
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient trifluoromethylation of C(sp2)–H functionalized α-oxoketene dithioacetals: a route to the regioselective synthesis of functionalized trifluoromethylated pyrazoles†
N. Sharmaa,
N. Kumarib,
T. S. Chundawatc,
S. Kumard and
S. Bhagat*a
aOrganic Synthesis Research Laboratory, Department of Chemistry, A. R. S. D. College, University of Delhi, New Delhi-110021, India. E-mail: sunitabhagat28@gmail.com
bInstitute of Nuclear Medicine & Allied Sciences, Defence Research & Development Organization, Brig. SK Mazumdar Marg, Delhi-110054, India
cDepartment of Applied Sciences, The Northcap University (formerly ITM University), Gurgaon-122001, Haryana, India
dPhysical Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, India
First published on 6th February 2017
Abstract
An operationally simple approach for the regioselective construction of diversely substituted trifluoromethylated pyrazoles via nucleophilic trifluoromethylation of iodo-substituted α-oxoketene dithioacetals is described. X-ray crystallographic studies confirmed the trifluoromethylation as well as formation of a regioselective cyclized product. Furthermore, trifluoromethylated pyrazoles bearing thiomethyl groups may allow further functionalization and are of considerable interest in medicinal chemistry.
Organofluorine compounds, particularly trifluoromethylated molecules have generated considerable attention in the field of organic chemistry due to their wide range of applications in material sciences as well as in pharmaceutical & agrochemical industries.1,2 The interesting medicinal properties that the trifluoromethyl group imparts are anticipated due to an increase in lipophilicity and metabolic stability.3,4 As naturally occurring trifluoromethylated compounds are not reported in the literature, tremendous research efforts from synthetic organic chemists are focused on the development of an efficient route for selective introduction of the trifluoromethyl group onto aromatic and heteroaromatic ring systems under mild and selective reaction conditions.5–7
Functionalized α-oxoketene dithioacetals have emerged as versatile intermediates for the synthesis of various heterocyclic compounds in the field of organic synthesis.8 Among them, α-halogenated oxoketene dithioacetals are of special interest due to their diverse applications as synthons in metal catalysed reactions.9 Despite their wide scope, synthetic utility of α-halogenated oxoketene dithioacetals has not been well documented. α-Iodinated ketene dithioacetals, to our knowledge, have been reported to be synthesized by iododecarboxylation which suffers from low efficiency and undesired side reactions.10 Furthermore, there are no reports on the utilization of these iodofunctionalized oxoketene dithioacetals in organic transformation reactions. In view of these reported limitations and our ongoing efforts to explore oxoketene dithioacetals for construction of N-heterocycles,11 we envisioned that the development of efficient iodination strategy as well as trifluoromethylation protocol of these α-iodo substituted dithioacetals would allow for a new access to multisubstituted trifluoromethylated oxoketene dithioacetals under mild reaction conditions. Moreover, elaborations on these trifluoromethylated synthons in cyclization reactions resulted in regioselective construction of biologically important functionalized trifluoromethylated pyrazoles.12,13 Here it should be noted that there are only few reports of trifluoromethylations of C(sp2)–H bond of oxoketene dithioacetals14 which are suffering from drawbacks such as use of oxidant, ligands and additives as well as longer reaction hours. Adding to this, using similar reaction conditions trifluoromethylated pyrazoles of completely different regioselectivity were obtained as compared to compounds reported by Yu et al.14a (Scheme 1).
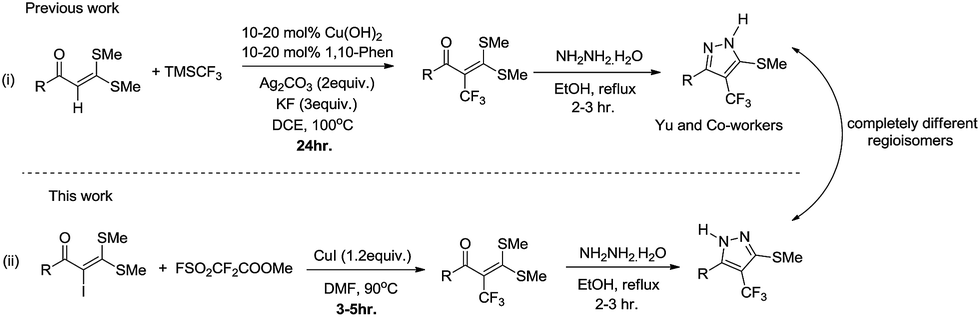 | ||
| Scheme 1 Functionalization of α-iodinated oxoketene dithioacetals. | ||
To probe the viability of the designed strategy, α-oxoketene dithioacetals15 were subjected to first direct C(sp2)–H iodination using N-iodosuccinimide as environmental friendly iodine source to get the desired compounds 2a–l in excellent yields. Formation of products was confirmed on the basis of 1H NMR and 13C NMR data. Disappearance of peak of vinylic proton at δ 6.4–6.8 due to substitution by iodine with co-presence of peak at δ 94–100 due to vinylic C–I confirmed the presence of iodine atom at α position.
