
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMorphology effects on singlet oxygen production and bacterial photoinactivation efficiency by different silica-protoporphyrin IX nanocomposites†
G. Zampini ab,
O. Planasb,
F. Marmottinia,
O. Gulíasb,
M. Agutb,
S. Nonell*b and
L. Latterini*a
ab,
O. Planasb,
F. Marmottinia,
O. Gulíasb,
M. Agutb,
S. Nonell*b and
L. Latterini*a
aDepartment of Chemistry, Biology and Biotechnology, University of Perugia, Via Elce di Sotto, 8, 06123 Perugia, Italy. E-mail: loredana.latterini@unipg.it
bInstitut Químic de Sarrià, Universitat Ramon Llull, Via Augusta 390, Barcelona 08017, Spain. E-mail: santi.nonell@iqs.url.edu
First published on 3rd March 2017
Abstract
Different silica-protoporphyrin IX (PpIX) nanocomposites have been synthesized to evaluate the dependence of singlet oxygen production and bacterial inactivation efficiency on the morphology of the nanomaterials. Modulation of the synthetic procedure allowed obtaining silica nanoparticles with different porosity which were characterized by TEM and spectroscopic analysis after PpIX adsorption. Time-resolved phosphorescence measurements on the different nanoporous samples revealed that the porosity of the nanoparticles plays a pivotal role on the singlet oxygen production and release from the nanoparticles. Thus, apart from the expected decay of singlet oxygen outside the silica matrix, a second component has been observed for the porous materials, attributed to the decay of singlet oxygen inside the pores. The relative efficiency of singlet oxygen production resulted to be higher for the sample with the greatest pores volume. The capability of the nanocomposites to inactivate bacteria was tested in vitro on Staphylococcus aureus strain. Interestingly, the efficiency for singlet oxygen production of the nanocomposites and their bacterial inactivation efficiency followed a different trend, indicating that the relative position of the photosensitizer and the superficial properties of the particles affect the antibacterial activity of the overall system.
Introduction
Antimicrobial photodynamic therapy (aPDT) is based on the use of a photosensitizer (PS), able to generate reactive oxygen species (ROS) through irradiation at specific wavelengths.1 It represents an effective treatment against antibiotic-resistant bacteria as ROS generated by PDT mechanism induce several damages on different levels of the cell,2,3 thus bacteria will not be easily able to develop resistance.4Particular interest has been focused on singlet oxygen (1O2), which is conveniently produced by energy transfer between the excited triplet state of a PS and molecular oxygen, normally present in the surrounding environment.1
The increase in 1O2 production efficiency is a crucial goal to achieve for an effective application of photodynamic systems. Hydrophobic natural PSs, such as protoporphyrin IX (PpIX), are potentially very promising but the extreme low solubility in physiological medium precludes their direct application in aPDT;5 indeed PpIX singlet excited states intersystem cross into triplet states with high quantum yields (ΦT = 0.63 ± 0.04),6 and the long-lived triplet states are quenched efficiently by molecular oxygen to generate 1O2 with a reported quantum yield values (ΦΔ) of 0.77 in CCl4 and 0.60 in aqueous/TX100 solution.7 However, the formation of ground-state dimers and aggregates is responsible for the lowering of 1O2 quantum yield, experimentally demonstrated by the reduction in ΦΔ value for increasing concentration of porphyrins in water, or with the increase in the ionic strength of the solution by the addition of NaCl.8
In the last decades, several micro and nano-heterogeneous systems, in particular silica-based nanomaterials, have been studied and developed9 to overcome the solubility and the stability problems of the PS;10 the increasing interest for this kind of nanomaterials arises from the huge benefits related to the great stability of a silica structure, conjugated with the versatility of the particle surface.11 Indeed, very often they are used as a vehicle-system,12–15 firstly because the solid and inorganic structure of the matrix acts as a protective barrier against species that can destroy or limit the activity of PSs,16–19 and secondly the limitations due to the lability or the insolubility of these molecules may be overcome.18,20
The interactions between the photosensitizers and silica nanovectors have been recently investigated, with the aim to identify the most promising system for PDT. Different systems have been reported, wherein the non-covalent and covalent encapsulation of PpIX in both compact and mesoporous silica nanoparticles have been analyzed.21,22
Rossi and coworkers demonstrated that the covalent linkage of PpIX within the silica network allowed a significant improvement of the ability to produce singlet oxygen, obtaining a value of singlet oxygen quantum yield up to 0.9.7
The synthetic approaches derived from the pioneering work of Prasad and Kopelman groups23,24 are based on the encapsulation of PpIX inside the core of silica nanoparticles, which were tested in PDT experiments both in vitro and in vivo.25,26 Vivero-Escoto et al. anchored PpIX to silica nanoparticles through the use of stimuli-responsive groups, in order to achieve the self-regulated delivery of the PS triggered by the biological system itself.27
Most of the strategies relay on the modulation of the interaction between PpIX and silica and on synthetic steps to tune such interactions. Limited studies have been presented to relate the morphology of silica nanoparticles to PDT performances; a detailed investigation on how the matrix nanostructures affect PpIX photophysical behavior and singlet oxygen production is concealing. However, the possibility to use the silica mesostructures to tune the chemical and photophysical behavior of PpIX represents an important simplification on the preparation procedures and possible scale-up. To achieve an improvement in smart nanomaterial development, it would be extremely important to connect the photophysical behavior to the photodynamic action of PS-loaded nanomaterials in physiological conditions. The anchoring on a solid nanosupport28,29 can inevitably modify not only the photophysics and the efficiency of a PS, but also the superficial properties of the materials, thus inducing eventual variations in the interactions with biological samples, such as bacteria.
Therefore, in this work, PpIX loaded on silica nanoparticles with different morphology have been analyzed through a systematic characterization of the ground, singlet and triplet excited states, evaluating how the morphology of the carrier affects the photophysical and photosensitizing pathways of the PS. Direct measurements of 1O2 luminescence have been performed and the correlation to bacterial inactivation has been investigated to assess the influence of the physical–chemical properties of the nanocomposites to their antimicrobial behavior.
