Department of Chemistry, University of Crete, Voutes Campus, Heraklion, Crete GR-71003, Greece. E-mail: demadis@uoc.gr
bDepartment of Chemistry, Western Michigan University, Kalamazoo, Michigan 49008-5413, USA. E-mail: gellert.mezei@wmich.edu
cCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: kirillov@tecnico.ulisboa.pt
First published on 23rd March 2017
Abstract
Herein, we report the use of diphosphonate building blocks and chelating auxiliary N,N-ligands to generate novel polymeric architectures. Specifically, we report new 1D and 2D coordination polymers incorporating three components: transition metal ions (Co2+, Cu2+, Mn2+ or Zn2+), diphosphonate ligands (methane-diphosphonate, MDPA, or 1,2-ethanediphosphonate, EDPA) and N,N-heterocyclic chelators (1,10-phenanthroline, phen, or 2,2′-bipyridine, bpy). Six compounds were isolated under mild synthesis (ambient temperature) conditions: [Cu2(phen)2(EDPA)2(H2O)4]∞ (1), [Co(phen)(EDPA)(H2O)2]∞ (1a), {[Cu(phen)(MDPA)]·H2O]}∞ (2), [Mn(bpy)(EDPA)(H2O)2]∞ (3), [Zn(bpy)(EDPA)]∞ (4), and, lastly, a discrete Ni2+ molecular derivative [Ni(phen)(H2O)4](EDPA) (5). Synthetic details, crystal structures, and intermolecular interactions (π–π stacking and hydrogen bonding) in 1–5 are discussed. Topological analyses and classification of the underlying metal–organic networks in 1–4 were performed, revealing the uninodal 1D chains with the 2C1 topology in 1–3 and the binodal 2D layers with the 3,4L13 topology in 4. In 1–3 and 5, multiple hydrogen bonds lead to the extension of the structures to give 3D H-bonded nets with the seh-4,6-C2/c topology in 1 and 3, 2D H-bonded layers with the 3,5L52 topology in 2, and a 3D H-bonded net with the 6,6T1 topology in 5. The catalytic activity of compounds 1 and 1a was evaluated in a single-step hydrocarboxylation of cyclic and linear C5–C8 alkanes to furnish the carboxylic acids with one more carbon atom. These reactions proceed in the presence of CO, K2S2O8, and H2O at 60 °C in MeCN/H2O medium. Compound 1 showed higher activity than 1a and was studied in detail. Substrate scope was investigated, revealing that cyclohexane and n-pentane are the most reactive among the cyclic and linear C5–C8 alkanes, and resulting in the total yields of carboxylic acids (based on substrate) of up to 43 and 36%, respectively. In the case of cycloalkane substrates, only one cycloalkanecarboxylic acid is produced, whereas a series of isomeric monocarboxylic acids is generated when using linear alkanes; an increased regioselectivity at the C(2) position of the hydrocarbon chain was also observed.
Introduction
Metal phosphonate chemistry is a rapidly expanding discipline under the umbrella of coordination polymers.1 Several attractive features of these materials have motivated the interest of researchers: (a) thermal stability,2,3 (b) resistance to oxidation and hydrolysis,4,5 (c) a wide selection of metal ion sources available for their syntheses,1 (d) an extensive phosphonate (and mixed phosphonate) ligand “toolbox” available for new synthetic systems,6–9 (e) access to the mono- (R–PO3H−), and bis-deprotonated (R–PO32−) forms of the phosphonate moiety (depending on pH),10–14 thus generating distinct reaction outcomes, and (f) the ability of the tetrahedral phosphonate group to create commonly unpredictable structural architectures.It is well established that there are several factors that influence the outcome of a synthetic procedure involving a metal ion and a ligand.15 More specifically, in metal phosphonate chemistry the following specific factors must be taken into account for the design of synthetic systems: (a) solution pH,16,17 (b) reactant molar ratios,18 (c) nature of the metal ion (charge, ionic radius),19 (d) number of phosphonate groups on the ligand,20 (e) reaction time,21 (f) presence of other “functional” moieties within the phosphonate ligand backbone e.g. amine,22 carboxylate,23–25 sulfonic groups,26,27 heterocyclic aromatic moieties,28–31 and (g) incorporation in the reaction system of a secondary ligand.32–42 Last but not least, the profound tendency of the phosphonate moiety to bridge metal ions, and thus to increase framework dimensionality must be highlighted.43–45
Incorporation of a secondary ligand into the reaction mixture can dramatically influence the synthesis outcome and the structure of the final product. Such ligands can either disrupt the “coordination polymer” architecture, by generating mononuclear complexes,46 or “decorate” or structurally alter the initial “metal phosphonate” structural motif. Such ligands have been called secondary auxiliary ligands (SALs) and they are usually neutral or anionic.38–41,47
Herein, we report that the use of diphosphonate building blocks along with chelating auxiliary ligands (2,2′-bipyridine and 1,10-phenanthroline) can generate novel polymeric architectures in metal phosphonate chemistry. Specifically, we report novel 1D and 2D coordination polymers or a 0D monomer incorporating three components: transition metal ions (Co2+, Cu2+, Mn2+, Zn2+, or Ni2+), phosphonate ligands (methane-diphosphonate, MDPA, or 1,2-ethanediphosphonate, EDPA) and N,N-heterocyclic chelators (2,2′-bipyridine, bpy, or 1,10-phenanthroline, phen), Fig. 1. Five compounds were isolated: [Cu2(phen)2(EDPA)2(H2O)4]∞ ([Cu(phen)(EDPA)], 1), [Co2(phen)2(EDPA)2(H2O)4]∞ ([Co(phen)(EDPA)], 1a), {[Cu(phen)(MDPA)]·H2O]}∞ ([Cu(phen)(MDPA)], 2), [Mn(bpy)(EDPA)(H2O)2]∞ ([Mn(bpy)(EDPA)], 3), [Zn(bpy)(EDPA)]∞ ([Zn(bpy)(EDPA)], 4), and [Ni(phen)(H2O)4](EDPA) ([Ni(phen)(EDPA)], 5). Synthetic details, crystal structures, and intermolecular interactions (π–π stacking and hydrogen bonding) are discussed. In addition, topological analysis and classification of the underlying metal–organic and/or H-bonded networks of 1–5 have been performed. Schematic structures of all ligands used (diphosphonates and SALs) are given in Fig. 1.
