
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a hierarchical nanoporous carbon material with controllable pore size and effective surface area for high-performance electrochemical capacitors†
Bing Hua,
Ling-Bin Kong *ab,
Long Kangb,
Kun Yana,
Tong Zhanga,
Kai Lia and
Yong-Chun Luob
*ab,
Long Kangb,
Kun Yana,
Tong Zhanga,
Kai Lia and
Yong-Chun Luob
aState Key Laboratory of Advanced Processing and Recycling of Non-ferrous Metals, Lanzhou University of Technology, Lanzhou 730050, P. R. China. E-mail: konglb@lut.cn; Fax: +86-931-2976578; Tel: +86-931-2976579
bSchool of Materials Science and Engineering, Lanzhou University of Technology, Lanzhou 730050, P. R. China
First published on 6th March 2017
Abstract
A simple carbonization procedure is proposed for the synthesis of hierarchical nanoporous carbons with controllable pore size and effective surface area as electrode materials for high-performance electrochemical double-layer capacitors. The procedure is based on the carbonization of interpenetrating polymer networks (IPNs) composed of cross-linked polystyrene (PS) and poly(methyl methacrylate) (PMMA). The as-obtained hierarchical nanoporous carbons (HNC-IPNs) have controllable pore size, interconnected pore structure, high specific surface area, excellent electrical conductivity and electrochemical stability with the different mass ratio of PS/PMMA. In addition, there is authentically an excellent linear relationship between effective specific surface area (E-SSA) and specific capacitance. Especially, the HNC-IPN-4 exhibits the highest specific surface area (SSA) of 1346 m2 g−1, relative high E-SSA of 603 m2 g−1, and excellent specific capacitance of 260 F g−1 under the current density of 0.5 A g−1 in 6 M KOH. Meanwhile, the HNC-IPN-4 exhibits a superior cycling performance without any degradation after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with the current density of 2 A g−1 as well as exhibits high capacitance retention, i.e., 96.0% of the initial specific capacitance after 20000 cycles.
000 cycles with the current density of 2 A g−1 as well as exhibits high capacitance retention, i.e., 96.0% of the initial specific capacitance after 20000 cycles.
1. Introduction
Porous carbon materials are attracting increasing interest in many energy storage systems such as electrochemical double-layer capacitors (EDLCs, known as supercapacitors).1–4 For advanced application, it is essential that the carbon materials possess a high specific surface area, large porosity, good electronic conductivity and controllable nanostructure.5–9 Nowadays, various approaches have been reported to synthesize porous carbon materials with large specific surface area, containing activation,10–13 direct carbonization of cross-linked/conjugated polymers,14,15 polymer blend and metal–organic frameworks,16–18 high-temperature chlorination of carbide materials,19–21 self-assembly of supramolecular complexes22,23 and nanocasting strategy with hard templates.24–26 Among these approach, activation and templates methods were resultful for programming and fabrication of multifarious microporous carbon with large specific surface area. However, it exhibits a severe drawback due to it seriously limits the transport of the electrolyte ions, resulting in a sharp reduction in EDLCs properties at large current densities.27 Meanwhile, numerous activators and template removal agents will cause contamination and corrode the fabrication facilities. Thus, the development of lager specific surface area while developing convenient and environment-friendly approach to design nanoporous carbon materials remains a formidable challenge.In addition, the template preparation approach (e.g. hard and soft templates) also supply an occasion for preparing mesoporous carbons. The hard templates include mesoporous silica materials,28–30 zeolites,31–33 metal–organic frameworks34,35 and so on. For example, Kong et al. synthesized a carbon/silica matrix which apply resorcinol–formaldehyde to serve as the precursor templated with 3-aminopropyl-triethoxysilane,36 then the HF or NaOH was used to remove the silica as hard template, which obtained the mesoporous carbon material. Dai and coworkers reported the ordered mesoporous carbon was synthesized by using soft template, which employs resorcinol–formaldehyde as carbon source and diblock copolymer polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) as a soft template.37Although these strategies supply several unexceptionable sequential mesoporous carbon with large specific surface area and superior mesoporous structure, but these strategies possess a serious drawback that the as-obtained mesoporous carbon with the unitary pore diameter. And above strategies are not universal to fabricate a battery of pore diameter controlled porous carbons by regulating the diameter of the unitary template in single synthetic pathway. Furthermore, the exact synthesis of templates with adjustable pore diameter was generally time-consuming, luxurious operating cost,38 comparative intricate,39 and highly hardness.40
Recently, polymer blend carbonization includes carbonizing composite polymers consisting of a carbon source and a resoluble polymer which is pyrolyzed to produce pores.41,42 Porous carbons with different pore sizes were obtained by using the preparation approach. For example, the phase-separate of polymer blend for thermosetting polymer (carbon source) and thermoplastics polymer (pore-former) such as poly(vinyl butyral),41 polyethylene oxide,42 poly(methyl methacrylate),43 polystyrene,44 and polyethylene,45 which were used to synthesize porous carbons by carbonizing the thermosetting polymers as well as pyrolyzing thermoplastic polymers. Nevertheless, these polymer blends frequently happen to macro-phase separation in the carbonization process of thermosetting polymers, which is closely related to the infirm intermolecular interaction between thermosetting polymers (carbon source) and thermoplastic polymers (pore-former), resulting in as-obtained carbon materials with separate pore structure, diminutive pore volume, and small specific surface area.
In consequence, comparing with traditional porous carbon material, the hierarchical nanoporous carbon (HNC) composed of united pore diameter which could be controlled upon a broad range of large scale, hierarchical porous carbons with explicit pore size can acquire minimized diffusive resistance to mass transport from macropores and large specific surface area for active site dispersion from micro/mesopores.46 For example, Wang et al. represent preparation of carbon stive with the hierarchical porous structure which integrates micropores, mesopores, and macropores.47 Fang et al. also research the diverse hierarchical nanostructured carbon materials for EDLC applications.48–51 The micropores were site for the charge accommodation in double electrical layer, the mesopores undertake enter closes for electrolyte ions transport as well as the macropores take on ion-buffering reservoirs. In previous studies, several accomplishments have been made in synthesis of HNC by template methods and post-activation combination approaches.52 Nevertheless, a majority of templates were costly and post-synthetic, removing of templates needs excess procedures, which are generally time-consuming and pernicious for the environment.
In this paper, we present a novel approach to develop a class of well-defined hierarchical nanoporous carbon from the PS/PMMA interpenetrating polymer networks (HNC-IPNs), which exhibit controllable pore size, advisable pore size distribution (PSD), interconnected pore structure, large specific surface area (SSA) relative high E-SSA and excellent electrochemical performance. As shown in Scheme 1, HNC-IPNs with controllable pore size and effective surface area were synthesized by a simple carbonization of PS/PMMA interpenetrating polymer networks (PS/PMMA-IPNs) without conventional templating and activation procedures, which were consisted of two polymer networks that were at lowest partly alters on a polymer scale without covalent bond each other. Following the process of carbonization, the high cross-linking density polymer network tended to shape carbon source as well as the relative low cross-linking density polymeric network broke up into gaseous produces, which remains pores on carbon matrix. In interpenetrating polymer networks, chain–chain reciprocity, cross-linking and inter-network involvement which impede chain segment campaign, leading to the nanophase separation with double phase continuity.53 The as-obtained HNC-IPNs possess the large SSA, relative high E-SSA, advisable pore size distribution, connected pore structure and controllable pore size ranging from micropores, mesopores to macropores. Compared to conventional synthesis approach of porous materials, the method can effectually settle these subsistent puzzles: (a) time-consuming and pernicious for the environment for synthesis and removing of hard template for templating approach and (b) uncontrollable pores diameter and separate pores structures for polymer blend method. Herein, IPN of cross-linked PS and PMMA was successfully synthesized by sequential polymerization. Unsurprisingly, the as-obtained HNC-IPNs possess controllable pore size, relative high E-SSA and excellent electrochemical properties for EDLCs. The straightforward and explicit approach is inexpensive, time-saving and environment-friendly (e.g. used one-off PS foam tableware likewise cloud be the appropriate raw materials for preparing cross-linked polystyrene). In conclusion, the robust method affords the new way for preparing HNC as well as opens the novel door for sustainable advanced material (e.g. the reuse of waste PS products).
