
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphitic carbon nitride with S and O codoping for enhanced visible light photocatalytic performance
Ran You,
Hailong Dou,
Lu Chen,
Shaohui Zheng and
Yongping Zhang *
*
Faculty of Materials and Energy, Southwest University, Chongqing 400715, China. E-mail: zhangyyping@yahoo.com
First published on 10th March 2017
Abstract
Graphitic carbon nitride (g-C3N4) shows great possibility to enhance its visible light photocatalytic performance by tuning its electronic structure and band gap via nonmetal element doping. S and O codoped g-C3N4 is synthesized by the polymerization of melamine and H2O2 bonded trithiocyanuric acid (TCA) at an elevated temperature and characterized as crimped nanosheets with mesoporous structures. The photocatalytic performance of S–O codoped g-C3N4 for RhB degradation increases 6 fold by enhancing visible light adsorption and decreasing its band gap compared to pristine g-C3N4 nanosheets. The substitution of the edge N with S and O dopant causes much more strongly delocalized HOMO and LUMO and increases the number of reactive sites, facilitating the migration of photogenerated electron/hole pairs.
1. Introduction
Nowadays humans consume even more fossil energy to meet insatiable demands for more high-quality products and high-efficiency transportation, leading to the ever-increasing problem of energy shortage and environmental deterioration. It is generally considered that one prospective strategy is to utilize solar resources effectively and efficiently. Semiconductor based photocatalysts provide promising approaches in water splitting and pollutant degradation using solar energy directly and have gained considerable attention from research experts in materials science and chemistry. Graphitic carbon nitride (g-C3N4) is composed of stacked 2D layers which are constructed from tri-s-triazine units connecting through planar amino groups. The g-C3N4 possesses excellent thermal and chemical stability, as well as an appropriate band structure and a band gap of 2.7 eV allowing it to be driven under visible light when serving as a photocatalyst for solar resource conversion.1–3 Therefore, ever since its emergence, g-C3N4 has attracted extensive attention in the field of visible light photocatalysis and pollutant degradation because of its unique atomic and electronic structures.Some intrinsic characteristics, such as small specific surface area and rapid recombination of the photogenerated electron–hole pair, restrict its application. Accordingly, many strategies have been adopted to improve the photocatalytic performance of g-C3N4 by means of the fabrication of nano/mesoporous structures with a soft or hard template,4–6 a heterojunction with other semiconductors,7–11 coupling with metal particles,12–19 and metal or non-metal doping,20–36 etc. Among these methods, doping with nonmetal elements showed great possibility to enhance the visible light photocatalysis of g-C3N4, since the electronic structure and band gap changed dramatically via atomic doping.20 Carbon and nitrogen self-doped g-C3N4 exhibited improved photoreactivity by forming delocalized π bonds to increase visible light absorption and electric conductivity.20,21 Many nonmetal heteroatoms, such as B,21–24 P,25–28 S,29–36 O,37–39 were introduced to improve the visible light photocatalytic activities for hydrogen generation and organic degradation compared with pristine g-C3N4. Experimental and theoretical studies showed that the dopants tune the electronic structure by forming localized states in the band gap, facilitating the transfer of photogenerated electron hole pairs. B-doped g-C3N4 was usually prepared by heating melamine and boron oxide.23,24 Different molecular structures, however, were deduced, such as C-NB2 groups in the linkage23 and B atoms replacing C atoms in the g-C3N4 frame.24 As for S-doped g-C3N4, Hong et al. reported that the S atoms substitute the C sites,29 while some researchers reported that the doped S forms the S–C bond in the triazine frame by replacing the N sites.33–35 Different preparation techniques may affect the substitution sites with dopant atoms, which leads to different electronic structures and subsequently affects the photocatalytic performance. Contradictions still exist about how to prepare, characterize and understand its mechanism. Therefore, further experimental and theoretical investigations are needed to gain deeper insight into the mechanism of photocatalytic performance via element doping. It is meaningful to explore a simple and effective approach to synthesize an efficient g-C3N4 photocatalyst via atomic doping with single or twofold elements and to study the dopant effect on the photocatalytic performance.
A valid pathway to improve the photocatalytic performance of g-C3N4 is by changing its electronic band structure and increasing the number of reactive sites via atomic doping. In this paper, we report a pathway to improve the photocatalytic performance by introducing S and O atoms into the g-C3N4 lattice. The experiments revealed that the S–O codoped g-C3N4 exhibits superior photoreactivity for RhB degradation under visible light irradiation. Density functional theory calculations showed that the S doping decreases the band gap and increases the number of reactive sites, facilitating the transfer of photogenerated electron–hole pairs. This work provides a facile way to modulate the intrinsic electronic and band structure of g-C3N4.
2. Experimental details
0.01 mol of melamine (M) powder was dissolved into 30 mL of water solution with 30% H2O2 in mass percentage. Then the solution was fully mixed inside a beaker and dried at 60 °C overnight. The obtained white substance was mixed with 0.01 mol of trithiocyanuric acid (TCA), and ground into a powder and then transferred to a silica boat with a cover and heated at 550 °C for 2 h in a nitrogen atmosphere. The obtained S–O doped g-C3N4 product was a yellow powder and was collected for further use. The S doped g-C3N4 was prepared in the same way without the H2O2 solution being involved in the preparation process. Scheme 1 illustrated the facile process for preparing the pristine (CN) and the doped g-C3N4 with S (S–CN) and S–O (S,O–CN) elements.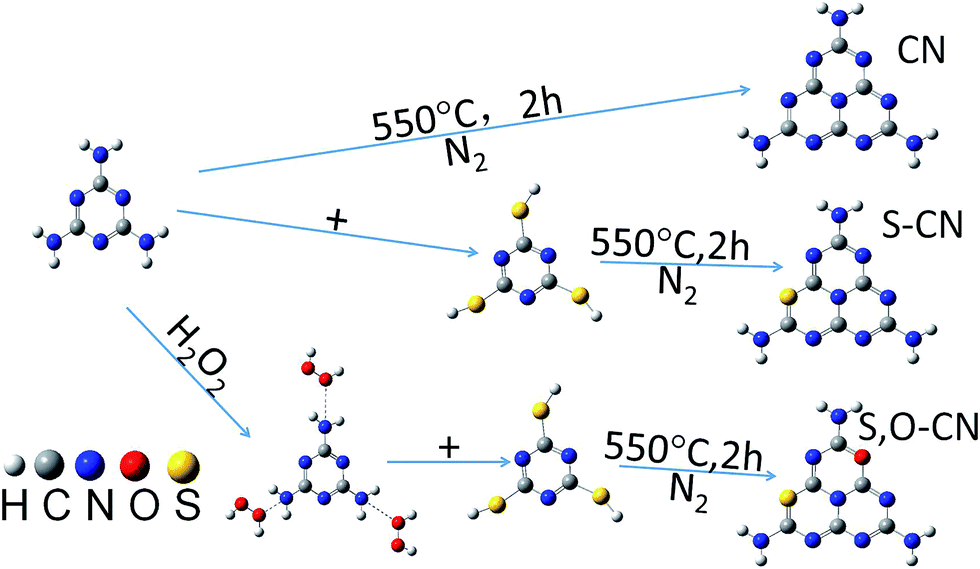 | ||
| Scheme 1 Facial preparation process of the pure and S–O doped g-C3N4. | ||
The surface morphology was observed by a scanning electron microscope (SEM, JSE-7800F, Jeol). X-ray diffraction (XRD) measurements were used to characterize the crystalline structures of the g-C3N4 by a diffractometer (ShimadzuXRD7000) with Cu Kα radiation (λ = 1.5418 Å). The vibrational information was measured using a Fourier transform infrared (FTIR, PerkinElmer) spectroscopy instrument in KBr pellets. X-ray photoelectron spectroscopy (XPS) was carried out to analyze the chemical state and composition on a VG ESCALAB 250 spectrometer with Al Kα radiation (hν = 1486.8 eV). Ultraviolet-visible (UV-vis) absorption spectra were performed on a U-3310 spectrophotometer (Hitachi, Japan) in the wavelength range of 300 nm to 800 nm. Photoluminescence (PL) spectra were carried out on an F-7000 fluorescence spectrophotometer (Hitachi, Japan) with an excitation wavelength at 273 nm using a 150 W Xe lamp as the excitation source.
