DOI:
10.1039/C7RA01006K
(Paper)
RSC Adv., 2017,
7, 16953-16962
Indolylbenzothiadiazoles with varying substituents on the indole ring: a systematic study on the self-recovering mechanochromic luminescence†
Received
23rd January 2017
, Accepted 10th March 2017
First published on 20th March 2017
Abstract
Self-recovering mechanochromic luminophores are able to autonomously regain their original color at room temperature after exposure to mechanical stimuli. In spite of recent extensive studies on mechanochromic luminophores, the generation of mechanochromic fluorophores with specific self-recovering behavior remains a challenging task owing to a lack of systematic studies on self-recovering mechanochromic luminophores. Herein, a series of self-recovering mechanochromic fluorophores with different solid-state emission colors and recovery times is described. The introduction of different substituents onto the indole ring of 4-(1H-indol-2-yl)-2,1,3-benzothiadiazole afforded control over the mechanochromic luminescence properties of the fluorophores. X-ray diffraction analyses revealed slipped stacks or no intermolecular stacking of the benzothiadiazole rings in the crystalline state of these dyes, which is indicative of the relative lability of the crystal structures upon exposure to mechanical stimuli. Remarkably, the fluorescence spectra after mechanical stimulus (grinding) were in good agreement with those of the molten fluorophores. Based on powder X-ray diffraction and DSC analyses of crystalline and amorphous samples, we propose an amorphization of the crystalline fluorophores to be responsible for the change of emission color in response to mechanical stimuli. Accordingly, the self-recovering nature should be rationalized in terms of a spontaneous recrystallization of the fluorophores from the amorphous states, which would be facilitated by unchanged partial crystalline phases in the immediate vicinity.
Introduction
Mechanochromic luminescent molecules change their solid-state emission color in response to a mechanical stimulus, and recover their original state by applying a second external stimulus. For this class of dyes, potential applications are conceivable in the context of advanced optical technologies such as pressure sensors, photoswitches, and anti-counterfeiting inks.1 In recent years, a considerable number of solid-state emissive organic dyes has been developed for optoelectronic applications, whereby the introduction of bulky substituents2 and aggregation-induced emission (AIE)3 may be used as effective strategies to overcome the concentration quenching caused by the intermolecular interactions of the fluorophores in the solid state. Notably, typical AIE-active molecules often also exhibit mechanochromic luminescence. The recent rapid growth in AIE-active molecules, has also led to the development of a growing number of mechanochromic fluorophores based on the structural modifications of typical fluorophores that are emissive in the solid state.4–9 However, the practical utility of most mechanochromic fluorophores reported so far suffers from the fact that these systems usually require external stimuli such as heat or exposure to solvents to recover their original color.1,4–9 Relatively few are known to recover autonomously at room temperature after exposure to a mechanical stimulus.10 In these reports, the structural variation of self-recovering mechanochromic fluorophores is also limited, and only very few compounds in each report exhibited a self-recovering nature. There is a lack of systematic study on the relationship between molecular structures and self-recovering properties of mechanochromic fluorophores. Recently, we have reported N-Boc-4-(3-methyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (Boc = tert-butoxycarbonyl) derivatives as a new class of non-AIE-type mechanochromic fluorophores.11 The fluorophores exhibited autonomously self-recovering mechanochromic luminescence, and the emission color, as well as the recovery time for the color change could be tuned via the substituents on the benzothiadiazole ring. The presence of a methyl group at the 3-position of the indole ring was indispensable to attain mechano-responsive properties, while the presence of the Boc group is important to achieve good fluorescence quantum yields in the solid state. However, the thermal instability of the Boc group hampered the detailed mechanistic study on the self-recovering properties of these fluorophores. Herein, we report the synthesis and emission properties of a series of N-substituted-4-(3-alkyl-1H-indol-2-yl)-2,1,3-benzothiadiazole derivatives (1). Seven new derivatives, which differ with respect to the substituents at the 3-position and at the N-atom of the indole ring, exhibited self-recovering mechanochromic luminescence properties with varying emission color and recovery time. Detailed studies on molten samples of these mechanochromic fluorophores offered deep insights into the self-recovering mechanochromic luminescence.
Results and discussion
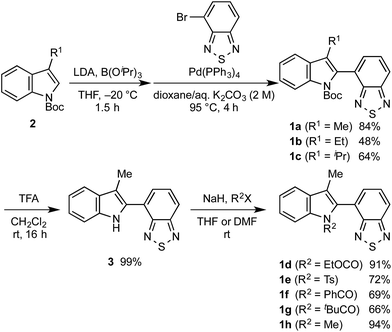
Following our previously reported procedure for the synthesis of N-Boc-indolylbenzothiadiazole 1a (R1 = Me),11 3-alkyl-substituted 1b (R1 = Et) and 1c (R1 = iPr) were obtained in 48% and 64% yield, respectively, from the Suzuki–Miyaura coupling between 4-bromo-2,1,3-benzothiadiazole and the corresponding N-Boc-3-alkylindole-2-boronic acid, which was prepared from the respective indole derivatives 2 (Scheme 1). Removal of the Boc group from 1a using trifluoroacetic acid (TFA) in dichloromethane afforded 4-(3-methyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (3) in 99% yield. N-Substituted indolylbenzothiadiazole derivatives 1d (R2 = EtOCO), 1e (R2 = Ts), 1f (R2 = PhCO), 1g (R2 = tBuCO), and 1h (R2 = Me) were subsequently synthesized in moderate to high yields by a consecutive treatment of 3 with sodium hydride and the corresponding halides R2X (1d–g: X = Cl, 1h: X = I).
 |
| | Scheme 1 Synthesis of mechanochromic fluorophores 1a–h. | |
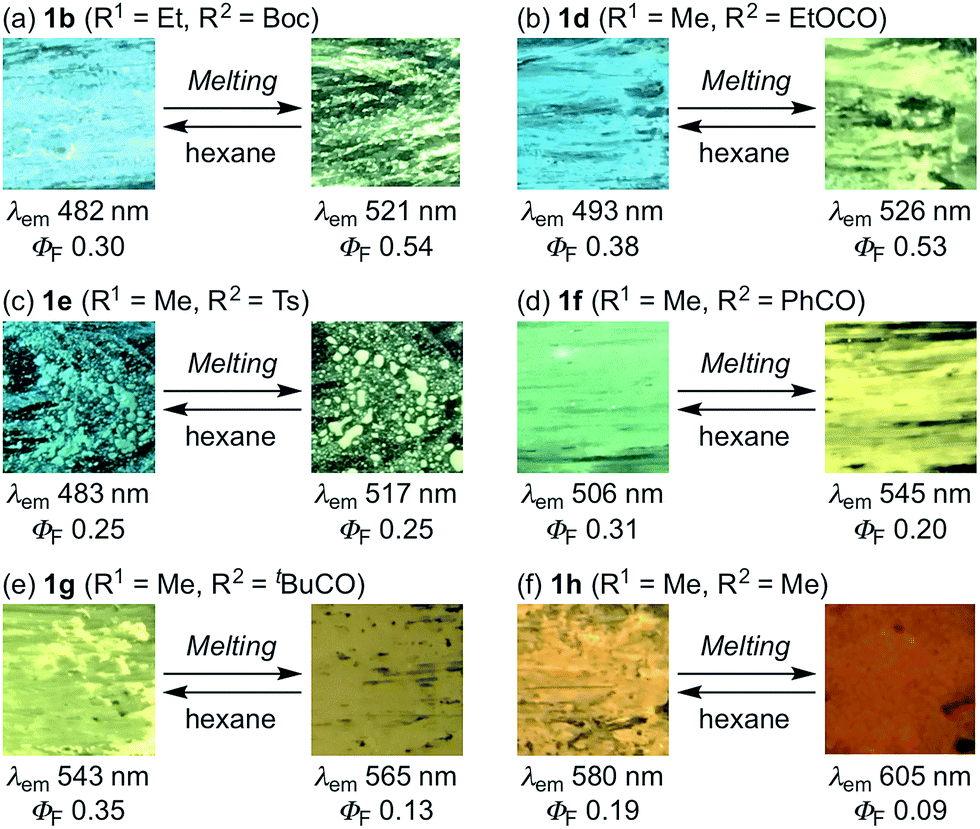
The photophysical properties of indolylbenzothiadiazoles 1a–h are summarized in Table 1. For 3-alkyl-substituted 1b and 1c in toluene, maximum absorption (λabs) and emission bands (λem) were similar to those of 3-methyl-substituted 1a. The replacement of the Boc group with an ethoxycarbonyl group had a negligible influence on the absorption and emission maxima of 1d in toluene. The maximum absorption and emission bands of 1e–h in toluene were bathochromically shifted, whereby the magnitude of the shift increased with a progressive reduction in the electron-withdrawing properties of the substituent on the N-atom of the indole moiety. The fluorescence quantum yields of 1e–h in toluene (ΦF = 0.06–0.10) decreased significantly compared to those of the carbamate derivatives 1a–d (ΦF = 0.36–0.56). The solid-state emission maximum of powdered 1b (λem = 482 nm, ΦF = 0.30) was hypsochromically shifted (Δλem = 39 nm), while its fluorescence quantum yield was slightly decreased compared to that of a toluene solution (λem = 521 nm, ΦF = 0.36). Similar to the case of 1b, a large hypsochromic shift (Δλem = 37 nm) was observed between the solid-state emission maxima of 1c (λem = 484 nm, ΦF = 0.37) and that in toluene (λem = 521 nm, ΦF = 0.36) under retention of the fluorescence quantum yield. In contrast, the hypsochromic shift of the solid-state emission for 1d (λem = 493 nm) was relatively moderate (Δλem = 31 nm), while the fluorescence quantum yield decreased substantially (ΔΦF = 0.18). Despite this decrease relative to the solution value, the fluorescence quantum yield for the solid-state emission of 1d (ΦF = 0.38) still represents a relatively good value. The emission maxima of 1e–h in the solid state (1e: 483 nm; 1f: 506 nm; 1g: 543 nm; 1h: 580 nm) were also hypsochromically shifted relative to those of the corresponding toluene solutions, and the fluorescence quantum yields of 1e–h afforded satisfactory values (1e: 0.25, 1f: 0.31, 1g: 0.35, 1h: 0.19).