Encouraged by the versatility of α-oxoketene dithioacetals to rapidly undergo substitution reactions,8e α-iodo substituted oxoketene dithioacetals were subjected to undergo trifluoromethylation reactions using relatively inexpensive, convenient but underutilized methyl fluorosulfonyl difluoroacetate (FSO2CF2COOMe)16 as reagent through transition metal catalysed cross coupling reaction in the presence of CuI.
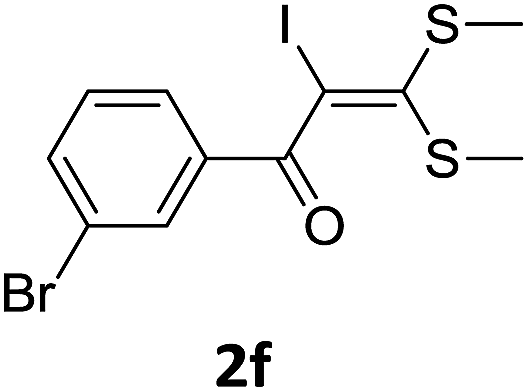
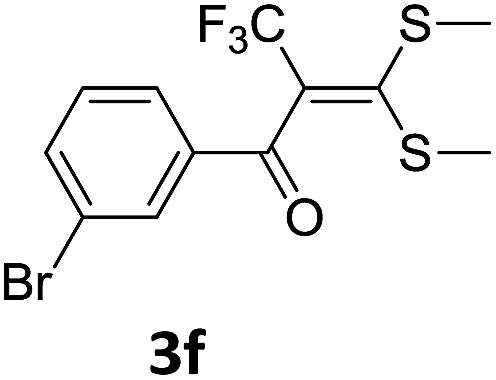
To identify the optimal conditions for the reaction, α-iodo oxoketene dithioacetal 2f was subjected to transition metal catalysed trifluoromethylation using methyl fluorosulfonyl difluoroacetate (FSO2CF2COOMe) and different reagents by varying solvents to yield 3f. When 1.5 eq. of FSO2CF2COOMe and 1.2 eq. of CuI were used in DMF as solvent at 60 °C, 40% yield of product 3f was observed (Table 1, entry 1). An increase of temperature to 80 °C resulted in the increase of yield to 60% (Table 1, entry 2). From the Table 1 it is clear that yield of desired compound 3f was maximum i.e. 86% when the reaction was heated at 90 °C (Table 1, entry 3). However no change in yield was observed when reaction temperature was increased from 90 °C to 110 °C (Table 1, entry 4). It should be noted that increase in equivalents of FSO2CF2COOMe and CuI to 2 eq. did not had any effect on yield of 3f (Table 1, entry 5). An examination of other copper source such as Cu(OAc)2 was not effective for this transformation and deiodination of 2f was observed (Table 1, entry 6). CuI was essential to the reaction because when KI was used in place of CuI or in absence of CuI desired product was not formed (Table 1, entries 7 & 8). By screening with polar organic solvents, such as n-BuOH, t-BuOH and PEG-400 there was no formation of desired product 3f (Table 1, entries 9–11).
|
|||||
|---|---|---|---|---|---|
| Entry | Source of CF3 | Reagent | Solvent | Temp (°C) | Yieldb (%) |
| a Reactions were performed using 2f (1 mmol), 1.5 equiv. of FSO2CF2COOMe, 1.2 equiv. of reagent 3 h unless otherwise stated.b Isolated yield.c Reaction were performed using FSO2CF2COOMe (2eq.), catalyst (2eq.). | |||||
| 1 | FSO2CF2COOMe | CuI | DMF | 60 | 40 |
| 2 | FSO2CF2COOMe | CuI | DMF | 80 | 60 |
| 3 | FSO2CF2COOMe | CuI | DMF | 90 | 86 |
| 4 | FSO2CF2COOMe | CuI | DMF | 110 | 86 |
| 5 | FSO2CF2COOMe | CuI | DMF | 90 | 86c |
| 6 | FSO2CF2COOMe | Cu(OAc)2 | DMF | 90 | — |
| 7 | FSO2CF2COOMe | KI | DMF | 90 | — |
| 8 | FSO2CF2COOMe | — | DMF | 90 | — |
| 9 | FSO2CF2COOMe | CuI | n-BuOH | 90 | — |
| 10 | FSO2CF2COOMe | CuI | t-BuOH | 90 | — |
| 11 | FSO2CF2COOMe | CuI | PEG-400 | 90 | — |
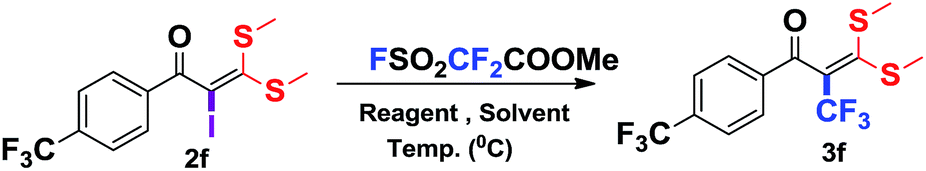
With the optimized conditions in hand (Table 1, entry 3), the scope of the reaction was explored considering both the yield and variety of substituents at α-iodo oxoketene dithioacetals (Table 2). The results shown in Table 2 suggested that the reaction of α-iodo oxoketene dithioacetals 2b–g bearing electron withdrawing substituents at both -m and -p positions could afford α-trifluoromethylated oxoketene dithioacetals 3a–g in good yields (entry 2–7).