 | ||
| Fig. 1 Schematic structures of the ligands used in this study. Abbreviations: MDPA = methane-diphosphonic acid, EDPA = 1,2-ethanediphosphonic acid, bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline. | ||
Furthermore, catalytic activity of compounds 1 and 1a has been studied in the mild hydrocarboxylation of cyclic and linear C5–C8 alkanes into carboxylic acids with one more carbon atom. The selection of alkane hydrocarboxylation as a model catalytic reaction has been governed by the following reasons. As main components of petroleum, alkanes are very abundant substrates for the synthesis of added-value organic products via various C–H functionalization protocols.48 However, the high inertness of alkanes prevents their broad application for a single-pot functionalization into valuable products. Thus, the development of new, selective, and efficient catalytic systems that are capable of transforming alkanes into desirable products under mild conditions and in a single-step is an important topic of modern chemistry.48,49 In this regard, the hydrocarboxylation of alkanes to carboxylic acids represents a particularly interesting albeit still poorly explored transformation,49 as it involves both the C–H activation and C–C formation steps, undergoes under mild conditions and in aqueous acetonitrile medium, and is preferably catalyzed by copper coordination polymers50 or multinuclear complexes.51
Experimental section
Materials and methods
All reagents were from commercial sources (CuCl2·2H2O Fluka, MnSO4·H2O Fischer, Zn(NO3)2·6H2O, Ni(NO3)2·6H2O and bpy Alfa-AESAR, EDPA and MDPA from Aldrich), and were used as received. Stock solutions of 1 M NaOH and HCl were used for pH adjustment. Ion exchange column-deionized (DI) water was used for all syntheses. The pH-meter used was a wTw pH315i set-up, equipped with a SeTix 41 electrode. All products reported herein gave satisfactory elemental analyses. Yields range from 60 to 80% based on the metal salt.Syntheses
X-ray crystallography
X-ray diffraction data were collected at room temperature from a single-crystal mounted atop a glass fiber with cyanoacrylate adhesive, with a Bruker SMART APEX II diffractometer using graphite-monochromated Mo-Kα (λ = 0.71073 Å) radiation. The structures were solved by employing SHELXTL direct methods and refined by full-matrix least squares on F2, using the APEX2 v2014.9-0 software package (Bruker AXS Inc.: Madison, WI, 2014). All non-H atoms were refined with independent anisotropic displacement parameters. Hydrogen atoms were placed at calculated positions and refined using a riding model, except for the water and phosphonic acid O–H hydrogens, which were located from the Fourier difference density maps and refined using a riding model with O–H distance restraints. Crystallographic details are summarized in Table 1, and important metric features (bond distances and angles) are found in Table 2. Hydrogen bonding interactions are given in Table 3.| Cu(phen)(MDPA) (2) | Mn(bpy)(EDPA) (3) | Zn(bpy)(EDPA) (4) | Ni(phen)(EDPA) (5) | |
|---|---|---|---|---|
| Formula 1 | C13H14CuN2O7P2 | C12H18MnN2O8P2 | C12H14N2O6P2Zn | C14H22N2NiO10P2 |
| Formula 2 | {[CuII(phen)(MDPA)]·H2O]}∞ | [MnII(bpy)(EDPA)(H2O)2]∞ | [ZnII(bpy)(EDPA)]∞ | [NiII(phen)(H2O)4](EDPA) |
| FW (g mol−1) | 435.74 | 435.16 | 409.56 | 498.98 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Orthorhombic |
| Space group | Cc | C2/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pnma |
| a (Å) | 22.3946(4) | 12.3487(3) | 5.5335(2) | 7.2046(1) |
| b (Å) | 4.8894(1) | 14.3471(4) | 10.6895(3) | 19.6744(2) |
| c (Å) | 16.9441(3) | 10.0005(3) | 12.6347(4) | 13.6407(1) |
| α (deg) | 90.000 | 90.000 | 95.967(2) | 90.000 |
| β (deg) | 122.616(1) | 107.578(1) | 90.237(2) | 90.000 |
| γ (deg) | 90.000 | 90.000 | 103.332(2) | 90.000 |
| V (Å3) | 1562.73(5) | 1689.04(8) | 722.96(4) | 1933.52(4) |
| Crystal size | 0.05 × 0.25 × 0.40 | 0.05 × 0.25 × 0.42 | 0.07 × 0.08 × 0.28 | 0.10 × 0.12 × 0.50 |
| Z | 4 | 4 | 2 | 4 |
| Dcalc (g cm−3) | 1.852 | 1.711 | 1.881 | 1.714 |
| μ (mm−1) | 1.645 | 1.016 | 1.953 | 1.227 |
| θ range (deg) | 2.16–30.56 | 2.24–29.70 | 1.62–28.84 | 1.82–28.70 |
| Reflns collected | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 347 347 |
23096 |
28580 |
37488 |
| Obsd reflns [I > 2σ(I)] | 4360 | 2088 | 3265 | 2250 |
| Data/restraints/parameters | 4637/6/238 | 2388/3/123 | 3780/0/214 | 2570/6/199 |
| GOF (on F2) | 1.043 | 1.043 | 1.057 | 1.067 |
| R factor [I > 2σ(I)] | R1 = 0.0220, wR2 = 0.0500 | R1 = 0.0257, wR2 = 0.0711 | R1 = 0.0269, wR2 = 0.0585 | R1 = 0.0243, wR2 = 0.0607 |
| R factor (all data) | R1 = 0.0251, wR2 = 0.0512 | R1 = 0.0321, wR2 = 0.0754 | R1 = 0.0347, wR2 = 0.0619 | R1 = 0.0302, wR2 = 0.0643 |
| Maximum peak/hole (e·Å−3) | 0.292/−0.257 | 0.291/−0.352 | 0.459/−0.392 | 0.483/−0.370 |