Table 1 Photophysical properties of indolylbenzothiadiazoles 1a–h
| Compd. |
Absorption in toluenea |
Fluorescence in tolueneb |
Solid-state fluorescenceb |
| λabs (nm) |
λem (nm) |
ΦFc |
λem (nm) |
ΦFc |
| Maximum absorption bands (λabs > 350 nm) in toluene (10−5 M). Maximum emission bands (λem) and absolute quantum yields (ΦF) in toluene (10−5 M) and in powdered form. Absolute quantum yields were measured using an integrating sphere. As the emission intensity was too weak, ΦF was measured by using a 10−4 M solution of 1h in toluene. |
| 1a |
383 |
523 |
0.43 |
485 |
0.46 |
| 1b |
380 |
521 |
0.36 |
482 |
0.30 |
| 1c |
380 |
521 |
0.36 |
484 |
0.37 |
| 1d |
380 |
524 |
0.56 |
493 |
0.38 |
| 1e |
371 |
519 |
0.10 |
483 |
0.25 |
| 1f |
391 |
539 |
0.09 |
506 |
0.31 |
| 1g |
412 |
560 |
0.08 |
543 |
0.35 |
| 1h |
432 |
603 |
0.06d |
580 |
0.19 |
With these solid-state emissive fluorophores in hand, we examined their mechano-responsive properties by grinding powdered samples with a spatula (Fig. 1 and S1–7†). The maximum emission wavelengths of 3-alkyl-substituted 1b and 1c were bathochromically shifted to 518 nm upon grinding, which is almost identical to the value for 1a (Fig. 1a–c). The times required for a recovery of the mechanically changed color was longer for 1b (∼10 min) than for 1c (∼2 min) and 1a (∼20 s). Similarly, the solid-state emission of N-ethoxycarbonyl derivative 1d changed to 525 nm upon grinding, and the original color was recovered within ∼1 min (Fig. 1d). Although the region and magnitude of the mechano-responsive emission shift of 1e were similar to those of 1a–d, a considerably longer time (∼1.5 h) was required to recover the original color (Fig. 1e). Green-emissive 1f (λem = 506 nm) and yellow-emissive 1g (λem = 543 nm) were by 38 and 21 nm bathochromically shifted, respectively, in response to mechanical stimuli (Fig. 1f and g). These derivatives also required long recovery times (1f: ∼2 h; 1g: ∼40 min). Upon grinding, the solid-state maximum emission wavelength of 1h was by 20 nm bathochromically shifted, while the recovery time of 1h (∼10 s) was the fastest observed for 1a–h.
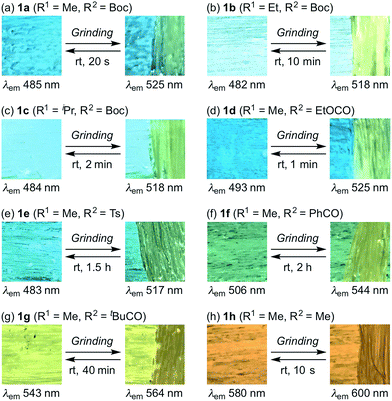 |
| | Fig. 1 Photographic images for the autonomously self-recovering mechanochromic luminescence of indolylbenzothiadiazoles 1a–h, which differ with respect to the substituents R1 and R2. | |
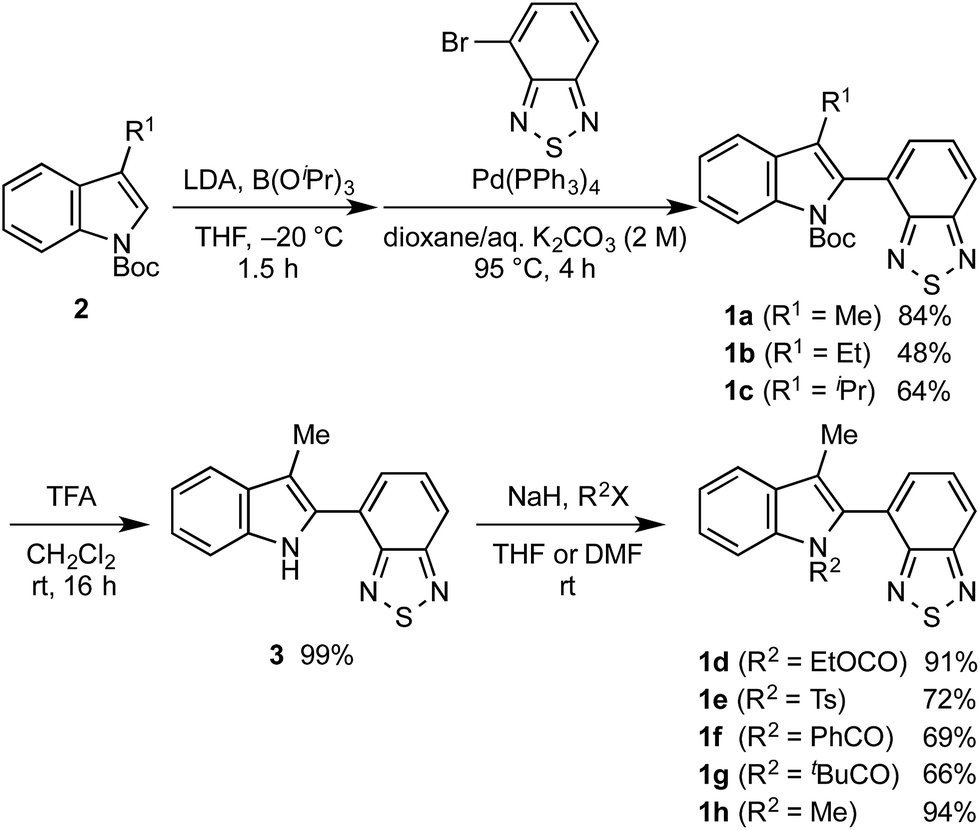
Powder X-ray diffraction (PXRD) analyses for powdered samples of 1b–h revealed that these were aggregates of microcrystals. Single-crystal X-ray diffraction analyses of 1b–h revealed crystal structures that are identical to those of the powdered samples, except for the case of 1e (Fig. 2 and S8–21†).12 As previously reported,11 1a exhibits anti-parallel intermolecular stacks of benzothiadiazole rings, whereby the tert-butyl groups of the Boc groups are located on the top and bottom of the stacked rings in the crystalline state. Notably, the stacked rings of 1a are offset due to the steric hindrance of the 3-methyl groups, which is different to the well-overlapping stacking of the corresponding non-substituted derivative (R1 = H) (Fig. S22†). Of 3-alkyl-substituted 1b and 1c, the latter exhibited a similar crystal structure to that of 1a (Fig. 2b). This structural similarity should account for the similar photophysical and mechanochromic properties of 1a and 1c in the