Experimental section
Materials
Protoporphyrin IX (PpIX, >97%) was obtained in diacid form from Frontier Scientific and used without further purification. Tetraethylorthosilicate (TEOS, 98%), (3-aminopropyl) triethoxysilane (APTES, ≥98%), hexadecyltrimethylammonium bromide (CTAB, 95%), hexadecyltrimethylammonium p-toluensulfonate (CTATos), triethanolamine (TEAH3, >98%), mesitylene (98%), Pluronic F-127 (EO106PO60EO106), hydrochloridric acid (HCl, 37%) and ammonium hydroxide solution (NH4OH, 28.0–30.0% NH3 basis), were all purchased from Sigma-Aldrich and used as received. Nanopure water (≤15.0 MΩ) from a Millipore Milli-Q gradient system, ethanol (96%) and methanol (anhydrous, 99.8%) from Sigma-Aldrich and N,N-dimethylformamide (DMF, ≥99%) from Acros Organics were used as solvents. Staphylococcus aureus CECT 239 were obtained from the Spanish Type Culture Collection (CECT).Synthetic procedures
The as-synthesized MPSSt were filtered and washed several times with water and ethanol. The removal of the surfactant was obtained through solvent extraction in a boiling acidic methanol for 6 hours. The extracted particles were washed several times with ethanol and then dried in air.32
The MPSLP thus obtained were centrifuged at 3000g for 15 minutes and washed several time with ethanol, followed by acidic extraction in methanol for at least 6 hours. The extracted particles were washed several time with ethanol and then dried in air.
In a typical synthesis procedure, a weighted amount of silica nanoparticles was suspended in ethanol and sonicated for 5 minutes to yield a final concentration of 5 mg mL−1. Then APTES (2 μL mL−1) was added and the mixture was stirred overnight at room temperature. The APTES-to-silica ratio was kept constant for all samples.
The amino-functionalized silica nanoparticles were obtained after centrifugation at 3000g for 20 minutes, followed by several washes with ethanol. Finally, the solids were dried under air at room temperature.
Each silica suspension was then centrifuged at 3000g for 20 minutes; the collected solid was washed twice with DMF and once with ethanol, and finally dried under air.
The concentration of PpIX on the silica surface was evaluated from the difference between the PpIX solution absorbance before (starting solution) and after (supernatant) the anchoring procedure (eqn S1†).
Physicochemical characterization of the nanoparticles
Size and morphology of all silica samples was investigated by a Philips 208 transmission electron microscope (TEM) operating at 80 kV. A small drop of the suspensions of amino-functionalized nanoparticles in ethanol was deposited on a 300 square mesh copper grid pre-coated with a Formvar film and evaporated under air at room temperature. The particle size distribution was evaluated from several micrographs by means of the image analyser software ImageJ (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2016). The zeta potential in aqueous suspensions was measured by a Nano-ZS Zetasizer (Malvern Instrument Ltd.). The samples were extensively sonicated prior to the measurements to ensure a complete dispersion of the nanoparticles.Nitrogen adsorption–desorption isotherms at 77 K were determined using a computer controlled Micromeritics (Norcross, GA, USA) ASAP 2010 apparatus. Prior to adsorption measurements, samples were outgassed at room temperature overnight. The specific surface area was determined by the Brunauer, Emmett and Teller (B.E.T.) technique;33 mesopore volume was analysed by the BJH method.34
Spectroscopic techniques
All spectroscopic measurements were carried out in spectroscopic grade DMF using a nanoparticle concentration of 2 mg mL−1 for all samples and after extensive sonication. Samples were sonicated before the measurements to ensure complete dispersion of the nanoparticles.Absorption spectra were recorded on a Varian Cary 8454 UV/Vis Diode Array and on a Varian Cary 6000i dual-beam spectrophotometers. Fluorescence emission spectra were recorded on a Jobin Yvon-Spex Fluoromax-4 spectrofluorometer. Time-resolved fluorescence emission decays were measured by use of PicoQuant Fluotime 200 time-correlated single photon counting system equipped with a red sensitive photomultiplier. Excitation was achieved using a 375 nm picosecond diode laser working at 10 MHz. Fluorescence lifetimes were determined using FluoFit 4.6.5 analysis software from PicoQuant. The specific 1O2 near-infrared phosphorescence decays at 1270 nm were recorded by means of a customized PicoQuant Fluotime 200 system described in detail elsewhere.35 Briefly, a 532 nm diode-pumped Nd:YAG laser (FTSS355-Q, Crystal Laser) working at 1 KHz repetition rate was used for excitation. Laser emission was filtered by a 1064 rugate notch filter (Edmund Optics) and an uncoated SKG-5 filter (CVI Laser Corportation) placed at the exit port of the laser to remove any residual component of its fundamental emission in the near-infrared. Sample luminescence was filtered by a 1100 long-pass filter (Edmund optics) and a narrow bandpass filter at 1270 nm (bk-1270-70-B, bk Interfernzoptik) to isolate the 1O2 emission. Photons emitted from the sample were detected using a TE-cooled near-IR sensitive photomultiplier tube assembly (H9170–45, Hamamatsu Photonics) and counted with a multichannel scaler (NanoHarp 250, PicoQuant GmbH). Time resolved-emission decays were fitted using non-linear regression function of Graph Pad Prism to recover the 1O2 and triplet lifetimes (τΔ and τT, respectively). The quantum yield of 1O2 production was derived from the amplitude of the signals.
Transient absorption experiments were carried out using a home-built nanosecond laser flash photolysis system. The second harmonic (532 nm) of a Continuum Surelite I-10 Nd:YAG laser (5 ns pulse width, 7.5 mJ per pulse) was directed to the sample and a 75 W short arc Xe lamp (USHIO) was used for probing the transient absorption in a right-angle geometry. Changes in the sample absorbance at different observation wavelengths were discriminated using a PTI 101 monochromator and detected using a Hamamatsu R928 photomultiplier. The signal was fed to a Lecroy WaveSurfer 454 oscilloscope for digitizing and averaging (typically 10 shot) and finally transferred to a PC computer for data storage and analysis. The energy of the laser pulsed was measured with a RJ 7610 energy meter from Laser Precision Corp. The system was controlled by a house-developed LKS software (LabView, National Instruments).