| Cu(phen)(MDPA) (2) | ||||
| Cu1–O1 1.958(2) | Cu1–N1 2.025(2) | P1–O1 1.511(2) | P1–O3 1.592(2) | P2–O5 1.500(2) |
| Cu1–O2 2.150(2) | Cu1–N2 2.026(2) | P1–O2 1.485(2) | P2–O4 1.510(2) | P2–O6 1.577(2) |
| Cu1–O4 1.947(2) | ||||
|
||||
| Mn(bpy)(EDPA) (3) | ||||
| Mn1–O1 2.105(1) | Mn1–N1 2.288(1) | P1–O1 1.495(1) | P1–O2 1.582(1) | P1–O3 1.513(1) |
| Mn1–O4 2.223(1) | ||||
|
||||
| Zn(bpy)(EDPA) (4) | ||||
| Zn1–O1 1.975(1) | Zn1–N1 2.174(2) | P1–O1 1.493(1) | P1–O3 1.511(2) | P2–O5 1.570 (2) |
| Zn1–O4 2.001(1) | Zn1–N2 2.152(2) | P1–O2 1.583(2) | P2–O4 1.494(1) | P2–O6 1.511(1) |
| Zn1–O6 1.976(1) | ||||
|
||||
| Ni(phen)(EDPA) (5) | ||||
| Ni1–O4 2.017(2) | Ni1–O6 2.058(1) | P1A–O1A 1.534(8) | P1A–O2A 1.595(7) | P1A–O3A 1.476(7) |
| Ni1–O5 2.047(1) | Ni1–N1 2.078(1) | P1B–O1B 1.467(5) | P1B–O2B 1.555(8) | P1B–O3B 1.558(7) |
| Compound | D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H–A (°) | Symmetry operator for A |
|---|---|---|---|---|---|---|
| Cu(phen)(MDPA) (2) | O3–H3O⋯O7# | 0.78(2) | 1.92(2) | 2.703(3) | 173(4) | x, y − 1, z |
| O6–H6O⋯O5# | 0.79(2) | 1.84(3) | 2.606(2) | 165(4) | x, y + 1, z | |
| O7–H7A⋯O5# | 0.82(3) | 1.93(3) | 2.746(3) | 172(4) | x, −y + 1, z − 1/2 | |
| O7–H7B⋯O3 | 0.78(2) | 2.17(3) | 2.937(3) | 169(4) | ||
| Mn(bpy)(EDPA) (3) | O2–H2⋯O3# | 0.804(15) | 1.760(15) | 2.5616(14) | 174(2) | −x, y, −z − 1/2 |
| O4–H4A⋯O3# | 0.785(15) | 1.934(15) | 2.7057(15) | 168(2) | −x − 1/2, −y + 1/2, −z | |
| O4–H4B⋯O2# | 0.791(15) | 2.025(16) | 2.7832(15) | 160(2) | −x, y, −z + 1/2 | |
| Zn(bpy)(EDPA) (4) | O5–H5⋯O3 | 0.72(3) | 1.92(3) | 2.629(2) | 168(3) | −x + 1, −y, −z + 1 |
| O2–H6⋯O3# | 0.80(2) | 1.87(2) | 2.662(2) | 174(2) | ||
| Ni(phen)(EDPA) (5) | O2A–H2A⋯O3A# | 0.82(2) | 1.87(3) | 2.581(6) | 146(5) | x − 1/2, y, −z + 3/2 |
| O4–H4A⋯O1A# | 0.83(2) | 1.95(2) | 2.756(2) | 165(2) | x + 1/2, −y + 3/2, −z + 3/2 | |
| O4–H4A⋯O1B# | 0.83(2) | 1.83(2) | 2.629(2) | 164(2) | x + 1/2, −y + 3/2, −z + 3/2 | |
| O5–H5A⋯O3A# | 0.80(2) | 2.12(2) | 2.809(3) | 144(2) | x, −y + 3/2, z | |
| O5–H5A⋯O1B# | 0.80(2) | 2.24(2) | 2.902(3) | 141(2) | x, −y + 3/2, z | |
| O5–H5B⋯O3A# | 0.82(2) | 1.89(2) | 2.695(4) | 168(2) | x − 1/2, −y + 3/2, −z + 3/2 | |
| O5–H5B⋯O3B# | 0.82(2) | 2.01(2) | 2.781(3) | 159(2) | x − 1/2, −y + 3/2, −z + 3/2 | |
| O6–H6O⋯O1A# | 0.81(1) | 1.93(1) | 2.714(2) | 164(2) | x − 1/2, −y + 3/2, −z + 3/2 | |
| O6–H6O⋯O1B# | 0.81(1) | 1.95(1) | 2.738(2) | 164(2) | x − 1/2, −y + 3/2, −z + 3/2 |
Catalytic hydrocarboxylation of alkanes
In a typical experiment, the reaction mixtures were prepared as follows: a stainless steel autoclave (20.0 mL) equipped with a Teflon-coated magnetic stir bar was filled with compound 1 or 1a (0.01 mmol), K2S2O8 (1.5 mmol), H2O (2.0 mL) and MeCN (4.0 mL; total solvent volume was 6.0 mL), and an alkane (1.0 mmol). Then, the autoclave was closed and flushed with CO three times to remove the air, and finally pressurized with 20 atm of CO. CAUTION: Due to the toxicity of CO, all operations should be carried out in a well-ventilated hood! The obtained reaction mixture was stirred for 4 h at 60 °C using a magnetic stirrer and an oil bath, whereupon it was cooled in an ice bath, degassed, opened, and transferred to a flask with tap. Diethyl ether (9.0 mL) and cycloheptanone (45 μL, typical GC internal standard) were added. In the case of cycloheptane hydrocarboxylation, cyclohexanone (45 μL) was used as a GC standard instead for cycloheptanone. The obtained mixture was vigorously stirred for 10 min using a magnetic stirrer. Then, the organic layer was analyzed by gas chromatography (internal standard method), revealing the formation of the corresponding monocarboxylic acids as the dominant products. The formation of oxidation products (cyclic alcohols and ketones) was also detected when using cycloalkanes as substrates. Assignment of peaks was made by comparison with chromatograms of authentic samples. Gas chromatography (GC) analyses were run on an Agilent Technologies 7820A series gas chromatograph (He as carrier gas) equipped with the FID detector and BP20/SGE (30 m × 0.22 mm × 0.25 μm) capillary column.Results and discussion
Synthetic considerations
All syntheses were carried out in acidic aqueous solutions (the pH varied in different synthetic systems) and all products were isolated as crystalline solids that precipitated out of solution at ambient conditions, without the addition of any other precipitant solvent. As reported earlier in several synthetic attempts,16,18–20,23–25 pH is a dominant factor for the reaction outcome.52 Specifically, the solution pH has to be within a range where the product precipitates/crystallizes and incorporates all three reaction components, i.e. metal ion, phosphonate ligand, and SAL. Regarding the phosphonic acid, the pH has to be sufficiently high so that the phosphonic acid moieties become partially or fully deprotonated. If the pH is too high, intractable amorphous products or mixtures of phases are isolated. In addition, due to the high affinity of a doubly-deprotonated phosphonate moiety (R–PO32−) for metal ions, the neutral pyridine-based SAL may be excluded from coordination. On the other hand, if the reaction pH is too low, the phosphonate ligand is mostly protonated and does not coordinate to the metal center. At the same time, a pyridine-based SAL can become protonated (due to the presence of basic N atoms on the ligand backbone) and can precipitate out of solution as salt. Therefore, selection of the proper pH for each synthesis is crucial for isolation of a single-phase crystalline product. Because of the idiosyncrasies of different synthetic systems, there are no specific guidelines to be followed. Based on past experience, we find a pH range of 3–5 to be appropriate for such three-component systems.Crystallographic studies