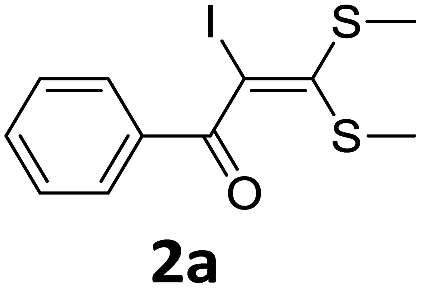
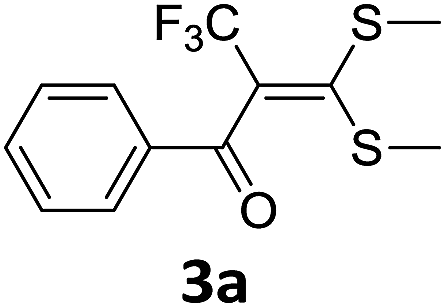
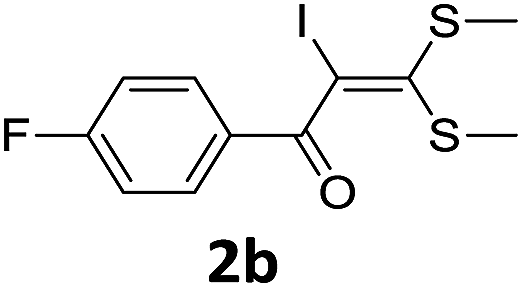
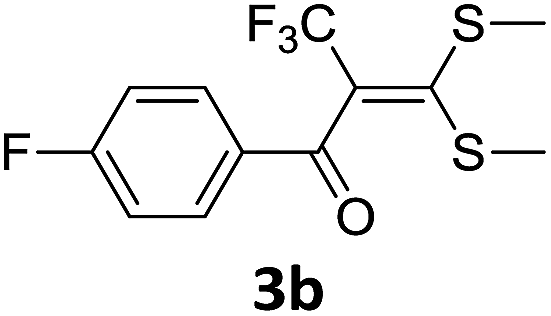
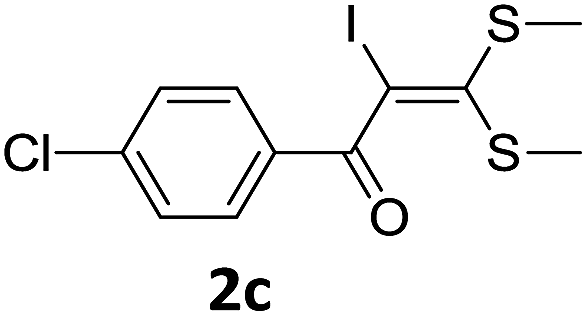
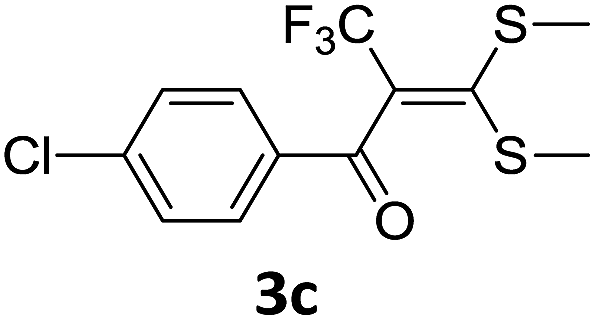
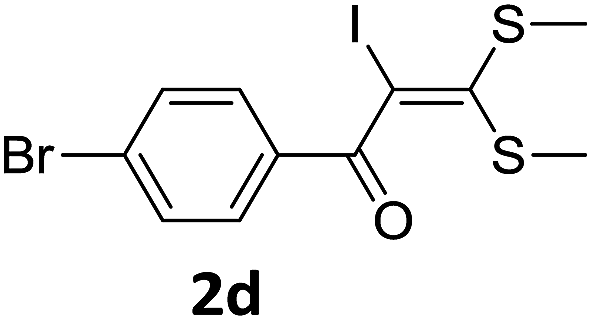
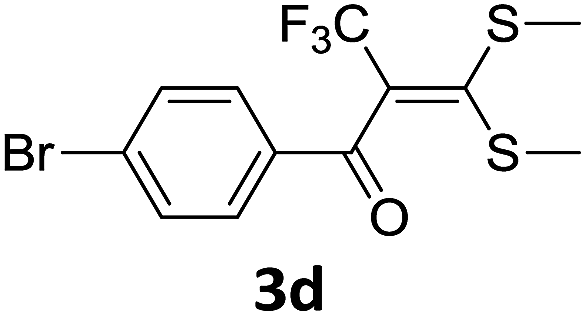
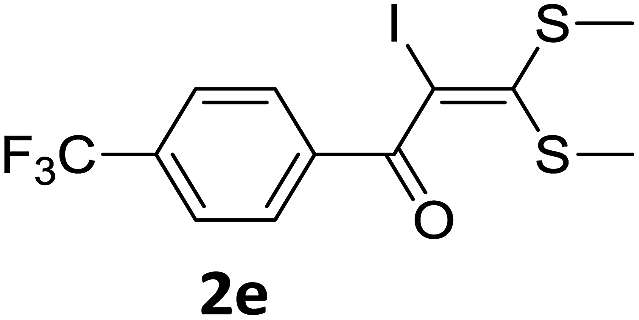
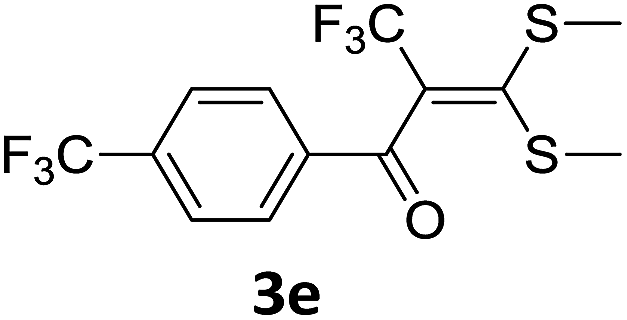
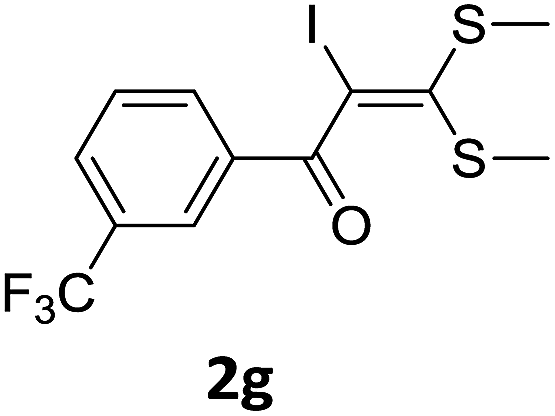
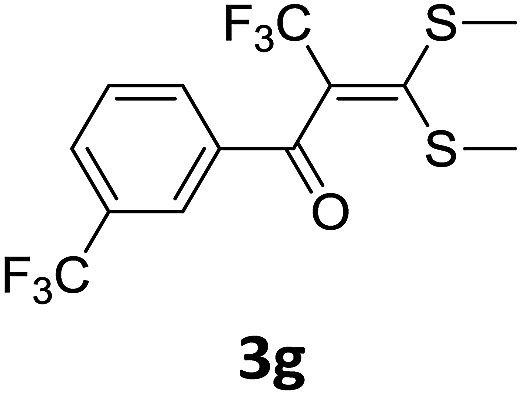
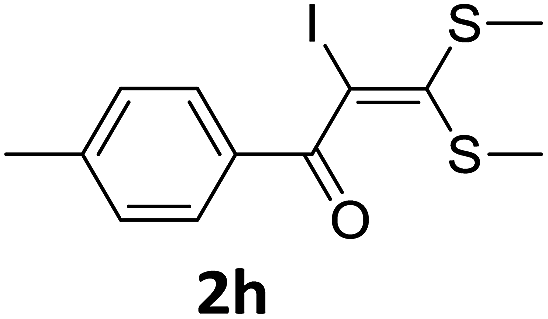
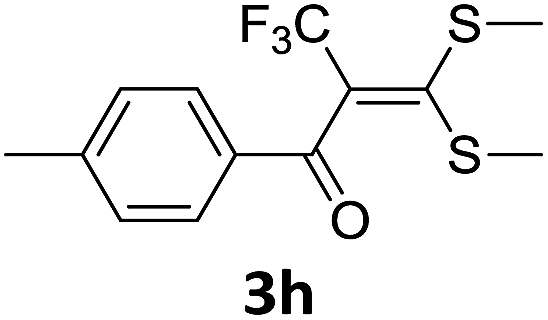
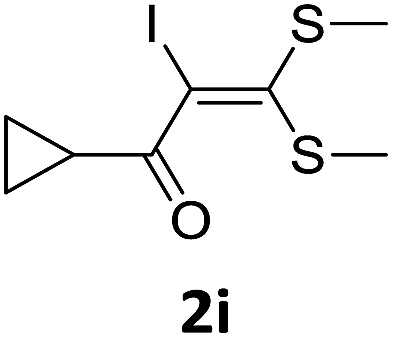
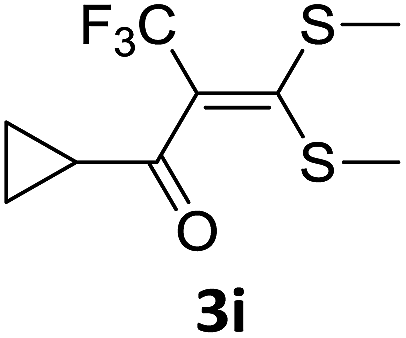
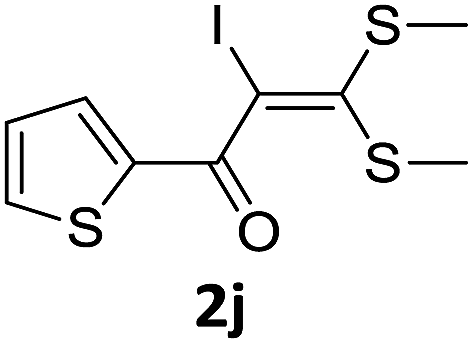