Bacterial inactivation tests
Results and discussion
Silica nanoparticles morphology and superficial properties
Fig. 1 shows the TEM images of the amino-functionalized nanostructures. Rapid inspection of the different contrast TEM images reveals that the prepared nanoparticles present four different types of morphologies: compact regular silica nanoparticles (CSNP, A), amorphous mesoporous silica nanoparticles (MPSAm, B), stellate mesoporous silica nanoparticles (MPSSt, C) and large-pore mesoporous silica nanoparticles (MPSLP, D). The mean diameter obtained for CSNP, MPSSt and MPSLP (see ESI, Fig. S1–S3†), was respectively 117 nm (σ = 7.1 nm), 83 nm (σ = 8.8 nm) and 94 nm (σ = 8.8 nm). It was not possible to accurately evaluate the size distribution for MPSAm due to the irregular nature of its structure, however nanostructures having tens of nm dimensions can be observed. | ||
| Fig. 1 TEM images of silica nanoparticles. (A) CSNP scale bar: 200 nm, (B) MPSAm scale bar: 100 nm, (C) MPSSt scale bar: 100 nm, (D) MPSLP scale bar: 200 nm. | ||
The control of nanoparticle's dimensions was achieved by the tight regulation of reactant's concentrations and synthetic conditions; the differential morphology of the silica nanoparticles is the result of templated synthetic strategies used for the sample preparation. In particular, CSNP were synthesized following the Stöber procedure with minor changes in the relative quantity of water, ethanol and ammonia, to achieve a good monodisperse size distribution for CSNP sample.30 The different pore shapes and morphologies for samples B, C and D (Fig. 1), were achieved by using different templating agents. Pluronic F127 yielded highly curved cage-like pores36 with small diameter,31 as in the case of MPSAm. Instead, the cationic surfactant cetyltrimethylammonium (CTA+) associated to tosilate (Tos−), a counterion with strong affinity to the polar head of the surfactant,32 led to the formation of a star-shaped (“stellate”) structure for MPSSt. Finally, the addition of mesitylene, a swelling agent, to the reaction mixture caused an increase in the dimensions of the pores37 without modifying their star shape (MPSLP). The achieved morphologies were not altered by amino-functionalization.
The surface area data of the nanosystems as well as zeta potential values in aqueous suspensions are summarized in Table S1.†
The adsorption and desorption isotherms of all the silica samples presented similar profiles, reflecting a mesoporous structure for all the samples; BET analysis of the data evidences that the surface area increases in the order CSNP, MPSAm, MPSSt and MPSLP, in agreement with the TEM findings. The evaluation of mesopore volume and mesopore size distribution was performed by BJH analysis of the adsorption and desorption data. The mesopore volume calculated from adsorption data for pores of dimensions between 20 Å and 400 Å is reported in Table S1 and in Fig. S5† the mesopore size distribution curves are showed. It is of interest to note that in the case of CSNP sample the calculated mesopore volume is 0.13 cm3 g−1 while the values obtained for others samples are included in the range 0.44–0.52 cm3 g−1. The pore size distribution curves put in evidence that, differently from the other samples, a wide maximum occur around a pore diameter of 100 Å in the case of MPSSt sample.
At the same time, a clear trend of the final zeta-potential values going from CSNP to MPSLP can be observed, which correlates with the surface area and pore-structuring of the nanomaterials. As porosity is increased, the zeta-potential diminishes. It is known that bare silica nanoparticles are generally negatively charged in water because of the presence of partly ionized silanol groups, whereas surface functionalization with terminal amino groups increases the value of the zeta potential.38,39 The results suggest that the pore size plays a fundamental role on the zeta potential values. This observation can be rationalized considering that APTES grafting has been carried out keeping constant