The crystal structures of compounds [Cu2(phen)2(EDPA)2(H2O)4]∞ (1)38 and [Co(phen)(EDPA)(H2O)2]∞ (1a)39 have been reported before. These two compounds are isostructural. The products we isolated from our synthetic methodology are the same as the ones previously reported, although the synthetic conditions were different. Hence, we will not present their crystal structures herein. However, we will discuss their topological analysis, vide infra.As a short comment, we would like to report that the structure of 1 is rather different from the previously reported [Cu(bpy)(EDPA)(H2O)]2·3H2O derivative.46 While the EDPA ligand is doubly deprotonated in both cases, only one of its two PO3H− groups is coordinated to the copper center in the case of [Cu(bpy)(EDPA)(H2O)]2·3H2O. In fact, this phosphonate group bridges two Cu atoms, leading to a dimeric structure, with two dangling phosphonate groups being hydrogen-bonded to the neighboring molecules. A molecular (dimeric) analog of 1 is also known, [Cu2(phen)2(EDPA)(H2O)2]·9H2O. This discrete copper(II) dimer was prepared by heating a mixture of CuSO4, EDPA and 1,10-phenanthroline in water, at pH 10 and 140 °C for 24 h. In this case, the diphosphonate ligand is completely deprotonated (bears a “4−” charge) and acts as a bridging moiety between two copper centers.53
 | ||
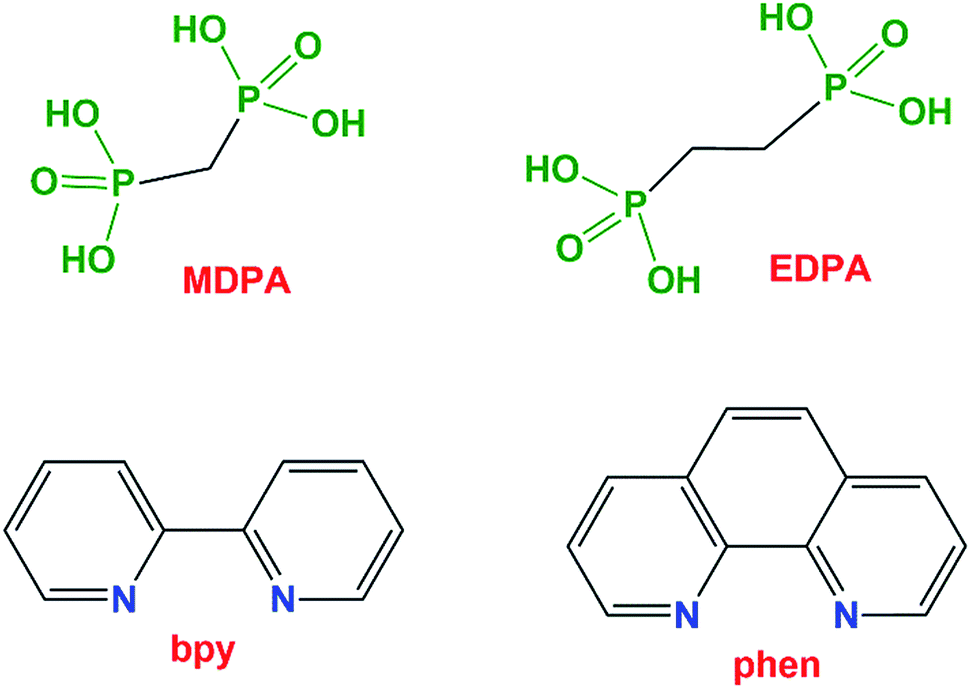
| Fig. 2 Crystal structure of Cu(phen)(MDPA) (2). Top image: 50% probability thermal ellipsoids (C–H hydrogens not shown for clarity). Symmetry code: (a) x, y + 1, z. Center images: linear 1D metal–organic chain and packing diagram (view along the b axis). Bottom image: coordination mode of the MDPA ligand. Atom color codes: Cu dark blue, P orange, O red, C black, N light blue, H white. | ||
 | ||
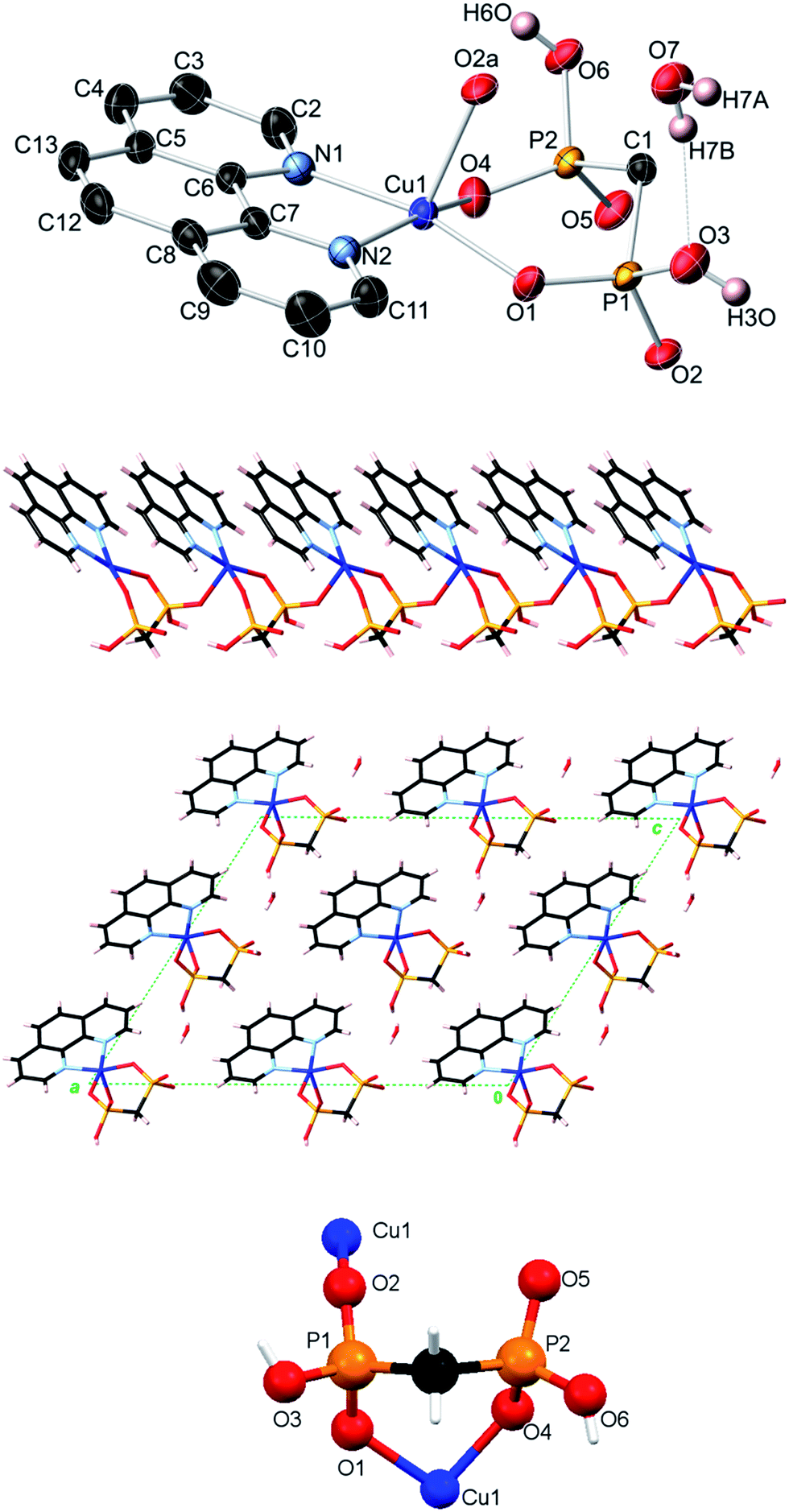
| Fig. 3 Crystal structure of Mn(bpy)(EDPA) (3). Top image: 50% probability thermal ellipsoids (C–H hydrogens not shown for clarity). Symmetry codes: (a) −x, y, −z + 1/2; (b) −x, −y + 1, −z; (c) x, −y + 1, z + 1/2. Center images: 2D sheet motif formed by interdigitated zigzag 1D metal–organic chains and packing diagram showing layered 2D sheet motifs (view along the c axis). Bottom image: coordination mode of the EDPA ligand. Atom color codes: Mn magenta, P orange, O red, C black, N light blue, H white. | ||
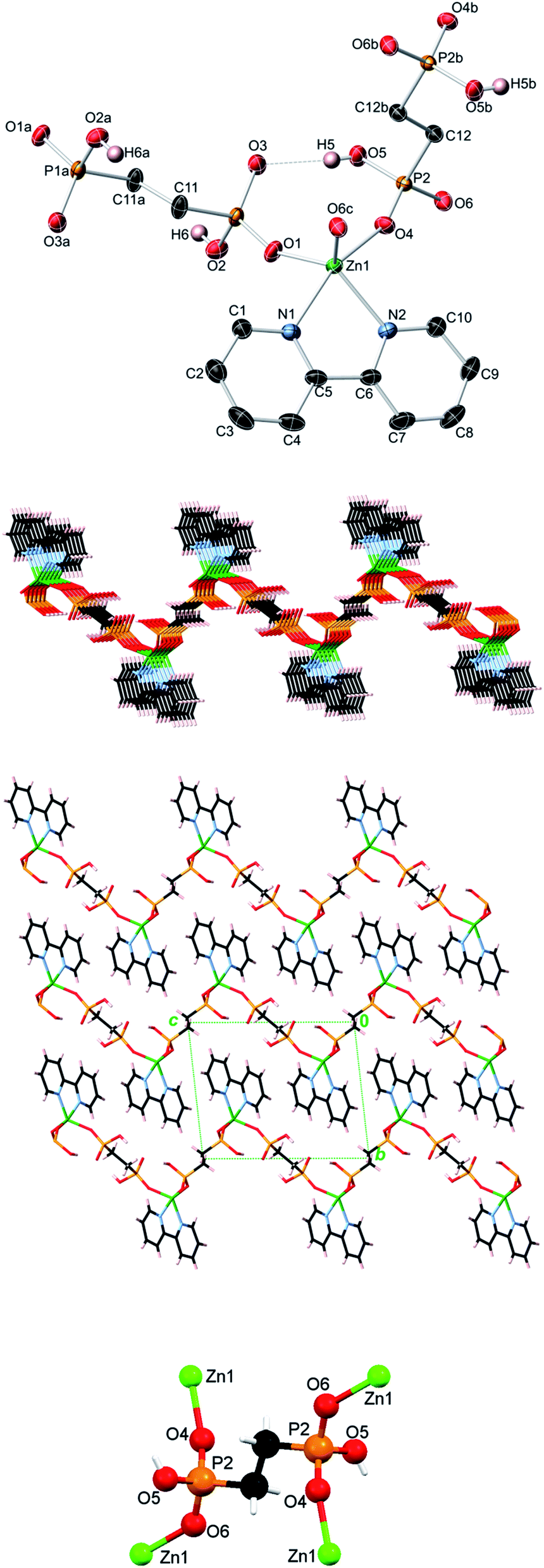 | ||
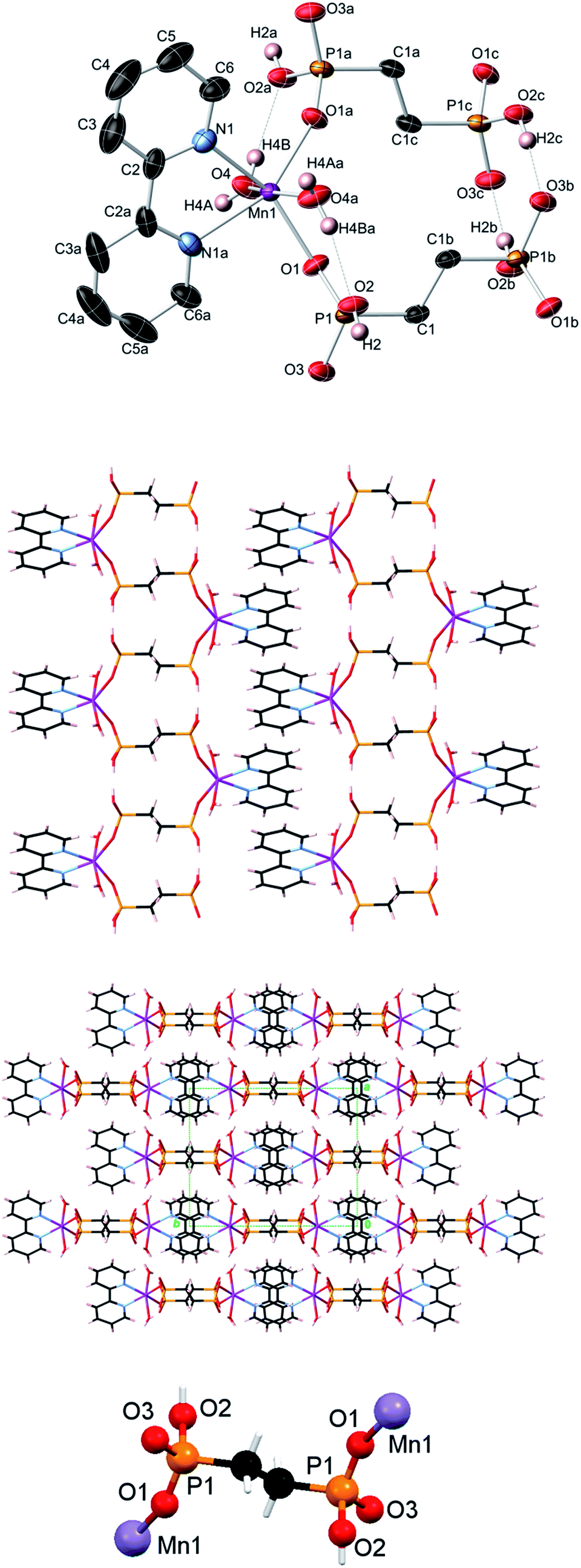
| Fig. 4 Crystal structure of Zn(bpy)(EDPA) (4). Top image: 50% probability thermal ellipsoids (C–H hydrogens not shown for clarity). Symmetry codes: (a) −x, −y, −z + 1; (b) −x + 1, −y, −z; (c) x − 1, y, z; (d) −x, −y, −z. Center images: undulating 2D metal–organic sheet and packing diagram showing the interdigitation of adjacent undulating 2D sheets (view along the a axis). Bottom image: coordination mode of the EDPA ligand. Atom color codes: Zn green, P orange, O red, C black, N light blue, H white. | ||
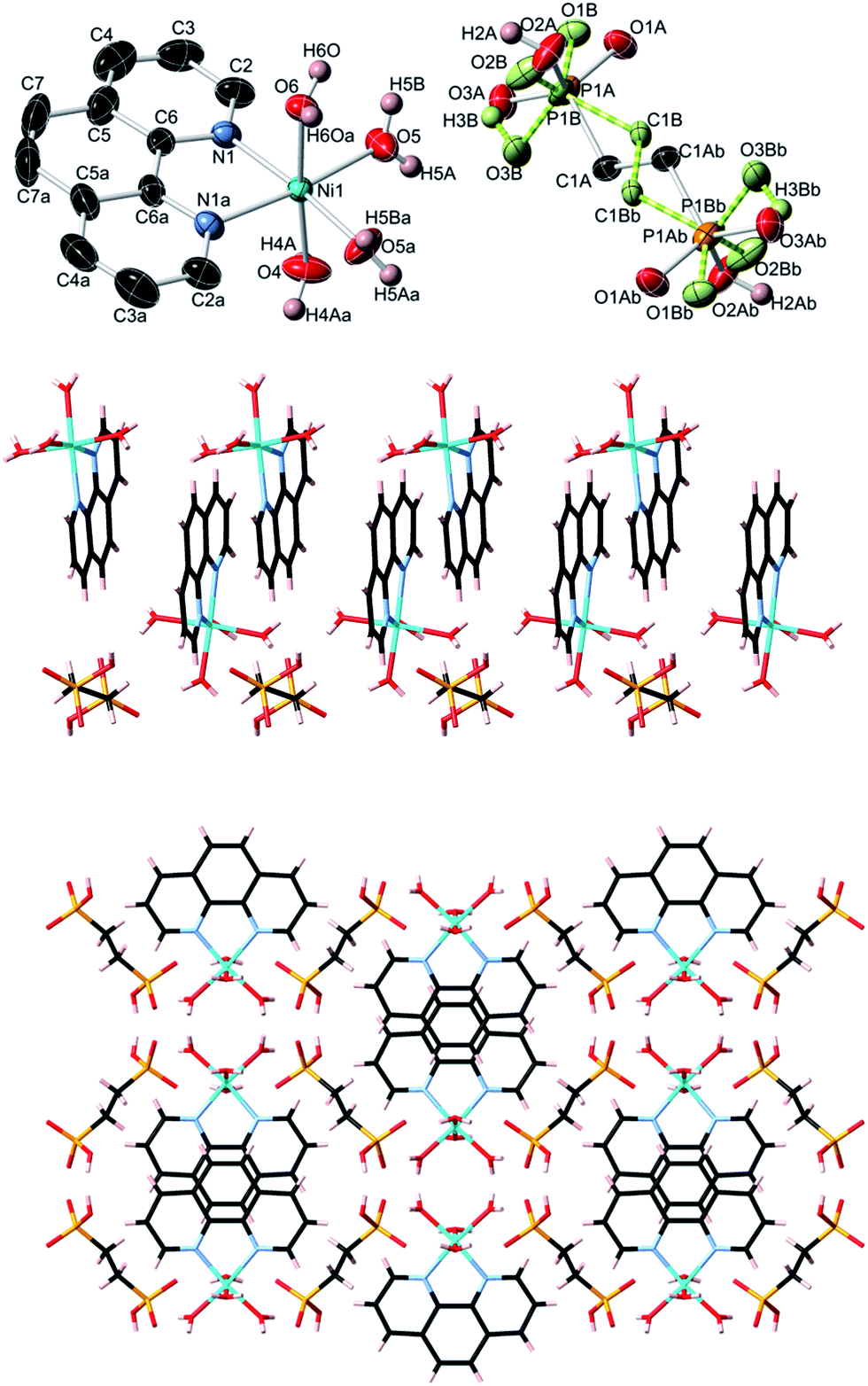 | ||
| Fig. 5 Crystal structure of Ni(phen)(EDPA) (5). Top: 50% probability thermal ellipsoids (C–H hydrogens not shown for clarity; one of the disordered EDPA ions is shown in green). Symmetry codes: (a) x, −y + 3/2, z; (b) −x, −y + 1, −z + 2. Center: 1D column showing π–π stacking between phen units. Bottom: packing diagram (view along the a axis). Atom color codes: Ni bright blue, P orange, O red, C black, N light blue, H white. | ||
Topological analysis of compounds 1–5
To get further insight into the crystal structures of 1–5, we have performed topological analysis and classification of both metal–organic and H-bonded networks, by following the concept of the simplified underlying net.54–57 Such simplified nets have been generated by reducing building blocks of 1–5 to their centroids maintaining their connectivity via coordination bonds or both coordination and hydrogen bonds. In the construction of underlying H-bonded nets, only the conventional D–H⋯A hydrogen bonds have been considered, wherein H⋯A < 2.50 Å, D⋯A < 3.50 Å, and ∠(D–H⋯A) > 120°; D and A stand for donor and acceptor atoms.54,55 The obtained underlying networks (Fig. 6–9) have been topologically analyzed and classified. We excluded compound 1a from the analysis, because it is isostructural to 1.The zigzag metal–organic chains of 1 can be topologically classified as a uninodal 2-connected net with a simple 2C1 topology driven by the [Cu(phen)(H2O)2] and EDPA nodes (Fig. 6a). These 1D chains are further extended by means of numerous hydrogen bonds into a very complex 3D H-bonded framework (Fig. 6b). It is composed of the 4-connected [Cu(phen)(H2O)2] and 6-connected EDPA nodes (Fig. 6b). The topological analysis of this net discloses the seh-4,6-C2/c topology54,55 described by the point symbol of (3.42.52.6)(32.42.52.64.74.8), wherein the (3.42.52.6) and (32.42.52.64.74.8) notations are those of the [Co(phen)(H2O)2] and EDPA nodes, respectively.