solid state. Conversely, the crystal structure of 1b is markedly different from that of 1a. The ethoxy group of 1b is oriented in the opposite direction with respect to the intramolecular benzothiadiazole ring, and only small parts of the benzothiadiazole rings are stacked. As a result, the difference between the recovery times of 1a (R1 = Me: ∼20 s) and 1b (R1 = Et: ∼10 min) is more pronounced than that between 1a and 1c (R1 = iPr: ∼2 min), even though the molecular structure of 1b is closer to that of 1a than that of 1c. The crystal structure of 1d is completely different, due to the exchange of a Boc group with an ethoxycarbonyl group, which resulted in an absence of intermolecular stacking for the benzothiadiazole rings (Fig. 2c). This structural difference between 1d and 1a should help to understand the aforementioned relatively limited hypsochromic shift and partial drop of fluorescence quantum yield for 1d in the solid state. N-Acyl-substituted 1f (R2 = PhCO) and 1g (R2 = tBuCO) also exhibited intermolecular antiparallel stacks of benzothiadiazole rings, wherein two benzene rings or tert-butyl groups of the acyl groups are located on either side of the stacked rings (Fig. 2d and e). However, the stacking structures of 1f and 1g are different from each other. The sulfur atoms of the benzothiadiazole rings are included in the stacking structure of 1f, whereas those of 1g protrude from the stacked rings, similar to those of 1a. Intermolecular stacks of N-methyl-substituted 1h also include the sulfur atoms, and the stacked rings of 1h share less overlapping area compared to those of 1f (Fig. 2f). Although antiparallel intermolecular stacking of the benzothiadiazole rings was observed in all cases except for 1d, the stacking modes are different. Nevertheless, all derivatives exhibited mechanochromic luminescence, suggesting that their crystal structures are relatively labile compared to the well-overlapping stacking structures of N-Boc-indolylbenzothiadiazoles, which do not contain a substituent at the 3-position (R1 = H) and hence not exhibit mechanochromic properties.11
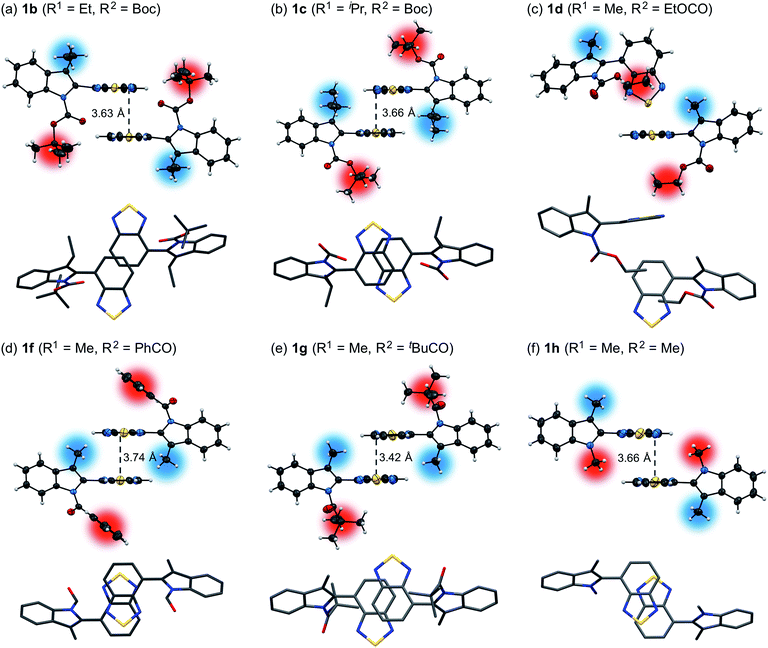 |
| | Fig. 2 Molecular structures of 1b (a), 1c (b), 1d (c), 1f (d), 1g (e), and 1h (f). Top rows: front view for two molecules in spatial proximity; atomic displacement parameters set at 30% (1d and 1f) or 50% (1b, 1c, 1g, and 1h) probability. Bottom rows: top view for two molecules in spatial proximity; all hydrogen atoms and phenyl groups (1f) were omitted for clarity. Color code: gray = C, red = O, blue = N, yellow = S, blurred blue circle = R1, blurred red circle = R2. | |
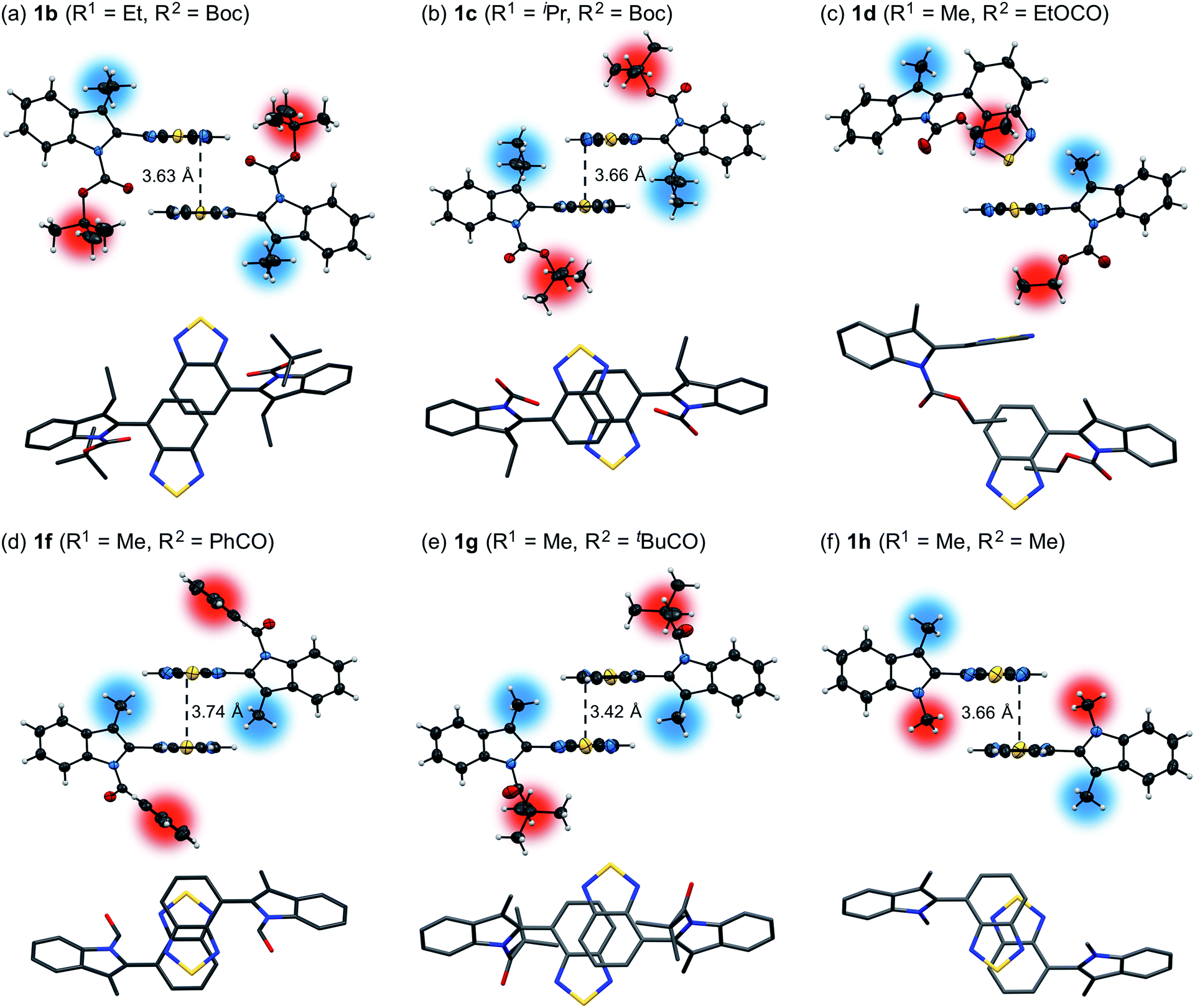
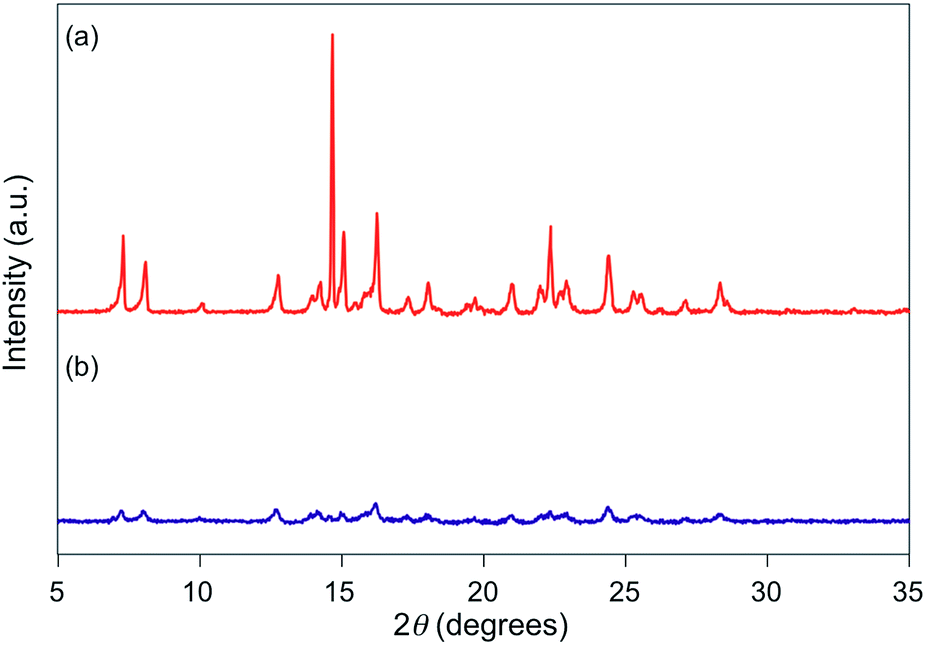
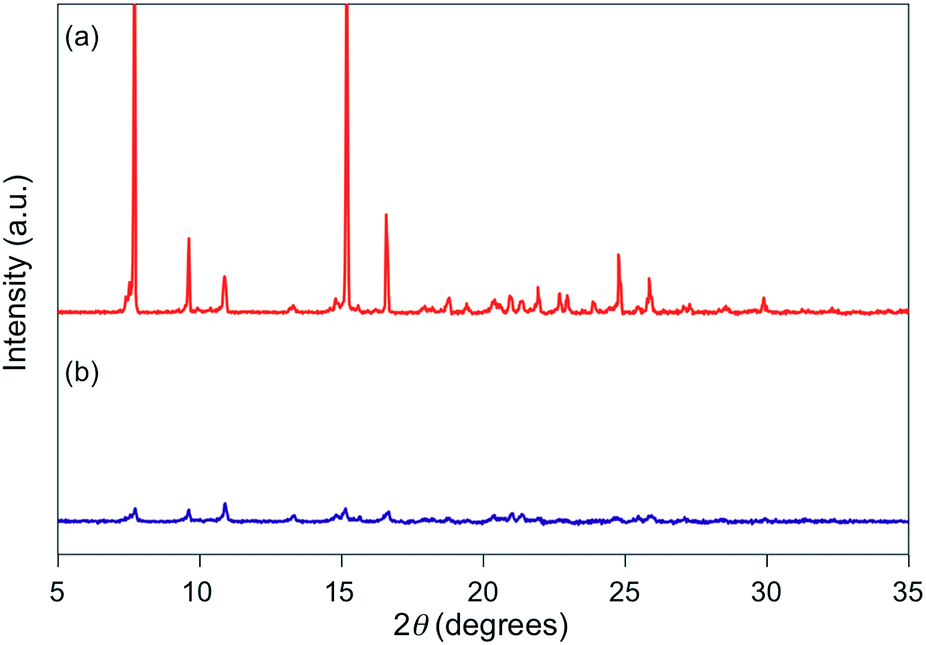
For 1e–g, PXRD analyses of the state following the mechanical stimulus were carried out, given that these derivatives exhibited relatively long recovery times. The intensity of the diffraction pattern of 1e significantly decreased after grinding (Fig. 3), indicating that the crystalline powder changed almost completely to an amorphous state. Similarly, crystalline powders of 1f and 1g also turned almost completely into amorphous states in response to the mechanical stimulus (Fig. 4 and 5). These results suggest that the mechano-responsive emission color change of these fluorophores is triggered by an amorphization of the crystals.