 | ||
| Scheme 1 Schematic illustration of fabrication procedure for hierarchical nanoporous carbon. | ||
2. Experimental
2.1 Materials
Styrene (St), carbon tetrachloride (CCl4), anhydrous aluminium chloride (AlCl3), hydrochloric acid (HCl), methyl methacrylate (MMA), ethylene glycol dimethacrylate (EGDMA), 2,2′-azobisisobutyronitrile (AIBN), N,N-dimethylformamide (DMF), ethanol were all supplied by Sigma-Aldrich. The whole chemical reagents used were chemically pure. 2,2′-Azobisisobutyronitrile (AIBN) was purified by recrystallization in ethanol. Methyl-methacrylate (MMA) was redistilled before use. The whole glass apparatus utilized were placed in the oven at 60 °C for a few hours.2.2 Synthesis of polystyrene (PS)
In a typical process, 0.5 g of polyvinyl alcohol (PVA) was added into 120 mL of deionized water in a three-neck flask. The mixture was refluxed with mechanical stir for 60 min at 75 °C. Then the initiator–monomer mixture of 0.25 g benzoyl peroxide (BPO) dissolved in 20.0 g of styrene (St) was added to the three-neck flask undergo suspension polymerization reaction. The mixture was refluxed with mechanical stir for 180 min at 85 °C. The as-obtained sample was filtered off, washed by 80–85 °C hot water, followed by drying at 60 °C.2.3 Synthesis of cross-linked polystyrene
In a typical process (Scheme S1†), 12.0 g of anhydrous aluminium chloride (AlCl3) was added into 100 mL of carbon tetrachloride (CCl4) in a three-neck flask. Then the mixture was refluxed with magnetic stir for 45 min at 75 °C. Afterwards, a solution of 5.0 g of polystyrene (PS) dissolved in 100 mL of CCl4 was added to undergo a Friedel–Crafts crosslinking reaction. After 60 min, 100 mL of ethanol–water solution (95 wt% ethanol/water = 80 mL/20 mL) was added tardily to stop reaction. The as-obtained sample was filtered off, washed by the 95 wt% ethanol/5 wt% dilute hydrochloric acid mixture (150 mL/50 mL) and deionized water, followed by drying at 110 °C for 12 h. The product was used the (I) network for subsequent preparation of IPN.2.4 Preparation of PS/PMMA-IPNs
For the PS/PMMA-IPNs preparation (Scheme S2†), the PS polymer (I) is swollen with the monomer methyl methacrylate (MMA) (II) containing the crosslinking agent of ethylene glycol dimethacrylate (EGDMA) (3% by weight with respect to MMA) and the initiator of 2,2-azobisisobutyronitrile (AIBN) (0.5% by weight with respect to MMA), and the second monomer (MMA) mixture is allowed to polymerize and crosslink in situ in 30 g of N,N-dimethylformamide (DMF) in the 100 mL three-necked flask. Then, this mixture was stirred and purged with nitrogen for 1 h and polymerized at 80 °C for 6 h. The as-obtained sample was transferred to a ceramic plate followed by evaporating solvent at 120 °C for 6 h in drying oven. The ultimate produce was the interpenetrating polymer networks of PS/PMMA (PS/PMMA-IPNs).2.5 Synthesis of HNC-IPNs
The resultant PS/PMMA-IPNs were grinded into fine powders. HNC-IPNs were prepared by carbonizing the PS/PMMA-IPNs powders at 900 °C under N2 atmosphere for 3 h with the heating rate of 2 °C min−1. The samples of HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 were obtained by pyrolyzing PS/PMMA-IPN with the mass ratio of 50/50, 40/60, 30/70, 20/80, and 10/90, respectively. For comparison, hierarchical nanoporous carbons were also pyrolysed from cross-linked PS and PS/PMMA polymer blends, and as-obtained carbons were named as HNC and HNC-B, respectively.2.6 Structural characterization
Fourier transform infrared (FTIR) spectra were measured using a Bruker Equinox 55 FTIR spectrophotometer. Samples were synthesized by casting pellets from sample/KBr mixtures. The thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were measured in the N2 flow with the heating rate of 10 °C min−1. The morphologies and structures of the samples were obtained using scanning electron microscopy (SEM) and transmission electron microscopy (TEM, 200 kV). Raman spectras were obtained by a Renishaw invia spectrometer with a 520 nm Ar-ion laser. Power X-ray diffraction (XRD) patterns were measured on the Bruker Focus D8 diffractometer with Cu Kα radiation (40 kV, λ = 0.15418 nm) between 10° and 90°. Pore structure was researched using the Micromeritics ASAP 2020 surface area and porosity analyzer. Specific surface areas were analyzed by Brunauer–Emmett–Teller (BET) theory. The pore size distribution (PSD) was calculated according to the nonlocal density function theory (NLDFT). The total pore volumes were calculated from the amount adsorbed at a relative pressure P/P0 of 0.99.2.7 Electrochemical characterization
HNC-IPN electrodes were obtained by mixing electroactive material powder (80 wt%), conducting graphite (7.5 wt%), acetylene black (7.5 wt%) and polytetrafluorethylene (PTFE, 5 wt% dispersion in water) in ethanol and then the mixture was press into a nickel foam current collector (surface area: 1 cm × 1 cm). The mass of electroactive material powder in each electrode is about 4 mg. The consequent nickel foam was pressed under 10 MPa, which were dried at 110 °C for 12 h in a drying oven.The EDLC properties were measured in 6 M KOH electrolyte on an electrochemical working station (CHI660E, Shanghai, China) which uses a normative three-electrode system with a saturated calomel reference electrode (SCE), a platinum foil counter electrode, and a working electrode. Cyclic voltammetry (CV) was obtained under different scan rate and galvanostatic charge–discharge (GCD) was measured at various current densities. Specific capacitances (Cm) were calculated according to the equation Cm = C/m = I × Δt/(m × ΔV), where Cm (F g−1), m (g), I (A), Δt (s), and ΔV (V) signify the specific capacitance, the mass of the working electrodes, the constant current density, discharging time and discharge potential, respectively. Electrochemical impedance spectroscopy (EIS) measurement was performed under frequency ranges from 105 to 10−2 Hz under an open circuit potential with an AC perturbation of 5 mV.