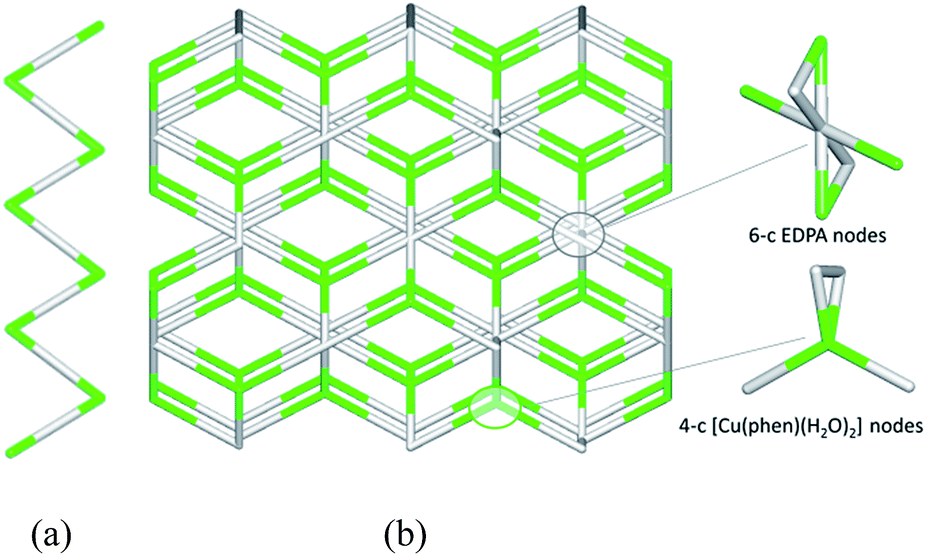 | ||
| Fig. 6 Topological representation of the underlying networks in 1: (a) 1D metal–organic chain and (b) 3D H-bonded net. (a) Uninodal 2-connected zigzag chain with the 2C1 topology (view along the c axis). (b) Binodal 4,6-connected net with the seh-4,6-C2/c topology (view along the c axis). Color codes: centroids of 4-connected [Cu(phen)(H2O)2] (green) and 6-connected EDPA (gray) nodes. | ||
It should be mentioned that a binodal 6,10-connected 3D H-bonded framework with the 6,10T9 topology, built from the [Cu(bpy)(EDPA)(H2O)]2 molecular nodes and (H2O)6 cluster nodes, has been earlier identified in a related discrete copper(II) dimer [Cu(bpy)(EDPA)(H2O)]2·3H2O.46 In the present case, the discrete dicopper(II) units are assembled into 2D layers, which are then extended into a 3D framework by H-bonding interactions involving water clusters from crystallization H2O molecules.46 In contrast, the 3D H-bonded net of [Cu(phen)(EDPA)(H2O)2]∞ (1) is directly assembled via H-bonds between adjacent 1D metal–organic chains.
The linear metal–organic chains of 2 are built from the 2-connected [Cu(phen)] and MDPA nodes (Fig. 7a), featuring the same 2C1 topology as in 1. These chains are reinforced by the intra-chain hydrogen bonds and extended to a 2D layer via H-bonding interactions with the water of crystallization. From the topological viewpoint, the resulting 2D H-bonded layer (Fig. 7b) is composed of the 5-connected MDPA and 3-connected H2O nodes and the 2-connected [Cu(phen)] linkers, thus leading to a binodal 3,5-connected net with a rare 3,5L52 topology.54,55,58 It is defined by the point symbol of (3.52)(32.53.64.7) with the (3.52) and (32.53.64.7) indices corresponding to the H2O and MDPA nodes, respectively. Although a few examples of compounds with the 3,5L52 topology have been identified, such Cu-based networks are limited to single cases.58
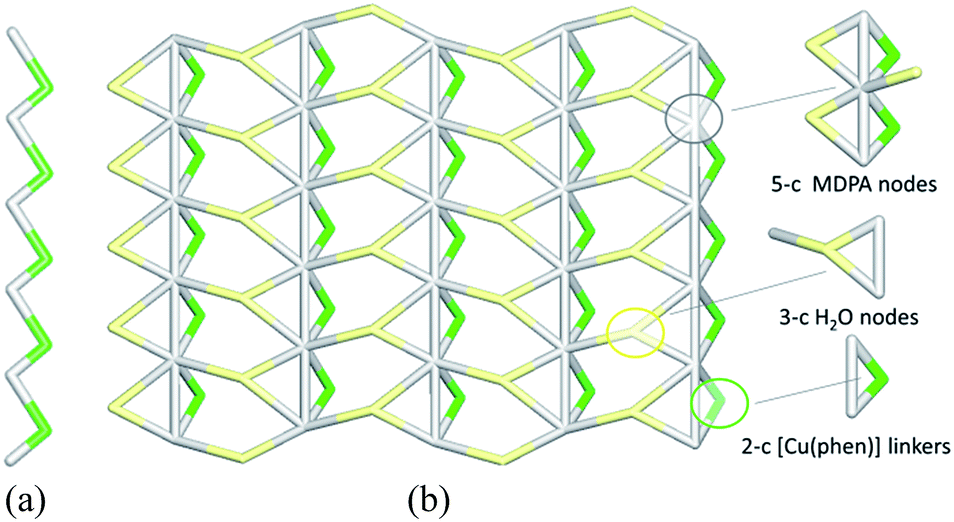 | ||
| Fig. 7 Topological representation of the underlying networks in 2: (a) 1D metal–organic chain and (b) 2D H-bonded layer (views along the a axis). (a) Uninodal 2-connected linear chain with the 2C1 topology. (b) Binodal 3,5-connected layer with the 3,5L52 topology. Color codes: (a) centroids of 2-connected [Cu(phen)] (green) and MDPA (gray) nodes; (b) centroids of 5-connected MDPA (gray) and 3-connected H2O (pale yellow) nodes, centroids of 2-connected [Cu(phen)] linkers (green). | ||
Compound 3 is isostructural to 1 and also possesses similar zigzag metal–organic chains with the 2C1 topology (see Fig. 6a for 1 as an example). These chains in 3 are assembled from the [Mn(bpy)(H2O)2] and EDPA blocks and are further extended into a 3D H-bonded framework, which is topologically similar to that of 1 (Fig. 6a).
In contrast to 1D metal–organic chains in 1–3 driven by the μ2-diphosphonates, compound 4 features a more complex 2D coordination network owing to the presence of both μ2- and μ4-EDPA building blocks. Hence, an underlying metal–organic 2D net in 4 is composed of the 3-connected [Zn(bpy)] and 4-connected μ4-EDPA nodes, as well as the 2-connected μ2-EDPA linkers (Fig. 8). This net can be classified as a binodal 3,4-connected layer with the 3,4L13 topology54,55 and the point symbol of (4.62)2(42.62.82), wherein the (4.62) and (42.62.82) indices concern the [Zn(bpy)] and μ4-EDPA nodes, respectively. The metal–organic layers in 4 are further reinforced by hydrogen bonds, although without extending an overall network dimensionality. A few Zn coordination polymers with the 3,4L13 topology have been reported.54,55,59–61
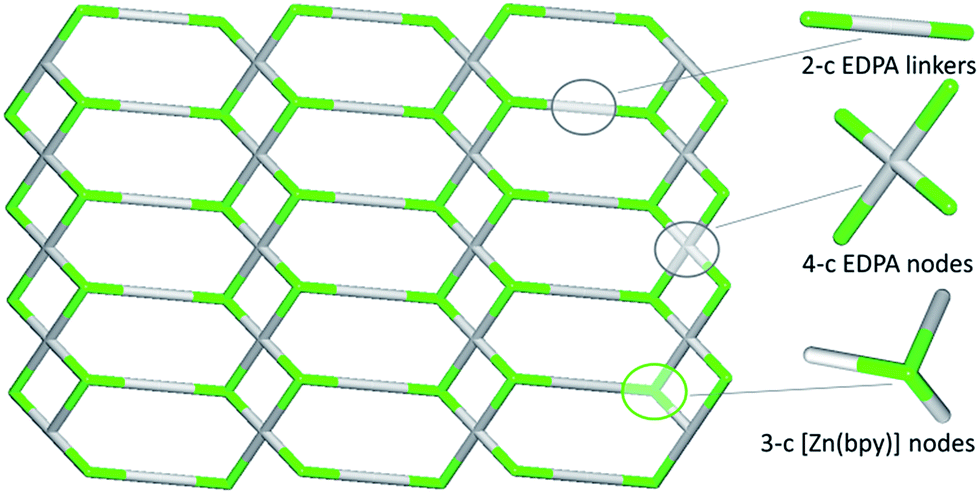 | ||
| Fig. 8 Topological representation of the underlying 2D metal–organic network in 4 showing a binodal 3,4-connected layer with the 3,4L13 topology (view along the b axis). Color codes: centroids of 3-connected [Zn(bpy)] nodes (green), centroids of 2- and 4-connected EDPA linkers and nodes (gray). | ||
In contrast to coordination polymers 1–4, the crystal structure of 5 is discrete and composed of a [Ni(phen)(H2O)4]2+ cation and an EDPA2− anion. However, both the cation and anion are multiply held together by strong hydrogen bonds, resulting in a 3D supramolecular network. This was simplified and analyzed topologically, revealing a binodal 6,6-connected underlying net (Fig. 9) with the 6,6T1 topology. It is defined by the point symbol of (413.62)(48.66.8), wherein the (413.62) and (48.66.8) indices are those of the [Ni(phen)(H2O)4] and EDPA nodes, respectively.