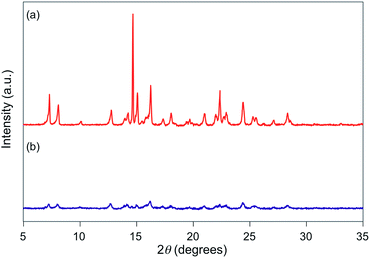 |
| | Fig. 3 PXRD patterns for powdered 1e (a) before and (b) after grinding. | |
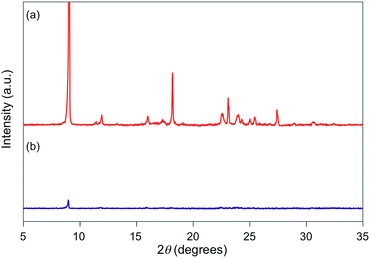 |
| | Fig. 4 PXRD patterns for powdered 1f (a) before and (b) after grinding. | |
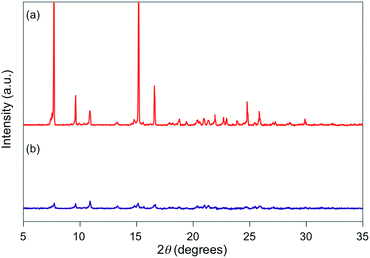 |
| | Fig. 5 PXRD patterns for powdered 1g (a) before and (b) after grinding. | |
An optimization of the molecular structures of 1b–h was carried out by density functional theory (DFT) calculations at the CAM-B3LYP/6-31G(d) level of theory,13 using the single-crystal X-ray diffraction structures as the initial geometry. In all cases, the HOMO is mainly localized on the indole ring and on a part of the benzothiadiazole moiety, whereas the LUMO is localized on the benzothiadiazole ring (Fig. S23†). Time-dependent DFT calculations on the optimized structures of 1b–h revealed that the maximum-wavelength absorption bands should be assigned to the HOMO → LUMO transition (Table S1 and Fig. S24†). Such an intramolecular charge-transfer transition would explain the large Stokes shifts and solvatochromic luminescence observed for 1b–h (Fig. S25–31†). Solvatochromic luminescence is usually caused by an efficient stabilization of excited states in polar solvents.14 The potentially solvatofluorochromic nature of 1b–h might be one reason for their change of emission color by amorphization, as the polarity surrounding the fluorophores should be different in the crystalline and amorphous state.
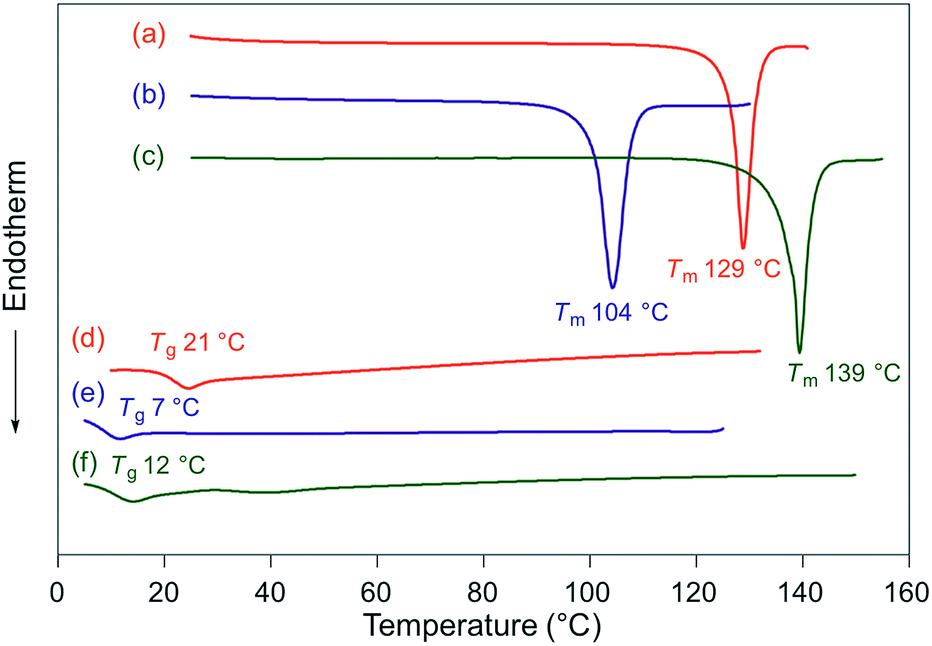
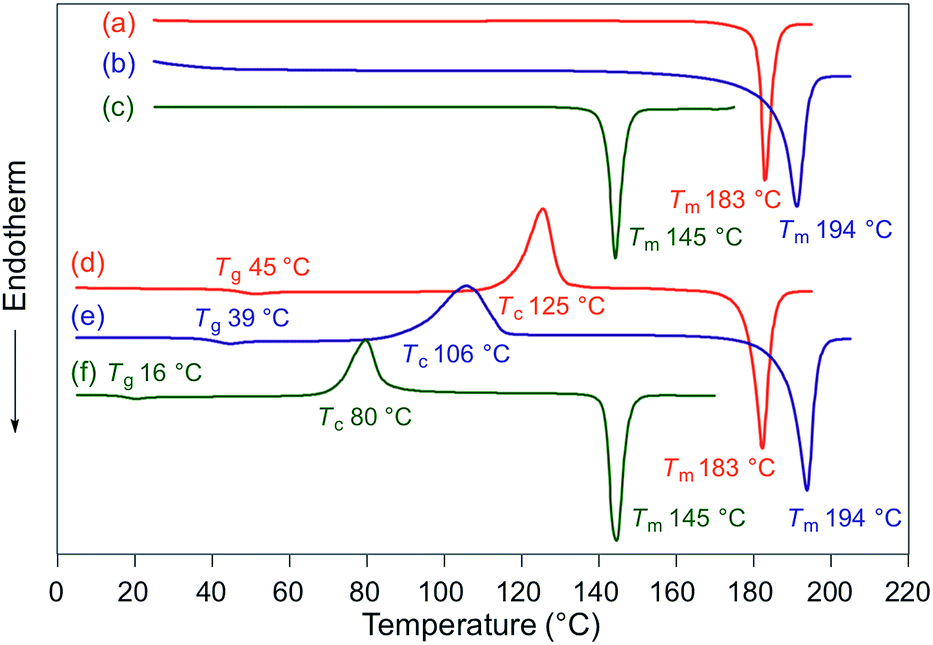
Fluorescence spectra of the molten mechanochromic fluorophores were measured to examine the change of emission color upon losing crystallinity. Although thermal annealing is a widely applied method to recover the original emission color after exposure to mechanical stimuli, there is a lack of systematic study on the relationship between the emission color of the molten and mechanically changed states.10f,15,16 N-Boc derivatives 1a and 1c decomposed upon heating prior to reaching their melting points, most likely due to the thermal instability of the Boc group. Derivatives 1b and 1d–h liquefied at temperatures above their melting points,17 and the fluorescence spectra were recorded after cooling the samples rapidly to room temperature. In all cases, bathochromic shifts of the emission maxima were observed upon melting (Fig. 6). Notably, the fluorescence spectra of the molten samples were in good agreement with those of the mechanically changed states (Fig. S1 and 3–7†). Differential scanning calorimetry (DSC) measurements were carried out on powdered and molten samples of 1b and 1d–h (Fig. 7 and 8). The DSC thermograms of all powdered crystalline samples showed only one endothermic peak that corresponds to their melting points, while transition peaks to another crystalline phase were not observed prior to melting. For the molten samples, glass-transition temperatures (Tg) were observed for 1b (21 °C), 1d (7 °C), and 1g (12 °C), which exhibit lower melting points (Tm; 1b: 129 °C, 1d: 104 °C, 1g: 139 °C), whereas Tg (1e: 45 °C, 1f: 39 °C, 1h: 16 °C) and cold-crystallization transition temperatures were observed for 1e, 1f, and 1h (Tc; 1e: 125 °C, 1f: 106 °C, 1h: 80 °C), which exhibit higher Tm values (1e: 183 °C, 1f: 194 °C, 1h: 145 °C). These results support the notion that mechanical stimuli cause a transition of the powdered crystalline state to a glass or supercooled liquid state with different emission properties. The relatively long recovery times for 1e and 1f might be explained as these are in glass states at room temperature.
