
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene sheets manipulated the thermal-stability of ultrasmall Pt nanoparticles supported on porous Fe2O3 nanocrystals against sintering†
Yunqian Dai *ab,
Xiaomian Qia,
Wanlin Fua,
Chengqian Huanga,
Shimei Wanga,
Jie Zhoua,
Tingying Helen Zengc and
Yueming Sun*a
*ab,
Xiaomian Qia,
Wanlin Fua,
Chengqian Huanga,
Shimei Wanga,
Jie Zhoua,
Tingying Helen Zengc and
Yueming Sun*a
aSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing, Jiangsu 211189, P. R. China. E-mail: daiy@seu.edu.cn; sun@seu.edu.cn
bState Key Laboratory of Silicon Materials, Zhejiang University, Hangzhou, Zhejiang, P. R. China
cAcademy for Advanced Research and Development, One Kendall Square, Cambridge, MA 02135, USA
First published on 14th March 2017
Abstract
Sintering of noble-metal nanoparticles (NPs) presents a major cause for catalyst deactivation as temperature rises. To date, finding simple strategies to develop sinter-resistant catalysts is still a daunting challenge. Herein, we report stable Pt nanoparticles (<3 nm) on porous Fe2O3 nanocrystals using wrinkled graphene sheets as a new stabilizing layer to manipulate their thermal-stability against sintering. Such a catalyst system allows the Pt NPs to achieve significant thermal stability against extremely severe thermal treatment up to 750 °C in both inert and oxidative atmosphere and retain strikingly remarkable activity; this is due to rhombohedral Fe2O3 nanocrystals ensuring the good dispersibility of Pt NPs across the entire surface, and the distinctive wrinkles on graphene sheets acting as new physical barriers. This study inspires a general approach of developing sinter-resistant catalysts with tunable compositions, to maximize the thermal stability and catalytic activity under harsh conditions.
1. Introduction
Noble-metal nanoparticles (NPs) are typically unstable and can easily experience a large thermal-dynamic driven force to sinter severely into a large species in a short period of time, at high temperatures, typically above 300 °C.1–3 The undesirable sintering of active noble-metal NPs has become a major cause for catalyst deactivation.4–6 To avoid aggregation, stabilizing mobile noble-metal NPs on a specific support, such as metal oxide (e.g., Fe2O3, CeO2 or TiO2) or carbon (e.g., graphene or carbon nanotubes), with extraordinarily high surface area is a popular approach to develop thermally stable noble-metal (e.g., Pt, Pd, or Au) NPs, by exploiting the so-called metal/support interaction.7–11 Moreover, the supports are also susceptible to a loss of surface area, as caused by irreversible aggregating during high-temperature operation, leading to a damaged or densified product. In addition, the inevitable coalescence of supports can induce and accelerate the coarsening of supported noble-metals, which are aggregated at their thermodynamically-preferred location, such as the interparticle pockets and grain boundaries in polycrystalline metal oxides.12–14New attempts to stabilize supported noble-metal NPs involve the formation of a physical barrier that inhibits the sintering of NPs by constructing a porous stabilizing layer of another metal oxide (e.g., SiO2, TiO2, Al2O3) on the top of the surface. Several successes illustrate the elegance of this concept, such as Pt/TiO2@SiO2, Pt/CeO2@SiO2, Pt/graphene@SiO2, Pt/Fe2O3@SiO2 and Pt/SiO2@TiO2 in triphasic configurations.15–18 The observed thermal-stability of metal NPs in fact comes at the expense of weaker catalytic activity compared to that of uncoated ones, as the stabilizing layer unavoidably occupies the same reactive sites on the NPs surface.15 In contrast with earlier reports, a recent breakthrough was achieved by selectively depositing an SiO2 layer onto the surface of substrate versus the surface of Pt NPs, demonstrating excellent thermal-stability of 3 nm-Pt NPs up to 700 °C in air on TiO2 nanofibers.19 Moreover, the thickness of the stabilizing layer should be carefully controlled to below 3 nm with molecular precision. In spite of these recent promising developments, the severe sintering issue has not been well-resolved. First, the stabilizing layer usually causes an unexpected induction time before starting catalytic reaction, and an additional pre-heating procedure at 500 °C is required before use.20 Second, the porous layer, like SiO2, is in fact unstable and densified during high-temperature operation, which reduces accessibility of the NPs surface and deactivates the coated Pt NPs in spite of unchanged small sizes.17 Third, the ultrasmall NPs (<3 nm) have ultrahigh chemical potential to aggregate easily even at room temperature, and their stabilization is rarely reported by coating a protective layer due to their lack of interaction with silicate oligomers.21,22 To overcome these limitations, exploring simple strategies to enhance dispersibility of uncovered ultrasmall noble-metal NPs on specific supports with a new stabilizing layer against sintering is still a daunting challenge.
In this regard, we demonstrate a new sinter resistant catalyst system consisting of uncoated Pt NPs (<3 nm) supported on metal-oxide nanocrystals; taking Fe2O3 as a proof-of-concept example, isolation is achieved by wrinkled graphene sheets as a stabilizing layer, referred as to Pt/Fe2O3/GO sheets. In this rationally-designed system, the facets in Fe2O3 NPs allow uncovered Pt NPs to be finely dispersed on their different surfaces, which slows the motion of Pt NPs against sintering with neighboring ones across different surfaces. The wrinkles of graphene sheets act as physical barriers and suppress the coalescence of Fe2O3 NPs, subsequently suppressing the coalescence of Pt NPs simultaneously at high operating temperatures. This new catalyst system demonstrates a maximized sinter resistance of ultrasmall Pt NPs against heat-treatment up to 750 °C in different atmospheres, retaining their remarkable activity.
2. Experimental
Chemicals and materials
Poly(vinyl pyrrolidone) (PVP, Mw ≈ 1.3 × 106 or 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), acetic acid (CH3COOH, glacial), iron(III) acetylacetonate (Fe(acac)3), chloroplatinic acid hydrate (H2PtCl6·xH2O, 99.995%) and sodium borohydride (NaBH4, 99.99%) were obtained from Sigma-Aldrich. Graphite powder (99.95%, 325 mesh) was obtained from Xfnano Materials Tech Co., Ltd. All other chemicals were provided by Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification. The water used in all experiments was filtered through a Millipore filtration system with a resistivity of 18 MΩ cm.
000), acetic acid (CH3COOH, glacial), iron(III) acetylacetonate (Fe(acac)3), chloroplatinic acid hydrate (H2PtCl6·xH2O, 99.995%) and sodium borohydride (NaBH4, 99.99%) were obtained from Sigma-Aldrich. Graphite powder (99.95%, 325 mesh) was obtained from Xfnano Materials Tech Co., Ltd. All other chemicals were provided by Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification. The water used in all experiments was filtered through a Millipore filtration system with a resistivity of 18 MΩ cm.
Fabrication of porous Fe2O3 nanocrystals
The Fe(acac)3/PVP composite nanofibers were used as the Fe2O3 precursor, and were prepared by electrospinning a solution containing 0.4 g of PVP (Mw ≈ 1.3 × 106), 3 mL of ethanol, 3.5 mL of acetic acid and 0.8 g of Fe(acac)3 under a flow rate of 0.3 mL h−1 at 17 kV. Furthermore, 16 mg of as-spun Fe(acac)3/PVP composite nanofibers was digested in 20 mL of acetic acid aqueous solution at a pH value of 2.8, followed by a hydrothermal reaction at 180 °C for 8 h in an autoclave. The red Fe2O3 nanocrystals were washed by water twice and then by ethanol one time and centrifuged at a rate of 13000 rpm for 7 min. The final products were redispersed in ethanol at a fixed concentration of 0.95 mg mL−1.
Fabrication of Pt/Fe2O3/GO catalyst system
The ultrasmall Pt nanoparticles (NPs) (<3 nm) were synthesized by a polyol method in EG. EG (4 mL) was added to a vial and pre-heated to 110 °C for 30 min. Then, 5.6 mg of PVP (Mw ≈ 55000) and 4.1 mg of H2PtCl6 were dissolved separately in 0.5 mL of EG and then added simultaneously into the system in 1 min. The reaction was continued at 110 °C for 1.5 h and the color of the reaction mixture changed to brown gray. Furthermore, 45 μL of Pt suspension was mixed with 2.6 mL of ethanol and 0.9 mL of water, and then it was added into 1 mL of Fe2O3 suspension, followed by gentle stirring for 2 h at room temperature. The as-prepared Pt/Fe2O3 nanocrystals were washed with ethanol three times and then redispersed in 2 mL of ethanol. Graphene oxide (GO) colloid was prepared by the modified Hummer's method.23 Moreover, 0.5 mL of GO (14.4 mg mL−1) was dispersed in 5 mL ethanol. Then, the Pt/Fe2O3 was immersed into the abovementioned GO solution with stirring for 2 h. The precipitates were washed with water three times.
Evaluation of thermal stability and catalytic activity of Pt nanoparticles
The Pt/Fe2O3/GO sheets were heat-treated in a tube furnace (Thermo Electron Corporation) at different temperatures to evaluate their thermal stability under nitrogen or air flow. We chose the reduction of 4-nitrophenol to 4-aminophenol by NaBH4 to estimate the catalytic activities. In a typical procedure, a given amount of the catalysts was introduced into 50 μL of 4-nitrophenol (7.4 mmol L−1) and 2.5 mL of water. Then, 50 μL of NaBH4 (2.4 mol L−1) was added to start the reaction. After 20 min, the solution was centrifuged immediately to quickly separate the catalyst from reactants prior to UV-Vis tests. The kinetic process of the reduction was monitored by measuring the adsorption of the solution at 400 nm as a function of time. The starting concentrations of 4-nitrophenol, NaBH4 and Pt were set at 1.42 × 10−4, 4.62 × 10−2 and 8.6 × 10−7 mol L−1, respectively.Characterizations
The transmission electron microscope (TEM) images were collected using a Tecnai G2 T20 (FEI) operated at 200 kV, and scanning electron microscope (SEM) images were taken using an Inspect F50 (FEI). The HAADF-STEM imaging and EDX analyses were performed using an FEI Titan 80–300 microscope with STEM detector operated at 300 kV. The crystal structure information was obtained via X-ray diffraction (Bruker, D8 advance using Cu-Kα radiation, λ = 1.5406 Å). X-ray photoelectron spectroscope (XPS) measurements were performed via Thermo Scientific K-Alpha using monochromatic Al Kα radiation. The Pt mass was determined by an inductively coupled plasma optical emission spectrometer (ICP-OES) (Optima 7300DV, Perkin Elmer Corporation). The UV-Vis spectra were recorded on a Cary 60 spectrophotometer. The ζ-potential was tested on a Zeta-potential Nano ZS90 at room temperature. Thermogravimetric analysis (TGA) was conducted on a Q5000 at a heating rate of 10 °C min−1 under N2 or air flow.3. Results and discussion
As illustrated in Scheme 1, a simple and wet-chemical protocol of fabricating this sinter-resistant catalyst system, involves three steps: (1) generation of well-defined Fe2O3 NPs by a hydrothermal method using iron salt/polymer composite nanofibers as the Fe2O3 precursor, (2) dispersion of Pt NPs onto the Fe2O3 surface with limited Pt NPs on each exposed facet, and (3) deposition of Pt/Fe2O3 NPs on graphene oxide (GO) sheets.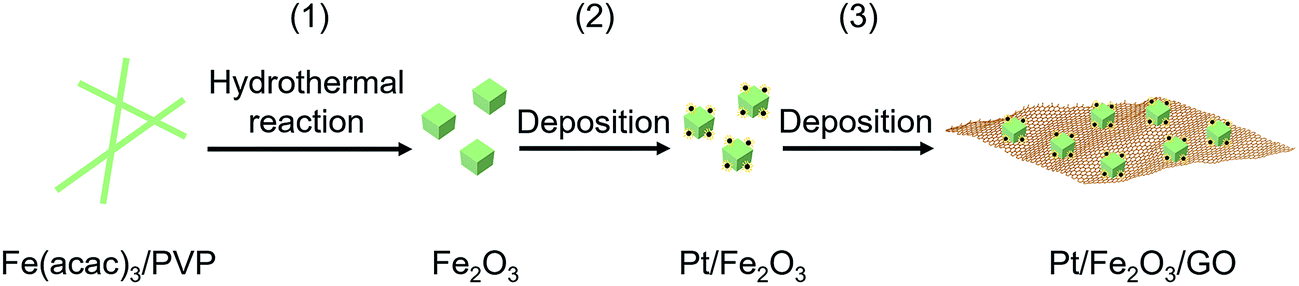 | ||
| Scheme 1 Schematic of a simple and wet-chemical protocol of the fabrication of the sinter-resistant catalyst system consisting of Pt/Fe2O3 stabilized on graphene oxide (GO) sheets. | ||
The porous Fe2O3 nanocrystals were prepared by electrospinning composite iron(III) acetylacetonate (Fe(acac)3)/PVP nanofibers, followed by a hydrothermal reaction. The Fe(acac)3 were dissolved in ethanol containing PVP as the electrospinning precursor, without generating complexation between each other. The electrospinning of this solution progressed in a stable and reliable manner and generated uniform Fe(acac)3/PVP nanofibers with 150 nm in diameter (Fig. 1A). The smooth nanofibers were homogeneous in the entire nanofiber in the absence of undesirable phase separation between Fe(acac)3 and PVP. Strikingly, the embedded Fe(acac)3 along the fibrous PVP matrix were readily digested in water without any stirring, despite its insolubility in water. This interesting phenomenon implies that the embedded Fe(acac)3 in polymer chains has been released from the water-soluble PVP matrix and become somewhat soluble in water. As observed the fibrous Fe2O3/PVP is endowed with a largely exposed surface that provides sufficient solid/liquid interface where the dissolution of PVP and release of Fe(acac)3 into water occurs. This strategy could be a promising way of dissolving solid precursors with weak solubility in specific solvents, allowing numerous unexplored starting materials as precursors and generating new nanocrystals with undisclosed composition and/or structures. This interesting result inspires us to conduct the crystal grown via the hydrothermal method in water at a pH value adjusted by acetic acid. As can be seen in representative TEM and SEM images (Fig. 1B and C), the monodispersed Fe2O3 NPs exhibited high purity in their morphology without agglomeration. Moreover, these Fe2O3 NPs presented a rhombohedral structure enclosed by six rhombic surfaces with sharp corners and edges around 30 nm. Their smooth surfaces were stabilized by PVP through the interaction between surface hydroxyl groups on Fe2O3 and the hydroxyl and/or lactim groups of PVP.24 This PVP sometimes has no essential effect on the nanocrystal morphologies,25 but its long polymer chains in fact act as block nanocages, slowing down the growth of larger Fe2O3 NPs at the expense of smaller ones, maintaining their uniform sizes. As depicted in the inset in Fig. 1D, this uniform rhombohedron exposes four equivalent {104} and two equivalent {114} facets as thermodynamically stable polyhedrons.24 The surface area of one typical Fe2O3 nanocrystal was estimated to be about 3814 nm2, by calculating the rhombohedron model using the measured geometric parameters, as shown in Fig. 1D. As a key observation, we note that the nanoscale holes, including pores and channels typically below 5 nm, form in the core inside the Fe2O3 in spite of smooth surfaces, as highlighted by white arrows in Fig. 1D. Their pore size distribution is summarized in Fig. S1,† and such porous structures were usually generated when sacrificial templates or pore generating agents were introduced, such as BF4−, F−, H2PO4−.26–28 Similar pore structures were recently observed but only in large Fe2O3 NPs with diameters of up to several hundred nanometers.24 Recently reported Fe2O3 polyhedrons of the same sizes as that of ours are rarely observed to exhibit pore structures.25 Evidently, embedding Fe(acac)3 in fibrous PVP matrix generated nanocrystals with novel precious structures without using complex sacrificial and/or etching reagents. Furthermore, our porous core also implied that the porous Fe2O3 was grown via a non-classical growth mechanism. One plausible formation mechanism of our new holes is the dissolution of core to form a recrystallized surface via a reversed crystal growth manner driven by the minimization of surface energy.26,29 The crystalline structure and composition of as-obtained products were further investigated by powder X-ray diffraction. The XRD patterns (Fig. 1E) of Fe2O3 can be indexed to rhombohedral alfa-Fe2O3 (hematite, JCPDS no. 33-0664), with good crystallinity.30,31 This thermodynamically most stable phase is beneficial to stabilizing noble-metal NPs at high temperatures. No detectable impurity phase, like metastable γ-Fe2O3 nor Fe3O4, demonstrates good phase purity of hematite in products. Based on the (104) diffraction peak, the average crystalline size of Fe2O3 was calculated to be around 30 nm using the Scherrer's equation, which is consistent with the TEM and SEM observations. The distinctive crystalline lattice fringes in the HRTEM image (Fig. 1F) further demonstrates the single-crystalline nature of the porous rhombohedron. It is worth noting that the observed lattice fringes usually cannot be assigned to the surface, except that the exposed facets were parallel to the electron beam in the TEM.24 The interplanar spacing of 4.6 and 4.1 Å correspond to the (003) and (101) lattice planes of Fe2O3, respectively, and the angle in the two labeled lattices is 57.3°. The lattice distance and angle values are also calculated to be identical to the theoretical values of d-spacing and dihedral angles between two indexed facets, using Miller indices and the cell parameters (a = 5.0356, c = 13.7489). This result excellently demonstrates that the structure morphologies of nanocrystals are considerably dominated by their growth conditions and methodologies.
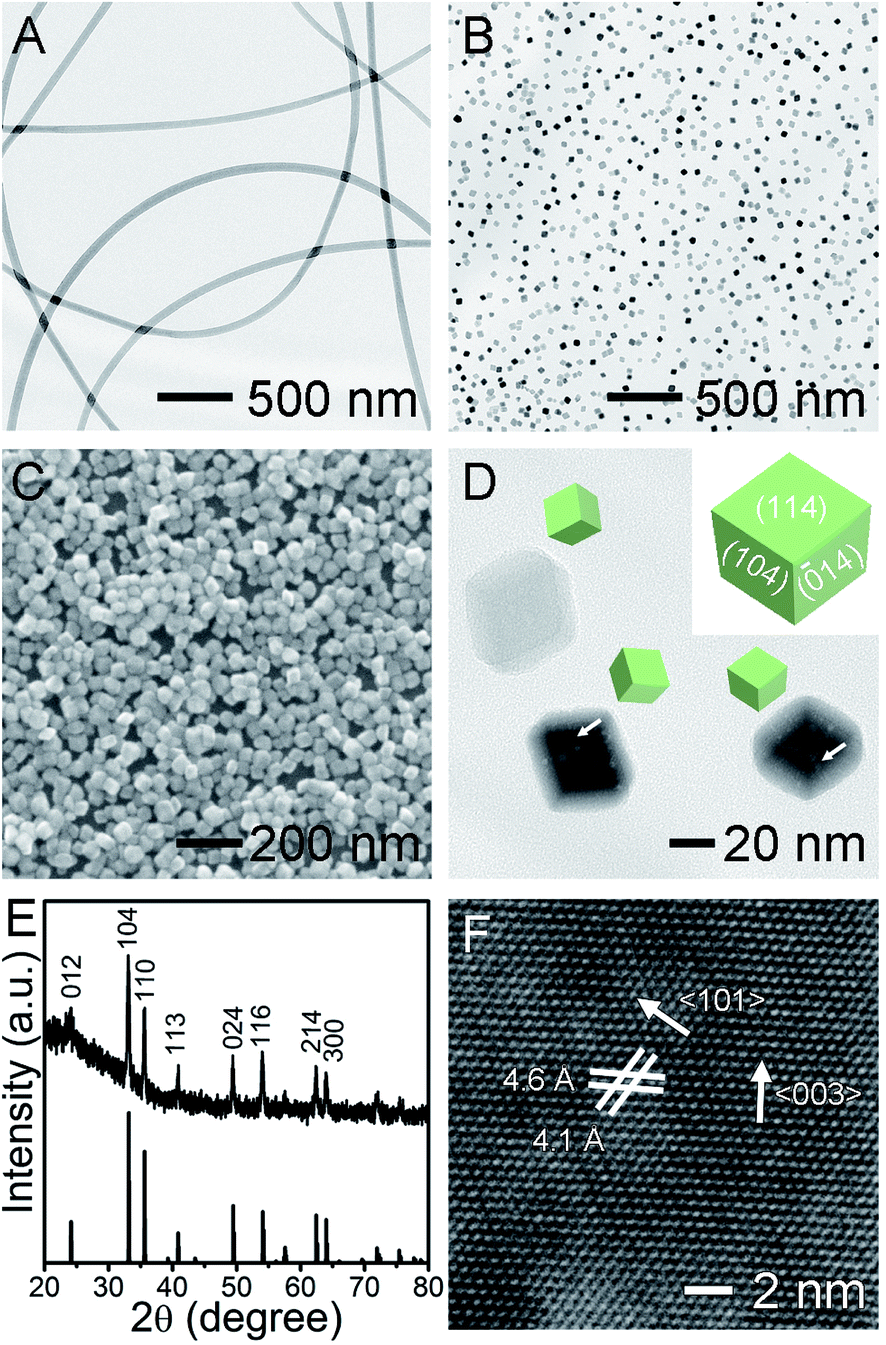 | ||
| Fig. 1 (A) TEM image of Fe(acac)3/PVP composite nanofibers fabricated by the electrospinning method. (B) TEM and (C) SEM images of Fe2O3 nanocrystals prepared by a hydrothermal reaction using Fe(acac)3/PVP composite nanofibers in (A) as precursor. (D) TEM image of the same sample in (B) at a higher magnification. The arrows highlight the nanoscale pores and channel inside the Fe2O3 nanocrystals. And the inset presents the structural model of a rhombohedron. (E) The XRD patterns of the Fe2O3 nanocrystals. All sharp diffraction peaks can be indexed to the hematite phase. (F) High-resolution TEM image of part of an individual hematite nanocrystal. | ||
The ultrasmall Pt NPs, with an average size of 2.5 ± 0.4 nm, were fabricated by a polyol method (Fig. S2†) and then supported on the Fe2O3 NPs via simply immersing them into Fe2O3 suspension. Unexpectedly, the Pt NPs failed to anchor on Fe2O3, even when prolonged immersion time and highly concentrated Pt were considered. ζ-Potential measurements uncover that the Pt NPs were positively charged (+4.41 mV, in ethanol), identical to that of Fe2O3 (+14.1 mV, in ethanol), possibly because their surfaces were both capped by PVP as stabilizers. Hence, we simply adjusted the surface charges of Pt by introducing 20 vol% of water into ethanol and obtained a negatively charged surface (ζ-potential of −17.1 mV), which exhibited strong affinity toward the Fe2O3 surface via electrical attraction. As expected, the Pt NPs could be readily decorated onto the surface of Fe2O3 supports (Fig. 2A), and they retained their individuality and tiny sizes well. Specifically, to get a fine dispersion, we controlled the average number of Pt at less than 3 per exposed facet of Fe2O3, and the Pt loading was determined to be 0.7 wt% in resulting Pt/Fe2O3, determined by ICP-OES. This success of tightly controlled loading on supports is beneficial for reducing noble metal consumption in an economic manner. HRTEM image in Fig. 2B enables a more specific view of one representative Pt/Fe2O3 NP, displaying excellent dispersibility of Pt NPs, which somehow prefer to anchor on six rhombic surfaces of Fe2O3 rather than the edges or corners, without preference for specific facets. The HAADF-STEM image of Pt/Fe2O3 in Fig. S3A† also demonstrates the homogeneous dispersion of Pt on Fe2O3 and the presence of pores in the Fe2O3 nanocrystals. A typical EDX spectrum of the Pt/Fe2O3 nanocrystals (Fig. S3B†) includes the Pt, Fe and O elements, confirming the presence of both Fe2O3 and Pt. This approach ensures not only the successful deposition of monodispersed Pt NPs on the Fe2O3 surfaces, but also the homogeneous dispersion of the Pt NPs across the entire faceted Fe2O3 nanocrystals. The Pt/Fe2O3 NPs were further supported on a stabilizing layer by simply mixing them with two-dimensional (2D) GO sheets in ethanol together, and the resultant products are referred to as Pt/Fe2O3/GO (Fig. 2C). The wrinkled surface texture in GO sheets is their distinctive feature, which is a result of minimizing their surface energies as observed in our previous studies.32,33 The wrinkled GO sheets act not only as a support with large areas to load a large number of Pt/Fe2O3 NPs, but also as a stabilizing layer supplying a physical barrier to separate individual Pt/Fe2O3 NPs from each other using their unique wrinkles, as highlighted by the arrows in Fig. 2C. As a result, the Pt/Fe2O3 NPs were highly isolated and were found to preferably be located around the wrinkles.
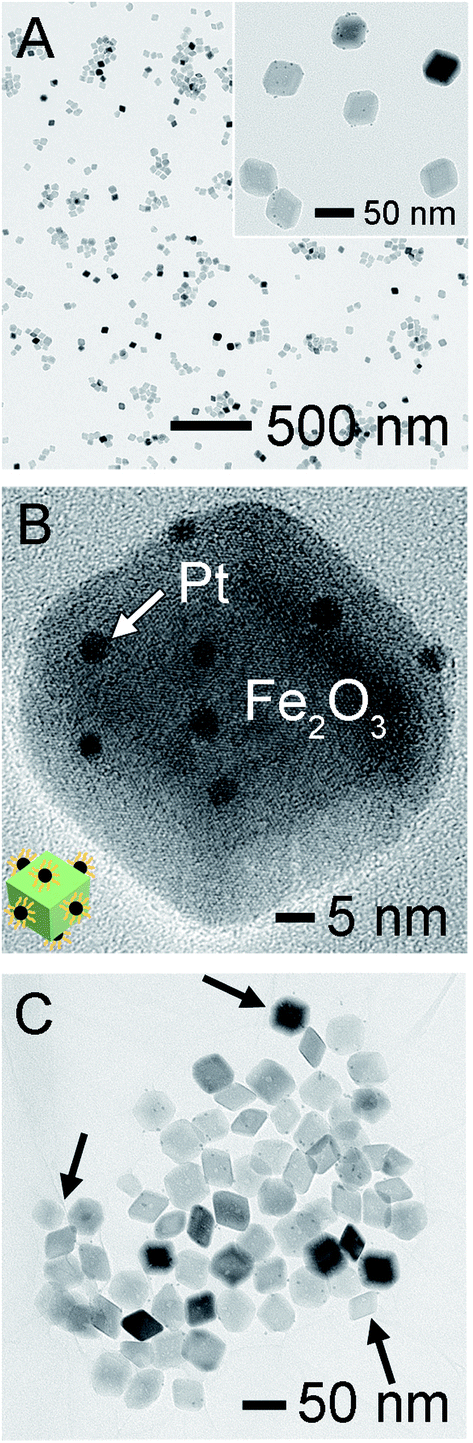 | ||
| Fig. 2 (A) TEM and (B) HRTEM images of Pt NPs finely dispersed on porous Fe2O3 rhombohedrons. The inset in (B) illustrates the schematic drawing of several Pt NPs across one rhombohedron surface. (C) TEM image of Pt/Fe2O3 on GO sheets. The arrows highlight the distinctive wrinkles acting as a space barrier to isolate the Pt/Fe2O3 NPs from each other. | ||
We carried out the thermal stability test of Pt/Fe2O3/GO sheets by aging at different temperatures in N2 for 2 h. The TEM images (Fig. 3A–E) and size analyses of Pt NPs (Fig. 3F and S4†) show that the sizes of supported Pt NPs in Pt/Fe2O3/GO sheets remain almost unchanged after heat-treatment up to 750 °C. The sintering of such Pt NPs had been observed at a heat-treatment temperature below 500 °C when they were homogeneously deposited on metal oxide, such as TiO2.14,19 The excellent thermal stability of Pt NPs up to 750 °C in our new ternary catalyst clearly demonstrated the superiority of this rational design. At 350 °C (Fig. 3A), the Fe2O3 kept their porous and rhombohedral structures, while the GO sheets became somewhat porous, possibly due to losing oxygen-containing groups (Fig. S5 and S6†). The wrinkles in GO sheets were highlighted by black arrows in Fig. 3A–E. Interestingly, the pores inside the Fe2O3 started to shrink without causing structural collapse, and the rhombohedral Fe2O3 converted to nearly spherical shapes under gradually elevated temperature up to 750 °C (Fig. 3B–D). This feature elucidates that porous Fe2O3 preferred densification at the pores inside, to the commonly observed coalescence with neighboring NPs, due to the efficient isolation by wrinkled GO sheets. The unexpected but notable sintering of Pt started at 850 °C (Fig. 3E), with size grown up to 4.1 ± 1.9 nm, and these appeared agglomerations may be induced by the fusion of Fe2O3 crystallites, regardless of occasionally observed GO sheets. As a result, the anti-sintering of Pt is found to be proportional to the suppressed coalescence of the Fe2O3 NPs, validating the function of nanometer-sized supports in stabilizing of supported noble-metal NPs. Evidently, these results prove that our new catalyst system presented thermal stability against sintering at a temperature as high as 750 °C. Furthermore, we used the polycrystalline α-Fe2O3 nanofibers as Pt supports for a simple comparison. In a sharp contrast, the Pt NPs exhibited weak thermal stability and severely sintered at 750 °C on porous Fe2O3 nanofibers (Fig. S7†), in spite of a much lower Pt loading of 0.3 wt%. This result unambiguously demonstrates the vital role of faceted Fe2O3 nanocrystals in stabilizing Pt under harsh conditions.
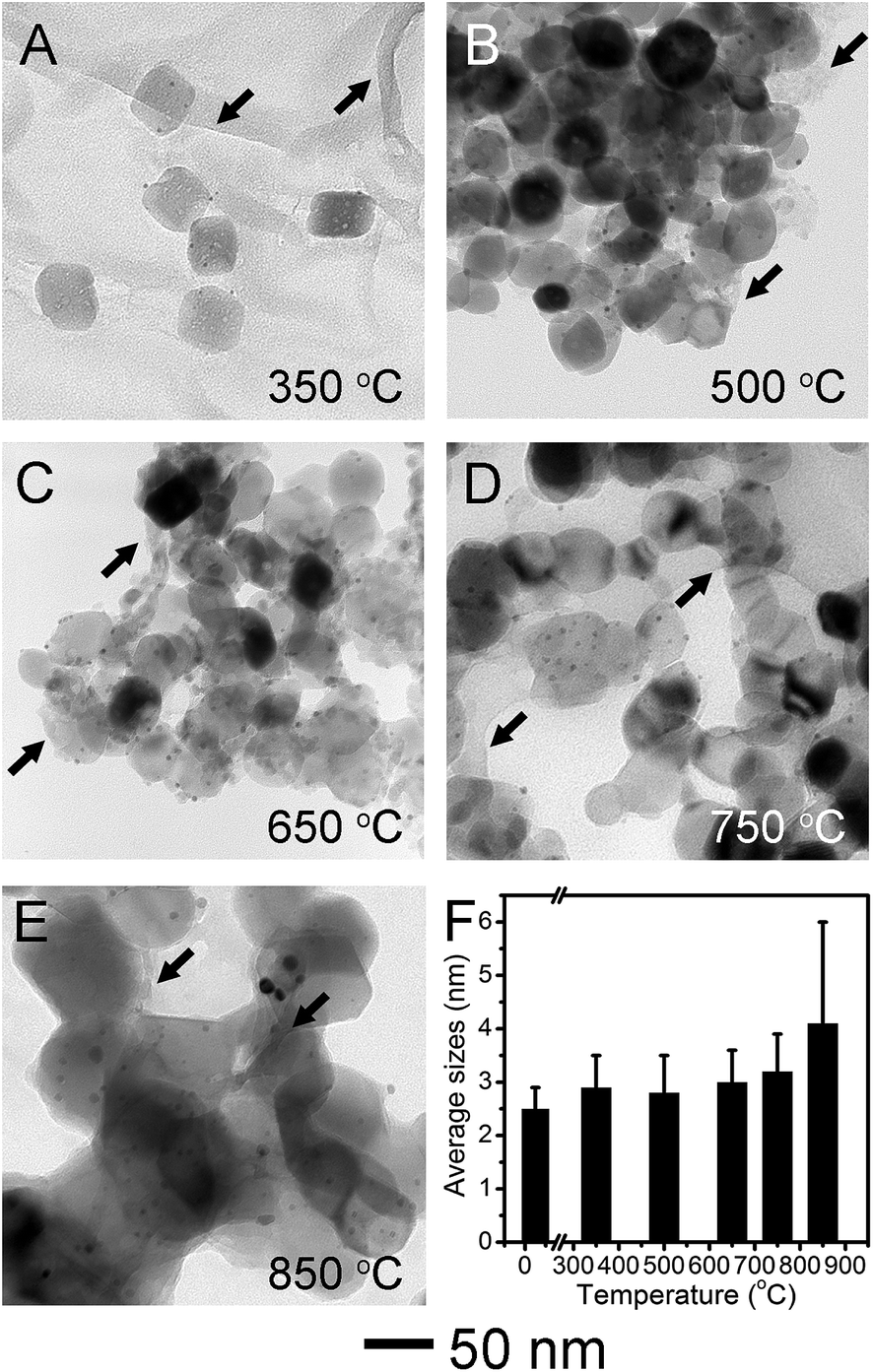 | ||
| Fig. 3 TEM images of the Pt/Fe2O3/GO sheets after being heat treated at (A) 350, (B) 500, (C) 650, (D) 750 and (E) 850 °C in N2 for 2 h. The black arrows highlight the wrinkles on GO sheets as a stabilizing layer. (F) The histogram of the average sizes of Pt in (A) to (E) versus temperatures, calculated by counting a minimum of 100 particles in TEM images. The values for standard deviation are included in the histogram. This new catalyst system presents excellent thermal-stability against sintering up to 750 °C. | ||
Interestingly, the thermal stability of supported Pt could be modulated by tuning the content of graphene sheets in the catalyst system (Fig. S8†). We reduced the amount of GO to half of pristine GO used in Pt/Fe2O3/GO (Fig. 2C) and conducted the same severe aging at 750 °C. The Fe2O3 NPs changed to spherical morphology and fused together, while the Pt NPs increased slightly to 3.9 ± 1.0 nm. In a sharp contrast, further decreasing the GO amount to 20% resulted in Fe2O3 dramatically aggregating to form polycrystalline in irregular shapes, causing the Pt NPs to grow to 4.5 ± 1.3 nm. In a sharp contrast, without GO sheets, the sintering of Pt NPs in Pt/Fe2O3 nanocrystals occurred at a relatively low temperature of 650 °C (Fig. S9†), and the Fe2O3 nanocrystals also coalesced together. This weaker thermal stability further confirmed that the wrinkled GO sheets enable the sinter-resistance of Pt by slowing the coalescence of Fe2O3 nanocrystals. In this regard, the facile modulated thermal stability of Pt NPs by GO sheets proves that this stabilizing layer is critical to resisting the undesirable sintering.
We also used XRD patterns (Fig. 4) to verify the sintering of Pt NPs, as two (111) and (200) peaks at 39.8° and 46.2° are commonly observed in the diffraction patterns of sintered Pt.19 Moreover, these two peaks did not show up in the XRD patterns of both fresh-made Pt/Fe2O3/GO sheets and heat-treated ones at 750 °C. The lack of these peaks can be ascribed to the ultrasmall size of Pt without detectable long-range periodicity and to the absence of sintering of Pt up to this temperature. The unchanged (104) peak of Fe2O3 at 750 °C also illustrates negligible change in size of Fe2O3 NPs at this temperature, which can be ascribed to its extraordinary thermodynamic stability. When the temperature was increased to 850 °C, two weak peaks of Pt appeared, implying the presence of Pt agglomerates. Moreover, a phase transformation of Fe2O3 to metastable γ-Fe2O3 from α-Fe2O3 proceeded simultaneously (JCPDS no. 39-1346).34 This irreversible phase change is also accountable for the growth of Pt sizes.
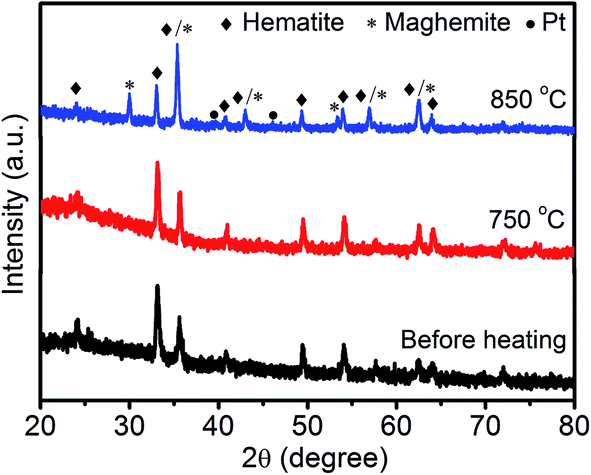 | ||
| Fig. 4 XRD patterns of the Pt/Fe2O3/GO catalysts before and after being heated at 750 °C or 850 °C for 2 h in N2 atmosphere. | ||
To investigate the chemical state of Pt nanoparticles, we conducted the XPS characterization to have a deeper insight of the surface feature of Pt and Pt/Fe2O3 interaction. High-resolution Pt 4f XPS spectrum (Fig. 5A) shows that the Pt NPs in this catalyst system are in metallic Pt0 state before heating, with distinctive Pt 4f7/2 and Pt 4f5/2 peaks located at 71.4 eV and 74.5 eV,30,31,35 respectively. Negligible chemical-shift of these two peaks reveals that Pt may interact weakly with Fe2O3 as the presence of PVP may shield their strong metal–support interaction at their interface sites. The supported Pt NPs (Fig. 5B–D) kept their metallic state of Pt(0) well during heat treatment. A noticeable up-shift of binding energy of about ∼0.3 eV emerged above 500 °C, at which temperature the PVP was selectively removed by thermal decomposition (Fig. S5†), proving that the Pt NPs interact with Fe2O3. This result demonstrates that the removal of surfactant could facilitate the metal to interact with supports directly via the so-called metal–support interaction, which further benefits the stabilization of metal on supports.
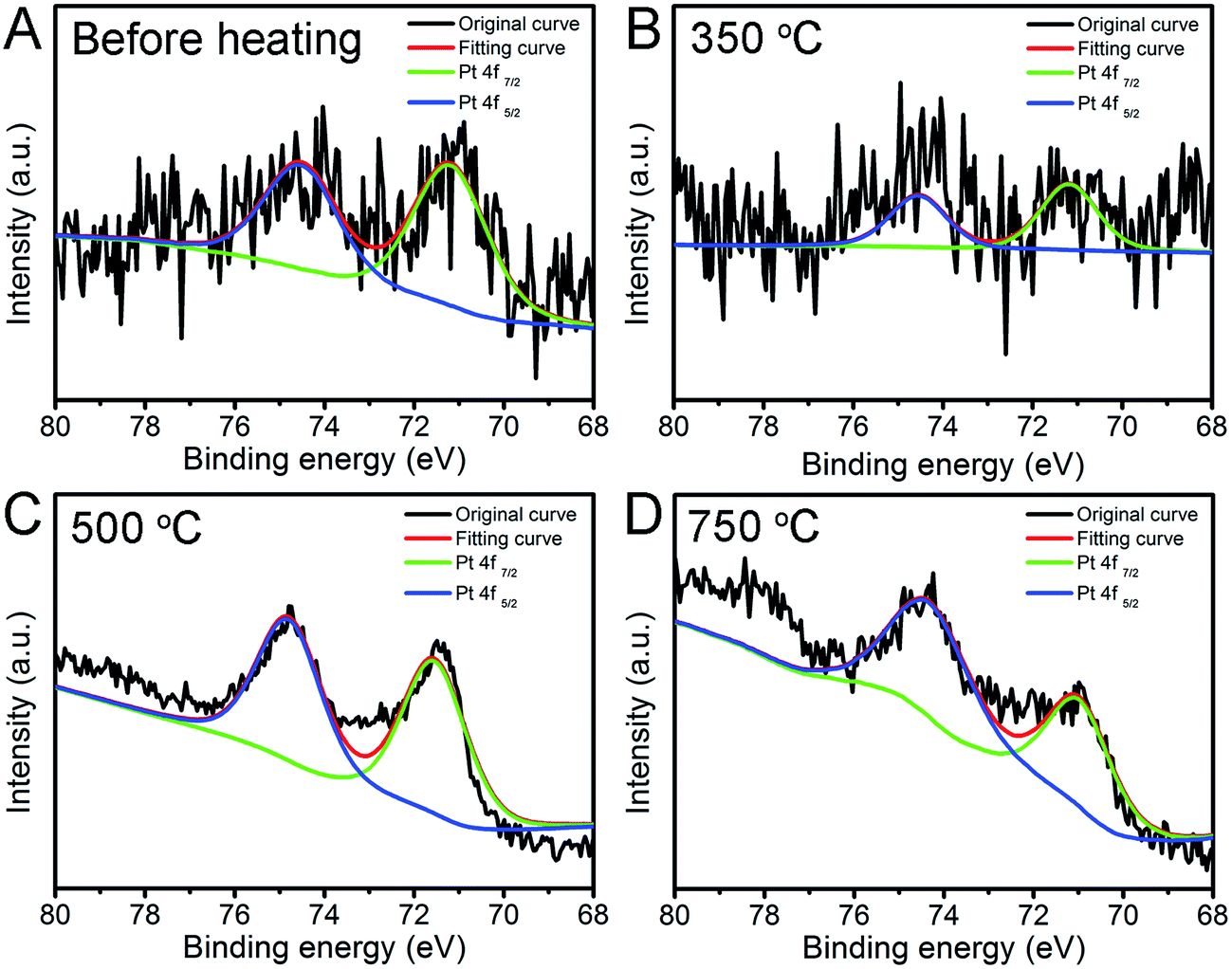 | ||
| Fig. 5 Pt 4f high resolution XPS spectra of Pt/Fe2O3/GO sheets (A) before and after being aged at (B) 350, (C) 500 and (D) 750 °C in N2 for 2 h. | ||
Moreover, even if the Pt/Fe2O3/GO sheets were put in an oxidative environment, such as air, this new catalyst also exhibited good thermal stability against sintering, enabling the supported Pt to present sizes of 3.3 ± 0.7 nm up to 750 °C (Fig. S10†). Despite the decomposition of GO in air at a high temperature (Fig. S11†), it can be reasonably deduced that it can still act as a space barrier before loss, slowing down the coalescence rate of Fe2O3 NPs. A deeper insight into the chemical state of Pt illustrates that metallic Pt0 was oxidized partially to PtO species (Fig. S12†) as a secondary metal-oxide phase in addition to support, providing stronger adhesion for stabilizing the Pt onto Fe2O3.6 Evidently, this thermal stability in ambient atmosphere further reveals the exceptional anti-sintering of Pt NPs in this new catalytic system under varied environments.
Taken together, the rationally designed new catalyst system exhibited excellent anti-sintering performance without coating protective shells, ensuring that the catalyst system has highly accessible surfaces available for reaction.
The success of stabilizing Pt on the Fe2O3 surface inspires us to quantitatively assess its catalytic performance after heat-treatment by choosing the hydrogenation of 4-nitrophenol to 4-aminophenol in borohydride as a case in point. This nitro-to-amino conversion is not only reliable in estimating the catalytic performance of the metal catalyst,36–38 but also non-replaceable for a large number of industrially important processes, such as for aniline and paracetamol productions.39 After adding the pre-aged Pt/Fe2O3/GO catalyst, a new peak at 315 nm for 4-aminophenol appeared (Fig. 6A). Fig. 6B shows that the intensity of the adsorption peak for 4-nitrophenol gradually dropped in a deceleration fashion at 400 nm as the catalytic reaction proceeded. This tendency allowed us to monitor the pseudo-first-order reaction conventionally by measuring the time-resolved UV-Vis spectra (Fig. 6C).38 We normalize the apparent rate constant by mass, referred to as Km, which reflects the intrinsic activity of the metal. The Km values as a function of heating temperatures are summarized in Fig. 6D, and the corresponding sizes of Pt are added for a better comparison. All heated Pt/Fe2O3/GO catalysts exhibited exceptional activities compared to that of free Pt NPs, which confirms that the supported Pt are highly active and accessible and the GO sheets allow good mass transfer during the entire reaction. Note that the Fe2O3/GO sheets as supports are inactive towards the reduction of 4-nitrophenol, and therefore are spectators in the catalytic reaction (Fig. S13†). A previous report reveals that the porous TiO2 allowed its anchored Pt NPs to retain only about 50% of activity after supporting,12 and the reason for this weaker activity is still unclear. It becomes evident, that both Fe2O3 NPs and GO sheets do not affect catalytic performances of Pt but benefit Pt by achieving remarkable activity in addition to stabilizing the supported Pt NPs in ultrasmall sizes. We observed an increase in the Km value of the Pt/Fe2O3/GO heated at 350 °C, and it reached its maximum of 10 s−1 mg−1, which is 6 times higher than that of free Pt, at an elevated temperature of 500 °C, while slightly decreasing to 7.9 s−1 mg−1 at 600 °C. The surprising catalytic activity of Pt NPs is attributed to the small sizes, the selective removal of PVP and good Pt/Fe2O3 interaction, which made a great contribution in accelerating the catalytic reactions. Moreover, the Km decreased to 4.2 s−1 mg−1, as the temperature increased to 750 °C, regardless of Pt having desirable unchanged sizes. The decline of remarkable activity is most likely due to the thermally-induced restructuring of Pt to its equilibrium shapes as the activity in noble metals are shape-dependent also. Notably, the bigger Pt NPs exhibit weak activity after being treated at 850 °C, but are still better than that of free Pt. These results clearly reveal that the intrinsic chemical activity and stability of a supported catalyst is greatly determined by its size, as well as the chemical environments, shape, and interaction with the support.
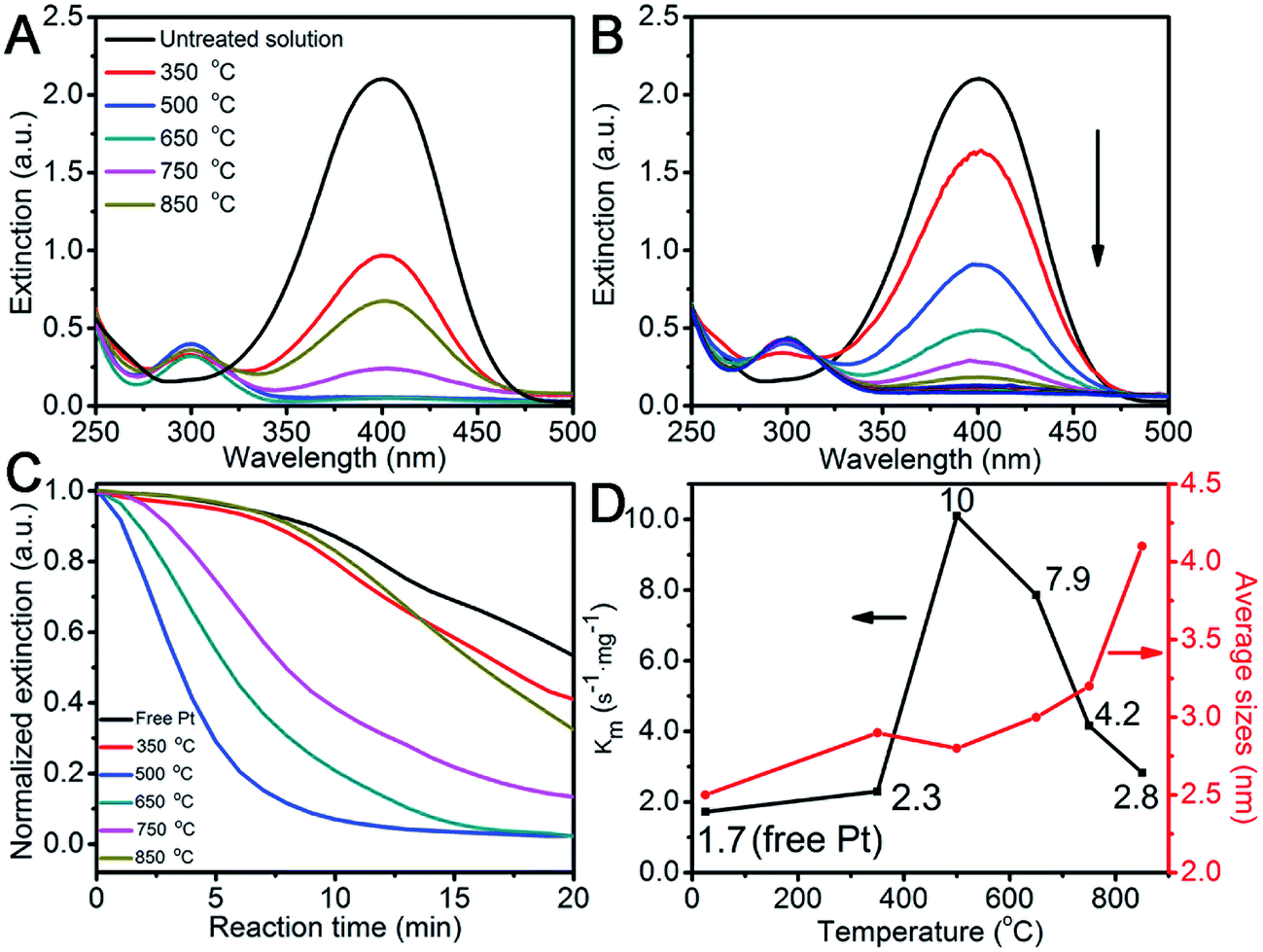 | ||
| Fig. 6 (A) Conversion of 4-nitrophenol to 4-aminophenol in the presence of Pt/Fe2O3/GO sheets pre-heated at varied temperatures. (B) A series of extinction spectra recorded at an interval of 2 min, presenting a gradually proceeding nitro-to-amino reduction. (C) The normalized extinction at 400 nm, and (D) a plot of reaction constant rate (Km) together with corresponding Pt sizes versus heat-treatment temperatures, proving that the supported Pt NPs have strikingly high reactivity even when being heated at an ultrahigh temperature up to 750 °C. | ||
4. Conclusions
In summary, we demonstrated that the thermal-stability of ultrasmall Pt NPs (<3 nm) on Fe2O3 nanocrystals was manipulated by a new stabilizing layer of GO sheets. The novel porous Fe2O3 nanocrystals in rhombohedral structure were grown through an unconventional reversal growth mechanism and endowed with six rhombic surfaces that suppressed finely dispersed Pt NPs from sintering with neighboring ones at a different surface. Furthermore, the flexible GO sheets isolate the Pt/Fe2O3 NPs from each other, using their distinctive wrinkles as physical barriers to slow the coalescence of Fe2O3 and thus the supported Pt. Such a catalyst presents strikingly excellent sinter-resistance up to 750 °C in both inert and oxidative environments. The success of stabilizing Pt NPs with a clean surface allows them to have extraordinary reactivity towards the nitro-to-amino conversion, and achieves a much better activity maximum to a 6 times higher Km value, compared with the pristine Pt NPs before heating. This strategy could be extendable to produce a variety of heterogeneous catalysts with high thermal stabilities in various catalytic reactions, and could open up an alternative avenue for a range of possible domains of applications.Acknowledgements
This study was financially supported by the National Basic Research Program (973 program, 2013CB932902), the National Natural Science Foundation of China (21201034 and 21310102005), the Science and Technology Support Program (Industry) Project of Jiangsu Province (BE 2013118), the Fundamental Research Funds for the Central Universities, the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Nanjing Science and Technology Committee (Platform No 2014-030002).References
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150 CrossRef CAS PubMed.
- C. T. Campbell and J. R. V. Sellers, Faraday Discuss., 2013, 162, 9 RSC.
- S. H. Joo, J. Y. Park, C. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126 CrossRef CAS PubMed.
- C. Du, Y. Guo, Y. Guo, X. Gong and G. Lu, J. Mater. Chem. A, 2015, 3, 23230 CAS.
- H. Liu, L. Zhang, N. Wang and D. S. Su, Angew. Chem., Int. Ed., 2014, 53, 12634 CAS.
- C. Han, P. Majumdar, E. E. Marinero, A. Aguilar-Tapia, R. Zanella, J. Greeley and V. Ortalan, Nano Lett., 2015, 15, 8141 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597 CrossRef CAS PubMed.
- M. Mahmoodinia, P. Åstrand and D. Chen, J. Phys. Chem. C, 2016, 120, 12452 CAS.
- L. Zhao, Z. Wang, J. Li, J. Zhang, X. Suia and L. Zhang, J. Mater. Chem. A, 2015, 3, 5313 CAS.
- H. Zhao, D. Wang, C. Gao, H. Liu, L. Han and Y. Yin, J. Mater. Chem. A, 2016, 4, 1366 CAS.
- P. Lu and Y. Xia, ACS Appl. Mater. Interfaces, 2013, 5, 6391 CAS.
- P. Lu, B. Qiao, N. Lu, D. C. Hyun, J. Wang, M. J. Kim, J. Liu and Y. Xia, Adv. Funct. Mater., 2015, 25, 4153 CrossRef CAS.
- K. Yoon, Y. Yang, P. Lu, D. Wan, H. Peng, K. S. Masias, P. T. Fanson, C. T. Campbell and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 9543 CrossRef CAS PubMed.
- Y. Dai, B. Lim, Y. Yang, C. M. Cobley, W. Li, E. C. Cho, B. Grayson, P. T. Fanson, C. T. Campbell, Y. Sun and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 8165 CrossRef CAS PubMed.
- A. S. Reddy, S. Kim, H. Y. Jeong, S. Jin, K. Qadir, K. Jung, C. H. Jung, J. Y. Yun, J. Y. Cheon, J. Yang, S. H. Joo, O. Terasakia and J. Y. Park, Chem. Commun., 2011, 47, 8412 RSC.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L. Wu, C. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2014, 53, 250 CrossRef CAS PubMed.
- P. Zhang, M. Chi, S. Sharma and E. McFarland, J. Mater. Chem., 2010, 20, 2013 RSC.
- P. Lu, C. T. Campbell and Y. Xia, Nano Lett., 2013, 13, 4957 CrossRef CAS PubMed.
- Z. Wang, H. Fu, D. Han and F. Gu, J. Mater. Chem. A, 2014, 2, 20374 CAS.
- T. Zhang, H. Zhao, S. He, K. Liu, H. Liu, Y. Yin and C. Gao, ACS Nano, 2014, 8, 7297 CrossRef CAS PubMed.
- W. Li, L. Kovarik, D. Mei, J. Liu, Y. Wang and C. H. F. Peden, Nat. Commun., 2013, 4, 2481 Search PubMed.
- Y. Dai, Y. Jing, J. Zeng, Q. Qi, C. Wang, D. Goldfeld, C. Xu, Y. Zheng and Y. Sun, J. Mater. Chem., 2011, 21, 18174 RSC.
- M. Lin, H. R. Tan, J. P. Y. Tan and S. Bai, J. Phys. Chem. C, 2013, 117, 11242 CAS.
- J. Ouyang, J. Pei, Q. Kuang, Z. Xie and L. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 12505 CAS.
- B. Xu, B. Huang, H. Cheng, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 6529 RSC.
- C. Wang, J. Shi, X. Cui, H. Wang, J. Wu, C. Zhang, L. Wang, B. Lv and Y. Xu, J. Mater. Chem. A, 2016, 4, 11000 CAS.
- C. Wang, S. Yang, W. Q. Fang, P. Liu, H. Zhao and H. G. Yang, Nano Lett., 2016, 16, 427 CrossRef CAS PubMed.
- W. Wu, S. Zhang, X. Xiao, J. Zhou, F. Ren, L. Sun and C. Jiang, ACS Appl. Mater. Interfaces, 2012, 4, 3602 CAS.
- Y. Li, H. Zhang, S. Wang, Y. Lin, Y. Chen, Z. Shi, N. Li, W. Wang and Z. Guo, J. Mater. Chem. A, 2016, 4, 11247 CAS.
- Z. Ruan, J. Wu, J. Huang, Z. Lin, Y. Li, Y. Liu, P. Cao, Y. Fang, J. Xie and G. Jiang, J. Mater. Chem. A, 2015, 3, 4595 CAS.
- Y. Dai, Y. Jing, J. Zeng, Q. Qi, C. Wang, D. Goldfeld, C. Xu, Y. Zheng and Y. Sun, J. Mater. Chem., 2011, 21, 18174 RSC.
- Y. Dai, Y. Sun, J. Yao, D. Ling, Y. Wang, H. Long, X. Wang, B. Lin, T. H. Zeng and Y. Sun, J. Mater. Chem. A, 2014, 2, 1060 CAS.
- H. J. Lee, W. Cho, E. Lim and M. Oh, Chem. Commun., 2014, 50, 5476 RSC.
- L. Chen, F. Li, B. Ni, J. Xu, Z. Fu and Y. Lu, RSC Adv., 2012, 2, 10057 RSC.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617 CrossRef CAS PubMed.
- R. He, Y. Wang, X. Wang, Z. Wang, G. Liu, W. Zhou, L. Wen, Q. Li, X. Wang, X. Chen, J. Zeng and J. G. Hou, Nat. Commun., 2014, 5, 4327 CAS.
- E. Menumerov, R. A. Hughes and S. Neretina, Nano Lett., 2016, 16, 7791 CrossRef CAS PubMed.
- X. Kong, Z. Sun, M. Chen, C. Chen and Q. Chen, Energy Environ. Sci., 2013, 6, 3260 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01188a |
| This journal is © The Royal Society of Chemistry 2017 |