3. Results and discussion
3.1 Fabrication of PS/PMMA interpenetrating polymer networks
Herein, the PS/PMMA interpenetrating polymer networks were synthesized successfully by sequential polymerization. Firstly, the preparation of cross-linked polystyrene (PS) by constructing inter-sphere –CO– cross-linking bridges. The product was used the (I) polymer network for subsequent preparation of IPNs (Scheme S1†). Subsequently, the cross-linked PS polymer (I) is swollen with the monomer methyl methacrylate (MMA) (II) containing the crosslinking agent of ethylene glycol dimethacrylate (EGDMA) and the initiator of 2,2-azobisisobutyronitrile (AIBN), and the second monomer (MMA) mixture is allowed to polymerize and crosslink in situ (Scheme S2†).As shown in the FTIR spectra (Fig. 1), carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching band at 1729 cm−1 for acrylate group of PMMA and conjugated carbonyl (–CO–) band at 1662 cm−1 for carbonyl group of cross-linked PS, while the presence of ester group of PMMA is confirmed by characteristic band at 1151 cm−1 and 1191 cm−1 and carbon–carbon band of benzene ring is observed at 1660 cm−1. As can be seen from the black line of FTIR spectra, the characteristic bands at 1729, 1151, and 1191 cm−1 can be detected, which illustrate a situation that PMMA is prepared by the monomer MMA polymerization in this polymer blend system. Hence, the above results determine that PS/PMMA interpenetrating polymer networks were successfully prepared through sequential interpenetrating blend.
O) stretching band at 1729 cm−1 for acrylate group of PMMA and conjugated carbonyl (–CO–) band at 1662 cm−1 for carbonyl group of cross-linked PS, while the presence of ester group of PMMA is confirmed by characteristic band at 1151 cm−1 and 1191 cm−1 and carbon–carbon band of benzene ring is observed at 1660 cm−1. As can be seen from the black line of FTIR spectra, the characteristic bands at 1729, 1151, and 1191 cm−1 can be detected, which illustrate a situation that PMMA is prepared by the monomer MMA polymerization in this polymer blend system. Hence, the above results determine that PS/PMMA interpenetrating polymer networks were successfully prepared through sequential interpenetrating blend.
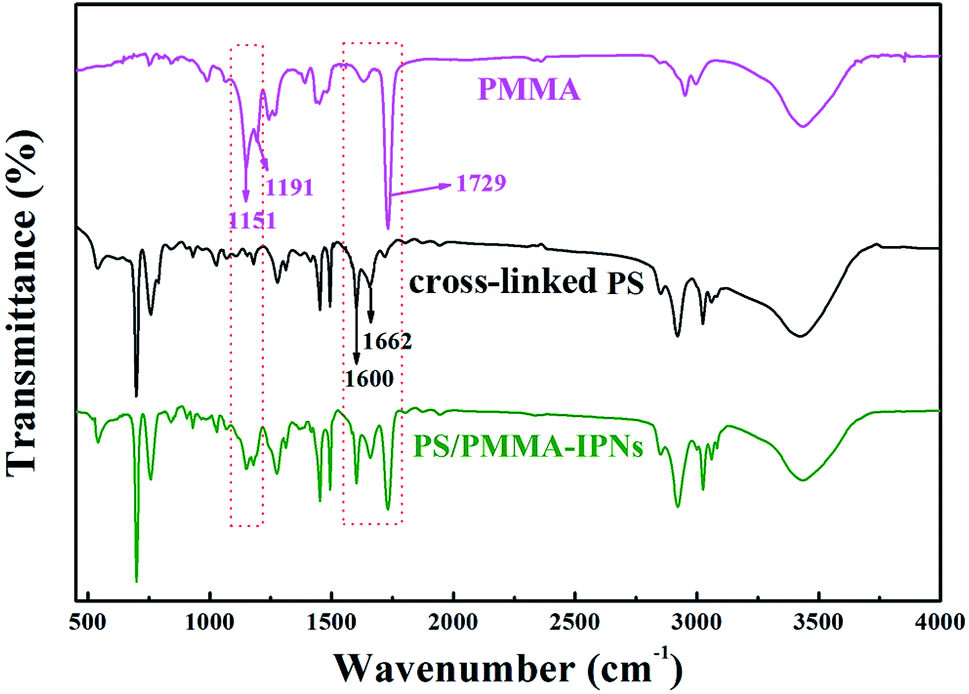 | ||
| Fig. 1 FTIR spectra of PMMA, cross-linked PS and PS/PMMA-IPNs. | ||
The decomposition of PS/PMMA-IPNs with the mass ratio of 20/80 is monitored by means of thermogravimetric analysis (TGA). The TGA curve in Fig. 2a clearly reveals four weight loss stages during the heating process. The first weight loss stage (∼8.48 wt%) is associated with the nano-network structure of cross-linked PS undergoes a sinter process, which is consistent with exothermic peak at 290–380 °C. Actually, PS is not an appropriate carbon source which is completely decomposed before 400 °C due to its linear structure. To solve the problem, the Friedel–Crafts reaction54 is used to construct –CO– crosslinking bridges between linear PS chains (Scheme S1†). The second mass loss stage (∼32.46 wt%) in temperature ranges from 400–450 °C because of decomposition of PMMA with low cross-linking density. Actually, the PMMA is completely decomposed at 410 °C because of low cross-linking density (see Fig. S1†). The third weight loss stage (∼14.06 wt%) is observed from the temperature range of 460–600 °C due to collapse of nano-network structure leading to form nonporous semi-carbonized PS. In the final stage, the weight loss is observed from 620–1000 °C, due to full carbonization of cross-linked PS for transformation into the partial graphitic structure. Hence, the cross-linked PS has a yield of 30.76 wt% at 900 °C, indicating that it has the excellent thermal stability and carbonizability characteristic. The microphase-separation behavior of PS/PMMA interpenetrating polymer networks is researched with DSC (Fig. 2b). Two glass transition temperatures (Tg) are obtained according to DSC curve: a wide endothermic peak at 127.52 °C and a sharp endothermic peak at 434.66 °C.