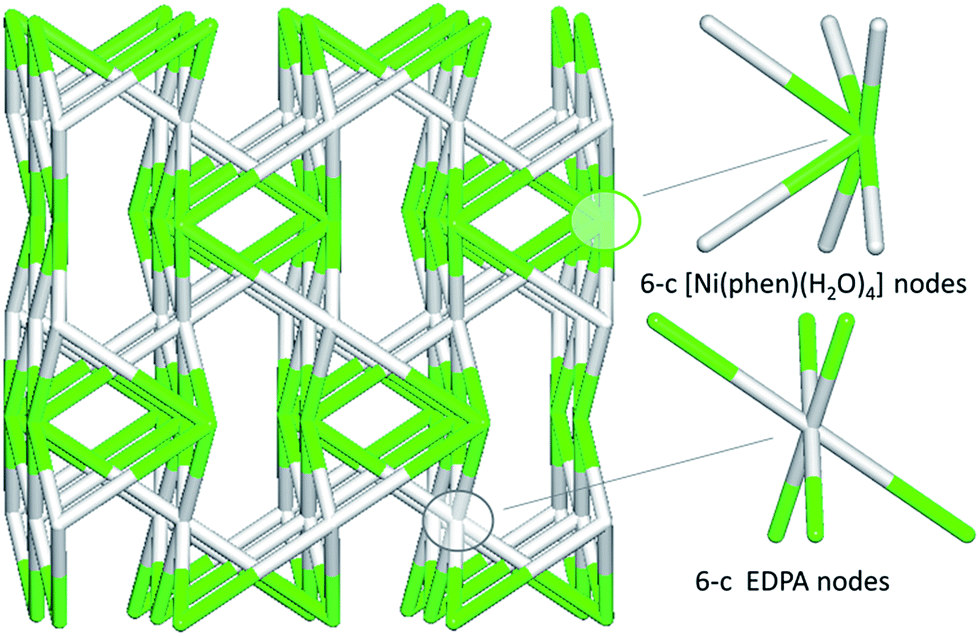 | ||
| Fig. 9 Topological representation of the underlying 3D supramolecular network in 5 showing a binodal 6,6-connected net with the 6,6T1 topology (view along the a axis). Color codes: centroids of 6-connected [Ni(phen)(H2O)4] nodes (green), centroids of 6-connected EDPA nodes (gray). | ||
Catalytic activity in hydrocarboxylation of alkanes
Catalytic activity of compounds 1 (Cu-phen-EDPA) or 1a (Co-phen-EDPA) was investigated in the hydrocarboxylation of various C5–C8 alkanes, including cyclic (cyclopentane, cyclohexane, cycloheptane, cyclooctane) and linear (n-pentane, n-hexane, n-heptane, n-octane) substrates. The hydrocarboxylation consists of reacting a Cn alkane with CO (carbonyl source), potassium peroxodisulfate (oxidant), and H2O (hydroxyl source) to form directly a Cn+1 carboxylic acid, Cn–COOH, or a mixture of isomeric carboxylic acids.49 The reactions proceed at 60 °C in water–acetonitrile medium and require the use of a homogeneous metal containing catalyst. The obtained results are summarized in Tables 4 and 5.Hence, the Cu-containing compound 1 catalyzes the hydrocarboxylation of cyclopentane and cyclohexane (Table 4, entries 1, 3) to give cyclopentanecarboxylic and cyclohexanecarboxylic acids in 26% and 43% yields, respectively; hereinafter, all the yields are based on alkane substrate. Under similar reaction conditions, the Co-containing compound 1a is less effective, resulting in 7% and 12% yields of C5H9COOH and C6H11COOH, respectively (Table 1, entries 2, 4). Given a much higher activity of 1, it was selected to further investigate the substrate scope. In fact, cycloheptane can also be converted into cycloheptanecarboxylic acid in up to 25% yield (entry 5), whereas in the case of the cyclooctane substrate only 10% yield of the corresponding acid product was detected (entry 6). In all cases, the hydrocarboxylation of cycloalkanes results in the formation of one monocarboxylic acid product due to the presence of a single type of carbon atom in their molecules. No formation of dicarboxylic acids was observed. However, the corresponding cyclic ketones and alcohols are also formed as a result of the competing partial cycloalkane oxidation reaction. The total yield of these oxidation products (ketone being formed in larger amounts than alcohol in all cases, Table 4) increases with the hydrocarbon size, namely from 1–3% for C5H10 and C6H12 to 9–13% for C7H14 and C8H16, respectively.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cycloalkane | Catalyst | Product yield,b % | |||
| Carboxylic acid | Ketone | Alcohol | Totalc | |||
| a Cyclic ketones and alcohols are also formed as products of cycloalkane oxidation. Reaction conditions (unless stated otherwise): cycloalkane (1.00 mmol), catalyst 1 or 1a (0.01 mmol), p(CO) = 20 atm, K2S2O8 (1.50 mmol), H2O (2.0 mL)/MeCN (4.0 mL), 60 °C, 4 h in an autoclave (20.0 mL capacity).b [Moles of product per mol of alkane] × 100%; determined by GC analysis.c Yield of all products. | ||||||
| 1 | C5H10 | 1 | 25.9 | 0.8 | 0.3 | 27.0 |
| 2 | C5H10 | 1a | 7.3 | 0.3 | 0.1 | 7.7 |
| 3 | C6H12 | 1 | 42.7 | 2.3 | 0.5 | 45.5 |
| 4 | C6H12 | 1a | 11.9 | 0.6 | 0.1 | 12.6 |
| 5 | C7H14 | 1 | 25.4 | 7.2 | 2.0 | 34.6 |
| 6 | C8H16 | 1 | 10.4 | 9.1 | 3.5 | 23.0 |
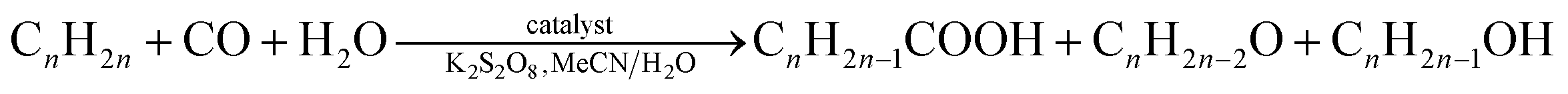
To further investigate the substrate scope, we studied the hydrocarboxylation of linear C5–C8 alkanes catalyzed by compound 1. Since linear alkanes contain both secondary (more reactive) and primary carbon atoms, a mixture of isomeric carboxylic acids is produced (Table 5). As expected, the branched carboxylic acids are formed as the main products, while the yields of the linear ones are negligible (∼1%). The highest total yield of acid products was observed for the n-pentane hydrocarboxylation (36%, Table 5, entry 1), followed by n-hexane (32%, entry 2), n-heptane (29%, entry 3), and n-octane (24%, entry 4). On increasing the length of the hydrocarbon chain, a regioselectivity parameter, C(1):C(2):C(3):C(4), that indicates the normalized (for the relative number of hydrogen atoms) reactivity of H atoms at different positions of linear alkane chains, gradually decreases from 1:30:26 for n-pentane to 1:18:16:16 for n-octane. In addition, the increased selectivity of the carbon atom at the second C(2) position of the hydrocarbon chain was observed in all the tested linear alkanes. Given a homogeneous character of the catalytic systems and complexity of the reaction mixtures, it is particularly difficult to recover the catalyst. This can potentially be overcome in future research by attaching the homogeneous catalysts onto a solid support.