The photocatalytic performance was evaluated by the degradation of Rhodamine B (RhB) in an aqueous solution under visible light irradiation using a 500 W Xe lamp as light source. 50 mg of photocatalyst was dispersed in RhB aqueous solution (50 mL, 10 ppm). Prior to irradiation, the suspension was magnetically stirred in a dark room for 1 h to reach adsorption–desorption equilibrium. During the irradiation process, 1 mL of the suspension was taken from the reaction cell at 30 min intervals for measuring the characteristic UV-vis absorption spectra after centrifugation. The maximum absorption peak was recorded and used to evaluate the concentration of RhB. The degradation rate of RhB can be calculated by definition:
| Degradation rate = (C0–C)/C0 |
3. Results and discussion
The SEM image of the pristine g-C3N4 nanosheets, shown in Fig. 1(a), indicates the g-C3N4 nanosheets appearing as a lamella structure with some crinkles. The S doped g-C3N4 in Fig. 1(b) appears as an elongated slate and crimped g-C3N4 sheets with irregular mesoporous structures after the M had reacted with TCA at an elevated temperature. This may be ascribed to the polymerization reaction of –SH in TCA and –NH in M facilitating the formation of larger g-C3N4 sheets. After the H2O2 hydrothermal treatment, the g-C3N4 sheets in Fig. 1(c) become longer and the lamella structures even more curved than those of the S doped g-C3N4 with irregular porous structures. The mesoporous structures may result from the decomposition of TCA during the polymerization process.16 The energy dispersive X-ray spectrum (EDS) analysis shows the composition of the S–O doped g-C3N4, comprising rich carbon (C) and nitrogen (N) with dispersed S and O elements. The overlapped C, N, S and O elemental EDS image (Fig. 1(d)) shows that the S and O elements are dispersed in g-C3N4 sheets. The EDS elemental mappings in Fig. 1(e–h) indicate clearly that the S and O elements are distributed discretely on the continuous C, N elemental background. | ||
| Fig. 1 The SEM images of (a) pristine g-C3N4 nanosheets, (b) S doped and (c) S–O codoped g-C3N4. Elemental EDS overlapped images of C, N, S and O (d) and mappings for C (e), N (f), S (g) and O (h). | ||
The crystal structures of the pristine g-C3N4 nanosheets, the S-doped and the S–O codped g-C3N4 were demonstrated by their XRD patterns. As shown in Fig. 2(a), the XRD pattern for the pristine g-C3N4 displays two distinct diffraction peaks located at 2θ of about 13.1° and 27.3°, which are in good accordance with the characteristic peaks of g-C3N4. These peaks correspond to the (100) and (002) crystal planes of g-C3N4 attributed to the lattice planes parallel to the c-axis and the stacking of the conjugated aromatic systems in the layered structure,15,29,31,38 respectively. With S and O doped g-C3N4, the 2θ position for the lattice peak (002) increases a little bit to 27.5° from the 27.3° of the pure g-C3N4. This indicates that the interplanar stacking distance becomes smaller after S and O doping, since the dopant elements of S and O have changed the localized electronic structures compared to the substituted N atoms. The stronger attraction between the g-C3N4 layers would result in shorter interplanar distances.38 The XRD results indicate that the S and O atoms have been incorporated into the g-C3N4 layer.
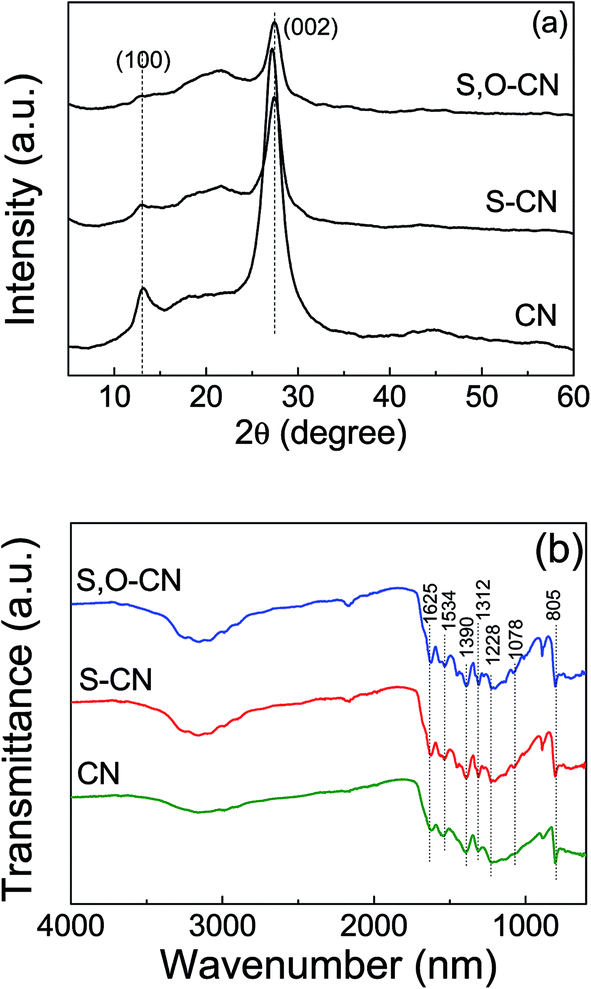 | ||
| Fig. 2 XRD patterns (a) and FTIR spectra (b) of pristine g-C3N4 nanosheets (CN), the S doped (S–CN) and S–O codoped g-C3N4 (S,O–CN). | ||