 |
| | Fig. 6 Photographic images for the luminescence of molten samples of indolylbenzothiadiazoles 1b and 1d–h. | |
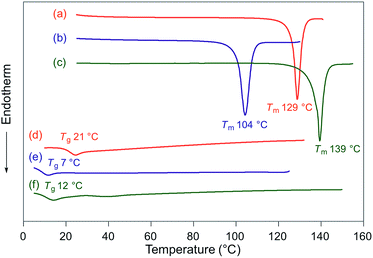 |
| | Fig. 7 DSC thermograms for powdered and molten samples of 1b (a and d), 1d (b and e), and 1g (c and f). Tm and Tg values are noted near the corresponding peaks and steps. | |
 |
| | Fig. 8 DSC thermograms for powdered and molten samples of 1e (a and d), 1f (b and e), and 1h (c and f). Tm, Tc, and Tg values are noted near the corresponding peaks and steps. | |
The molten samples required considerably longer times (several hours to several days) to recover the original emission color at room temperature. Meanwhile, the original emission color was immediately recovered upon scratching hexane suspensions of molten samples with a spatula followed by natural evaporation (Fig. 6). The stability of the molten amorphous samples should be explained by the complete loss of crystallinity, in contrast to the amorphous state created by mechanical stimuli. The recrystallization of the mechanically created amorphous phases should be facilitated by the presence of unchanged crystalline phases in close spatial proximity, which should lead to a fast recovery of the emission color at room temperature. The difference in recovery time would be associated with the degree of crystallinity of the crystal structure, or the stabilization of the amorphous phase by polarized substituents.
Conclusion
A series of self-recovering mechanochromic fluorophores was obtained by introducing different substituents on the indole ring of indolylbenzothiadiazole. Although the mechanochromic luminescence for 1a–e was observed in the blue to green region irrespective of their structural differences, the times required for the recovery of the original color varied from 20 s to 1.5 h. For 1f–h, which contain benzoyl, pivaloyl, or methyl groups at N-atom of the indole ring, respectively, bathochromically shifted solid-state emission colors relative to 1a–e were observed. The recovery time increased upon introduction of acyl groups, and shortened by the presence of the N-methyl group. All these mechanochromic fluorophores are crystalline powders, and the PXRD analyses for 1e–g, which exhibit relatively long recovery times, indicated a loss of crystallinity in response to mechanical stimuli. Moreover, the fluorescence spectra of samples of 1b and 1d–h after grinding showed good agreement with those for the molten amorphous state, whereas a considerably longer time was required for the molten samples to recover their crystallinity at room temperature. The DSC analyses for the molten samples exhibited that the Tg values for 1e and 1f are above room temperature, which might explain the long recovery times of these fluorophores. These results suggest that the present mechanism of self-recovering mechanochromic luminescence is based on an amorphization of the crystals upon grinding, followed by spontaneous recrystallization of the amorphous phase, which should be facilitated by the presence of unchanged crystalline phases in the immediate vicinity. Self-recovering mechanochromic fluorophores should potentially find applications in mechano-sensors that may provide information on the time-lapse after exposure to a mechanical stimulus or in anti-counterfeiting inks that may temporarily change their emission color upon crumpling. The present study should offer guidelines to further the development of such smart materials.
Experimental
General
All air-sensitive experiments were carried out under an atmosphere of argon unless otherwise noted. IR spectra were recorded on a Nicolet iS10 FT-IR spectrometer. 1H and 13C NMR spectra were recorded on a Bruker DRX-300 or DRX-500 spectrometer using tetramethylsilane as an internal standard. UV-vis absorption and fluorescence spectra were measured on a JASCO V560 spectrophotometer and a JASCO FP-8300 or FP-8500 fluorescence spectrometer, respectively. The absolute fluorescence quantum yields were determined using a 100 mm ϕ integrating sphere JASCO ILF-835. A miniature fiber-optic spectrometer (FLAME-S-XR1-ES, Ocean Optics) was used for the measurements of mechanochromic luminescence. PXRD measurements were performed on a Rigaku RINT-2500 system using CuKα radiation. Melting points were determined on a Stuart melting point apparatus SMP3 and are uncorrected. DSC data were recorded on a Seiko Instruments DSC-6100 equipped with a liquid nitrogen cooling unit (heating rate: 10 °C min−1). High-resolution mass spectra (HRMS) were recorded on a JEOL JMS-600 mass spectrometer (EI) and a Hitachi Nano Frontier LD spectrometer (ESI). Elemental analyses were carried out on a Vario EL III Elemental analyzer. TLC analyses were done on silica-gel 60 F254-precoated aluminum backed sheets (E. Merck). Wakogel C-200 and Silica gel 60 N (spherical, neutral, 63–210 μm) were used for column chromatography. N-Boc-3-methyl-1H-indole (2a),18 N-Boc-3-ethyl-1H-indole (2b),19 3-isopropyl-1H-indole,20 4-bromo-2,1,3-benzothiadiazole,21 and tert-butyl 2-(2,1,3-benzothiadiazol-4-yl)-3-methyl-1H-indole-1-carboxylate (1a)11 were synthesized according to the literature procedure. The spectroscopic grade solvents for UV-vis absorption and fluorescence measurements were purchased from Kanto Chemical Co., Inc. and Wako Pure Chemical Industries, Ltd.
Synthesis of N-Boc-3-isopropyl-1H-indole (2c)
To a stirred solution of 3-isopropyl-1H-indole (480 mg, 3.0 mmol) and DMAP (37 mg, 0.30 mmol) in dichloromethane (30 mL) was added di-tert-butyl dicarbonate (790 mg, 3.6 mmol), and the mixture was stirred at room temperature for 24 h. After water was added to the mixture, the organic layer was separated and the aqueous layer was extracted with dichloromethane three times. The combined organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, crude product was purified with silica-gel column chromatography (hexane/ethyl acetate = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give N-Boc-3-isopropyl-1H-indole (2c, 765 mg, 98%) as colorless oil.
:1) to give N-Boc-3-isopropyl-1H-indole (2c, 765 mg, 98%) as colorless oil.
N-Boc-3-isopropyl-1H-indole (2c). Colorless oil; IR (neat): νmax 3051, 2966, 2931, 2872, 2391, 2360, 1732, 1609, 1568, 1455, 1422, 1371, 1340, 1309, 1298, 1255, 1221, 1158, 1108, 1085, 1064, 1021, 1000, 935, 858, 835, 804, 765, 745, 668 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.13 (br s, 1H), 7.55 (d, J = 7.9 Hz, 1H), 7.33 (br s, 1H), 7.31–7.27 (m, 1H), 7.24–7.19 (m, 1H), 3.12–3.09 (m, 1H), 1.65 (s, 9H), 1.34 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3): δ (ppm) 149.9, 135.7, 130.1, 128.0, 124.1, 122.1, 120.5, 119.3, 115.3, 83.2, 28.2, 25.2, 22.6; anal. calcd for C16H21NO2: C, 74.10; H, 8.16; N, 5.40. Found: C, 73.72; H, 8.32; N, 5.45.
Synthesis of N-Boc-indolylbenzothiadiazole derivatives 1b and 1c
Typical experimental procedure (1b, Scheme 1). To a stirred solution of N-Boc-3-ethylindole (245 mg, 1.0 mmol) and triisopropyl borate (282 mg, 1.5 mmol) in THF (4.0 mL) was added a solution of lithium diisopropyl amide, prepared from diisopropylamine (350 μL, 2.5 mmol) and butyllithium (2.6 M in hexane, 960 μL, 2.5 mmol) in THF (2.0 mL), through a syringe at −20 °C, and the mixture was stirred at the same temperature for 1.5 h. Saturated ammonium chloride solution and water were added to the reaction mixture. The organic layer was separated and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, crude product was dissolved in dioxane (6.0 mL). To the solution was added 4-bromo-2,1,3-benzothiadiazole (129 mg, 0.6 mmol) and 2 M aqueous K2CO3 (0.6 mL), and the mixture was degassed under ultrasonic irradiation. To the mixture was added Pd(PPh3)4 (139 mg, 0.12 mmol), and the mixture was further degassed under ultrasonic irradiation. After the mixture was stirred at 95 °C for 4 h, water and dichloromethane were added to the mixture. The organic layer was separated and the aqueous layer was extracted with dichloromethane three times. The combined organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, crude product was purified with silica-gel column chromatography (hexane/diethyl ether = 10:1) to give tert-butyl-2-(2,1,3-benzothiadiazol-4-yl)-3-ethyl-1H-indole-1-carboxylate (1b, 109 mg, 48%) as yellow solid.