|
||||
|---|---|---|---|---|
| Entry | Alkane | Carboxylic acidb (yield, %) | Total yieldc (%) | Regioselectivity C(1):C(2):C(3):C(4)d |
| a Reaction conditions (unless stated otherwise): alkane (1.00 mmol), catalyst 1 (0.01 mmol), p(CO) = 20 atm, K2S2O8 (1.50 mmol), H2O (2.0 mL)/MeCN (4.0 mL), 60 °C, 4 h in an autoclave (20.0 mL capacity).b [Moles of acid per mol of alkane] × 100%; determined by GC analysis.c Yield of all acid products.d Regioselectivity parameter C(1):C(2):C(3):C(4) means the normalized (for the relative number of hydrogen atoms) reactivity of H atoms at different positions of linear alkane chains. |
||||
| 1 | n-C5H12 | C2H5CH(COOH)C2H5 (10.6) | 35.8 | 1:30:26 |
| C3H7CH(COOH)CH3 (24.0) | ||||
| n-C5H11COOH (1.2) | ||||
| 2 | n-C6H14 | C3H7CH(COOH)C2H5 (15.1) | 32.4 | 1:24:22 |
| C4H9CH(COOH)CH3 (16.3) | ||||
| n-C6H13COOH (1.0) | ||||
| 3 | n-C7H16 | C3H7CH(COOH)C3H7 (5.3) | 29.1 | 1:21:18:18 |
| C4H9CH(COOH)C2H5 (10.5) | ||||
| C5H11CH(COOH)CH3 (12.4) | ||||
| n-C7H15COOH (0.9) | ||||
| 4 | n-C8H18 | C4H9CH(COOH)C3H7 (7.4) | 23.7 | 1:18:16:16 |
| C5H11CH(COOH)C2H5 (7.6) | ||||
| C6H13CH(COOH)CH3 (8.0) | ||||
| n-C8H17COOH (0.7) | ||||
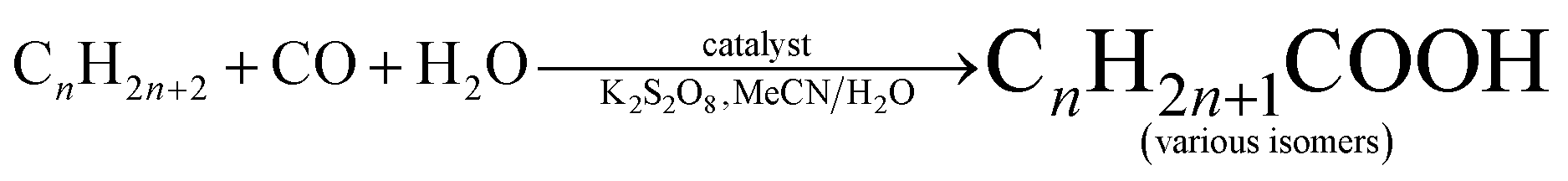
Following a previous approach49 and guided by the selectivity parameters observed herein, a simplified free-radical mechanism for the hydrocarboxylation of alkanes can be proposed (Fig. 10). The first principal step (step a) involves the formation of alkyl radicals via the abstraction of a hydrogen atom from the alkane by SO4˙− (sulfate radicals are generated from K2S2O8). Then, alkyl radicals R˙ are rapidly carbonylated by CO to give acyl radicals RCO˙ (step b). These are further oxidized by CuII to acyl cations RCO+ (step c), whereas the regeneration of the CuII species occurs upon oxidation of CuI by K2S2O8 (step c′). The acyl cations are finally hydrolyzed by water to form carboxylic acids RCOOH (step d).
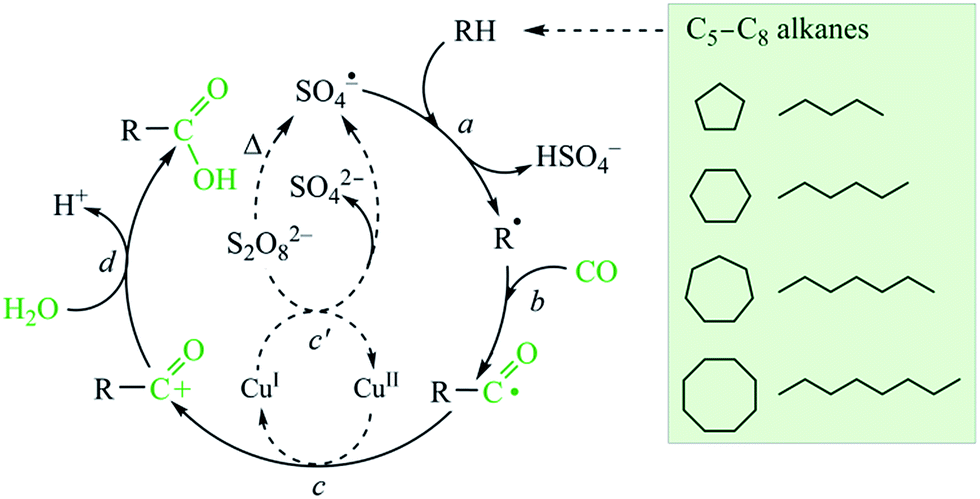 | ||
| Fig. 10 Simplified free-radical mechanism for the hydrocarboxylation of alkanes. | ||
Conclusions
The present paper continues our efforts for structural mapping of metal phosphonate materials that incorporate a metal ion, a phosphonate ligand, and a secondary auxiliary ligand (the chelating bpy and phen, in this case).The conclusions of this work are summarized below:
(a) Chelating N-heterocyclic ligands, such as 2,2′-bipyridine and 1,10-phenanthroline can be used to “decorate” a metal-phosphonate coordination network, imparting dramatic changes to the dimensionality and overall structure.
(b) Small structural alterations of the phosphonate ligand backbone impart significant changes to the product. This is exemplified in compounds 1 and 2. Coordination polymer 1 contains EDPA, and 2 contains MDPA, a structural “analog” of EDPA having one –CH2– less than EDPA. Compound 1 features a 1D zigzag chain that is formed by the bridging action of EDPA (each end phosphonate coordinates Cu in a terminal fashion). However, 2 forms a linear 1D chain in which MDPA chelates a Cu center (by the two end phosphonates) and simultaneously bridges (through one of the phosphonates) a neighboring Cu center.
(c) The two rather similar SALs, bpy and phen, can chelate metal centers in the same fashion; however, they form different π–π interactions that influence the supramolecular structure of the materials.
(d) The coordination geometry of the M2+ centers apparently affects the final structure. The Cu2+ center in 1 is octahedral, whereas it is square pyramidal in 2. The Mn2+ center is octahedral in 3 and the Zn2+ center is trigonal bipyramidal in 4.
(e) The hydrogen bonding interactions can play a significant role in the structure of each compound. The presence of water molecules (either metal-coordinated or in the lattice) enriches the hydrogen-bonding patterns.
(f) An additional factor that plays a role in the crystal packing of the discussed compounds is the π–π stacking interactions between the bpy or phen aromatic rings.
(g) Both the metal–organic and/or H-bonded networks in 1–5 were analyzed from the topological viewpoint. This analysis has disclosed the simple 2C1 topology of 1D coordination chains in 1–3 and the 3,4L13 topology of the 2D metal–organic layers in 4. Besides, the structures of 1–3 and 5 are further extended [1D → 3D (1, 3), 1D → 2D (2), 0D → 3D (5)] by numerous hydrogen bonds, resulting in the generation of H-bonded nets with the seh-4,6-C2/c topology in 1 and 3, the 3,5L52 topology in 2, and the 6,6T1 topology in 5. Hence, the current study also contributes to the topological classification of metal–organic and H-bonded networks.
(h) Furthermore, some of the obtained coordination compounds were tested as homogeneous catalysts for the mild hydrocarboxylation of alkanes, by CO, K2S2O8, and H2O to give carboxylic acids with one more carbon atom. In fact, compound 1 efficiently catalyzes the hydrocarboxylation of various cyclic (cyclopentane, cyclohexane, cycloheptane, cyclooctane) and linear (n-pentane, n-hexane, n-heptane, n-octane) alkanes. The investigation of the substrate scope revealed that the highest yields of products are observed when using cyclohexane (43% of C6H11COOH) and n-pentane (36% of isomeric C5H11COOH). These product yields are very good in the field of mild oxidative functionalization of alkanes, considering their extremely high inertness and the rather mild reaction conditions employed (60 °C, aqueous medium, absence of a strong acid).48,49
Acknowledgements
This research has been co-financed by the European Union (European Social Fund – ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) – Research Funding Program: “Heracleitus II: Investing in knowledge society” through the European Social Fund. MVK and AMK acknowledge the Foundation for Science and Technology (FCT), Portugal (IF/01395/2013/CP1163/CT005, UID/QUI/00100/2013). GM acknowledges support from the National Science Foundation under Grant No. CHE-1404730.Notes and references
- Metal Phosphonate Chemistry: From Synthesis to Applications, ed. A. Clearfield and K. D. Demadis, Royal Society of Chemistry, London, 2012, and references therein Search PubMed.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, Catal. Sci. Technol., 2015, 5, 4258 CAS.
- T. Hoffmann, P. Friedel, C. Harnisch, L. Häußler and D. Pospiech, J. Anal. Appl. Pyrolysis, 2012, 96, 43 CrossRef CAS.
- K. D. Demadis, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 167 CrossRef CAS.
- K. D. Demadis and A. Ketsetzi, Sep. Sci. Technol., 2007, 42, 1639 CrossRef CAS.
- J. Zon, P. Garczarek, and M. Bialek, in Metal Phosphonate Chemistry: From Synthesis to Applications, ed. A. Clearfield and K. D. Demadis, Royal Society of Chemistry, London, 2012, p. 170 Search PubMed.
- P. Kafarski and J. Zon, in Aminophosphonic and Aminophosphinic Acids, V. P. Kukhar and H. R. Hudson, John Wiley and Sons, New York, 2000, p. 33 Search PubMed.
- L. Delain-Bioton, J.-F. Lohier, D. Villemin, J. Sopková-de Oliveira Santos, G. B. Hix and P.-A. Jaffres, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o47 CAS.
- D. Villemin, B. Moreau, A. Elbilali, M.-A. Didi, M. Kaid and P.-A. Jaffres, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 2511 CrossRef CAS.
- M. B. Tomson, A. T. Kan and J. E. Oddo, Langmuir, 1994, 10, 1442 CrossRef CAS.
- T. Ichikawa and K. Sawada, Bull. Chem. Soc. Jpn., 1997, 70, 2111 CrossRef CAS.
- R. M. Cigala, M. Cordaro, F. Crea, C. De Stefano, V. Fracassetti, M. Marchesi, D. Milea and S. Sammartano, Ind. Eng. Chem. Res., 2014, 53, 9544 CrossRef CAS.
- V. Deluchat, B. Serpaud, E. Alves, C. Caullet and L.-C. Bollinger, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 109, 209 Search PubMed.