The FTIR spectra of the pristine g-C3N4 nanosheets, the S-doped and the S–O codped g-C3N4 are depicted in Fig. 2(b), in which the typical characteristic peaks at 1228, 1312, 1390, 1534, and 1625 cm−1 can be assigned to the stretching modes of the aromatic C–N heterocycle and the peak at 805 cm−1 is ascribed to the breathing mode of the triazine units. This demonstrates that the original graphitic C–N network was kept intact. The peak at 1228 cm−1 corresponds to aromatic C–N, C–O and might represent S–O stretching vibrations.34 Compared to the pristine g-C3N4, a new peak at 1078 cm−1 appears in the FTIR spectra for the O doped and S–O codoped g-C3N4, which can be attributed to the stretching mode of the C–O or C–S bond, which confirms that the oxygen and sulfur are doped into the g-C3N4 lattice. This is consistent with the FTIR results of the stretching vibration of C–O reported by Ma et al.26 and Seredych et al.34
XPS measurement was carried out to further investigate the chemical state and element valence changes of the pristine g-C3N4 and the g-C3N4 after S and S–O doping. Fig. 3 shows the high-resolution XPS spectra of C, N, O and S elements of the pristine g-C3N4, and the S-doped, and S–O codoped g-C3N4. The high-resolution C 1s XPS spectra of the three samples are shown in Fig. 3(a). For the pristine g-C3N4 nanosheets, the peak centered at about 288.4 eV (C1) is typically attributed to the sp2 C atoms bonded to N-containing aromatic skeleton rings (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) coordination. The peak at 285.48 eV (C2) is attributed to C–NH2 bonded in the triazine ring of the reactant intermediary product.21,23 The peak at 284.6 eV (C3) could be attributed to adventitious carbon contamination (C–C and CC).15,38 The O-doped g-C3N4 shows a newly generated peak at 288.86 eV ascribed to the C–OC bond, which indicates that O atoms could be directly bonded to sp2-hybridized carbon in the g-C3N4 layer. The N 1s spectra for pristine g-C3N4 in Fig. 3(b) can be mainly decomposed to three typical peaks located at about 398.4 eV (N1), 399.7 eV (N2), and 401.0 eV (N3), which could be attributed to the sp2-hybridized aromatic N atoms bonded to carbon atoms (C–NC), sp3-hybridized N atoms of N(–C)3 and terminal amino functions (C–NH2), respectively.23,38 The S atomic dopant does not change the chemical shift of the C 1s spectrum since the element S has similar electronegativity compared to the C atom. The N 1s spectra remain unchanged since S and O elements substitute the N atoms in the g-C3N4 lattice.
N) coordination. The peak at 285.48 eV (C2) is attributed to C–NH2 bonded in the triazine ring of the reactant intermediary product.21,23 The peak at 284.6 eV (C3) could be attributed to adventitious carbon contamination (C–C and CC).15,38 The O-doped g-C3N4 shows a newly generated peak at 288.86 eV ascribed to the C–OC bond, which indicates that O atoms could be directly bonded to sp2-hybridized carbon in the g-C3N4 layer. The N 1s spectra for pristine g-C3N4 in Fig. 3(b) can be mainly decomposed to three typical peaks located at about 398.4 eV (N1), 399.7 eV (N2), and 401.0 eV (N3), which could be attributed to the sp2-hybridized aromatic N atoms bonded to carbon atoms (C–NC), sp3-hybridized N atoms of N(–C)3 and terminal amino functions (C–NH2), respectively.23,38 The S atomic dopant does not change the chemical shift of the C 1s spectrum since the element S has similar electronegativity compared to the C atom. The N 1s spectra remain unchanged since S and O elements substitute the N atoms in the g-C3N4 lattice.
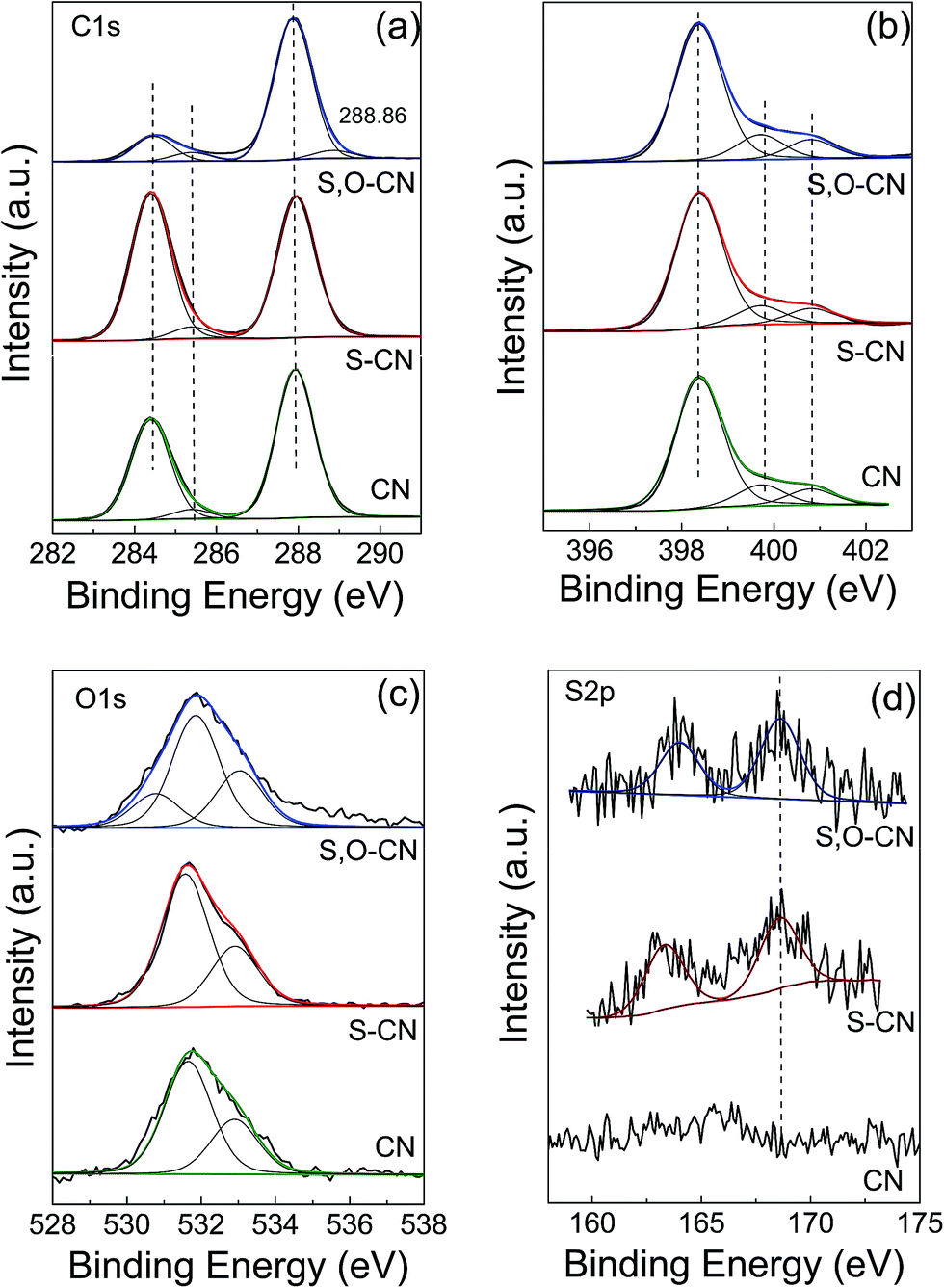 | ||
| Fig. 3 XPS high resolution spectra for C 1s (a) and N 1s (b), O 1s (c) and S 2p (d) of the pristine g-C3N4 nanosheets (CN) and S doped g-C3N4 (S–CN) and S–O codoped g-C3N4 (S,O–CN). | ||