tert-Butyl 2-(2,1,3-benzothiadiazol-4-yl)-3-ethyl-1H-indole-1-carboxylate (1b). Yellow solid; mp 128.0–128.6 °C; IR (KBr): νmax 2969, 1732, 1601, 1538, 1454, 1411, 1392, 1369, 1332, 1286, 1254, 1223, 1149, 1116, 1089, 1018, 905, 878, 846, 815, 796, 763, 743, 705 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.31 (dt, J = 8.3, 0.9 Hz, 1H), 8.04 (dd, J = 8.8, 1.1 Hz, 1H), 7.69 (dd, J = 8.8, 6.8 Hz, 1H), 7.63 (ddd, J = 7.2, 1.3, 0.9 Hz, 1H), 7.56 (dd, J = 6.8, 1.1 Hz, 1H), 7.39 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 7.30 (td, J = 7.2, 0.9 Hz, 1H), 2.68 (dt, J = 21.8, 7.6 Hz, 1H), 2.53 (dt, J = 21.8, 7.6 Hz, 1H), 1.20 (t, J = 7.6 Hz, 3H), 0.97 (s, 9H); 13C NMR (126 MHz, CDCl3): δ (ppm) 155.0, 154.9, 149.9, 136.9, 130.3, 129.3, 128.7, 128.3, 125.0, 124.3, 122.6, 121.0, 119.2, 115.7, 82.4, 27.4, 17.8, 15.3; anal. calcd for C21H21N3O2S: C, 66.47; H, 5.58; N, 11.07; S, 8.45. Found: C, 66.20; H, 5.76; N, 10.88; S, 8.40. Crystal data for 1b (CCDC 1523441†): C21H21N3O2S, M = 379.48, triclinic, a = 9.401(5) Å, b = 10.660(5) Å, c = 11.058(5) Å, α = 107.936(5)°, β = 98.134(3)°, γ = 110.380(3)°, V = 949.1(8) Å3, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 2, Dc = 1.328 g cm−3, F(000) = 400.00, T = 223(1) K, μ(Mo-Kα) = 1.917 cm−1, 6870 reflections measured, 4148 independent (Rint = 0.0206). The final refinement converged to R1 = 0.0463 for I > 2.0σ(I), wR2 = 0.1320 for all data.
(no. 2), Z = 2, Dc = 1.328 g cm−3, F(000) = 400.00, T = 223(1) K, μ(Mo-Kα) = 1.917 cm−1, 6870 reflections measured, 4148 independent (Rint = 0.0206). The final refinement converged to R1 = 0.0463 for I > 2.0σ(I), wR2 = 0.1320 for all data.
tert-Butyl 2-(2,1,3-benzothiadiazol-4-yl)-3-isopropyl-1H-indole-1-carboxylate (1c). Yellow solid; mp > 145 °C decomp.; IR (KBr): νmax 3075, 3015, 2973, 2931, 2873, 1721, 1600, 1536, 1456, 1417, 1366, 1331, 1274, 1236, 1210, 1157, 1110, 1087, 999, 903, 868, 851, 818, 811, 769, 754 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.34 (dt, J = 8.3, 1.0 Hz, 1H), 8.05 (dd, J = 8.8, 1.0 Hz, 1H), 7.80 (ddd, J = 7.9, 1.3, 1.0 Hz, 1H), 7.69 (dd, J = 8.8, 6.7 Hz, 1H), 7.52 (dd, J = 6.7, 1.0 Hz, 1H), 7.37 (ddd, J = 8.3, 7.3, 1.3 Hz, 1H), 7.27 (ddd, J = 7.9, 7.3, 1.0 Hz, 1H), 2.93 (sept, J = 7.3 Hz, 1H), 1.42 (d, J = 7.3 Hz, 3H), 1.26 (d, J = 7.3 Hz, 3H), 0.95 (s, 9H); 13C NMR (126 MHz, CDCl3): δ (ppm) 155.1, 154.9, 149.8, 137.2, 129.3, 129.24, 129.20, 128.6, 128.0, 127.7, 124.6, 122.2, 121.1, 120.8, 115.8, 82.4, 27.4, 26.3, 22.6, 22.3; anal. calcd for C22H23N3O2S: C, 67.15; H, 5.89; N, 10.68; S, 8.15. Found: C, 66.75; H, 5.93; N, 10.66; S, 8.32. Crystal data for 1c (CCDC 1523442†): C22H23N3O2S, M = 393.50, triclinic, a = 9.38360 Å, b = 9.5084(5) Å, c = 12.6685(3) Å, α = 84.707(12)°, β = 69.198(7)°, γ = 77.736(12)°, V = 1032.35(9) Å3, space group P (no. 2), Z = 2, Dc = 1.266 g cm−3, F(000) = 416.00, T = 223(1) K, μ(Mo-Kα) = 1.787 cm−1, 7509 reflections measured, 4499 independent (Rint = 0.0196). The final refinement converged to R1 = 0.0455 for I > 2.0σ(I), wR2 = 0.1229 for all data.
Synthesis of 4-(3-methyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (3)
To a stirred solution of 1a (219 mg, 0.6 mmol) in dichloromethane (9.8 mL) was added trifluoroacetic acid (2.2 mL) at 0 °C, and the mixture was stirred at room temperature for 16 h. After the mixture was cooled to 0 °C, saturated aqueous sodium hydrogen carbonate solution was added to the mixture. The organic layer was separated and the aqueous layer was extracted with dichloromethane three times. The combined organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, crude product was purified with silica-gel column chromatography (hexane/diethyl ether = 1:1) to give 4-(3-methyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (3, 157 mg, 99%) as red-orange solid.
4-(3-Methyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (3). Red-orange solid; mp 123–124 °C; IR (KBr): νmax 3482, 3381, 3054, 2963, 2861, 1645, 1588, 1541, 1523, 1481, 1463, 1445, 1435, 1394, 1380, 1367, 1332, 1309, 1250, 1238, 1184, 1163, 1154, 1127, 1108, 1051, 904, 884, 858, 841, 807, 791, 746, 740, 687 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 10.56 (br s, 1H), 7.94 (d, J = 7.6 Hz, 1H), 7.90 (dd, J = 8.8, 0.9 Hz, 1H), 7.65–7.70 (m, 2H), 7.47 (dt, J = 8.1, 0.9 Hz, 1H), 7.26 (ddd, J = 8.1, 7.9, 1.0 Hz, 1H), 7.15 (ddd, J = 7.9, 7.6, 0.9 Hz, 1H), 2.65 (s, 3H); 13C NMR (75.5 MHz, CDCl3): δ (ppm) 155.6, 152.8, 136.0, 130.1, 129.9, 129.3, 126.5, 125.7, 123.1, 119.5, 119.3, 119.0, 111.7, 111.2, 11.5; HRMS-EI (m/z): [M]+ calcd for C15H11N3S, 265.0674; found, 265.0668.
Synthesis of N-substituted-indolylbenzothiadiazole derivatives 1d–h
Typical experimental procedure (1d, Scheme 1). To a stirred suspension of sodium hydride (60% mineral oil dispersion, 24 mg, 0.6 mmol), washed with hexane prior to use, in THF (3.0 mL), 4-(3-methyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (3, 80 mg, 0.30 mmol) in THF (3.0 mL) was added dropwise through a syringe at 0 °C. The reaction mixture was stirred at room temperature for 1 h. To the mixture was added dropwise a solution of ethyl chloroformate (65 mg, 0.6 mmol) in THF (3.0 mL) through a syringe. After the reaction mixture was stirred at room temperature for 24 h, water was added to the reaction mixture at 0 °C. The organic layer was separated and the aqueous layer was extracted with dichloromethane three times. The combined organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, crude product was purified with silica-gel column chromatography (hexane/diethyl ether = 1:1) to give ethyl 2-(2,1,3-benzothiadiazol-4-yl)-3-methyl-1H-indole-1-carboxylate (1d, 92 mg, 91%) as yellow solid.
Ethyl 2-(2,1,3-benzothiadiazol-4-yl)-3-methyl-1H-indole-1-carboxylate (1d). Yellow solid; mp 97–98 °C; IR (KBr): νmax 3049, 2978, 2930, 2912, 2869, 1733, 1601, 1538, 1479, 1456, 1412, 1379, 1328, 1320, 1302, 1281, 1226, 1198, 1161, 1128, 1117, 1025, 889, 848, 830, 810, 742, 704, 676 cm−1; 1H NMR (300 MHz, CDCl3): δ (ppm) 8.26 (dt, J = 8.3, 1.0 Hz, 1H), 8.04 (dd, J = 8.7, 0.9 Hz, 1H), 7.70 (dd, J = 8.7, 7.0 Hz, 1H), 7.62–7.55 (m, 2H), 7.41 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 7.33 (ddd, J = 8.2, 7.2, 1.0 Hz, 1H), 4.04–3.87 (m, 2H), 2.21 (s, 3H), 0.68 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.82, 154.77, 151.4, 136.4, 130.9, 130.5, 129.3, 129.2, 127.7, 125.3, 122.9, 121.1, 119.2, 118.6, 115.5, 62.4, 13.3, 9.4; HRMS-ESI (m/z): [M + H]+ calcd for C18H16N3O2S, 338.0958; found, 338.0963; crystal data for 1d (CCDC 1523443†): C18H15N3O2S, M = 337.40, monoclinic, a = 11.028(7) Å, b = 14.215(8) Å, c = 11.554(7) Å, β = 114.243(7)°, V = 1651.5(16) Å3, space group P21/c (no. 14), Z = 4, Dc = 1.357 g cm−3, F(000) = 704.00, T = 223(1) K, μ(Mo-Kα) = 2.111 cm−1, 12326 reflections measured, 3762 independent (Rint = 0.0526). The final refinement converged to R1 = 0.0724 for I > 2.0σ(I), wR2 = 0.2121 for all data.