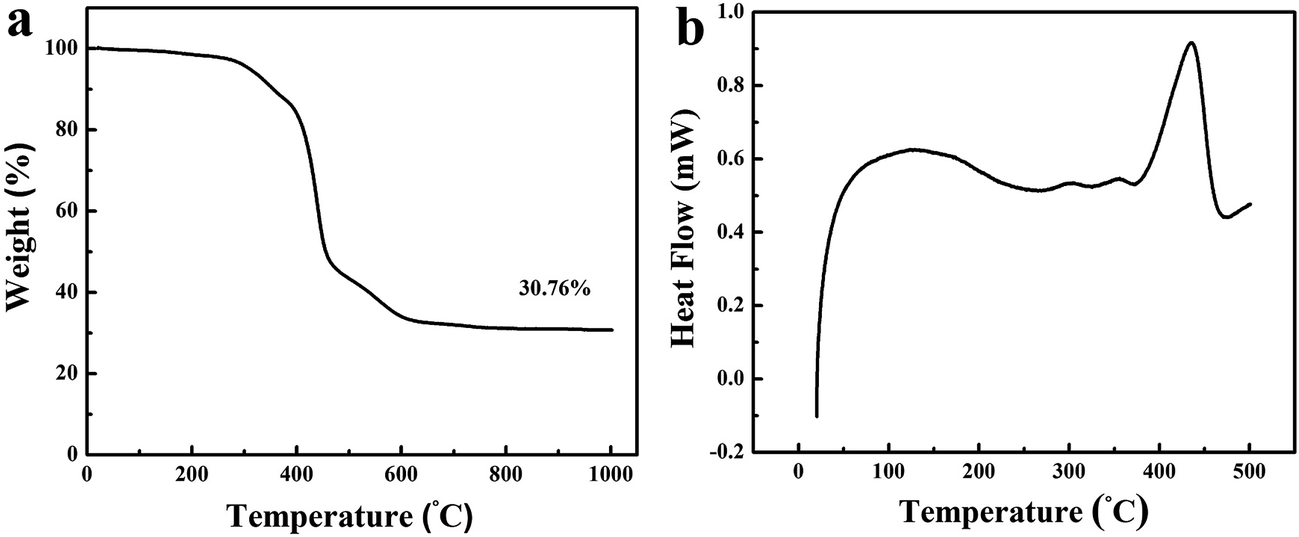 | ||
| Fig. 2 (a) TGA and (b) DSC curves of the PS/PMMA-IPNs with the mass ratio of 20/80. | ||
3.2 Synthesis and nanostructure control of HNC-IPNs
The nanoporous structure of HNC and HNC-IPN-4 are revealed by SEM and TEM (Fig. 3 and S2†), which are excellent tools for the examination of nanostructure morphology. Fig. 3b shows the HNC-IPN-4 possesses a continuous sponge-like branched framework, which is consisted of connected nanoparticle entity unit. The nanoparticle entity unit themselves involve micropores as well as the decomposition of PMMA leave pores in carbon matrix to form mesopores and macropores ranging from several to hundreds of nanometers. For comparison, HNC only possess carbon blocks and few macropores in the carbon matrix (Fig. 3a). The TEM image of HNC-IPN-4 displays the mesopores structure, which is attribute to the leaving of sacrificial network, i.e., PMMA pyrolyzed at 900 °C (Fig. 3c). The HRTEM image of HNC-IPN-4 reveals abundant and uniform micropores, which is attribute to nanoparticle units themselves contain micropores, i.e., cross-linked PS pyrolyzed at 900 °C (Fig. 3d). In addition, Fig. 3d also shows partial graphitic structure, which will result in an enhanced electrical conductivity. The electron diffraction diagram of HNC-IPN-4, shown as an inset, achieved carbon sample is an approximate crystalline material. Nevertheless, as a comparison, tiny pores can be observed in the TEM images of HNC (Fig. S2†). | ||
| Fig. 3 SEM images of (a) HNC and (b) HNC-IPN-4; TEM image of (c) HNC-IPN-4 synthesized PS/PMMA interpenetrating polymer networks pyrolysis; HRTEM image of (d) HNC-IPN-4, and the electron diffraction pattern (shown as an inset). Scale bars, 100 nm (a and b), 50 nm (c), 5 nm (d). | ||
Nitrogen adsorption–desorption measurement was implemented to investigate the pore features of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 (Fig. 4). From Fig. 4a, it can be seen that the nitrogen adsorption–desorption isotherm of all samples exhibit features of type I/IV based on the category of IUPAC. At low relative pressure, a lofty nitrogen uptake attests the presence of enormous micropores within nano-particles. At high relative pressure, the hysteresis loops indicate the existence of mesopores. Herein, the inter-network entanglement, chain–chain interaction and cross-link hinder chain segments activity of IPNs, leading to nanophase separation with double phases seriality,53 whose domain size is approximately many hundreds of nanometer. Accomplish the following carbonization process, the as-obtained carbon possess connected pore structure ranging from micropore to mesopore. It was demonstrated by PSD of HNC-IPNs, shown in Fig. 4b, which was estimated by the DFT model from isotherms absorption branches. These PSD curves of HNC-IPNs indicate the presence of micro/mesopore, nevertheless, the PSD of mesopore is much broader. Compared with HNC-IPNs, the HNC without IPN shows a pore size of 1.48, 2.04, and 39.8 nm, respectively, which indicates the nanoparticle entity unit themselves possess micropore, and meso/macropores were formed due to their compact and loose aggregation. The HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 display the micropore sizes of 0.39, 0.5, 0.84, and 1.17 nm, respectively, due to nanophase separation, and the mesopore sizes of 2.02, 37.17, and 50 nm (Fig. 4c), which are derived from the removal of sacrificial network (PMMA) during the carbonization process. In addition, HNC-IPN-3 and HNC-IPN-4 display the mesopore sizes of 9.3 and 17.19 nm (Fig. 4c), respectively. The outcome reveals that the mesopore sizes of HNC-IPNs were controlled by regulating the relative mass ratio of PS/PMMA. The synthesis conditions and pore structure parameters of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 are presented in Table 1. As can be seen, the SSAs of products from HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, to HNC-IPN-4 increase from 209, 476, 573, 1021, to 1346 m2 g−1 as well as the pore volumes increase from 0.10, 0.27, 0.30, 0.65, to 0.88 cm3 g−1. And the specific surface area and the pore volumes of HNC-IPN-5 were 578 and 0.30 cm3 g−1. The above results indicate that the specific surface areas and the pore volumes of HNC-IPNs were primarily augmented and then decreased with the enhancement of the mass of PMMA. Hence, HNC-IPNs possess the adjustable pore size, the specific surface area, and pore volumes. Therein HNC-IPN-4 exhibits the highest specific surface area and pore volume, which indicates that the method of IPNs carbonization is feasible for preparing HNC. For comparison, the HNC-B was obtained by carbonizing the PS/PMMA polymer blends with the mass ratio of 20/80, and the nitrogen adsorption–desorption isotherm and pore size distribution were shown as Fig. S4a and b,† respectively. Compared with HNC-IPN-4, the HNC-B with specific surface areas and pore volumes of 608 m2 g−1 and 0.35 cm3 g−1 were lower than it (see Table S1†). As shown in Fig. 4d, the SSA and PSD measurements which are based on the nitrogen adsorption experiment datum of slit/cylindrical NL-DFT model.55,56 Insert vertical dashed line reveals positive ions size for KOH electrolyte. Note the positive ion size is 0.662 nm for K+ (hydrated radii for K+ is about 0.331 nm).57 Hence, the positive ion size is applied to estimation and model following. Effective specific surface areas (E-SSA) of HNC-IPNs were defined as cumulative SSA of pore diameter beyond positive ion size for KOH electrolyte. Table 2 demonstrate and compare the particular cumulative SSA, E-SSA and specific capacitance. From Fig. 4e, the relationship between cumulative SSA and E-SSA with specific capacitance of HNC-IPNs which reveals the specific capacitance is concerned in E-SSA (not BET SSA) of HNC-IPNs for KOH electrolyte. Certain conclusions can be obtained from these data. It is momentous to notice that PSD is similarly momentous if not more momentous to confirm E-SSA and both large total SSA and the suitable PSD require to attain large E-SSA. Such as HNC-IPN-3 and HNC-IPN-4, which possess close total cumulative SSA (1172 m2 g−1 and 1300 m2 g−1, respectively), nonetheless the various PSD lead to disparate E-SSA (408 m2 g−1 and 603 m2 g−1, respectively). A distinct linear relationship is between specific capacitance and E-SSA (Fig. 4f). The coefficient of determination is 0.998 and the slope of fitting line is 0.249, which reveals the excellent linear relationship between E-SSA and specific capacitance, indicating that specific capacitances were able to be calculated immediately using E-SSA without manufacture of equipment, quickening up the materials programming and screening.