In the O 1s spectrum for the pristine g-C3N4 in Fig. 3(c), the peak at 532.88 eV is ascribed to –O− in water or chemisorbed oxygen species. And the peak at 531.68 eV could be ascribed to OC in carbonyl or OS in sulfoxide residual introduced in the preparation process.34 After O doping, a new peak at 530.78 eV appeared, which can be attributed to the formation of C–O and N–C–O in the g-C3N4 lattice. There is also a new peak at a higher binding energy of 288.86 eV in the C 1s spectrum ascribed to the C–OC bond. This result indicates that the O atom is bonded to the sp2-C by substituting the N atom. The peak at 531.68 eV increases slightly compared to that of the pristine g-C3N4, since the intermediate sulfoxide products increase after reacting with TCA. As for the S and S–O doped g-C3N4, an S 2p peak located at 164.0 eV can be reasonably assigned to C–S bonds formed in the g-C3N4 lattice via substituting N. The peak at 168.6 eV is ascribed to SO in the intermediate product of sulfoxide resulting from the decomposing TCA.34,38 By removing the adventitious carbon contamination, the C/N atomic ratio is 0.74 for the pristine g-C3N4, which is fairly close to the stoichiometric value of g-C3N4. The C/N atomic ratio for the S, O codoped g-C3N4 is about 0.76, which is slightly larger that that of the pristine g-C3N4. XPS results show that the S and O atoms have been doped into the g-C3N4 lattice and may preferentially substitute N atoms. For the S–O codoped g-C3N4, the S content is about 0.08% and the O content is about 0.15%.
The photoabsorption property of the S–O codoped g-C3N4, as shown in Fig. 4(a), was investigated by measuring the UV-vis absorbance spectra in the range of 300 to 800 nm. There is a sharp absorption edge for the pristine g-C3N4 nanosheets at around 460 nm indexing to a band gap energy of about 2.7 eV, which is firmly associated with the photocatalytic property in visible light.34,38 The Kubelka–Munk plots in Fig. 4(b) show that the adsorption edge is red shifted with lower bonding energy of 2.4 eV for the S-doped g-C3N4 and 2.3 eV for the S–O codped g-C3N4. The absorption intensity is remarkably enhanced in the visible region after the S and O doping.37 The absorption peaks around 300–400 nm are assigned to π–π* transitions in the conjugated ring systems, including heterocyclic aromatics. The features near 500 nm are due to n–π* transitions involving lone pairs on the edge N atoms of the triazine rings and the intensity increases for the S, O codoped g-C3N4 compared with the pristine g-C3N4. SEM images have shown that S and O doping causes the g-C3N4 sheets to curl up. The crimped structure facilitates the n–π* transitions. These results reveal that the S–O doping g-C3N4 composites could significantly promote the optical absorption performance and enhance the utilized efficiency of solar light, which subsequently results in its promotion of the photocatalytic activity.37
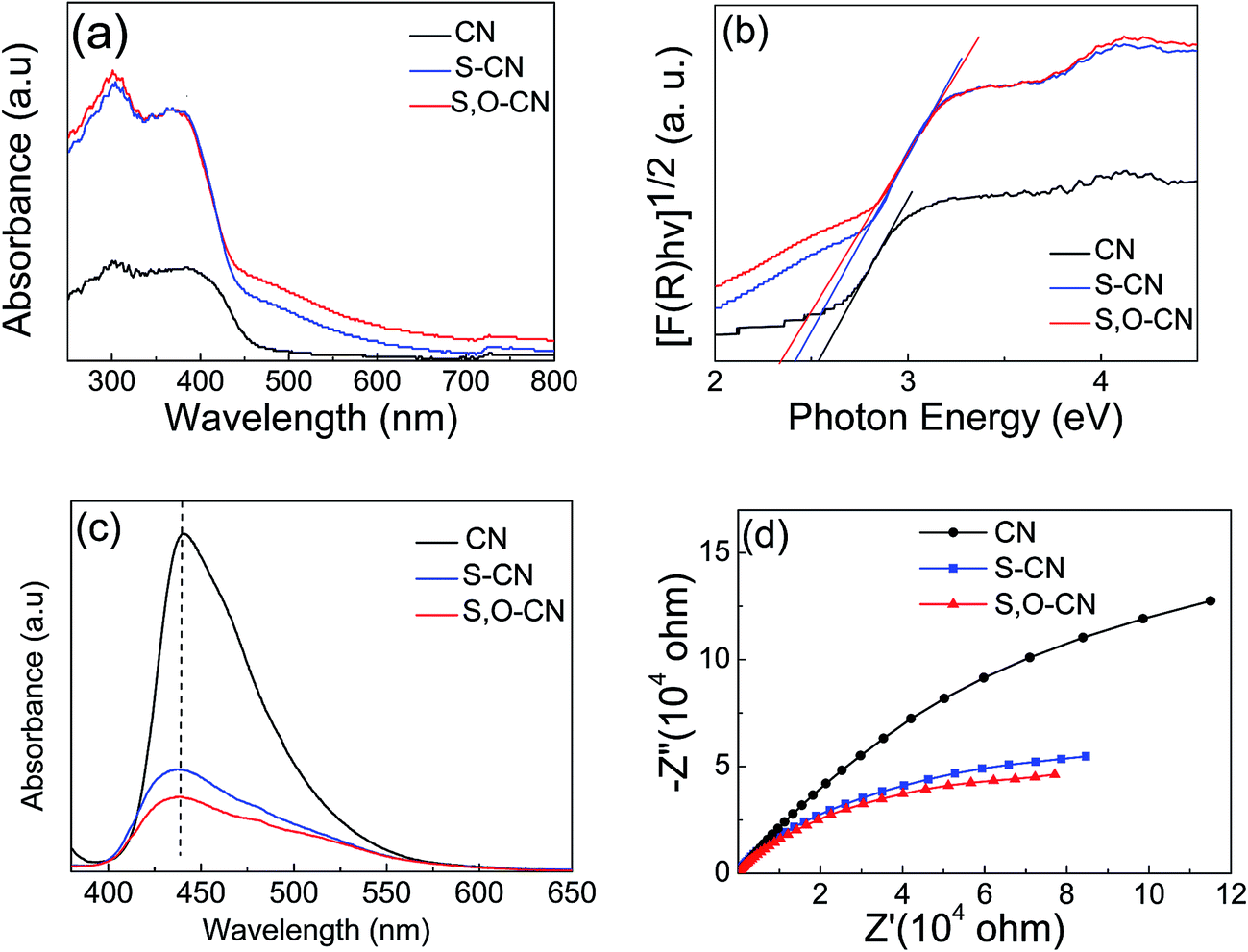 | ||
| Fig. 4 (a) UV-vis absorbance spectra, (b) corresponding Kubelka–Munk plots, (c) PL and (d) EIS spectra of the pure g-C3N4 nanosheets and S doped g-C3N4 and S–O codoped g-C3N4. | ||
Photoluminescence (PL) spectra were carried out to investigate the recombination/separation of photoinduced charge carriers in the pristine g-C3N4 nanosheets and the S–O codoped g-C3N4 under the excitation wavelength of 273 nm. The measured PL spectra, shown in Fig. 4(c), showed that all of the samples exhibit a main emission peak appearing at about 440 nm, which is consistent with the reported value in the literature.36,37 Compared to the pristine g-C3N4, the S–O doped g-C3N4 shows weaker PL intensity, revealing the lower recombination probability of photoinduced electrons and holes,37 which could give rise to a higher photocatalytic activity.