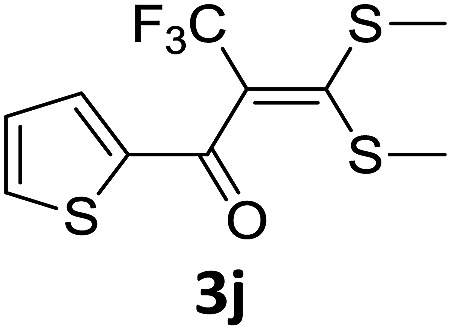
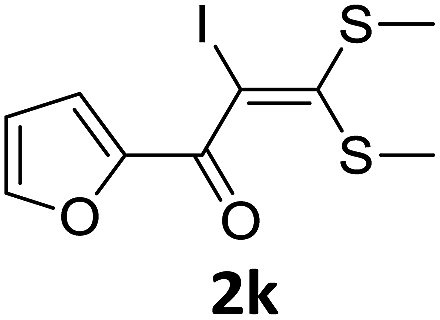
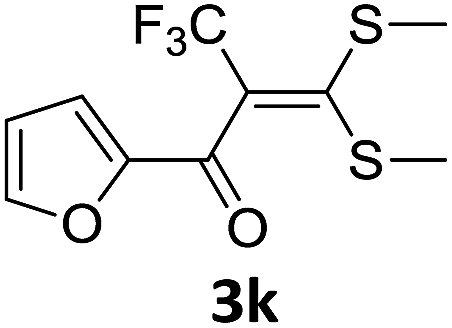
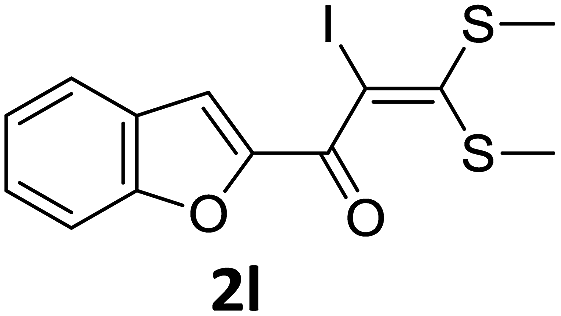
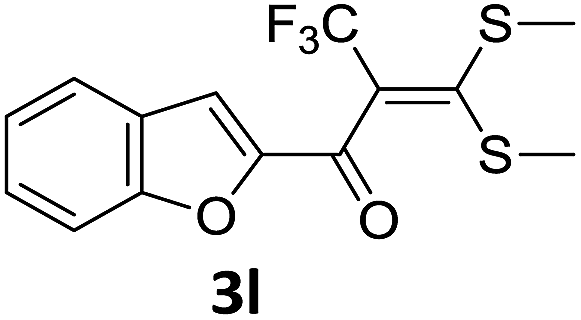
For substrates with electron releasing group on aromatic ring, effective conversion was observed only with –pCH3 as substituent (entry 8).
However, α-iodo oxoketene dithioacetals substituted with –OCH3 at -o or -p positions were proven ineffective. This may be anticipated due to inability of nucleophilic substitution reaction to take place due to increased electron density at vinylic position through conjugation. Cyclopropyl substituted α-trifluoromethylated oxoketene dithioacetals were obtained in 86% yield (entry 9). Furthermore, when a heteroaromatic nucleus was introduced in substrates 2j–l, the reaction provided the desired products 3j–l in good to excellent yields (entries 10–12). Structural assignments have been made on the basis of 1H NMR, 13C NMR and HRMS data. In 13C NMR data of compounds 3a–l, disappearance of peak at δ 94–100 with simultaneous appearance of peaks at δ 122 and δ 119 which corresponds to vinylic carbon as well as carbon atom of –CF3 group was observed. HRMS data of each compound showed M+1 peak. The structure of the product 3l was determined by single crystal X-ray diffraction (Fig. 1). The structures of the other products were concluded by analogy.