 | ||
| Fig. 4 (a) The nitrogen adsorption–desorption isotherms, (b) pore size distributions corresponding to the HNC and HNC-IPNs prepared at different synthesis condition, (c) mesopore and macropore size distributions of six samples, (d) the distribution of cumulative surface area with pore width for HNC-IPNs and the insert vertical dashed line represent hydrated diameter of K+, (e) relationship between the specific capacitance and effective specific surface area, (f) linear relationship between specific capacitance and effective specific surface area. The violet points demonstrated the effective specific surface area and specific capacitance data of HNC-IPNs in KOH electrolytes. | ||
| Sample | PS/PMMA (mg mg−1) | SBETa (m2 g−1) | Smicb (m2 g−1) | Smesoc (m2 g−1) | Vtotald (cm3 g−1) | Vmice (cm3 g−1) | Vmic/Vtotal (%) | Daf (nm) | Cgh (F g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Specific surface areas were estimated by BET approach.b Micropores surface area.c Mesopores surface area.d Total pore volume measured at P/P0 = 0.99.e Micropore volume.f The average pore size was estimated from adsorption isotherms according to DFT model.h The galvanostatic capacitance estimated from discharge curve at under the current density of 0.5 A g−1 in 6 M KOH solution. | |||||||||
| HNC | 100/0 | 209 | 166 | 43 | 0.10 | 0.07 | 70.0 | 1.9 | 85 |
| HNC-IPN-1 | 50/50 | 476 | 433 | 43 | 0.27 | 0.19 | 70.0 | 2.3 | 162 |
| HNC-IPN-2 | 40/60 | 573 | 445 | 128 | 0.34 | 0.20 | 58.8 | 2.4 | 175 |
| HNC-IPN-3 | 30/70 | 1021 | 901 | 119 | 0.65 | 0.41 | 63.1 | 2.5 | 210 |
| HNC-IPN-4 | 20/80 | 1346 | 1123 | 222 | 0.88 | 0.51 | 57.9 | 2.6 | 260 |
| HNC-IPN-5 | 10/90 | 578 | 522 | 55 | 0.30 | 0.23 | 76.7 | 2.1 | 190 |
| Product | Cumulative SSA (m2 g−1) | E-SSA for KOHb (m2 g−1) | Capacitance in KOHc (F g−1) |
|---|---|---|---|
| a E-SSA of HNC-IPNs in KOH electrolyte was defined as the cumulative SSA of pore with size beyond positive ion size in KOH electrolyte.b The hydrated diameter of K+ is 0.662 nm.c The specific capacitance was estimated from galvanostatic discharge curve under a current density of 0.5 A g−1. | |||
| HNC-IPN-1 | 646 | 207 | 162 |
| HNC-IPN-2 | 717 | 268 | 175 |
| HNC-IPN-3 | 1172 | 408 | 210 |
| HNC-IPN-4 | 1300 | 603 | 260 |
| HNC-IPN-5 | 771 | 334 | 190 |
The obtained carbon sample used the XRD patterns to characterize the structural aspects (Fig. 5a). Two broad peaks located at around 2θ = 23° and 44°, which are indexed to carbon (002) and (100) diffraction planes, respectively. The (100) peaks represent planes of amorphous carbon, which signify the becoming of interlayer condensation of carbon materials, accounting for graphitic structures were progress to mild degree. The broad and weak (002) peaks indicate a graphite-like microcrystalline structure. Furthermore, the values of interlayer distance (0.38 nm) were estimated by Bragg equation, larger than which of representative graphite (0.334 nm),58 standing for the random combination of graphitic and turbostratic stacking.59
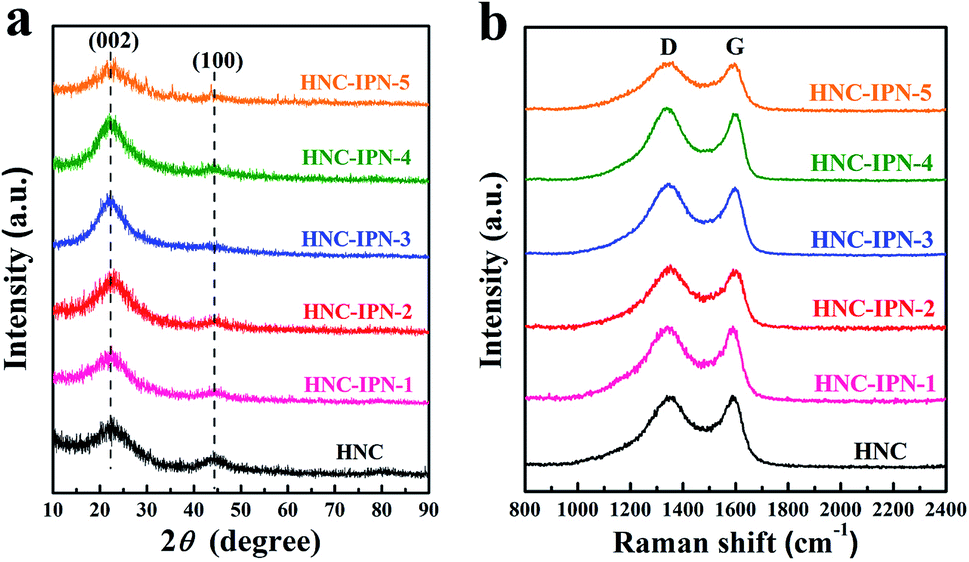 | ||
| Fig. 5 XRD profiles (a) and Raman spectrums (b) of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5. | ||
In order to further research structural characteristic for the obtained carbon sample, which has been characterized by employing the Raman spectroscopy. As shown in Fig. 5b, two peaks around 1342 cm−1 and 1593 cm−1 can be denoted as D (disordered) peak and G (graphitic) peak, respectively.60 The D band is related to the presence of disordered carbon structure and crystal defect, owing to the phonons near the Brillouin zone boundary active in small crystallites or on the boundaries of larger crystallites, while the G band is attributed to “in-plane” zone-centre atomic vibrations of large graphite crystallites.61 The microcrystalline planar crystal size La can be calculated using the empirical formula found by Tuinstra and Koenig (La = 4.35IG/ID (nm), where IG and ID are integrated intensity of G peak and D peak, respectively). For these carbon samples, La of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 are 2.06, 2.25, 2.27, 2.29, 2.31, and 2.28 nm (Table S2†), respectively, indicating that carbon samples display a graphite-like microcrystalline structure, in accord with the results of XRD. The degree of graphitization is associated with the relative intensity ratio of D and G band (ID/IG), and the higher the ID/IG ratio, the smaller the degree of graphitization. Herein, the estimated ID/IG value of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 is 1.05, 1.03, 1.03, 1.02, 0.98, and 1.01 respectively, which implies carbon materials are primarily partial graphite layers combined with disordered carbon. A high ID/IG intensity ratio of these carbon samples manifests the generation of plenty of defect.
3.3 Electrochemical performance
Herein, so as to investigate the influence of HNC-IPNs on electrochemical capacitance performances, electrochemical impedance spectroscopy (EIS) survey was executed to illuminate kinetic on electrode/electrolyte interface, which manifest the contribution of nanoporous structure for EDLCs.62,63 The Nyquist plots of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 were obtained in 6 M KOH solution in ranges from 105 to 10−2 Hz, shown in Fig. 6. The Nyquist plots contain the vertical lines in the low-frequency region, almost 45° line in the intermediate frequency region, and the quasi-semicircle at high frequency region. At the low-frequency, the Nyquist plots of HNC electrode materials stray from vertical lines, which indicates the electrolyte ions cloud can not enter into pores, due to abundant micropores limit ions entering into porous carbons (Fig. 4b). Nevertheless, a series of HNC-IPNs show vertical lines, which manifests that the samples of HNC-IPNs possess excellent capacitance behavior due to their porous property. The intermediate frequency displays the 45° line, indicating the feature of electrolyte ions into the pores of carbon electrode material. At high frequency, the quasi-semicircle indicates the feature of materials porosity and thickness on ions migration rate from electrolyte inside to carbon electrode and the intercept in the real axis of the Nyquist plots shows the internal resistance (Rb) of electroactive material.64 Normally, the smaller value of Rb signifies good conductivity of HNC-IPNs, suggesting the rapid ion transfer in interface of electrode/electrolyte. The Rb of HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 are 0.557, 0.606, 0.551, 0.541, and 0.671 Ω, respectively. It is easily found that the obtained materials possess smaller values, indicating that the partial graphitic-structure carbon materials have the excellent electrolyte transfer to the interface between electrolyte and electrode of HNC-IPNs, which is in accord with XRD patterns and Raman spectrums (Fig. 5). In addition, the Rb of HNC-IPN-4 possesses minimum value, which indicates the sample of HNC-IPN-4 has better conductivity than others. In order to further investigate carbonization of interpenetrating polymer networks which is different from the polymer blends, the sample of HNC-B was prepared by pyrolyzing PS/PMMA polymer blend with the mass ratio of 20/80. Compared with HNC-IPN-4, the EIS curves of HNC-B reveal the worse diffusive resistance (Fig. S5c†). Meanwhile, the spectra of HNC-IPNs display higher angles than 45°, which reveals the compatibility of the HNC-IPNs as electrode materials for EDLCs on account of their effective pores which can be convenient for the transport of the electrolyte ions and excellence of HNC-IPNs as carbon electrode material for EDLCs. | ||
| Fig. 6 Nyquist plot of HNC and HNC-IPNs in 6 M KOH solution measured in the frequency range of 105 to 10−2 Hz at open circuit potential with an alternate current amplitude of 5 mV. | ||
Fig. 7a displays CV cures of HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 at the scan rate of 5 mV s−1 in 6 M KOH electrolyte. The HNC pyrolyzed from cross-linked PS was measured as contrast, however, the CV curve of HNC is deviated from rectangular shape (see Fig. S3a†), which is ascribed to the existence of plentiful micropores which a strict ions entering into carbon electrode materials and lower E-SSA (Table 2). Nevertheless, the CV cures of HNC-IPNs possess the quasi-rectangular shapes with the potential window from −1.0 to 0 V, indicating an optimal double-layer capacitance behavior with excellent charge transport as well as easiest ions propagation in carbon electrodes.65 Favorable rectangular shapes of HNC-IPNs also mean that these carbon electrode materials possess the rapid charging/discharging characteristic.66 Generally, the specific capacitance are varies as CV profile covered areas at the uniform scan rates and potentials.67 Distinctly, the HNC-IPN-4 possesses the largest rectangular area with good shapes, which reveals excellent electrochemical properties as the carbon electrode materials in 6 M KOH solution, it should be attributed to ideal PSD and relative high E-SSA (see Table 2 and Fig. 4b). The galvanostatic charge–discharge (GCD) experiment further evaluates the capacitive performance at a current density of 0.5 A g−1 with potential window from −1 to 0 V. As shown in Fig. 7b, the GCD curve of these carbon electrodes are entire perfectly linear as well as nearly symmetric with the piecemeal slop transformation, which exhibits the excellent coulombic efficiency as well as an optimal double-layer capacitance behavior. The specific capacitances of HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, HNC-IPN-4, and HNC-IPN-5 electrode materials are 162, 175, 210, 260, and 190 F g−1 at 0.5 A g−1 from discharge curves, respectively. They are much higher than 85 F g−1 of HNC (Fig. 7b and S3b†) due to the formation of massive mesopores, reasonable PSD, and relative large E-SSA. Especially, the as-obtained HNC-IPN-4 exhibits the highest specific capacitance of 260 F g−1 because of more abundant effective pores than others and higher E-SSA of 603 m2 g−1.
 | ||
| Fig. 7 Electrochemical performance of HNC and HNC-IPNs prepared at different synthesis condition in 6 M KOH aqueous solution: (a) CV curves measured under the scan rate of 5 mV s−1 with potential window from −1 to 0 V; (b) GCD curves measured under the current density of 0.5 A g−1. | ||
The CV cures of the HNC-IPN-4 as carbon electrode materials at various scan rates in 6 M KOH electrolyte. At low scan rates from 5 to 100 mV s−1 (Fig. 8a), HNC-IPN-4 exhibits good rectangular shapes without obvious redox peaks, which indicates HNC-IPN-4 possesses optimal double-layer capacitance behavior, due to electrolyte ions easy to enter into carbon nanoporous structures for the becoming of electric double-layer. Even under high scan rates (Fig. 8b), HNC-IPN-4 possesses a correspondingly excellent quasi-rectangular shape when scan rates were increased to 200–500 mV s−1, due to its effective pore, reasonable broad PSD and relative high E-SSA for diffusion of the electrolyte ions, which implies that HNC-IPN-4 can satisfy rapid charge–discharge power. In order to further investigate carbonization of interpenetrating polymer networks which is different from the polymer blends, the sample of HNC-B was prepared by pyrolyzing PS/PMMA polymer blend with the mass ratio of 20/80. Compared with HNC-IPN-4, the as-synthesized HNC-B material possesses a smaller rectangular curve than HNC-IPNs samples (see Fig. S5a†), which is ascribed to the existence of abundant micropores (the microporosity of 80%), limiting ion transportation into the electrode material. What is more, in the high scan rates, HNC-B displays a distorting rectangular CV shape due to the movement of ions resistance in little pores, which influences the becoming of electric double-layer. The GCD curves of HNC-IPN-4 under different current densities (Fig. 8c and d). It is noteworthy that HNC-IPN-4 still maintain typical triangle-shaped curves, indicating that HNC-IPN-4 as electrode material possesses excellent coulombic efficiency and optimal electric double-layer capacitive behavior. Furthermore, extreme slight ohmic drops are observed under the high current densities of 10 to 50 A g−1, which suggests that HNC-IPN-4 has the supernal small equivalent series resistance and efficiency for charge–discharge.68 The as-obtained HNC-IPN-4 possess nicer capacitive behavior under various current densities, the values of specific capacitance which estimated according to discharge curve are 260, 232, 220, 185, 182, 168, 153, and 135 F g−1 under current densities of 0.5, 1, 2, 5, 10, 20, 30, and 50 A g−1. By contrast, the specific capacitance values of HNC-B which calculated according to discharge curves are 189, 172, 162, 159, 156, 150, 143, and 128 F g−1 under current densities of 0.5, 1, 2, 3, 4, 5, 10, and 20 A g−1 (Fig. S5b†), which are much lower than HNC-IPN-4, due to the effective pore quantity of NHC-B are less than HNC-IPN-4. The change of specific capacitance under different current densities of 0.5 to 50 A g −1 was estimated as shown in Fig. 8e. HNC-IPN-4 exhibits 70%, 64.4%, 58.8%, and 51.9% capacitance retention under 10, 20, 30, and 50 A g−1, respectively, which reveals excellent retention rate capabilities, indicating that consecutive nanoporous structures supplied an effective approach which electrolyte ion easily enter into the carbon matrix (Scheme 1). Meanwhile, the excellent rate capabilities of HNC-IPN-4 also can be explained by low resistance values (Fig. 6). The HNC, HNC-IPN-1, HNC-IPN-2, HNC-IPN-3, and HNC-IPN-5 possess 55.3%, 61.7%, 62.9%, 72.2%, and 67.8% capacitance retention at 10 A g−1 (Fig. S6†), respectively, which are lower than HNC-IPN-4. In the meantime, the carbon material of HNC-B shows the worse capacitance retention capability of 67.7% than HNC-IPN-4 (shown in Fig. S5d†), due to the existence of abundant micropores (the microporosity of 80%), which a strict ion transportation into the electrode material. In order to ulteriorly comprehend electrochemical properties of HNC-IPN electrode materials, the cycling stability of HNC-IPN-4 was researched at a current density of 2 A g−1 in 6 M KOH electrolyte for investigating specific capacitances retention. As shown in Fig. 8f, HNC-IPN-4 possesses a superior cycling stability without degradation after 10000 cycles as well as exhibits high capacitance retention, i.e., 96.0% of the initial specific capacitance after 20000 cycles (Fig. S7†). Moreover, the almost triangular charge–discharge curves (Fig. S7,† inset) which reveals HNC-IPN-4 carbon material providing superior charge propagation and stable properties. In conclusion, the excellent cycling stability can be mainly ascribed to the appropriate reasonable micro/mesopore size distribution with high SSA and E-SSA, the continuous porous carbon framework and partial graphitization structures for EDLCs in the cycling course. As comparison with other carbon materials, HNC-IPN-4 possesses more superior electrochemical performance, such as specific capacitance, Rb, and cycling stability (Table 3). All the above electrochemical measures demonstrate that the HNC-IPN-4 material possesses the excellent capacitive properties and immense underlying application for EDLCs.
 | ||
| Fig. 8 Electrochemical performance of HNC-IPN-4 in 6 M KOH aqueous solution: (a) CV curves measured under low scan rates from 5 to 100 mV s−1; (b) CV curves measured under high scan rates from 200 to 500 mV s−1; (c) GCD curves measured under low current densities from 0.5 to 5 A g−1; (d) GCD curves measured under high current densities from 10 to 50 A g−1; (e) specific capacitances of the HNC-IPN-4 under different current densities ranging from 0.5 to 50 A g−1; (f) cycling performance of the HNC-IPN-4 electrode. | ||
| Sample | Specific capacitance (F g−1) | Rb (Ω) | Cycling stability (%) | Ref. |
|---|---|---|---|---|
| HNC-IPN-4 | 260 (0.5 A g−1) | 0.541 | 100 (10000 cycles) |
This work |
| HPC | 250 (0.05 A g−1) | — | 97.3 (5000 cycles) | 69 |
| HPC4.2-16.8-6C | 194 (0.1 A g−1) | 1.150 | 93.2 (1000 cycles) | 70 |
| C-IPN | 252 (0.5 A g−1) | 0.84 | 97.9 (5000 cycles) | 71 |
| aGA-0.5 | 204 (0.2 A g−1) | 0.42 | 92 (10000 cycles) |
72 |
| PCF | 246.5 (0.5 A g−1) | 0.179 | 97 (10000 cycles) |
73 |
| G-aC | 280 (1 A g−1) | — | 97.9 (5000 cycles) | 74 |
4. Conclusions
In summary, a novel and cost-effective procedure for preparation of hierarchical nanoporous carbon with controllable pore size and effective surface area was successfully developed by directly carbonizing PS/PMMA interpenetrating polymer networks. This preparation approach is attractive due to successful employment of PS-derived white pollution (e.g., disposable foam tableware) with –CO– cross-linking bridges as the carbon source. The mesopore size ratio of HNC-IPNs are increased following the augment of the mass of the (II) polymer networks (PMMA) as pore-former, which easily forms ideal electric double-layer. The as-obtained porous carbon materials with the local graphitization structure exhibit high SSA of 476–1346 m2 g−1, relative high E-SSA of 207–603 m2 g−1 and high pore volumes of 0.27–0.88 cm3 g−1. The above conclusion implies that the control of pore size can be at large scale. The well-controlled pore structure of the HNC-IPNs supplies robust underlying application as electrode material for EDLCs. Especially, the as-synthesized HNC-IPN-4 possesses the highest specific capacitance of 260 F g−1 at 0.5 A g−1, which is ascribed to advisable PSD and relative high E-SSA of 603 m2 g−1. What is more, there is the excellent linear relationship between E-SSA and specific capacitance, which indicates that the specific capacitance calculated immediately according to the E-SSA without manufacture of equipment, quickening up the materials programming and screening. Furthermore, HNC-IPN-4 also exhibits a good rate 51.9% capacitance retention under various current densities increased from 0.5 to 50 A g−1 as well as the excellent cycling stability of 100% capacitance retention after 10000 cycles under the current density of 2 A g−1, meanwhile, 96.0% of the initial specific capacitance after 20000 cycles. The synthesis of hierarchical nanoporous carbon from PS/PMMA-IPNs (HNC-IPNs) is facile and time-saving, which can unfold new occasion to develop a class of novel remarkable properties hierarchical nanoporous carbons suited to multifarious positive adhibition oriented to adsorption, energy, environment and so on.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 51362018) and the Foundation for Innovation Groups of Basic Research in Gansu Province (No. 1606RJIA322).References
- D. S. Su and R. Schlög, ChemSusChem, 2010, 3, 136 CrossRef CAS PubMed.
- A. Ghosh and Y. H. Lee, ChemSusChem, 2012, 5, 480 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797 RSC.
- L. Hao, X. Li and L. Zhi, Adv. Mater., 2013, 25, 3899 CrossRef CAS PubMed.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677 CrossRef CAS.
- A. Stein, Z. Y. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265 CrossRef CAS.
- M. X. Liu, J. S. Qian, Y. H. Zhao, D. Z. Zhu, L. H. Gan and L. W. Chen, J. Mater. Chem. A, 2015, 3, 11517 CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, 28 CrossRef PubMed.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. L. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace and M. Thommes, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Funct. Mater., 2012, 22, 827 CrossRef CAS.
- Y. R. Liang, B. M. Wu, D. C. Wu, F. Xu, Z. H. Li, J. W. Luo, H. Zhong, R. W. Fu and K. Matyjaszewski, J. Mater. Chem., 2011, 21, 14424 RSC.
- H. Zhong, F. Xu, Z. H. Li, R. W. Fu and D. C. Wu, Nanoscale, 2013, 5, 4678 RSC.
- Z. H. Xiang, Y. H. Xue, D. P. Cao, L. Huang, J. F. Chen and L. M. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433 CrossRef CAS PubMed.
- Z. H. Li, D. C. Wu, Y. R. Liang, R. W. Fu and R. W. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 4805 CrossRef CAS PubMed.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 2864 CrossRef CAS PubMed.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita and H. Shioyama, J. Am. Chem. Soc., 2011, 133, 11854 CrossRef CAS PubMed.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Porter, P. Simon and P. L. Taberna, Science, 2006, 313, 1760 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P. L. Tabema, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730 CrossRef CAS PubMed.
- J. T. Lee, Y. Y. Zhao, S. Thieme and G. Yushin, Adv. Mater., 2013, 25, 4573 CrossRef CAS PubMed.
- Y. Wan, Y. F. Shi and D. Y. Zhao, Chem. Mater., 2008, 20, 932 CrossRef CAS.
- J. Liu, T. Y. Yang, D. W. Wang, M. Liu, D. Y. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 Search PubMed.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677 CrossRef CAS.
- A. H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, J. Electrochem. Soc., 2008, 155, 531 CrossRef.
- L. C. Sang, A. Vinu and M. O. Coppens, J. Mater. Chem., 2011, 21, 7410 RSC.
- H. Lu, W. Dai, M. Zheng, N. Li, G. Ji and J. Cao, J. Power Sources, 2012, 209, 243 CrossRef CAS.
- Z. Chen, J. Wen, C. Yan, L. Rice, H. Sohn and M. Shen, Adv. Energy Mater., 2011, 1, 551 CrossRef CAS.
- A. Garsuch and O. Klepel, Carbon, 2005, 43, 2330 CrossRef CAS.
- H. Itoi, H. Nishihara, T. Kogure and T. Kyotani, J. Am. Chem. Soc., 2011, 133, 1165 CrossRef CAS PubMed.
- A. Kajdos, A. Kvit, F. Jones, J. Jagiello and G. Yushin, J. Am. Chem. Soc., 2010, 132, 3252 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456 CrossRef CAS.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita and H. Shioyama, J. Am. Chem. Soc., 2011, 133, 11854 CrossRef CAS PubMed.
- Y. Kong, Y. Zhong, X. Shen, S. Cui, M. Yang, K. Teng and J. Zhang, J. Non-Cryst. Solids, 2012, 358, 3150 CrossRef CAS.
- C. D. Liang, K. L. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785 CrossRef CAS PubMed.
- H. Y. Liu, K. P. Wang and H. S. Teng, Carbon, 2005, 43, 559 CrossRef CAS.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520 RSC.
- C. M. Yang, B. Zibrowius, W. Schmidt and F. Schüth, Chem. Mater., 2003, 15, 3739 CrossRef CAS.
- J. Ozaki, N. Endo, W. Ohizumi, K. Igarashi, M. Nakahara and A. Oya, Carbon, 1997, 35, 1031 CrossRef CAS.
- T. Horikawa, K. Ogawa, K. Mizuno, J. Hayashi and K. Muroyama, Carbon, 2003, 41, 465 CrossRef CAS.
- M. Yamazaki, M. Teduka, K. Ikeda and S. Ichihara, J. Mater. Chem., 2003, 13, 975 RSC.
- N. Patel, K. Okabe and A. Oya, Carbon, 2002, 40, 315 CrossRef CAS.
- D. Hulicova and A. Oya, Carbon, 2003, 41, 1443 CrossRef CAS.
- C. Zou, D. C. Wu, M. Z. Li, Q. C. Zeng, F. Xu, Z. Y. Huang and R. W. Fu, J. Mater. Chem., 2010, 20, 731 RSC.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS PubMed.
- B. Z. Fang, A. Bonakdarpour, M. S. Kim, J. H. Kim, D. P. Wilkinson and J. S. Yu, Microporous Mesoporous Mater., 2013, 182, 1 CrossRef CAS.
- B. Z. Fang, J. H. Kim, M. S. Kim, A. Bonakdarpour, A. Lam, D. P. Wilkinson and J. S. Yu, J. Mater. Chem., 2012, 22, 19031 RSC.
- B. Z. Fang, J. H. Kim, M. S. Kim and J. S. Yu, Acc. Chem. Res., 2013, 46, 1397 CrossRef CAS PubMed.
- B. Z. Fang and L. Binder, J. Power Sources, 2006, 163, 616 CrossRef CAS.
- W. Xing, C. C. Huang, S. P. Zhuo, X. Yuan, G. Q. Wang, D. HulicovaJurcakova, Z. F. Yan and G. Q. Lu, Carbon, 2009, 47, 1715 CrossRef CAS.
- L. H. Sperling, Adv. Chem., 1991, 1, 3 Search PubMed.
- Z. H. Li, D. C. Wu, Y. Liang, F. Xu and R. Fu, Nanoscale, 2013, 5, 10824 RSC.
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- O. Barbieri, M. Hahn, A. Herzog and R. Kotz, Carbon, 2005, 43, 1303 CrossRef CAS.
- E. R. Nightingale Jr, J. Phys. Chem., 1959, 63, 1381 CrossRef.
- K. T. Cho, S. B. Lee and J. W. Lee, J. Phys. Chem. C, 2014, 118, 9357 CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2013, 56, 155 CrossRef CAS.
- D. C. Wu, H. C. Dong, J. Pietrasik, E. K. Kim, C. M. Hui, M. J. Zhong, M. Jaroniec, T. Kowalewski and K. Matyjaszewski, Chem. Mater., 2011, 23, 2024 CrossRef CAS.
- F. Xu, Z. W. Tang, S. Q. Huang, L. Y. Chen, Y. R. Liang, W. C. Mai, H. Zhong, R. W. Fu and D. C. Wu, Nat. Commun., 2015, 6, 7221 CrossRef PubMed.
- S. H. Aboutalebi, A. T. Chidembo, M. Salari, K. Konstantinov, D. Wexler, H. K. Liu and S. X. Dou, Energy Environ. Sci., 2011, 4, 1855 CAS.
- S. Chen, J. J. Duan, M. Jaroniec and S. Z. Qiao, J. Mater. Chem. A, 2013, 1, 9409 CAS.
- M. Itagaki, Y. Hatada, I. Shitanda and K. Watanabe, Electrochim. Acta, 2010, 55, 6255 CrossRef CAS.
- D. D. Zhou, Y. J. Du, Y. F. Song, Y. G. Wang, C. X. Wang and Y. Y. Xia, J. Mater. Chem. A, 2013, 1, 1192 CAS.
- M. X. Liu, L. H. Gan, W. Xiong, F. Zhao, X. Fan, D. Zhu and L. W. Chen, Energy Fuels, 2013, 27, 1168 CrossRef CAS.
- B. Xu, F. Wu, R. Chen, G. Cao, S. Chen and Y. Yang, J. Power Sources, 2010, 195, 2118 CrossRef CAS.
- D. C. Guo, J. Mi, G. P. Hao, W. Dong, G. Xiong, W. C. Li and A. H. Lu, Energy Environ. Sci., 2013, 6, 652 CAS.
- R. Ruiz-Rosas, M. J. Valero-Romero, D. Salinas-Torres, J. Rodríguez-Mirasol, T. Cordero, E. Morallón and D. Cazorla-Amorós, ChemSusChem, 2014, 7, 1458 CrossRef CAS PubMed.
- X. J. He, N. Zhao, J. S. Qiu, N. Xiao, M. X. Yu, C. Yu, X. Y. Zhang and M. D. Zheng, J. Mater. Chem. A, 2013, 1, 9440 CAS.
- J. Zhang, X. F. Zhong, H. B. Chen, Y. Gao and H. M. Li, Electrochim. Acta, 2014, 148, 203 CrossRef CAS.
- X. X. Sun, P. Cheng, H. J. Wang, H. Xu, L. Q. Dang, Z. H. Liu and Z. B. Lei, Carbon, 2015, 92, 1 CrossRef CAS.
- F. Zhang, T. Y. Liu, G. H. Hou, L. Yue, R. F. Guan and Y. Li, Nano Res., 2016, 9, 2875 CrossRef CAS.
- J. L. Huang, J. Y. Wang, C. W. Wang, H. N. Zhang, C. X. Lu and J. Z. Wang, Chem. Mater., 2015, 27, 2107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01151b |
| This journal is © The Royal Society of Chemistry 2017 |