4-(3-Methyl-1-tosyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (1e). According to the typical experimental procedure, the reaction of 3 with p-toluenesulfonyl chloride in the presence of sodium hydride was carried out in DMF at room temperature for 16 h to give 1e in 72% yield. Yellow solid; mp 177.5–178.3 °C; IR (KBr): νmax 3067, 2975, 2921, 1597, 1537, 1452, 1371, 1360, 1222, 1188, 1173, 1132, 1121, 1089, 1018, 951, 900, 830, 820, 811, 757, 683, 667 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.30 (dt, J = 8.2, 1.0 Hz, 1H), 8.11 (dd, J = 8.8, 1.0 Hz, 1H), 7.73 (dd, J = 8.8, 6.9 Hz, 1H), 7.61 (dd, J = 6.9, 1.0 Hz, 1H), 7.50 (ddd, J = 7.5, 1.3, 1.0 Hz, 1H), 7.40 (ddd, J = 8.2, 7.2, 1.3 Hz, 1H), 7.32 (ddd, J = 7.5, 7.2, 1.0 Hz, 1H), 7.27 (d, J = 8.2 Hz, 2H), 6.96 (d, J = 8.2 Hz, 2H), 2.26 (s, 3H), 2.02 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.63, 154.62, 144.4, 137.4, 135.1, 132.2, 131.5, 131.3, 129.2, 128.8, 126.7, 125.38, 125.35, 123.8, 122.1, 121.5, 119.4, 115.9, 21.5, 9.7; HRMS-ESI (m/z): [M + H]+ calcd for C22H18N3O2S2, 420.0835; found, 420.0844; crystal data for 1e (CCDC 1523444†): C22H17N3O2S2·CHCl3, M = 538.89, triclinic, a = 11.305(8) Å, b = 11.319(8) Å, c = 11.788(8) Å, α = 109.674(6)°, β = 98.760(4)°, γ = 115.528(4)°, V = 1200.7(14) Å3, space group P (no. 2), Z = 2, Dc = 1.490 g cm−3, F(000) = 552.00, T = 223(1) K, μ(Mo-Kα) = 5.820 cm−1, 8608 reflections measured, 5216 independent (Rint = 0.0363). The final refinement converged to R1 = 0.0658 for I > 2.0σ(I), wR2 = 0.1911 for all data.
2-(2,1,3-Benzothiadiazol-4-yl)-3-methyl-1H-indol-1-yl(phenyl)methanone (1f). According to the typical experimental procedure, the reaction of 3 with benzoyl chloride in the presence of sodium hydride was carried out in DMF at room temperature for 16 h to give 1f in 69% yield. Yellow solid; mp 191.2–191.6 °C; IR (KBr): νmax 3052, 2913, 2858, 1673, 1598, 1598, 1535, 1473, 1453, 1390, 1351, 1331, 1230, 1201, 1152, 1051, 1024, 902, 874, 853, 829, 812, 761, 748, 716, 697, 669 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 7.80–7.76 (m, 1H), 7.69–7.64 (m, 2H), 7.54–7.52 (m, 2H), 7.44 (d, J = 7.3 Hz, 2H), 7.34–7.28 (m, 2H), 7.21 (t, J = 9.1 Hz, 1H), 7.05 (t, J = 7.9 Hz, 2H), 2.33 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) 169.6, 154.6, 153.6, 137.3, 135.1, 131.94, 131.88, 130.4, 130.1, 129.2, 129.0, 127.5, 126.6, 125.1, 122.9, 121.0, 119.3, 118.9, 114.6, 9.7; HRMS-ESI (m/z): [M + H]+ calcd for C22H16N3OS, 370.1009; found, 370.1021; crystal data for 1f (CCDC 1523445†): C22H15N3OS, M = 369.44, monoclinic, a = 20.828(7) Å, b = 11.825(4) Å, c = 15.926(5) Å, β = 111.203(4)°, V = 3657(2) Å3, space group C2/c (no. 15), Z = 8, Dc = 1.342 g cm−3, F(000) = 1536.00, T = 223(1) K, μ(Mo-Kα) = 1.937 cm−1, 13776 reflections measured, 4172 independent (Rint = 0.0591). The final refinement converged to R1 = 0.0843 for I > 2.0σ(I), wR2 = 0.2476 for all data.
2-(2,1,3-Benzothiadiazol-4-yl)-3-methyl-1H-indol-1-yl-2,2-dimethylpropan-1-one (1g). According to the typical experimental procedure, the reaction of 3 with pivaloyl chloride in the presence of sodium hydride was carried out in DMF at room temperature for 16 h to give 1g in 66% yield. Yellow-orange solid; mp 135.6–137.2 °C; IR (KBr): νmax 3065, 2970, 2927, 2864, 1715, 1475, 1449, 1327, 1299, 1220, 1172, 1126, 908, 856, 841, 823, 810, 776, 756, 750, 638, 619 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.06 (dd, J = 8.9, 1.1 Hz, 1H), 7.71 (dd, J = 8.7, 6.8 Hz, 1H), 7.64 (dd, J = 7.8, 0.9 Hz, 1H), 7.60 (dd, J = 6.9, 0.9 Hz, 1H), 7.53 (dd, J = 8.2, 0.9 Hz, 1H), 7.33 (ddd, J = 8.2, 7.8, 0.9 Hz, 1H), 7.25 (td, J = 8.2, 0.9 Hz, 1H), 2.28 (s, 3H), 0.96 (s, 9H); 13C NMR (126 MHz, CDCl3): δ (ppm) 184.9, 155.1, 153.5, 136.4, 131.1, 130.5, 129.4, 129.3, 127.1, 124.3, 121.4, 121.3, 119.4, 116.6, 112.3, 43.9, 27.9, 10.0; HRMS-ESI (m/z): [M + H]+ calcd for C20H20N3OS, 350.1322; found, 350.1332. Crystal data for 1g (CCDC 1523446†): C20H19N3OS, M = 349.45, monoclinic, a = 12.119(5) Å, b = 13.780(5) Å, c = 21.310(8) Å, β = 98.941(7)°, V = 3516(2) Å3, space group P21/n (no. 14), Z = 8, Dc = 1.320 g cm−3, F(000) = 1472.00, T = 223(1) K, μ(Mo-Kα) = 1.968 cm−1, 27090 reflections measured, 8018 independent (Rint = 0.0360). The final refinement converged to R1 = 0.0506 for I > 2.0σ(I), wR2 = 0.1268 for all data.
4-(1,3-Dimethyl-1H-indol-2-yl)-2,1,3-benzothiadiazole (1h). According to the typical experimental procedure, the reaction of 3 with methyl iodide in the presence of sodium hydride was carried out in THF at room temperature for 16 h to give 1h in 94% yield. Orange solid; mp 140.2–140.8 °C; IR (KBr): νmax 3044, 2921, 2904, 1592, 1558, 1470, 1362, 1329, 1312, 1268, 1244, 1174, 1158, 1128, 1007, 951, 896, 855, 828, 758, 741 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.10 (dd, J = 8.8, 1.0 Hz, 1H), 7.74 (dd, J = 8.8, 6.8 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.61 (dd, J = 6.8, 1.0 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.30 (dd, J = 7.9, 7.3 Hz, 1H), 7.17 (dd, J = 8.2, 7.3 Hz, 1H), 3.57 (s, 3H), 2.27 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) 155.1, 154.4, 137.7, 133.2, 131.7, 129.2, 128.4, 125.9, 122.3, 121.5, 119.23, 119.16, 110.9, 109.4, 31.5, 9.6; anal. calcd for C16H13N3S: C, 68.79; H, 4.69; N, 15.04; S, 11.48. Found: C, 68.40; H, 4.76; N, 14.96; S, 11.48. Crystal data for 1h (CCDC 1523447†): C16H13N3S, M = 279.36, triclinic, a = 8.367(3) Å, b = 8.840(3) Å, c = 10.153(3) Å, α = 80.099(13)°, β = 67.010(11)°, γ = 82.285(14)°, V = 679.2(4) Å3, space group P (no. 2), Z = 2, Dc = 1.366 g cm−3, F(000) = 292.00, T = 223(1) K, μ(Mo-Kα) = 2.305 cm−1, 4962 reflections measured, 2968 independent (Rint = 0.0181). The final refinement converged to R1 = 0.0610 for I > 2.0σ(I), wR2 = 0.1786 for all data.
Theoretical calculations
The theoretical calculations were performed using Gaussian 09 program.22 Molecular geometry of 1b was optimized using density functional theory (DFT) calculations at the CAM-B3LYP/6-31G(d) level of theory. The single-crystal X-ray structure of 1b was used as a starting point. The long-range corrected hybrid functional CAM-B3LYP was used as CAM-B3LYP often provides better results in time-dependent density functional theory (TD-DFT) calculations than conventional B3LYP.23 The HOMO and LUMO energy levels of 1b are −6.90 eV and −1.00 eV, respectively. The lowest 6 singlet–singlet transitions of 1b were computed by the TD-DFT calculation at the CAM-B3LYP/6-31G(d) level of theory. The calculated first excited state, which consists of the transition from HOMO to LUMO (0.669), is 336 nm (3.69 eV) with oscillator strength of 0.187. The theoretical calculations of other compounds 1c–h were also carried out as described for 1b (Table S1†).