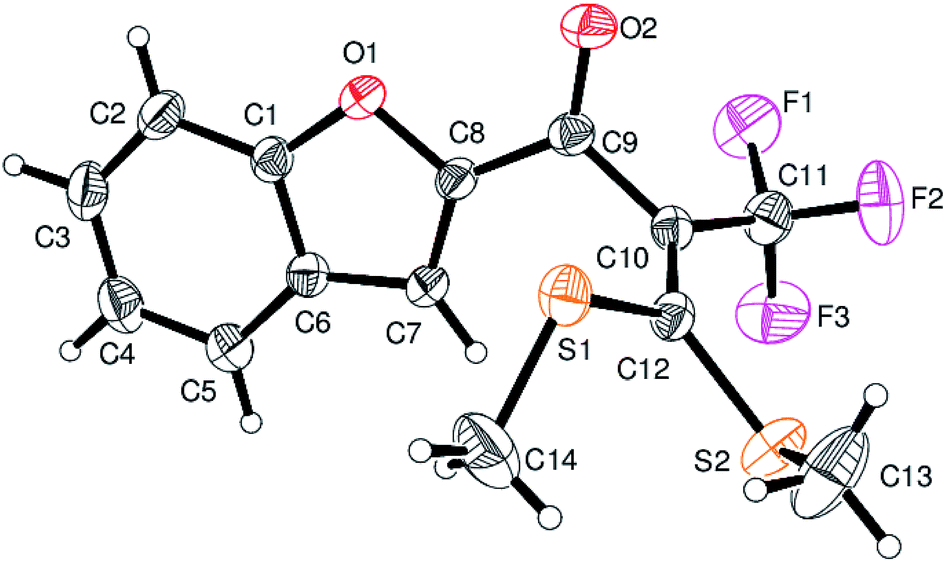 | ||
| Fig. 1 ORTEP drawings of compound 3l. | ||
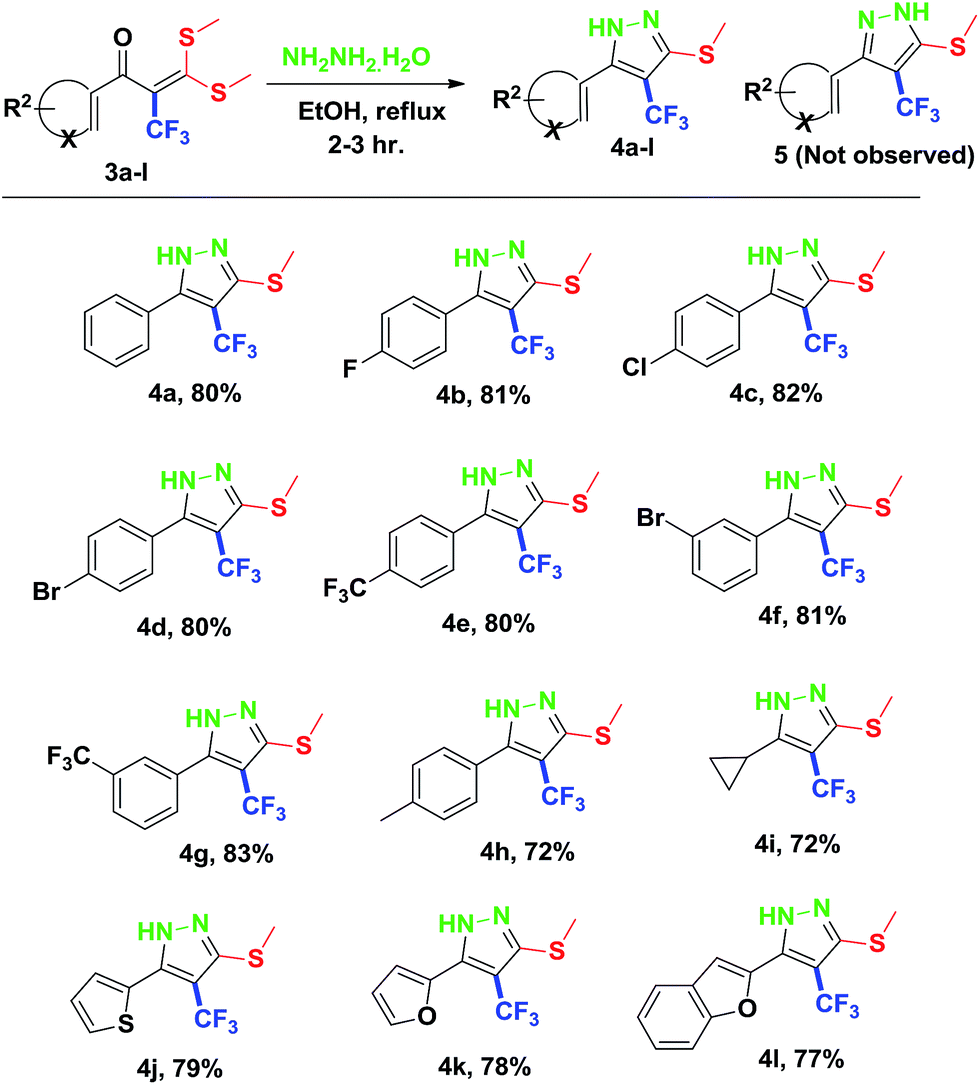
Finally, we demonstrated the further applicability of α-trifluoromethylated oxoketene dithioacetals 3a–l in cyclization reactions, regioselective construction of aromatic/heteroaromatic multisubstituted 1H-pyrazoles 4a–l was achieved in 72–83% yield by cyclization with hydrazine hydrate (Table 3). Formation of the regioselective cyclized product was confirmed on the basis of 1H NMR and X-ray crystallographic studies of 4i (Fig. 2). Structural assignments of 4a–l have been made on the basis of IR, 1H NMR, 13C NMR, 19F NMR and HRMS data. IR spectra of the products revealed absorption peaks for secondary amino groups (in the region 3170–3460 cm1) while no peak for ketocarbonyl group around 1670–1690 was observed which confirmed that –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O was utilized in cyclization reaction. In the 1H NMR spectra, disappearance of one of the characteristic peak of –SMe group with appearance of upfield shift of –NH protons as broad singlet at δ 7.8–9.3 ppm as compared to –NH protons of literature reported compound 5 (ref. 14a) which appeared at δ 11.9–12.2 ppm supported the formation of cyclized pyrazole ring of 4(a–l) regioselectively instead of compound 5. Disappearance of peaks in 13C NMR for carbonyl carbon at δ 175.5–195.5 ppm and –SCH3 carbon at 14.2–14.9 ppm confirms that the cyclized product was formed. Appearance of a singlet at δ −53.0–55.0 in 19F NMR data confirmed the presence of –CF3 group in the desired cyclized compound.
O was utilized in cyclization reaction. In the 1H NMR spectra, disappearance of one of the characteristic peak of –SMe group with appearance of upfield shift of –NH protons as broad singlet at δ 7.8–9.3 ppm as compared to –NH protons of literature reported compound 5 (ref. 14a) which appeared at δ 11.9–12.2 