the ratio of silica and APTES concentrations for all samples. The larger are the pores, the higher is the exposed surface (inner and outer the pores). Moreover, the samples bearing larger pores have a higher surface concentration of silanol moieties, resulting in lower zeta potentials. As a result, the surface of the nanoparticles with larger structures is on average, less covered by positively charged amino groups in the nanoparticles with larger pores, yielding to a more negative zeta potential value. Accordingly, for CSNP, in which no pores are present, the most positive zeta potential value is obtained.
Silica–PpIX conjugates
PpIX was electrostatically attached onto the different amino-functionalized silica nanostructures suspended in DMF by acid–base reaction of its carboxylic acid groups with the nanoparticle amino groups, and the samples PpIX-CSNP, PpIX-MPSAm, PpIX-MPSSt and PpIX-MPSLP were obtained. The concentration of PpIX (nmolPpIX/mgsilica) on the different silica samples was quantified through spectrophotometric measurements and is given in Table S1.† The concentration is the highest for CSNPs, and roughly equal for all mesoporous nanomaterials, independent of the pore size. Conjugation of PpIX did not modify the zeta potentials to a great extent (Table S1†).Photophysical properties of silica–PpIX conjugates
To evaluate the effect of the silica matrix on the photophysical behavior of PpIX, each sample was re-suspended in DMF and their absorption and fluorescence spectra were recorded. The scattering corrected absorption spectra of PpIX-loaded nanoconstructs are shown in Fig. 2a. For all samples, the classical shape of PpIX absorption is observed, with an intense Soret band centered at 406 nm and several Q bands in the visible range. However, an additional band at 675 nm, attributed to aggregated species40–42 can be detected for all samples, albeit with different intensities.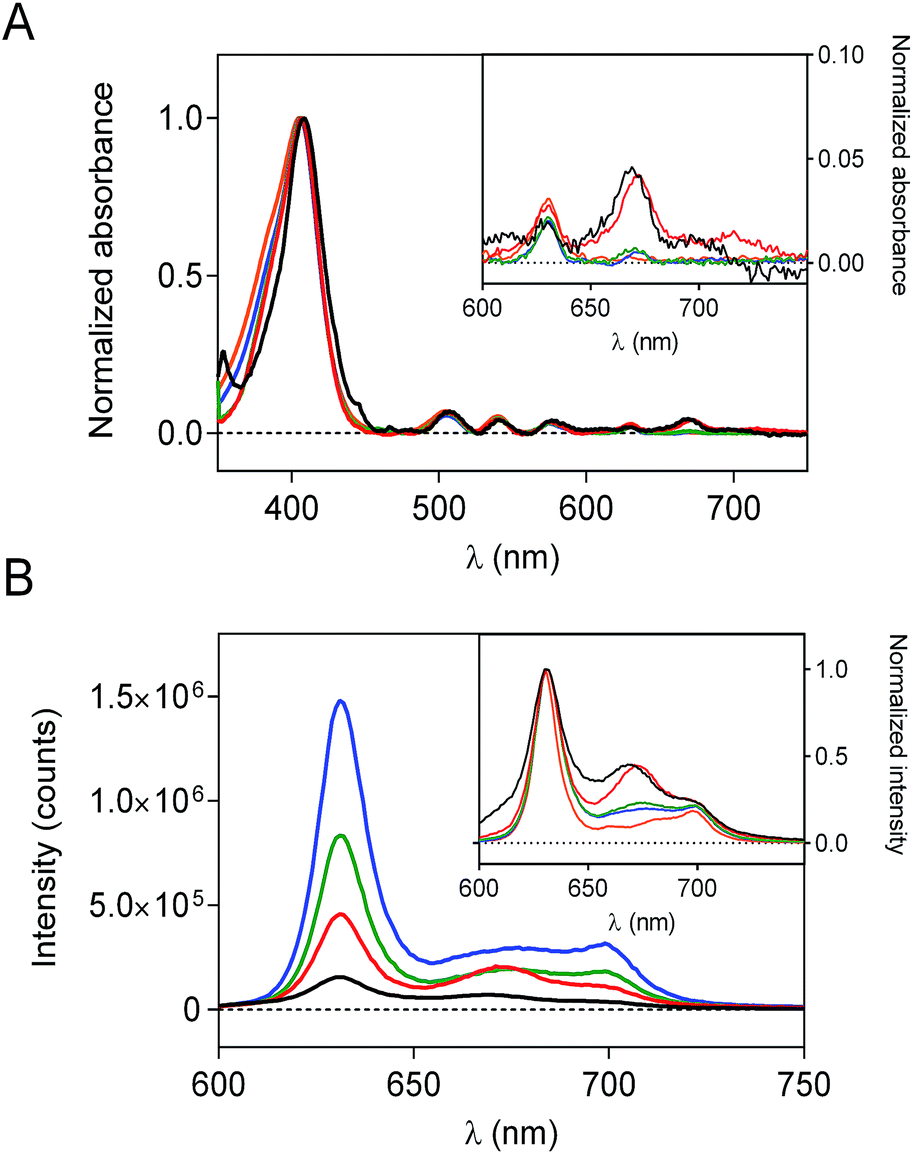 | ||
| Fig. 2 (A) Normalized absorption spectra of different silica nanoconjugate suspensions (black line PpIX-CSNP, red line PpIX-MPSAm, green line PpIX-MPSSt and blue line PpIX-MPSLP) in concentration of 2 mg mL−1 in DMF. Inset: magnification between 650–700 nm. (B) Overlaid fluorescence emission spectra (λexc = 532 nm); normalized fluorescence emission spectra, including PpIX emission in DMF, is shown as an inset for comparison purposes. | ||
Fluorescence spectra (Fig. 2b) likewise show the typical shape of PpIX but a new band at 680 nm is observed, whose relative intensity varies from sample to sample. Porphyrins have a strong tendency to aggregate and dimers are known to show red-shifted fluorescence relative to monomers, while higher-order aggregates are non-fluorescent.41 The observed spectra are consistent with the presence of both fluorescent dimers and non-fluorescent higher-order aggregates. Thus, PpIX-MPSLP displays the highest fluorescence intensity while PpIX-CNSP shows the lowest, despite their respective PpIX loading show the opposite trend (Table S1†). This indicates that PpIX is most aggregated in CNSP, consistent with its lowest specific surface.
The aggregation phenomenon was further confirmed by time-resolved fluorescence spectroscopy (Fig. 3). For each suspension, two contributions to the fluorescence decay were detected: a long-lived component of 14 ns, with the spectral features of monomeric PpIX in DMF,43 and a shorter-lived component of about 5 ns, attributed to the fluorescent dimers.44
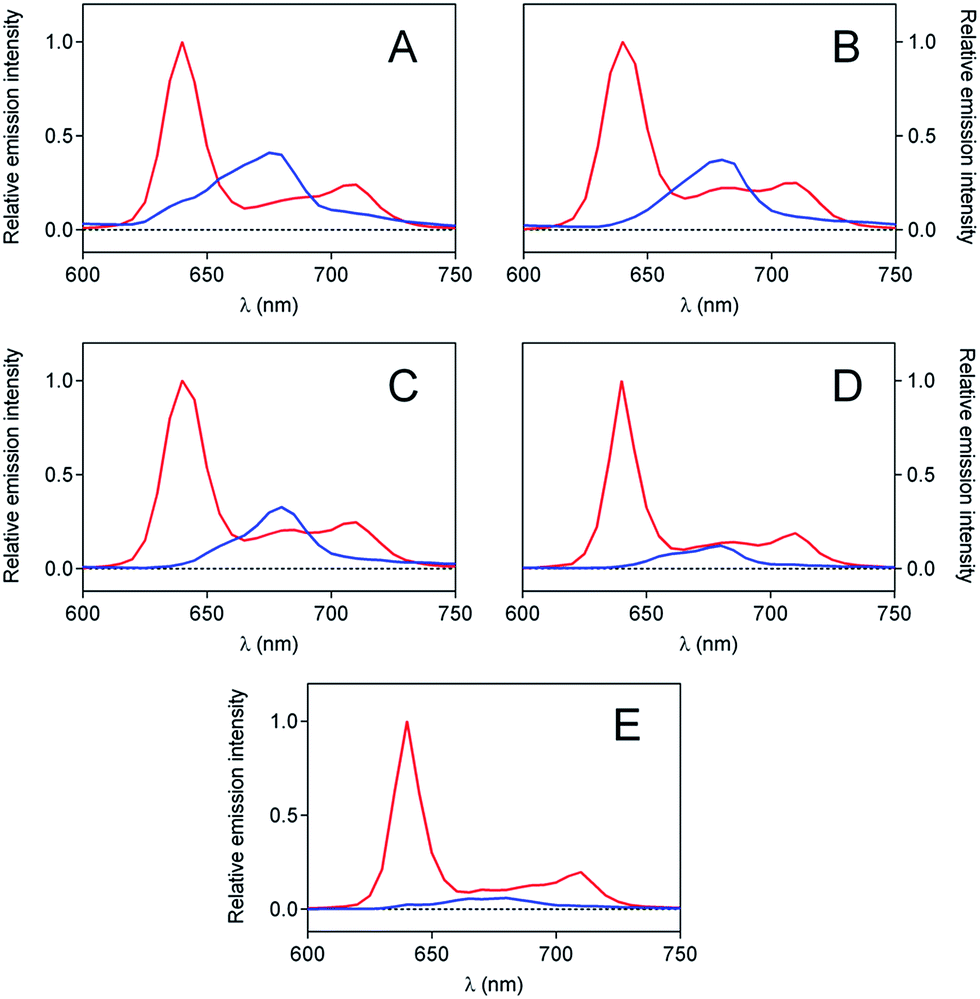 | ||
| Fig. 3 Time-resolved fluorescence spectra of (A) PpIX-CSNP, (B) PpIX-MPSAm, (C) PpIX-MPSSt, (D) PpIX-MPSLP,and (E) PpIX following the excitation at 532 nm. 14 ns component is indicated with a red line and 5 ns in blue. | ||