Electrochemical impedance spectra (EIS) of the pristine g-C3N4 and the g-C3N4 doped with O and S were measured to understand the photocatalytic mechanism. The arc on the EIS Nyquist plot indicates the charge transfer resistance. Generally speaking, the smaller the arc radius, the lower the charge transfer resistance will be.15,37 As shown in Fig. 4(d), the Nyquist plots of all the O and S doped g-C3N4 samples demonstrate a smaller arc radius attributed to the reduced electronic resistance and increased electronic conductivity by doping with O and S elements compared to the pristine g-C3N4. Similarly, the arc radius for S–O codoped g-C3N4 is smallest in all three samples, which is associated with the highest efficiency of the charge separation.36 Moreover, it is vital that the change trend of the arc radius for g-C3N4 samples is roughly consistent with the results displayed in the PL spectra.
The photocatalytic performance of S–O doped g-C3N4 was evaluated by RhB degradation under visible light irradiation (Fig. 5(a)). After 3 h of irradiation with visible light, about 48% of the RhB is degraded in presence of the S doped g-C3N4, compared to only 20% of the RhB being decomposed in the presence of the pristine g-C3N4. As for the S–O codoped g-C3N4, approximately 75% of RhB is decomposed after 3 h of visible light irradiation. Fig. 5(b) shows the first-order reaction kinetics for the RhB degradation, which is expressed as −ln(C/C0) = kt, where k is the reaction rate, and t is the reaction time. The photocatalytic performance of the S–O doped g-C3N4 is 6 times that of the pristine g-C3N4 calculated from the results in Fig. 5(b). The stability of the S–O codoped g-C3N4 was evaluated by recycle experiments for RhB degradation. After 3 h of visible light irradiation, the catalysts were retrieved by centrifugation and a subsequent drying process and used for the next RhB degradation test. The photocatalytic performance was investigated in fresh material and after three recycles, as shown in Fig. 5(c). No obvious difference was observed after three recycles, indicating that the S–O codoped g-C3N4 is relatively stable under visible light irradiation.
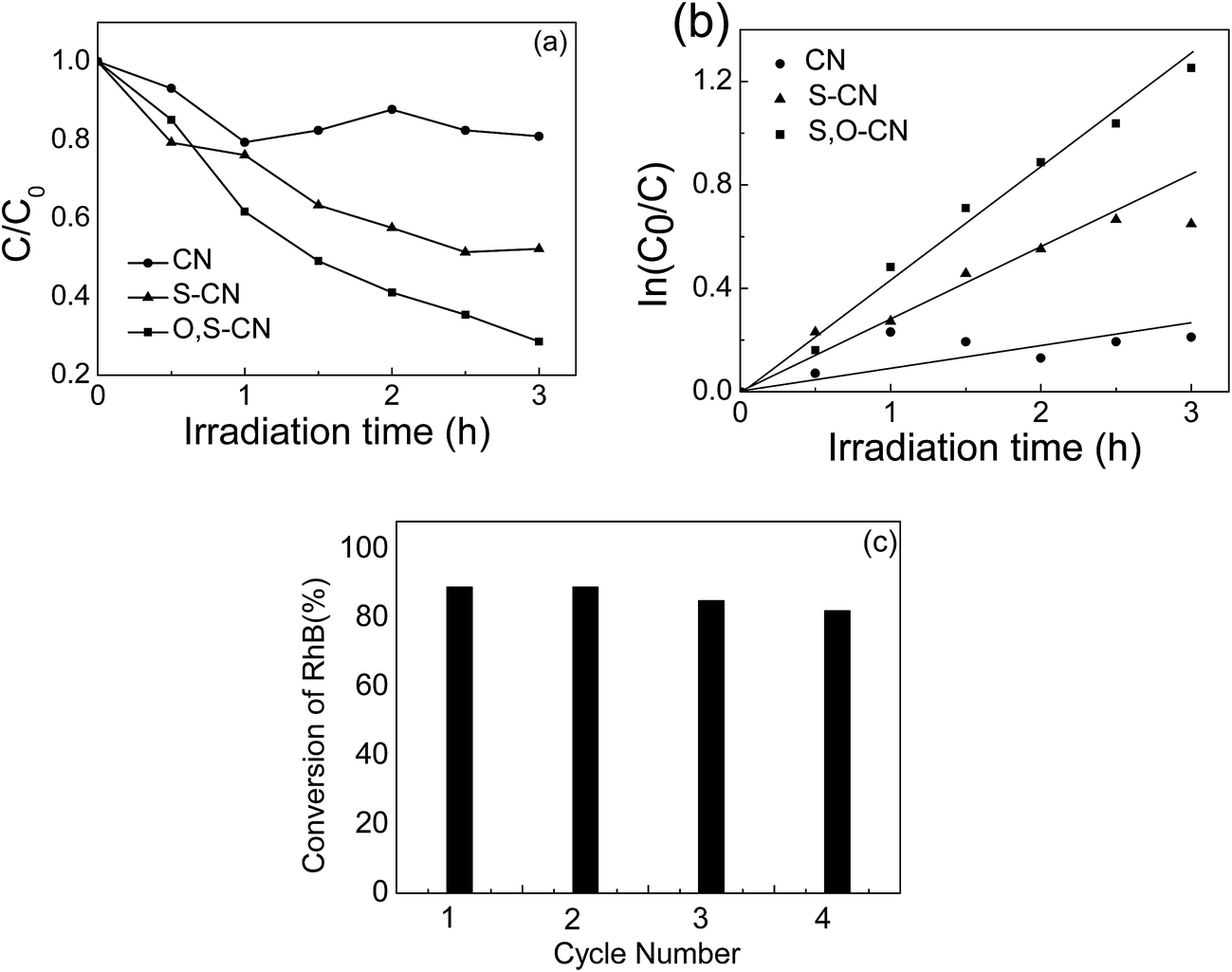 | ||
| Fig. 5 (a) Photocatalytic performances and (b) corresponding first-order reaction kinetics in the degradation of RhB under visible light irradiation for the pristine g-C3N4, the S doped and S–O codoped g-C3N4. (c) Stability test of the S–O codoped g-C3N4. | ||
A theoretical investigation was carried out to further understand the effect of different atomic doping on the electrical structure of the g-C3N4 layer. We performed density functional theory (DFT) calculations using the Gaussian09 software package for the O and S doped monolayer g-C3N4. DFT B3LYP/6-31G(d) level of theory was used to optimize the geometry of g-C3N4 and to calculate the natural orbital population distribution. The calculation of HOMO and LUMO, and density of state were based on the DFT B3PW91/6-31G(d) level.