ppm supported the formation of cyclized pyrazole ring of 4(a–l) regioselectively instead of compound 5. Disappearance of peaks in 13C NMR for carbonyl carbon at δ 175.5–195.5 ppm and –SCH3 carbon at 14.2–14.9 ppm confirms that the cyclized product was formed. Appearance of a singlet at δ −53.0–55.0 in 19F NMR data confirmed the presence of –CF3 group in the desired cyclized compound.
| a Unless otherwise specified, reactions were performed using 1 equiv. of 3, 1.2 equiv. of NH2NH2·H2O in EtOH. |
|---|
 |
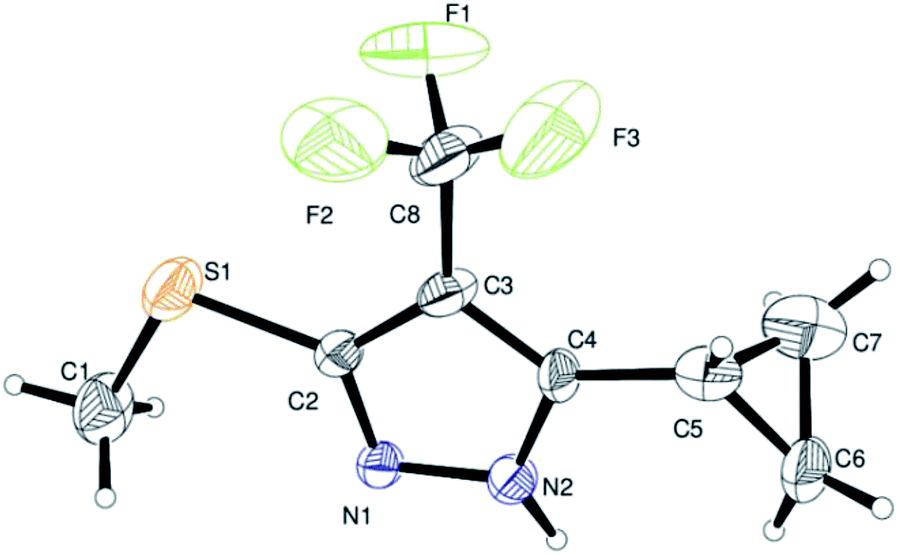 | ||
| Fig. 2 ORTEP drawings of compound 4i. | ||
In conclusion, we have developed a mild, ligand free approach for the trifluoromethylation of readily synthesized α-iodo oxoketene dithioacetals in excellent yields. The reaction was well tolerant towards a wide variety of substituents. The simplicity and generality of this method makes it attractive for the introduction of CF3 group into functionally diverse α-iodo substituted oxoketene dithioacetals. The resultant trifluoromethylated synthons were further utilized as bifunctional building blocks in cyclocondensation reaction for the exclusive regioselective synthesis of valuable trifluoromethylated pyrazoles. These five membered heterocycles also has the flexibility to offer further diversification of the thiomethyl moiety by employing arylboronic acids in the substitution reactions.17
Acknowledgements
The authors are thankful to SERB, Department of Science & Technology, India for providing financial support and USIC, University of Delhi for providing instrumentation facilities. NS and NK are thankful to DST for INSPIRE Fellowship and UGC for SRF respectively.Notes and references
- (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- S. K. Ritter, Chem. Eng. News, 2012, 90, 10 Search PubMed; M. Shimizu and T. Hiyama, Angew. Chem. Int. Ed., 2005, 44, 214 (Angew. Chem., 2005, 117, 218) CrossRef CAS PubMed.