Acknowledgements
This work was partly supported by the Futaba Electronic Memorial Foundation. The authors are grateful to Mr Shinji Ishihara (Instrumental Analysis Center, Yokohama National University) for EI- and ESI-HRMS analyses and elemental analyses.
References
- For recent reviews, see:
(a) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed;
(b) Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811 CrossRef CAS PubMed.
- For a review, see: M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516 CrossRef CAS PubMed.
- For a review, see: J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- For recent examples based on tetraphenylethenes, see:
(a) Z. Zhuang, P. Shen, S. Ding, W. Luo, B. He, H. Nie, B. Wang, T. Huang, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Chem. Commun., 2016, 52, 10842 RSC;
(b) X. Fang, Y.-M. Zhang, K. Chang, Z. Liu, X. Su, H. Chen, S. X.-A. Zhang, Y. Liu and C. Wu, Chem. Mater., 2016, 28, 6628 CrossRef CAS;
(c) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519 CrossRef CAS PubMed;
(d) Z. He, L. Zhang, J. Mei, T. Zhang, J. W. Y. Lam, Z. Shuai, Y. Q. Dong and B. Z. Tang, Chem. Mater., 2015, 27, 6601 CrossRef CAS;
(e) B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236 RSC.
- For recent examples based on distyrylarene derivatives, see:
(a) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330 CrossRef CAS PubMed;
(b) Y. Xiong, X. Yan, Y. Ma, Y. Li, G. Yin and L. Chen, Chem. Commun., 2015, 51, 3403 RSC;
(c) G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933 CAS;
(d) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CAS;
(e) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed.
- For recent examples based on triarylamines, see:
(a) J. Zhang, Z. Chen, L. Yang, F. Hu, G.-A. Yu, J. Yin and S.-H. Liu, Dyes Pigm., 2017, 136, 168 CrossRef CAS;
(b) Y. Chen, Y. Ling, L. Ding, C. Xiang and G. Zhou, J. Mater. Chem. C, 2016, 4, 8496 RSC;
(c) Z.-F. Chang, L.-M. Jing, Y.-Y. Liu, J.-J. Liu, Y.-C. Ye, Y. S. Zhao, S.-C. Yuan and J.-L. Wang, J. Mater. Chem. C, 2016, 4, 8407 RSC;
(d) Y. Lei, Y. Liu, Y. Guo, J. Chen, X. Huang, W. Gao, L. Qian, H. Wu, M. Liu and Y. Cheng, J. Phys. Chem. C, 2015, 119, 23138 CrossRef CAS;
(e) P. S. Hariharan, N. S. Venkataramanan, D. Moon and S. P. Anthony, J. Phys. Chem. C, 2015, 119, 9460 CrossRef CAS.
- For recent examples based on boron difluorides, see:
(a) M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 4, 5314 RSC;
(b) S. Wang, R. Tan, Y. Li, Q. Li and S. Xiao, Dyes Pigm., 2016, 132, 342 CrossRef CAS;
(c) W. A. Morris, M. Kolpaczynska and C. L. Fraser, J. Phys. Chem. C, 2016, 120, 22539 CrossRef CAS;
(d) T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242 CrossRef CAS PubMed;
(e) Z. Zhang, Z. Wu, J. Sun, B. Yao, G. Zhang, P. Xue and R. Lu, J. Mater. Chem. C, 2015, 3, 4921 RSC.
- For recent examples based on other fluorophores, see:
(a) S. Ito, T. Yamada and M. Asami, ChemPlusChem, 2016, 81, 1272 CrossRef;
(b) Y. Sagara, Y. C. Simon, N. Tamaoki and C. Weder, Chem. Commun., 2016, 52, 5694 RSC;
(c) B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Chem. Sci., 2016, 7, 2201 RSC;
(d) Y. Matsunaga and J.-S. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985 CrossRef CAS PubMed;
(e) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181 CrossRef CAS PubMed;
(f) M. Tanioka, S. Kamino, A. Muranaka, Y. Ooyama, H. Ota, Y. Shirasaki, J. Horigome, M. Ueda, M. Uchiyama, D. Sawada and S. Enomoto, J. Am. Chem. Soc., 2015, 137, 6436 CrossRef CAS PubMed;
(g) Y. Jiang, D. Gindre, M. Allain, P. Liu, C. Cabanetos and J. Roncali, Adv. Mater., 2015, 27, 4285 CrossRef CAS PubMed;
(h) R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922 RSC;
(i) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed.
- Mechanochromic luminescence has also been observed for some liquid-crystalline compounds and metal complexes. For recent examples, see:
(a) B. Huitorel, Q. Benito, A. Fargues, A. Garcia, T. Gacoin, J.-P. Boilot, S. Perruchas and F. Camerel, Chem. Mater., 2016, 28, 8190 CrossRef CAS;
(b) O. Toma, M. Allain, F. Meinardi, A. Forni, C. Botta and N. Mercier, Angew. Chem., Int. Ed., 2016, 55, 7998 CrossRef CAS PubMed;
(c) T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252 CrossRef CAS PubMed;
(d) M. Mitani, S. Ogata, S. Yamane, M. Yoshio, M. Hasegawa and T. Kato, J. Mater. Chem. C, 2016, 4, 2752 RSC;
(e) S. Yagai, T. Seki, H. Aonuma, K. Kawaguchi, T. Karatsu, T. Okura, A. Sakon, H. Uekusa and H. Ito, Chem. Mater., 2016, 28, 234 CrossRef CAS;
(f) Q. Benito, X. F. Le Goff, S. Maron, A. Fargues, A. Garcia, C. Martineau, F. Taulelle, S. Kahlal, T. Gacoin, J.-P. Boilot and S. Perruchas, J. Am. Chem. Soc., 2014, 136, 11311 CrossRef CAS PubMed;
(g) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS PubMed.
- For self-recovering mechanochromic organic luminophores, see:
(a) H. Wu, C. Hang, X. Li, L. Yin, M. Zhu, J. Zhang, Y. Zhou, H. Ågren, Q. Zhang and L. Zhu, Chem. Commun., 2017, 53, 2661 RSC;
(b) D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Y. Zhang, H. Yan and Q. Zhao, Chem. Commun., 2016, 52, 12494 RSC;
(c) K. C. Naeem, A. Subhakumari, S. Varughese and V. C. Nair, J. Mater. Chem. C, 2015, 3, 10225 RSC;
(d) Y. Lin, G. Chen, L. Zhao, W. Z. Yuan, Y. Zhang and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 112 RSC;
(e) L. Bu, M. Sun, D. Zhang, W. Liu, Y. Wang, M. Zheng, S. Xue and W. Yang, J. Mater. Chem. C, 2013, 1, 2028 RSC;
(f) K. Mizuguchi, H. Kageyama and H. Nakano, Mater. Lett., 2011, 65, 2658 CrossRef CAS;
(g) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed . See also: ref. 4b, 6e and 9d.
- S. Ito, T. Yamada, T. Taguchi, Y. Yamaguchi and M. Asami, Chem.–Asian J., 2016, 11, 1963 CrossRef CAS PubMed.
- Single crystals of 1e contained chloroform molecules. The calculated powder diffraction patterns for these single crystals were thus different from the results of the PXRD analysis on the powdered sample.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- P. Suppan, J. Photochem. Photobiol., A, 1990, 50, 293 CrossRef CAS.
- The emission maximum of a tetraphenylethylene derivative after grinding was different from the emission maximum of its molten form; see: X. Zhou, H. Li, Z. Chi, X. Zhang, J. Zhang, B. Xu, Y. Zhang, S. Liu and J. Xu, New J. Chem., 2012, 36, 685 RSC.
- The emission maxima of two diphenyldibenzofulvene derivatives after grinding were almost
identical to those of their molten form, although the emission intensity decreased considerably; see: X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261 CrossRef CAS PubMed.
- The melting point of 1b (128.0–128.6 °C) was below the decomposition temperature of the Boc group (>140 °C).
- G. Özüduru, T. Schubach and M. M. K. Boysen, Org. Lett., 2012, 14, 4990 CrossRef PubMed.
- R. D. Clark and A. Jahangir, Lateral Lithiation Reactions Promoted by Heteroaromatic Substituents; Organic Reactions, Wiley, New York, 1995, vol. 47, pp. 1–314 Search PubMed.
- Y.-M. Su, Y. Hou, F. Yin, Y.-M. Xu, Y. Li, X. Zheng and X.-S. Wang, Org. Lett., 2014, 16, 2958 CrossRef CAS PubMed.
- J. Guo, S. Wang, N. Dai, Y. N. Teo and E. T. Kool, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3493 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, photographs of the solvatochromic luminescence, PXRD data, X-ray diffraction analyses, and theoretical calculations. CCDC 1523441 (1b), 1523442 (1c), 1523443 (1d), 1523444 (1e), 1523445 (1f), 1523446 (1g), and 1523447 (1h). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01006k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Tomohiro Taguchi,
Takeshi Yamada,
Takashi Ubukata,
Yoshitaka Yamaguchi and
Masatoshi Asami
*,
Tomohiro Taguchi,
Takeshi Yamada,
Takashi Ubukata,
Yoshitaka Yamaguchi and
Masatoshi Asami