Further analysis was conducted to characterize the photophysical behavior of PpIX triplet state, both conjugated and unconjugated to silica, in air and in argon-saturated solution (Fig. S6 and Table S2†). All transient absorption signals could be satisfactorily fitted with a monoexponential decay function. In air-saturated samples, a triplet lifetime of about 0.4 μs was found for all the samples. However, when oxygen was removed from the suspensions through extensive argon bubbling, large differences were observed between all samples with a clear trend: the larger the pore, the longer the triplet lifetime becomes, suggesting that porous silica nanoparticles protect PpIX from self-quenching.45
Singlet oxygen measurements
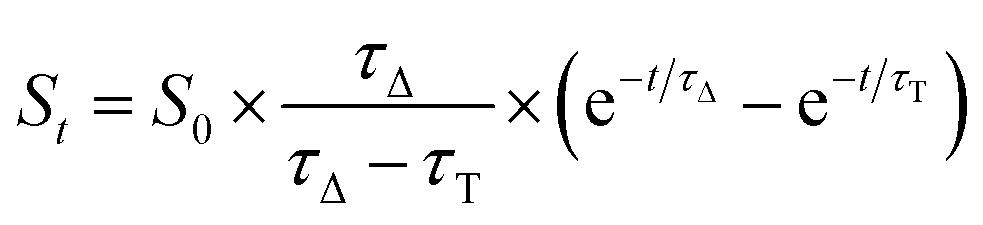 | (1) |
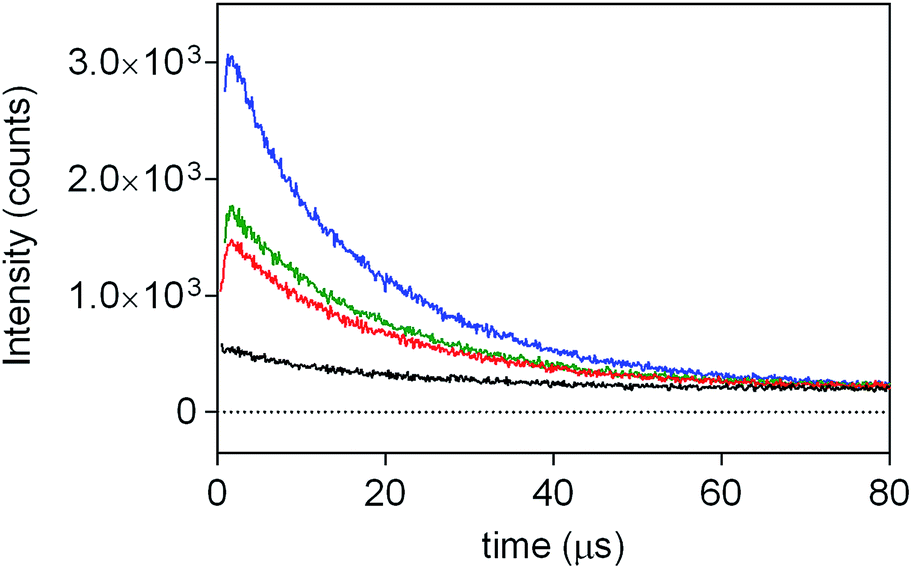 | ||
| Fig. 4 Time-resolved 1O2 phosphorescence spectra of PpIX-CSNP (black line), PpIX-MPSAm (red line), PpIX-MPSSt (green line) and PpIX-MPSLP (blue line) acquired at 1270 nm with an excitation wavelength of 532 nm. | ||
In contrast, singlet oxygen luminescence signals from mesoporous nanomaterials required the introduction of an additional decay component to adequately reproduce the experimental traces (eqn (2)).
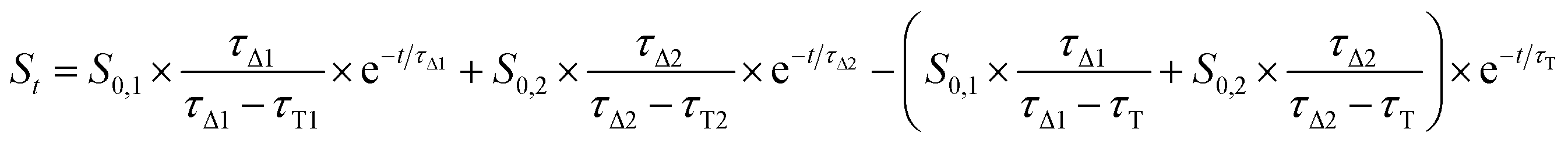 | (2) |
Results from fittings are summarized in Table 1. PpIX and PpIX-CSNP showed a growth lifetime of 0.4 and 0.5 μs and a decay lifetime of 22 and 20 μs, respectively. These lifetimes match those observed for molecular PS in DMF.46 In mesoporous nanomaterials, a similar rise lifetime was observed (0.3–0.5 μs) along with two decay components of 3 μs and 19–20 μs. In order to assign the observed lifetimes, phosphorescence decays were also recorded under oxygen-saturated conditions. A clear shortening of the rise component was observed while the decay components remained almost unaffected (Fig. S7 and Table S3†). Taken together with the results of the transient absorption experiments, these data indicate that the rise component corresponds to the formation of 1O2 from the triplet state of PpIX and the decay components to the decay of 1O2.
| Conjugate | S0,1/cnts | S0,2/cnts | τT/μs | τΔ1/μs | τΔ2/μs |
|---|---|---|---|---|---|
| PpIX | N/A | — | 0.4 | 22 | — |
| PpIX-CSNP | 341 | — | 0.5 | 19.9 | — |
| PpIX-MPSAm | 1291 (95%) | 71 (5%) | 0.4 | 19.5 | 3.0 |
| PpIX-MPSSt | 1515 (92%) | 125 (8%) | 0.3 | 19.5 | 3.0 |
| PpIX-MPSLP | 2584 (76%) | 792 (24%) | 0.5 | 19.2 | 3.1 |
In order to explain these results, it is worth recalling that while in compact nanoparticles only the external surface is available for anchoring the PS, the porosity of the mesoporous nanoparticles may enable the localization of PpIX both on the external surface and inside the silica pores. It can therefore be proposed that two populations of 1O2 might exist; one formed at the silica–solvent interface and able to escape and relax in the outer bulk solvent and another that decays inside the silica pores. The external 1O2 population decays therefore with a lifetime of 20 μs, as in the case of PpIX-CSNP. The inner-1O2 population, trapped in the silica structures, shows a much faster decay, probably due to a combination of three different effects: (i) an increase in the collision frequency of 1O2 with the walls of the pores,47 (ii) quenching of 1O2 by free amino groups deriving from APTES functionalization,48,49 or (iii) quenching by OH groups of the surface silanols and of strongly hydrogen-bonded water molecules remaining from the nanoparticle preparation procedure.50
This interpretation was confirmed by a second series of experiments in which PpIX-MPSLP was re-suspended in D2O. It is well known that OD groups, produced by isotopic exchange of the trapped water molecules and surface silanols with D2O, are far worse quenchers of 1O2 than OH groups.51 One would therefore expect that the 1O2 lifetime inside the pores would now be closer to that in the external bulk solvent. Consistent with this expectation, the phosphorescence decay could be satisfactorily fitted by a biexponential model with a 3 μs rise time and 53 μs for the single decay component (Fig. S8†). One can notice that the determined 1O2 decay time value (53 μs) is slightly shorter than the reported lifetime of 1O2 in D2O, (67 μs),52 indicating that silica still exerts some quenching effect.