Fig. 6 shows the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for the (001) lattice plane of the g-C3N4 monolayer. The calculated HOMO for the pristine g-C3N4 suggests that the edge N atoms provide the sites for oxidation of water to O2, whereas the LUMO indicates that the C and the inner N atoms are the preferred reduction sites to form H2.40 The migration of the photogenerated e−/h+ pairs is not efficient due to the differently localized HOMO and LUMO. No HOMO and LUMO present on the bridge N atoms inhibits the carrier migration from one heptazine unit to another and this reduces the photocatalytic performance. The substitution of the edge N with O causes slightly more strongly delocalized HOMO and LUMO compared to the pristine g-C3N4 monolayer. The dispersion of the HOMO and LUMO distribution can enhance the carrier mobility. The substitution of the edge N with S causes more strongly delocalized HOMO and LUMO, and thus increases the number of reactive sites.36,40 It is noted that the bridge N can act as a channel to connect the adjacent heptazine unit and facilitate the migration of photogenerated electron/hole pairs. The geometry for the S doped g-C3N4 monolayer is inclined to deform and become crimped, which is consistent with the morphology shown in the SEM image.
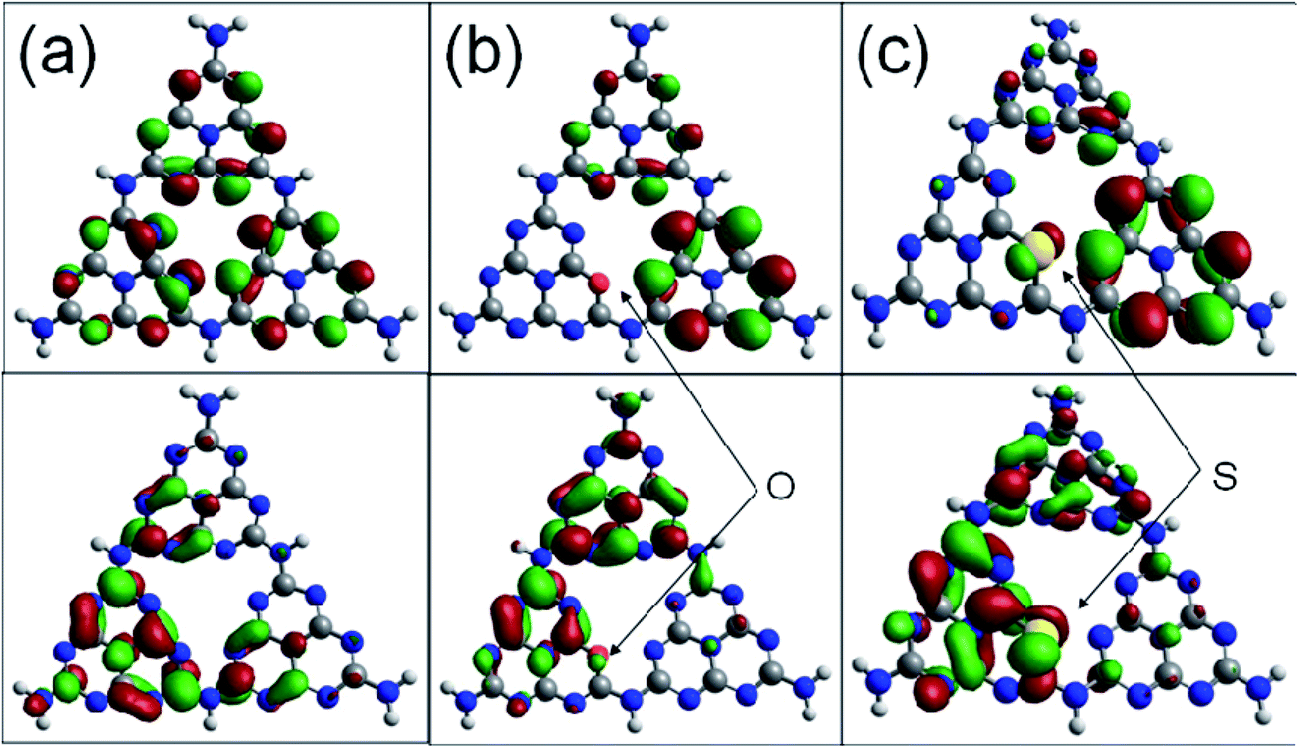 | ||
| Fig. 6 Calculated HOMO (top) and LUMO (bottom) of the pristine (a), O doped (b) and S doped (c) g-C3N4 monolayer. The isosurface is taken at a value of 0.003e/Bohr3. Carbon atoms are in grey and nitrogen in blue. | ||
The total and partial density of state (DOS) for the pristine g-C3N4 are shown in Fig. 7(a). The DOS in the valance bond is mainly contributed by the carbon and nitrogen atoms, while the DOS in the conductance bond is from the nitrogen atoms. The band gap narrows upon the S and O doping and the Fermi level is shifted to the conducting band, as shown in Fig. 7(b) and (c). The intensity and wave profile of the partial DOS for C and N are similar in the ranges of −20 to −12 eV and −5 eV to the Fermi level for the g-C3N4 monolayer, which indicates that the orbitals of C and N hybridized. The S dopant contributes more DOS compared with the O dopant and the partial DOS of S elements near the Fermi level is stronger that that of O elements.36,40
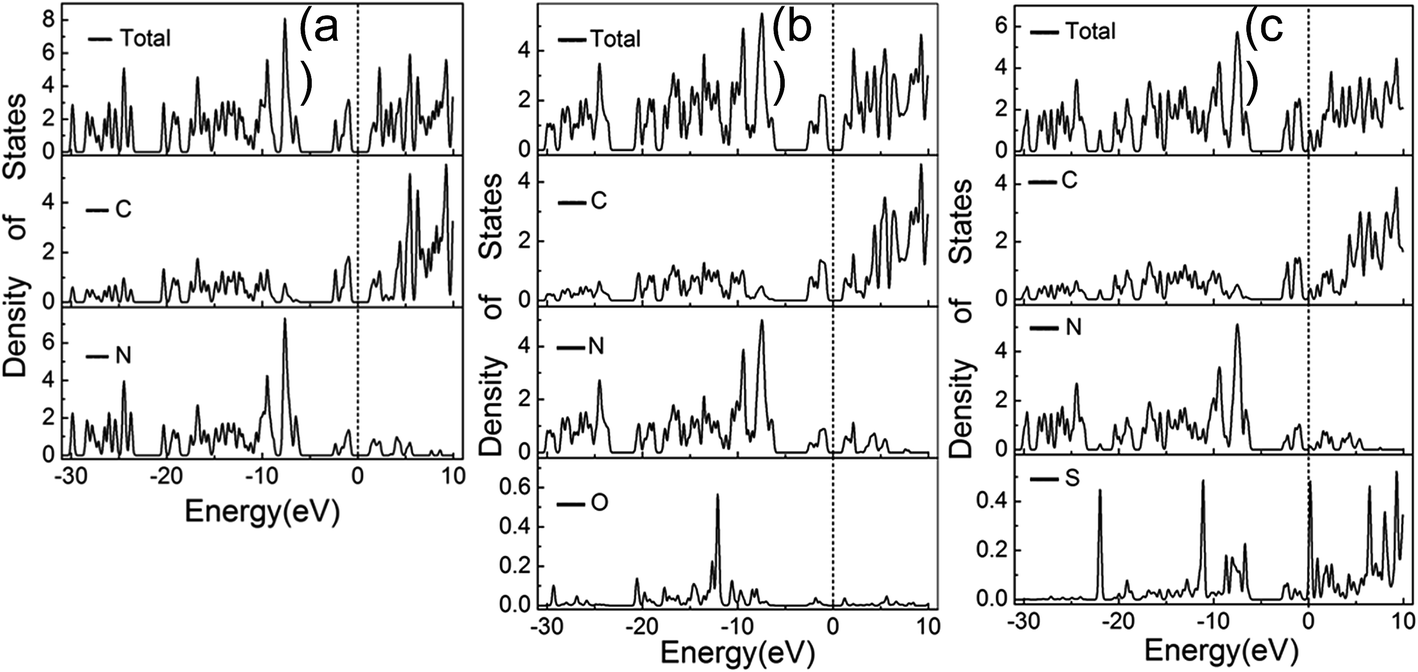 | ||
| Fig. 7 Calculated total and partial DOS plots of C, N, O and S elements for the pristine (a), O doped (b) and (c) S doped g-C3N4 monolayer. | ||
The calculated natural orbital population distribution showed that the valence electron configuration of the carbon atom is 2s0.74–0.762p2.60–2.61. The valence electron of the carbon atoms alters imperceptibly after the O and S doping, and the modification disappears far away from the dopant atoms. The valence electron configurations of the doped O and S are 2s1.592p4.89, 3s1.673p3.95, respectively. The electron orbitals of the doped O and S have hybridized with those of other atoms, covalently binding with adjacent atoms. The valence electron divergence between the dopant atoms and the adjacent intrinsic atoms yields a new energy band in the g-C3N4 monolayer, thus changing its photocatalytic performance.
Finally, a clearer picture can be drawn by taking above-mentioned experimental and theoretical strands into consideration. The semiconductor photocatalysis process involves the generation, the separation and transfer, and the recombination of photoinduced charge carriers under visible light excitation. Non-metal element doping causes slightly delocalized HOMO and LUMO, and tunes the semiconductor band structure and the band gap as well. XPS and XRD results showed that the O and S atoms substitute the N atoms in the g-C3N4 lattice by forming O–C and S–C bonds and leaving the N 1s spectra unchanged after O and S doping. The O and S doping