- K. Popov, H. Rönkkömäki and L. H. J. Lajunen, Pure Appl. Chem., 2001, 73, 1641 CrossRef CAS.
- M. Dua, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282 CrossRef.
- (a) K. D. Demadis, M. Papadaki, R. G. Raptis and H. Zhao, Chem. Mater., 2008, 20, 4835 CrossRef CAS; (b) R. M. P. Colodrero, A. Cabeza, P. Olivera-Pastor, J. Rius, D. Choquesillo-Lazarte, J. M. García-Ruiz, M. Papadaki, K. D. Demadis and M. A. G. Aranda, Cryst. Growth Des., 2011, 11, 1713 CrossRef CAS.
- M. Feyand, A. Hübner, A. Rothkirch, D. S. Wragg and N. Stock, Inorg. Chem., 2012, 51, 12540 CrossRef CAS PubMed.
- S. Lodhia, A. Turner, M. Papadaki, K. D. Demadis and G. B. Hix, Cryst. Growth Des., 2009, 9, 1811 CAS.
- (a) K. D. Demadis, S. D. Katarachia, H. Zhao, R. G. Raptis and P. Baran, Cryst. Growth Des., 2006, 6, 836 CrossRef CAS; (b) K. D. Demadis, E. Barouda, H. Zhao and R. G. Raptis, Polyhedron, 2009, 28, 3361 CrossRef CAS.
- R. M. P. Colodrero, A. Cabeza, P. Olivera-Pastor, A. Infantes-Molina, E. Barouda, K. D. Demadis and M. A. G. Aranda, Chem.–Eur. J., 2009, 15, 6612 CrossRef CAS PubMed.
- R. F. Mendes, P. Silva, M. M. Antunes, A. A. Valente and F. A. Almeida Paz, Chem. Commun., 2015, 51, 10807 RSC.
- C. Schmidt, M. I. Feyand, A. Rothkirch and N. Stock, J. Solid State Chem., 2012, 188, 44 CrossRef CAS.
- K. D. Demadis, J. D. Sallis, R. G. Raptis and P. Baran, J. Am. Chem. Soc., 2001, 123, 10129 CrossRef CAS PubMed.
- N. Stavgianoudaki, K. E. Papathanasiou, R. M. P. Colodrero, D. Choquesillo-Lazarte, J. M. Garcia-Ruiz, A. Cabeza, M. A. G. Aranda and K. D. Demadis, CrystEngComm, 2012, 14, 5385 RSC.
- (a) K. D. Demadis, M. Papadaki, M. A. G. Aranda, A. Cabeza, P. Olivera-Pastor and Y. Sanakis, Cryst. Growth Des., 2010, 10, 357 CrossRef CAS; (b) K. D. Demadis, E. Armakola, K. E. Papathanasiou, G. Mezei and A. M. Kirillov, Cryst. Growth Des., 2014, 14, 5234 CrossRef CAS.
- Z.-Y. Du, H.-R. Wen and Y.-R. Xie, J. Mol. Struct., 2008, 891, 272 CrossRef CAS.
- Z.-Y. Du, A. V. Prosvirin and J.-G. Mao, Inorg. Chem., 2007, 46, 9884 CrossRef CAS PubMed.
- F. Zhai, M. Deng, Y. Ling, Z. Chen, L. Weng and Y. Zhou, Inorg. Chim. Acta, 2013, 402, 104 CrossRef CAS.
- K.-R. Ma, Y. Zhang, Y.-H. Kan, X.-J. Yang and M.-H. Cong, Synth. Met., 2013, 182, 40 CrossRef CAS.
- C. Li, C.-Q. Jiao, Z.-G. Sun, K. Chen, C.-L. Wang, Y. Y. Zhu, J. Zhu, Y. Zhao, M.-J. Zheng, S.-H. Sun, W. Chu and H. Tian, CrystEngComm, 2012, 14, 5479 RSC.
- R. Fu, S. Hu and X. Wu, CrystEngComm, 2012, 14, 3478 RSC.
- V. Chandrasekhar, R. Azhakar, T. Senapati, P. Thilagar, S. Ghosh, S. Verma, R. Boomishankar, A. Steiner and P. Kogerler, Dalton Trans., 2008, 1150 RSC.
- V. Chandrasekhar, T. Senapati and R. Clérac, Eur. J. Inorg. Chem., 2009, 1640 CrossRef CAS.
- R. P. Doyle, M. Nieuwenhuyzen and P. E. Kruger, Dalton Trans., 2005, 3745 RSC.
- P. E. Kruger, R. P. Doyle, M. Julve, F. Lloret and M. Nieuwenhuyzen, Inorg. Chem., 2001, 40, 1726 CrossRef CAS PubMed.
- H.-H. Song, L.-M. Zheng, Z. Wang, C.-H. Yan and X.-Q. Xin, Inorg. Chem., 2001, 40, 5024 CrossRef CAS PubMed.
- H.-H. Song, L.-M. Zheng, C.-H. Lin, S.-L. Wang, X.-Q. Xin and S. Gao, Chem. Mater., 1999, 11, 2382 CrossRef CAS.
- K.-J. Lin, S.-J. Fu, C.-Y. Cheng, W.-H. Chen and H.-M. Kao, Angew. Chem., 2004, 116, 4282 CrossRef.
- S.-J. Fu, C.-Y. Cheng and K.-J. Lin, Cryst. Growth Des., 2007, 7, 1381 CAS.
- (a) L.-M. Zheng, H.-H. Song, C.-H. Lin, S.-L. Wang, Z. Hu, Z. Yu and X.-Q. Xin, Inorg. Chem., 1999, 38, 4618 CrossRef CAS PubMed; (b) P. Yin, S. Gao, Z.-M. Wang, C.-H. Yan, L.-M. Zheng and X.-Q. Xin, Inorg. Chem., 2005, 44, 2761 CrossRef CAS PubMed; (c) L.-M. Zheng, H.-H. Song, C.-Y. Duan and X.-Q. Xin, Inorg. Chem., 1999, 38, 5061 CrossRef CAS PubMed.
- H. Tian, Y.-Y. Zhu, Z.-G. Sun, F. Tong, J. Zhu, W. Chu, S.-H. Sun and M.-J. Zheng, New J. Chem., 2013, 37, 212 RSC.
- S.-F. Zhang, Z. Liu, X.-G. Yang, Q. Yu and B.-R. Hou, Inorg. Chem. Commun., 2008, 11, 604 CrossRef CAS.
- A. Clearfield, Dalton Trans., 2008, 6089 RSC.
- G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430 RSC.
- E. Barouda, K. D. Demadis, S. Freeman, F. Jones and M. I. Ogden, Cryst. Growth Des., 2007, 7, 321 CAS.
- K. D. Demadis, A. Panera, Z. Anagnostou, D. Varouhas, A. M. Kirillov and I. Cisarova, Cryst. Growth Des., 2013, 13, 4480 CAS.
- (a) J.-G. Mao, in Metal Phosphonate Chemistry: From Synthesis to Applications, ed. A. Clearfield and K. D. Demadis, Royal Society of Chemistry, London, 2012, p. 133 Search PubMed; (b) Q. Li and J. Qian, RSC Adv., 2014, 4, 32391 RSC.
- (a) A. E. Shilov and G. B. Shulpin, Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes, Kluwer Acad. Publ., Dordrecht, 2000 Search PubMed; (b) G. B. Shul'pin, Catalysts, 2016, 6, 50 CrossRef; (c) G. A. Olah and A. Molnar, Hydrocarbon Chemistry, Wiley, 2003 CrossRef; (d) Alkane C–H Activation by Single-Site Metal Catalysis, ed. P. J. Pérez, Springer, 2012 Search PubMed.
- A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Coord. Chem. Rev., 2012, 256, 2741 CrossRef CAS.
- (a) M. V. Kirillova, A. M. Kirillov, A. N. C. Martins, C. Graiff, A. Tiripicchio and A. J. L. Pombeiro, Inorg. Chem., 2012, 51, 5224 CrossRef CAS PubMed; (b) A. M. Kirillov, Y. Y. Karabach, M. V. Kirillova, M. Haukka and A. J. L. Pombeiro, Cryst. Growth Des., 2012, 12, 1069 CrossRef CAS.
- (a) T. A. Fernandes, V. André, A. M. Kirillov and M. V. Kirillova, J. Mol. Catal. A: Chem., 2017, 426, 357 CrossRef CAS; (b) A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Adv. Inorg. Chem., 2013, 65, 1 CrossRef CAS.
- L.-S. Long, CrystEngComm, 2010, 12, 1354 RSC.
- R. C. Finn and J. Zubieta, Inorg. Chim. Acta, 2002, 332, 191 CrossRef CAS.
- V. A. Blatov, IUCr Computing Commission Newsletter, 2006, 7, 4 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CAS.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 CrossRef CAS PubMed.
- C.-S. Liu, E. C. Sanudo, M. Hu, L.-M. Zhou, L.-Q. Guo, S.-T. Ma, L.-J. Gao and S.-M. Fang, CrystEngComm, 2010, 12, 853 RSC.
- N. B. Shustova, B. D. McCarthy and M. Dinca, J. Am. Chem. Soc., 2011, 133, 20126 CrossRef CAS PubMed.
- A. M. Kutasi, D. R. Turner, P. Jensen, B. Moubaraki, S. R. Batten and K. S. Murray, Inorg. Chem., 2011, 50, 6673 CrossRef CAS PubMed.
- Z. Hulvey, J. D. Furman, S. A. Turner, M. Tang and A. K. Cheetham, Cryst. Growth Des., 2010, 10, 2041 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA traces, calculated and measured XRD powder patterns for all compounds. CCDC 1435205–1435207 and 1521331. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00738h |
| This journal is © The Royal Society of Chemistry 2017 |