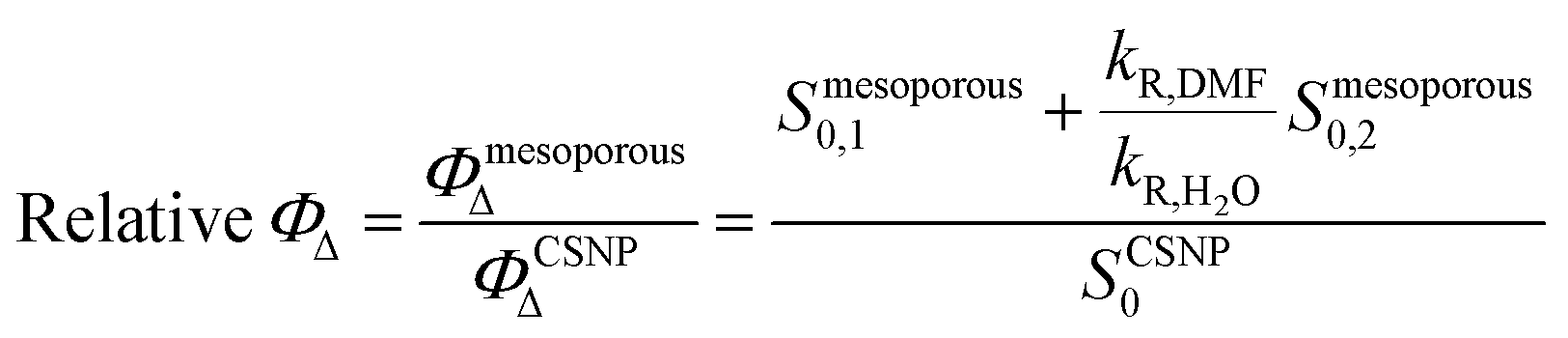 | (3) |
As shown in Table 2, the mesoporous materials have a higher efficiency in the photosensitization of 1O2. In particular, PpIX-MPSAm, PpIX-MPSSt and PpIX-MPSLP display a relative efficiency about 4.4, 5.5 and 14.6 times higher than PpIX-CSNP, respectively, with PpIX-MPSLP presenting the highest value. However, in the mesoporous materials, the two populations of 1O2 molecules, kinetically detected (Table 1), might show different effectiveness to photoinactivate bacteria. The relative contribution of the two populations can be estimated from the amplitude of the components corrected for the radiative rate constants as detailed in eqn (4) and is also given in Table 2.
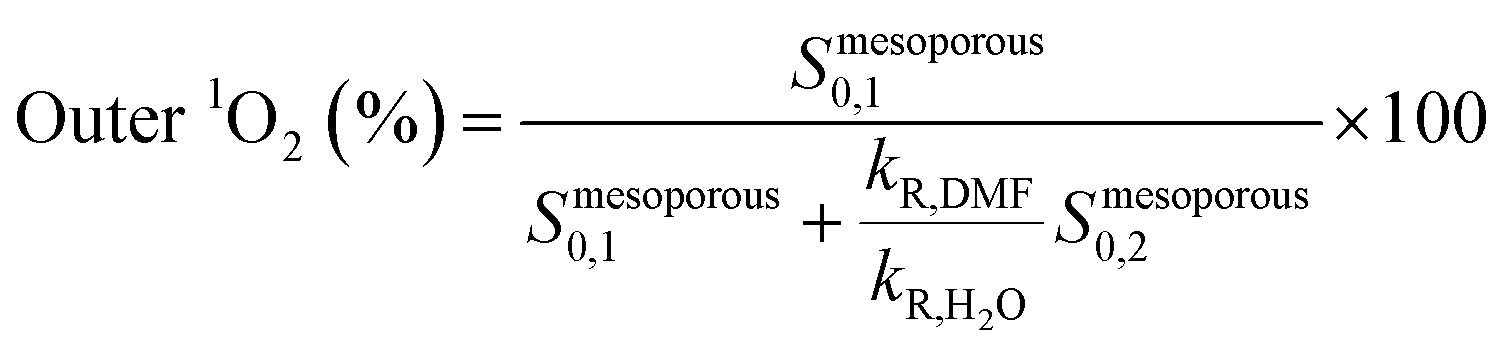 | (4) |
| Sample | Loading nmolPpIX/mgsilica | Relative 1O2 efficiency (% outer population) |
|---|---|---|
| PpIX-CSNP | 1.01 | 1 (100%) |
| PpIX-MPSAm | 0.74 | 4.4 (86%) |
| PpIX-MPSSt | 0.60 | 5.5 (81%) |
| PpIX-MPSLP | 0.64 | 14.6 (52%) |
Bacterial inactivation
The photoinduced antibacterial property of PpIX-doped silica nanoparticles was tested to correlate this activity with the morphology and 1O2 production capacity. Staphylococcus aureus was chosen as a model Gram positive bacteria, and the concentration of all silica samples was kept constant (see Table S4 in the ESI†) to properly compare the efficiency in bacterial killing.As expected, neither bare silica nanoparticles nor any of the PpIX–silica conjugates showed any remarkable dark toxicity towards S. aureus (Fig. 5). Moreover, bare silica nanoparticles, with no PpIX adsorbed, were non-toxic even at the highest light doses tested (12 J cm−2).