has narrowed the band gap by the UV-vis spectra, and promotes the optical absorption performance. In addition, the PL and EIS spectra confirm the higher separation and transfer efficiency of the photoinduced charge carriers. The DFT calculations show that dispersion of the HOMO and LUMO distribution can enhance the carrier mobility. For the pristine g-C3N4, no HOMO and LUMO are present on the bridge N atoms, thus suppressing the carrier migration from one heptazine unit to another and reducing the photocatalytic performance. The substitution of the edge N with S causes more strongly delocalized HOMO and LUMO, thus increasing the number of reactive sites.33,36 It is noted that the bridge N can act as a channel to connect the adjacent heptazine unit and facilitate the migration of photogenerated electron/hole pairs by increasing the density of state around the bridge N atoms. The g-C3N4 layers doped with S and O result in an integrated effect under visible light irradiation, having largely promoted the separation and suppressed the recombination of photoexcited charge carriers, thus achieving a higher photocatalytic activity than that of the pristine g-C3N4.
4. Conclusion
In summary, the doped S and O atoms substituting the lattice N atoms facilitate the separation and transfer of the photoinduced electron–hole pairs. The crimped structure resulting from the S and O doping enhanced UV-vis absorption by boosting the n–π* transition. The photocatalytic performance for RhB degradation increases 6 fold compared to the pristine g-C3N4 nanosheets. The more strongly delocalized HOMO and LUMO and narrower band gap by S and O doping facilitate the photoexcitation and migration of photoinduce charge carriers, thus enhancing the photocatalytic performance.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21173170).References
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability?, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Cora, S. Firth, J. A. Darr and P. F. McMillan, H2 and O2 evolution from water half-splitting reactions by graphitic carbon nitride materials, J. Phys. Chem. C, 2013, 117, 7178–7185 CAS.
- H. Yan, Soft-templating synthesis of mesoporous graphitic carbon nitride with enhanced photocatalytic H2 evolution under visible light, Chem. Commun., 2012, 48, 3430–3432 RSC.
- J. Zhang, F. Guo and X. Wang, An optimized and general synthetic strategy for fabrication of polymeric carbon nitride nanoarchitectures, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- P. Li, W. Zhang, Y. Zhang, Y. Sun and F. Dong, (NH4)2SO4-assisted polycondensation of dicyandiamide for porous g-C3N4 with enhanced photocatalytic NO removal, RSC Adv., 2016, 6, 96334–96338 RSC.
- Y. Tian, B. Chang, J. Lu, J. Fu, F. Xi and X. Dong, Hydrothermal synthesis of graphitic carbon nitride-Bi2WO6 heterojunctions with enhanced visible light photocatalytic activities, ACS Appl. Mater. Interfaces, 2013, 5, 7079–7085 CAS.
- L. Ye, D. Wang and S. Chen, Fabrication and enhanced photoelectrochemical performance of MoS2/S-doped g-C3N4 heterojunction film, ACS Appl. Mater. Interfaces, 2016, 8, 5280–5289 CAS.
- M. Xu, L. Han and S. Dong, Facile fabrication of highly efficient g-C3N4/Ag2O heterostructured photocatalysts with enhanced visible-light photocatalytic activity, ACS Appl. Mater. Interfaces, 2013, 5, 12533–12540 CAS.
- S. Kumar, T. Surendar, A. Baruah and V. Shanker, Synthesis of a novel and stable g-C3N4–Ag3PO4 hybrid nanocomposite photocatalyst and study of the photocatalytic activity under visible light irradiation, J. Mater. Chem. A, 2013, 1, 5333–5340 CAS.
- S. Kumar, A. Baruah, S. Tonda, B. Kumar, V. Shanker and B. Sreedhar, Cost-effective and eco-friendly synthesis of novel and stable N-doped ZnO/g-C3N4 core-shell nanoplates with excellent visible-light responsive photocatalysis, Nanoscale, 2014, 6, 4830–4842 RSC.