- (a) T. Billard and B. R. Langlois, Eur. J. Org. Chem., 2007, 6, 891 CrossRef; (b) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135 RSC.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617 CrossRef CAS.
- P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef CAS PubMed.
- (a) Q. Yang, P. Wu, J. Chen and Z. Yu, Chem. Commun., 2014, 50, 6337 RSC; (b) Y. Li, X. Xu, J. Tan, C. Xia, D. Zhang and Q. Liu, J. Am. Chem. Soc., 2011, 133, 1775 CrossRef CAS PubMed; (c) R. K. Dieter, Tetrahedron, 1986, 42, 3029 CrossRef CAS; (d) H. Junjappa, H. Ila and C. V. Asokan, Tetrahedron, 1990, 46, 5423 CrossRef CAS; (e) l. Pan, X. Bi and Q. liu, Chem. Soc. Rev., 2013, 42, 1251 RSC.
- A. De Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Weinheim, Wiley-VCH Verlag GmbH & Co. KGaA, 2004 Search PubMed.
- M. Wang, X. Xiu Xu, Q. Liu, L. Xiong, B. Yang and L. -Xun Gao, Synth. Commun., 2002, 32, 3437 CrossRef CAS.
- (a) N. Sharma, T. S. Chundawat, S. C. Mohapatra and S. Bhagat, Synthesis, 2016, 48, 4495–4508 CrossRef CAS; (b) N. Sharma, T. S. Chundawat, S. C. Mohapatra and S. Bhagat, RSC Adv., 2013, 3, 16336 RSC.
- (a) T. N. Glasnov, K. Groschner and C. O. Kappe, ChemMedChem, 2009, 43, 645 Search PubMed; (b) C. Lamberth, Heterocycles, 2007, 71, 1467 CrossRef CAS; (c) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Doctor, M. J. Greveto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, S. A. Gregory, C. M. Icoboldt, W. E. Perkus, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS PubMed.
- (a) B. D. Maxwell, J. Labelled Compd. Radiopharm., 2000, 43, 645 CrossRef CAS; (b) F.-G. Zhang, Y. Wei, Y.-P. Yi, J. Nie and J.-A. Ma, Org. Lett., 2014, 16, 3122 CrossRef CAS PubMed; (c) E. Y. Slobodyanyuk, O. S. Artamonov, O. V. Shishkin and P. V. Mykhailiuk, Eur. J. Org. Chem., 2014, 2487 CrossRef CAS; (d) T.-R. Li, S.-W. Duan, W. Ding, Y.-Y. Liu, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2014, 79, 2296 CrossRef CAS PubMed.
- (a) Z. Mao, F. Huang, H. Yu, J. Chen, Z. Yu and Z. Xu, Chem.–Eur. J., 2014, 20, 3439 CrossRef CAS PubMed; (b) C. Xu, J. Liu, W. Ming, Y. Liu, J. Liu, M. Wang and Q. liu, Chem.–Eur. J., 2013, 19, 9104 CrossRef CAS PubMed.
- X. Yang, F. Hu, H. Di, D. li, X. Cheng, X. Kan, X. Zou and Q. Zhang, Org. Biomol. Chem., 2014, 12, 8947 CAS.
- (a) Q.-Y. Chen and S.-W. Wu, J. Chem. Soc., Chem. Commun., 1989, 705 RSC; (b) W. Yu, X.-H. Xu and F.-L. Qing, Org. Lett., 2016, 18, 5130 CrossRef CAS PubMed; (c) S. L. Clarke and G. P. Mcglacken, Chem.–Eur. J., 2016, 22, 1 CrossRef.
- W. Jin, H. Yu and Z. Yu, Tetrahedron Lett., 2011, 52, 5884 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C NMR & HRMS data for target compounds. X-ray crystallographic data of compounds 3l with CCDC no. 1051488 and 4i with CCDC no. 1051460. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01130j |
| This journal is © The Royal Society of Chemistry 2017 |