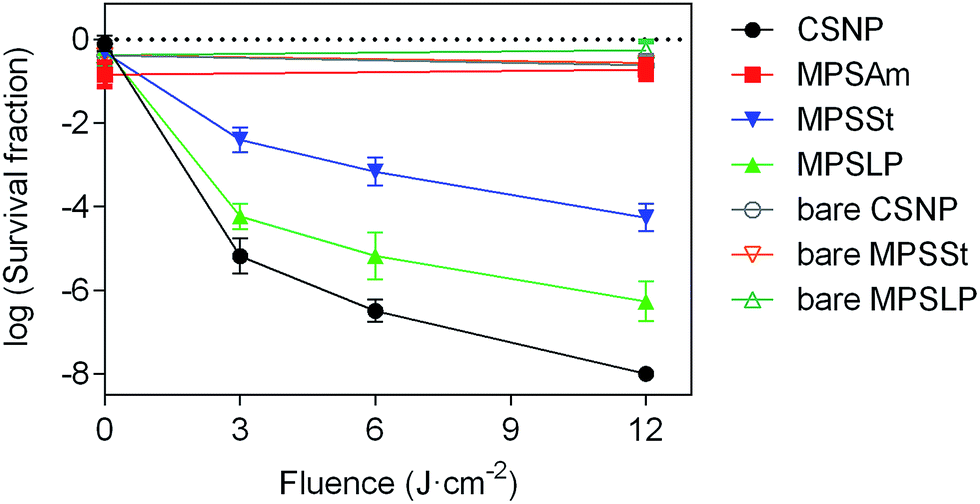 | ||
| Fig. 5 Survival curves of S. aureus incubated with different silica nanoconjugates; the closed symbols are used to indicate the systems with PpIX (in solution or anchored onto silica surface), whereas the open symbols are the control experiments (bare silica nanoparticles with no photosensitizer adsorbed). The irradiation was performed at 420 nm. | ||
The efficacy of bacterial photoinactivation varied dramatically for the different PpIX-loaded samples, highlighting the importance of the nanoparticle structure. PpIX-MPSAm, which has an undefined geometry and irregular structure, does not develop any photo-induced antibacterial activities; indeed for this sample a decrease in bacterial population comparable to the one measured in the dark, has been observed. Thus, this material was not considered worth of further scrutiny.
Among the samples with well-defined nanostructures, the highest reduction in the survival of S. aureus was achieved upon irradiation of PpIX-CSNP; the result can be quite surprising since this nanomaterial shows the lowest 1O2 production efficacy (Table 2). A possible explanation for such apparent contradiction is that its positive zeta potential enables the closest proximity to the bacteria, thereby facilitating the delivery of 1O2 and/or PpIX to the bacteria.53–55
Among the mesoporous nanomaterials, those with large pores (PpIX-MPSLP) showed higher antibacterial efficacy than those with small pores (PpIX-MPSSt), which correlates with the respective 1O2 quantum yields and the extent of PpIX aggregation. One could probably expect that if their zeta potentials had been as positive as those of CSNP, their antibacterial efficacy would be higher.
Therefore, the effort to direct the synthesis toward the construction of nanomaterials with definite geometry is crucial to maximize the exploitation of their properties. These results underline that the increase in ΦΔ is not necessarily the ultimately goal for achieving maximum efficiency in bacterial photoinactivation, factors such as zeta potential and morphology appear at least as crucial. Therefore, for a complete evaluation of new systems, photophysical studies should be correlated with the nanoparticle structure using physicochemical and biological assays to ascertain the effective improvement of the overall efficiency.
Conclusions
In this work, deeper insight on the photophysical behavior, singlet oxygen production efficiency and antimicrobial activity of PpIX-doped silica nanoparticles has been achieved through correlation of the different silica morphology with the optical and photophysical properties of PpIX–silica conjugate.From our results, it can be derived that the silica nanoparticles' morphology and PpIX localization in the inorganic matrix cause major changes in the photophysical behavior of PpIX, in the photosensitization of 1O2 and in the bacterial photoinactivation efficiency.
Acknowledgements
Financial support for this research was obtained from the Spanish Ministerio de Economía y Competitividad (Grant No. CTQ2013-48767-C3-1-R) and the University of Perugia. G.Z. thanks the support of the University of Perugia for the Erasmus fellowship. O.P. thanks the European Social Funds and the SUR del DEC de la Generalitat de Catalunya for his predoctoral fellowship (grant No. 2016 FI_B2_00100).Notes and references
- D. R. Kearns, Chem. Rev., 1971, 71, 395–427 CrossRef CAS.
- H. Pelicano, D. Carney and P. Huang, Drug Resist. Updates, 2004, 7, 97–110 CrossRef CAS PubMed.
- R. Dosselli, R. Ruiz-González, F. Moret, V. Agnolon, C. Compagnin, M. Mognato, V. Sella, M. Agut, S. Nonell, M. Gobbo and E. Reddi, J. Med. Chem., 2014, 57, 1403–1415 CrossRef CAS PubMed.
- F. Giuliani, M. Martinelli, A. Cocchi, D. Arbia, L. Fantetti and G. Roncucci, Antimicrob. Agents Chemother., 2010, 54, 637–642 CrossRef CAS PubMed.
- H. Ding, B. D. Sumer, C. W. Kessinger, Y. Dong, G. Huang, D. A. Boothman and J. Gao, J. Controlled Release, 2011, 151, 271–277 CrossRef CAS PubMed.
- C. B. Nielsen, J. S. Forster, P. R. Ogilby and S. B. Nielsen, J. Phys. Chem. A, 2005, 109, 3875–3879 CrossRef CAS PubMed.
- L. M. Rossi, P. R. Silva, L. L. R. Vono, a U. Fernandes, D. B. Tada and M. S. Baptista, Langmuir, 2008, 24, 12534–12538 CrossRef CAS PubMed.
- D. G. Fresnadillo and S. Lacombe, in Singlet Oxygen: Applications in Biosciences and Nanosciences, 2016, vol. 1, pp. 105–143 Search PubMed.
- J. L. Vivero-Escoto, in Photodynamic Therapy: Fundamentals, Applications and Health Outcomes, 2015, pp. 1–35 Search PubMed.
- Y. Hsia, M. Sivasubramanian, N.-T. Chen and L.-W. Lo, in Nanomaterials for Tumor Targeting Theranostics: A Proactive Clinical Perspective, 2016, pp. 143–176 Search PubMed.
- V. Ambrogi, A. Donnadio, D. Pietrella, L. Latterini, F. A. Proietti, F. Marmottini, G. Padeletti, S. Kaciulis, S. Giovagnoli and M. Ricci, J. Mater. Chem. B, 2014, 2, 6054 RSC.
- V. Ambrogi, L. Perioli, C. Pagano, L. Latterini, F. Marmottini, M. Ricci and C. Rossi, Microporous Mesoporous Mater., 2012, 147, 343–349 CrossRef.
- L. Latterini and L. Tarpani, J. Phys. Chem. C, 2011, 115, 21098–21104 CAS.
- Z. Chu, S. Zhang, C. Yin, G. Lin and Q. Li, Biomater. Sci., 2014, 2, 827–832 RSC.
- J. Vivero-Escoto and M. Elnagheeb, Nanomaterials, 2015, 5, 2302–2316 CrossRef CAS.
- M. Manzano, V. Aina, C. O. Areán, F. Balas, V. Cauda, M. Colilla, M. R. Delgado and M. Vallet-Regí, Chem. Eng. J., 2008, 137, 30–37 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S.-Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- C. Barbé, J. Bartlett, L. Kong, K. Finnie, H. Q. Lin, M. Larkin, S. Calleja, A. Bush and G. Calleja, Adv. Mater., 2004, 16, 1959–1966 CrossRef.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- F. Figueira, J. A. S. Cavaleiro and J. P. C. Tomé, J. Porphyrins Phthalocyanines, 2011, 15, 517–533 CrossRef CAS.
- P. Couleaud, V. Morosini, C. Frochot, S. Richeter, L. Raehma and J.-O. Durand, Nanoscale, 2010, 2, 1083–1095 RSC.
- I. Roy, T. Y. Ohulchanskyy, H. E. Pudavar, E. J. Bergey, A. R. Oseroff, J. Morgan, T. J. Dougherty and P. N. Prasad, J. Am. Chem. Soc., 2003, 125, 7860–7865 CrossRef CAS PubMed.
- F. Yan and R. Kopelman, Photochem. Photobiol., 2003, 78, 587–591 CrossRef CAS PubMed.
- V. Simon, C. Devaux, A. Darmon, T. Donnet, E. Thiénot, M. Germain, J. Honnorat, A. Duval, A. Pottier, E. Borghi, L. Levy and J. Marill, Photochem. Photobiol., 2010, 86, 213–222 CrossRef CAS PubMed.
- J. Qian, A. Gharibi and S. He, J. Biomed. Opt., 2015, 14, 14012 CrossRef PubMed.
- J. L. Vivero-Escoto and D. L. Vega, RSC Adv., 2014, 4, 14400 RSC.
- L. Latterini and M. Amelia, Langmuir, 2009, 25, 4767–4773 CrossRef CAS PubMed.
- G. Alberto, I. Miletto, G. Viscardi, G. Caputo, L. Latterini, S. Coluccia and G. Martra, J. Phys. Chem. C, 2009, 113, 21048–21053 CAS.
- R. Lindberg, J. Sjoblom and G. Sundholm, Colloids Surf., A, 1995, 99, 79–88 CrossRef CAS.
- K. Suzuki, K. Ikari and H. Imai, J. Am. Chem. Soc., 2004, 126, 462–463 CrossRef CAS PubMed.
- K. Zhang, L.-L. Xu, J.-G. Jiang, N. Calin, K.-F. Lam, S.-J. Zhang, H.-H. Wu, G.-D. Wu, B. Albela, L. Bonneviot and P. Wu, J. Am. Chem. Soc., 2013, 135, 2427–2430 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- A. Jiménez-Banzo, X. Ragàs, P. Kapusta and S. Nonell, Photochem. Photobiol. Sci., 2008, 7, 1003–1010 Search PubMed.
- K. J. Klabunde and R. M. Richards, Nanoscale Materials in Chemistry, John Wiley & Sons, 2nd edn, 2009 Search PubMed.
- C. Robertson, R. Beanland, S. a. Boden, A. L. Hector, R. J. Kashtiban, J. Sloan, D. C. Smith and A. Walcarius, Phys. Chem. Chem. Phys., 2015, 17, 4763–4770 RSC.
- T. Schiestel, H. Brunner and G. E. M. Tovar, J. Nanosci. Nanotechnol., 2004, 4, 504–511 CrossRef CAS PubMed.
- R. Selvaggi, L. Tarpani, A. Santuari, S. Giovagnoli and L. Latterini, Appl. Catal., B, 2015, 168, 363–369 CrossRef.
- C. Aggelidou, T. A. Theodossiou and K. Yannakopoulou, Photochem. Photobiol., 2013, 89, 1011–1019 CrossRef CAS PubMed.
- L. Monsù Scolaro, M. Castriciano, A. Romeo, S. Patanè, E. Cefalì and M. Allegrini, J. Phys. Chem. B, 2002, 106, 2453–2459 CrossRef.
- A. A. Sivash, Z. Masinovský and G. I. Lozovaya, BioSystems, 1991, 25, 131–140 CrossRef CAS PubMed.
- M. A. Jhonsi, A. Kathiravan and R. Renganathan, Spectrochim. Acta, Part A, 2008, 71, 1507–1511 CrossRef PubMed.
- F. Ricchelli, J. Photochem. Photobiol., B, 1995, 29, 109–118 CrossRef CAS.
- M. A. Filatov, S. Baluschev and K. Landfester, Chem. Soc. Rev., 2016, 45, 4668–4689 RSC.
- S. Oelckers, T. Hanke and B. Roder, J. Photochem. Photobiol., A, 2000, 132, 29–32 CrossRef CAS.
- K. K. Iu and J. K. Thomas, J. Photochem. Photobiol., A, 1993, 71, 55–60 CrossRef CAS.
- R. H. Young, R. L. Martin, D. Feriozi, D. Brewer and R. Kayser, Photochem. Photobiol., 1973, 17, 233–244 CrossRef CAS.
- B. M. Monroe, J. Phys. Chem., 1977, 81, 1861–1864 CrossRef CAS.
- B. Cojocaru, M. Laferrière, E. Carbonell, V. Parvulescu, H. García and J. C. Scaiano, Langmuir, 2008, 24, 4478–4481 CrossRef CAS PubMed.
- M. Hild and R. Schmidt, J. Phys. Chem. A, 1999, 103, 6091–6096 CrossRef CAS.
- E. Boix-Garriga, B. Rodríguez-Amigo, O. Planas and S. Nonell, in Singlet Oxygen: Applications in Biosciences and Nanosciences, Royal Society of Chemistry, 2016, vol. 1, pp. 23–46 Search PubMed.
- B. Gottenbos, D. W. Grijpma, H. C. van der Mei, J. Feijen and H. J. Busscher, J. Antimicrob. Chemother., 2001, 48, 7–13 CrossRef CAS PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, 1–16 Search PubMed.
- A. M. El Badawy, R. G. Silva, B. Morris, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2011, 45, 283–287 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00784a |
| This journal is © The Royal Society of Chemistry 2017 |