- J. Liu, S. Xie, Z. Geng, K. Huang, L. Fan, W. Zhou, L. Qiu, D. Gao, L. Ji, L. Duan, L. Lu, W. Li, S. Bai, Z. Liu, W. Chen, S. Feng and Y. Zhang, Carbon nitride supramolecular hybrid material enabled high-efficiency photocatalytic water treatments, Nano Lett., 2016, 16, 6568–6575 CrossRef CAS PubMed.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Co-monomer control of carbon nitride semiconductors to optimize hydrogen evolution with visible light, Angew. Chem., Int. Ed., 2012, 124, 3237–3241 CrossRef.
- K. Tian, W.-J. Liu and H. Jiang, Comparative investigation on photoreactivity and mechanism of biogenic and chemosythetic Ag/C3N4 composites under visible light irradiation, ACS Sustainable Chem. Eng., 2015, 3, 269–276 CrossRef CAS.
- L. Chen, Y. H. Man, Z. Q. Chen and Y. P. Zhang, Ag/g-C3N4 layered composites with enhanced visible light photocatalytic performance, Mater. Res. Express, 2016, 3, 115003 CrossRef.
- L. Ge, C. Han, J. Liu and Y. Li, Enhanced visible light photocatalytic activity of novel polymeric g-C3N4 loaded with Ag nanoparticles, Appl. Catal., A, 2011, 409, 215–222 CrossRef.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Photocatalytic reduction of CO2 into hydrocarbon solar fuels over g-C3N4–Pt nanocomposite photocatalysts, Phys. Chem. Chem. Phys., 2014, 16, 11492–11501 RSC.
- Z. Ni, F. Dong, H. Huang and Y. Zhang, New insights into how Pd nanoparticles influenced the photocatalytic oxidation and reduction ability of g-C3N4 nanosheets, Catal. Sci. Technol., 2016, 6, 6448–6458 CAS.
- G. Jiang, X. Li, M. Lan, T. Shen, X. Lv, F. Dong and S. Zhang, Monodisperse bismuth nanoparticles decorated graphitic carbon nitride: Enhanced visible-light-response photocatalytic NO removal and reaction pathway, Appl. Catal., B, 2017, 205, 532–540 CrossRef CAS.
- G. Dong, K. Zhao and L. Zhang, Carbon self-doping induced high electronic conductivity and photoreactivity of g-C3N4, Chem. Commun., 2012, 48, 6178–6180 RSC.
- J. Fang, H. Fan, M. Li and C. Long, Nitrogen self-doped graphitic carbon nitride as efficient visible light photocatalyst for hydrogen evolution, J. Mater. Chem. A, 2015, 3, 13819–13826 CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Photodegradation of rhodamine B and methyl orange over boron-doped g-C3N4 under visible light irradiation, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- D. Gao, Y. Liu, P. Liu, M. Si and D. Xue, Atomically thin B doped g-C3N4 nanosheets: high-temperature ferromagnetism and calculated half-metallicity, Sci. Rep., 2016, 6, 35768 CrossRef CAS PubMed.
- N. Sagara, S. Kamimura, T. Tsubota and T. Ohno, Photoelectrochemical CO2 reduction by a p-type boron-doped g-C3N4 electrode under visible light, Appl. Catal., B, 2016, 192, 193–198 CrossRef CAS.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, Phosphorus-doped carbon nitride solid: enhanced electrical conductivity and photocurrent generation, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Phosphorus-doped graphitic carbon nitrides grown in situ on carbon-fiber paper: flexible and reversible oxygen electrodes, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- S. Hu, L. Ma, J. You, F. Li, Z. Fan, F. Wang, D. Liu and J. Gui, A simple and efficient method to prepare a phosphorus modified g-C3N4 visible light photocatalyst, RSC Adv., 2014, 4, 21657–21663 RSC.
- Y. Zhou, L. Zhang, J. Liu, X. Fan, B. Wang, M. Wang, W. Ren, J. Wang, M. Li and J. Shi, Brand new P-doped g-C3N4enhanced photocatalytic activity for H2 evolution and rhodamine B degradation under visible light, J. Mater. Chem. A, 2015, 3, 3862–3867 CAS.
- J. Hong, X. Xia, Y. Wang and R. Xu, Mesoporous carbon nitride with in situ sulfur doping for enhanced photocatalytic hydrogen evolution from water under visible light, J. Mater. Chem., 2012, 22, 15006 RSC.
- L. Ye and S. Chen, Fabrication and high visible-light-driven photocurrent response of g-C3N4 films: the role of thiourea, Appl. Surf. Sci., 2016, 389, 1076–1083 CrossRef CAS.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H. Cheng, Unique electronic structure induced high photoreactivity of sulfur-doped graphitic C3N4, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- L. Cao, R. Wang and D. Wang, Synthesis and characterization of sulfur self-doped g-C3N4 with efficient visible-light photocatalytic activity, Mater. Lett., 2015, 149, 50–53 CrossRef CAS.
- J. Chen, Z. Hong, Y. Chen, B. Lin and B. Gao, One-step synthesis of sulfur-doped and nitrogen-deficient g-C3N4 photocatalyst for enhanced hydrogen evolution under visible light, Mater. Lett., 2015, 145, 129–132 CrossRef CAS.
- M. Seredych and T. J. Bandosz, Nitrogen enrichment of S-doped nanoporous carbon by g-C3N4: insight into photosensitivity enhancement, Carbon, 2016, 107, 895–906 CrossRef CAS.
- J. Hong, X. Xia, Y. Wang and R. Xu, Mesoporous carbon nitride with in situ sulfur doping for enhanced photocatalytic hydrogen evolution from water under visible light, J. Mater. Chem., 2012, 22, 1500615012 Search PubMed.
- S. Stolbov and S. Zuluaga, Sulfur doping effects on the electronic and geometric structures of graphitic carbon nitride photocatalyst: insight from first principles, J. Phys.: Condens. Matter, 2013, 25, 085507 CrossRef PubMed.
- Z. F. Huang, J. Song, L. Pan, Z. Wang, X. Q. Zhang, J. Zou, W. Mi, X. W. Zhang and L. Wang, Carbon nitride with simultaneous porous network and O-doping for efficient solar-energy-driven hydrogen evolution, Nano Energy, 2015, 12, 646–656 CrossRef CAS.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity, Chem. Commun., 2012, 48, 12017–12019 RSC.
- H. Ma, Y. Li, S. Li and N. Liu, Novel P–O codoped g-C3N4 with large specific surface area: hydrothermal synthesis assisted dissolution-precipitation process and their visible light activity under anoxic conditions, Appl. Surf. Sci., 2015, 357, 131–138 CrossRef CAS.
- J. Cui, S. Liang, X. Wang and J. Zhang, First principle modeling of oxygen-doped monolayer graphitic carbon nitride, Mater. Chem. Phys., 2015, 161, 194–200 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |