
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrostatic polypyridine–ruthenium(II)⋯decatungstate dyads: structures, characterizations and photodegradation of dye†
Shugai Wanga,
Songzhu Xinga,
Zhuolin Shia,
Jiachen Hea,
Qiuxia Han *ab and
Mingxue Li*a
*ab and
Mingxue Li*a
aHenan Key Laboratory of Polyoxometalate Chemistry, Institute of Molecular and Crystal Engineering, School of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, People’s Republic of China. E-mail: qiuxia_han@163.com; limingxue@henu.edu.cn; Fax: +86-371-23881589
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China
First published on 24th March 2017
Abstract
Two hybrids, [Ru(bpy)2(CH3CN)2]2[W10O32]·2CH3CN (RuW-1) and [Ru(bpy)3]2[W10O32] (RuW-2) (bpy = 2,2′-bipyridine), were synthesized by the reaction of (2,2′-dipyridyl)ruthenium complexes and decatungstate, under hydrothermal conditions, and characterized by IR, UV, fluorescence and transient absorption spectroscopy. The influences of the coordination environment of the centre ion, Ru(II), are compared and discussed in detail on the basis of analyzing their photophysical and photochemical properties. Kinetics experiments for the photodegradation of Rhodamine B (RhB) dye were followed with spectrophotometric analysis showing an absorbance decrease at 544 nm resulting from the cleavage of the aromatic ring of RhB. UV spectroscopy indicated that the degradation proceeds with a pseudo-first-order rate constant in the range of 10−3 to 10−2 s−1. These results demonstrate that the hybrids have effective activity and reusability for the photodegradation of RhB.
Introduction
With the development of industries such as petrochemical engineering, plastics, coking, printing, pesticide, dyeing and so on, the contamination of water caused by refractory organic pollutants has raised serious environmental problems.1 Organic dyes are a typical class of pollutant with common characteristics such as toxicity, complicated composition and high chemical oxygen consumption. Consequently, the field of wastewater treatment is attracting much more attention from experts around the world and becoming an appealing challenge.Photocatalytic technology has been considered an efficient, nontoxic and environment friendly method for decomposing organic dyes in wastewater.2 Decatungstate (W10O324−) is an attractive candidate for photocatalysis in the polyoxometalate (POM) family, for which it can easily absorb the photon energy products to furnish an excited state.3 Due to the strong oxidising properties of the charge transfer excited state, the excited intermediate can not only directly oxidize the target pollutant, but also react with water, oxygen or other electron donors to generate an ˙OH and ˙O2− radical, and then induce the oxidation process of the reactant molecules.4 Recently, W10O324− has been applied as an effective green homogeneous catalyst for the photochemical degradation of pollutants undergoing mineralization upon photolysis with near visible and UV light.5 However, at present, the technical challenges remain for the practical application, including less photocatalytic activity, low efficiency in solar energy utilization, difficulty with reusability and recyclability, and so on.5f,6 In addition to these, the mechanism of photooxidation is still in an immature stage due to lack of identification of the active intermediate.
Desired heterogeneous catalysts for the photodegradation of a dye require multi-functional systems that combine light absorption, charge separation, and catalytic conversion. One promising approach is the combination of a photosensitizer and POM anions in one molecule to develop donor–acceptor dyad systems, in which the charge-separation state has a long lifetime and does not lose energy by sequential electron transfer. Generally, light-driven POM catalysts can be put into two categories of structure: non-covalent systems, typically combining a cationic photosensitizer with the POM anion,7 and covalent systems, typically covalently linking the photosensitizer and POM anion.7f,8
Bipyridineruthenium complexes are excellent photosensitizers from the viewpoint of the luminance and electrochemistry properties, which have been extensively investigated for applications in inorganic and materials chemistry. The hybrids assembled from bipyridineruthenium complexes and POM anions via electrostatic interactions have been well developed. Although this approach is highly popular, it can be hampered by uncontrolled electrostatic aggregation, leading to colloid formation and precipitation. As far as we know, there are only four reports on the structurally-characterized hybrid compounds [Ru(2,2′-bpy)3]2[W10O32]·3DMSO,9 KNa[Ru(2,2′-bpy)3]2[H2W12O40]·8H2O,10 K6[Ru(pzc)3]2[SiW12O40]·12H2O,10 and [Ru(bpy)3][KPW12O40].11
Herein we report two hybrids [Ru(bpy)2(CH3CN)2]2[W10O32]·2CH3CN (RuW-1) and [Ru(bpy)3]2[W10O32] (RuW-2), which are assembled via electrostatic interactions. In this work, particular attention is devoted to a comparison of their photophysical and electron-transfer properties, which may be influenced by the coordination environment of the centre ion, Ru(II). In the hybrids, the bipyridineruthenium complexes are expected to transfer electrons to W10O324− through electrostatic interactions, thus they can be used as photocatalysts under visible light irradiation.
Experimental
Materials and methods
All chemicals were of reagent grade quality, obtained from commercial sources, and used without further purification. [Ru(bpy)2]Cl2·6H2O,12 [Ru(bpy)3]I2 (ref. 13) and (NBu4)4W10O32 (ref. 14) were prepared by the literature procedures.The elemental analyses (EA) of C, H and N were performed on a Vario EL III elemental analyzer. The inductively coupled plasma (ICP) spectroscopic analyses of Ru and W were performed on a Jarrel-Ash Model J-A1100 spectrometer. The infrared spectra (IR) were recorded from a sample powder palletized with KBr on a Nicolet170 SXFT-IR spectrometer over a range of 4000–400 cm−1. The UV-Vis absorption spectra were collected on a Hitachi Model U-4100, a UV-Vis spectrometer from 200 to 900 nm with a 60 mm-diameter integrating sphere at room temperature.The fluorescence spectra were recorded on an Edinburgh Model FS920 luminescence spectrometer. The nanosecond time-resolved transient difference absorption spectra were recorded on a LP 920 laser flashphotolysis spectrometer (Edinburgh Instruments, Livingston, UK). The sample solutions were purged with N2 for 15 min before measurement. The samples were excited with a 350 nanosecond pulsed laser, and the transient signals were recorded on a Tektronix TDS 3012B oscilloscope.
Synthesis
Crystallography
The data for the hybrid RuW-1 were collected on a Bruker SMART APEX CCD diffractometer with graphite-monochromated Mo-Kα (λ = 0.71073 Å) using the SMART and SAINT programs.15,16 A routine Lorentz polarization and multi-scan absorption correction were applied to the intensity data. The structure was determined and the heavy atoms were found by direct methods using the SHELXTL-97 program package.17 The remaining atoms were found from successive full-matrix least-squares refinements on the F2 and Fourier syntheses. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms within the ligand backbones were fixed geometrically at their positions and allowed to ride on the parent atoms. The crystal data, experimental details, and refinement results are listed in Table 1.| RuW-1 | |
|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2; w = 1/[σ2(Fo2) + (xP)2 + yP], P = (Fo2 + 2Fc2)/3, where x = 0.0457, y = 0 for RuW-1. | |
| Empirical formula | C52H50N14O32Ru2W10 |
| CCDC | 1530386 |
| Formula weight (g mol−1) | 3423.7 |
| T (K) | 273 (2) |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 10.689 (2) |
| b (Å) | 13.960 (2) |
| c (Å) | 14.063 (2) |
| α (deg) | 70.1 (1) |
| β (deg) | 70.63 (1) |
| γ (deg) | 89.66 (1) |
| V (Å3) | 1848.6 (5) |
| Z | 1 |
| Dc (g cm−3) | 3.07527 |
| μ (mm−1) | 15.971 |
| −12 ≤ h ≤ 12 | |
| Limiting indices | −16 ≤ k ≤ 16 |
| −16 ≤ l ≤ 16 | |
| Rint | 0.0373 |
| Restrains/parameters | 2/484 |
| θ Range (°) | 1.64–25.00 |
| Goodness-of-fit on F2 | 1.001 |
| R1, wR2 [I > 2σ(I)] | 0.0330, 0.0796 |
| R1a, wR2b[all data] | 0.0474, 0.0851 |
Catalysis
The photocatalytic activities of the catalysts RuW-1 and RuW-2 were tested using RhB as the target though the following approach. 5 mL of RhB solution (2 × 10−5 mol L−1, 9.6 mg L−1) was added into a quartz test tube. Then, 10 mg of catalyst RuW-1 was suspended in the RhB solution, and the suspension was ultrasonically dispersed for 5 min. The RhB solution was then irradiated under a CHF-XM35-500W xenon lamp with intense stirring. The temperature was kept at 20 °C by the circulation of cool water. The degradation of RhB was followed by monitoring the decrease in the absorbance at 544 nm due to the cleavage of the aromatic ring of the RhB dye. The reaction mixture was withdrawn at regular intervals to study the progress of the reaction. In a typical operation, 30 μL of solution was taken out with the help of a micro syringe and then injected into 3 mL of water solution. The concentration of RhB is calculated by a calibration curve. The degradation efficiency (%) can be calculated as efficiency% = C0 − Ct/C0 × 100%, where C0 is the initial concentration of RhB and Ct is the terminal concentration after given time intervals.Results and discussion
Structural description
The structure of [Ru(bpy)3]2[W10O32]·3DMSO has been reported previously, in which bipyridineruthenium complexes and decatungstate were combined by electrostatic attraction and C–H⋯O hydrogen bonds to produce a cation radical salt.9Herein, we mainly describe the structure of the hybrid RuW-1. The hybrid RuW-1 crystallizes in a space group P. As shown in Fig. 1a, the RuW-1 consists of one [W10O32]4− anion, two [Ru(bpy)2(CH3CN)2]2+ cations and two free CH3CN molecules. In the decatungstate anion, the two W5O18 units are bonded mirror-symmetrically through four corner-sharing oxygen atoms with the formation of an empty octahedral space. In the W5O18 unit, the five distorted WO6 octahedra are bonded via common edges with one oxygen atom common to each of them. In the [Ru(bpy)2(CH3CN)2]2+ fragment, the crystallographically independent Ru(II) ion adopts a distorted octahedral geometry and coordinates to the four N atoms from the two 2,2′-bipyridine ligands and the two N atoms from the two acetonitrile groups with Ru–N distances of 2.04(1)–2.062(1) Å. It is particularly worth mentioning that the stronger field (relative to bpy) C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N ligands are introduced into RuW-1, which will influence the electron configuration of the centre ion, Ru(II), and cause further differences in the photophysical and photochemical properties between RuW-1 and RuW-2.
N ligands are introduced into RuW-1, which will influence the electron configuration of the centre ion, Ru(II), and cause further differences in the photophysical and photochemical properties between RuW-1 and RuW-2.
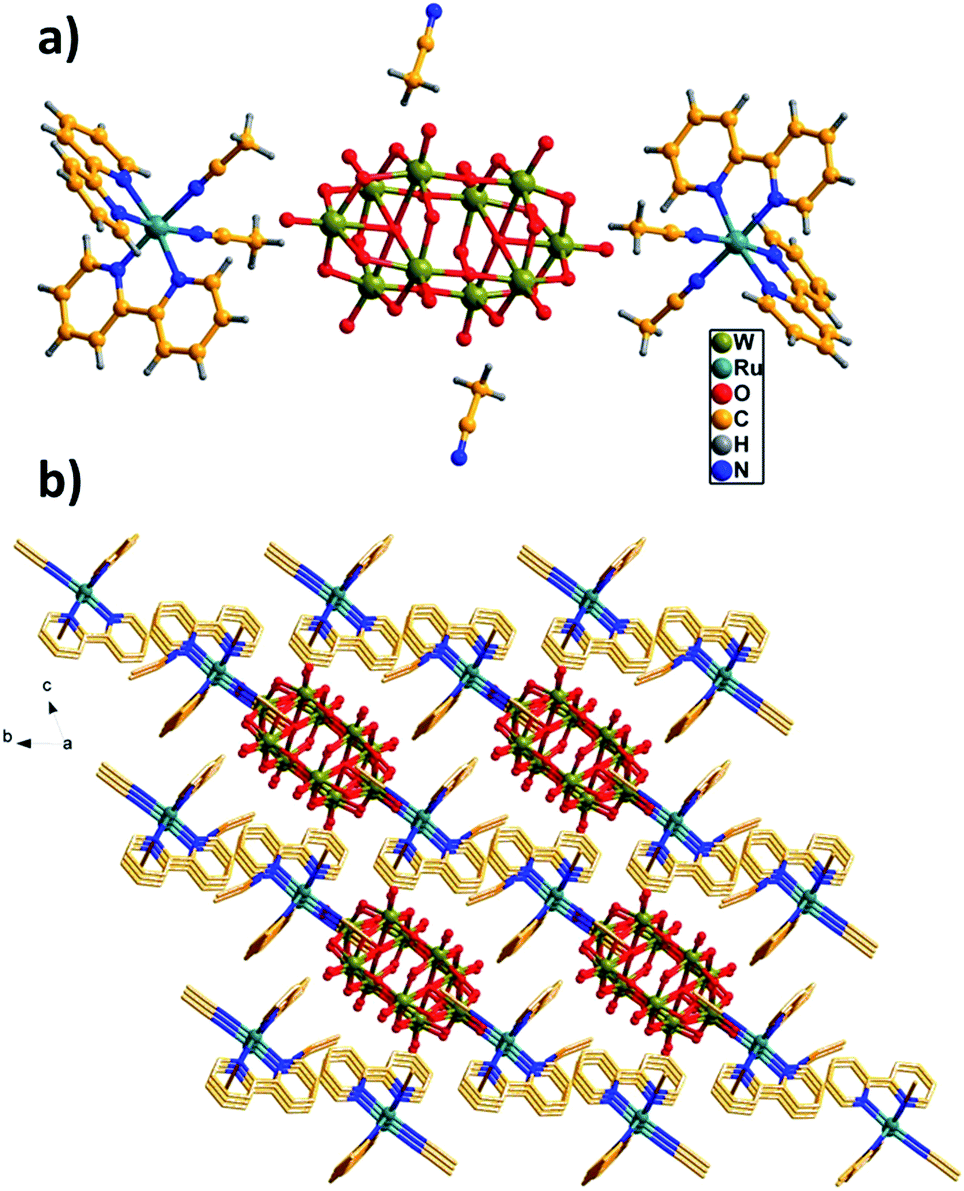 | ||
| Fig. 1 (a) Unit cell structure of RuW-1. (b) π–π interactions of the bpy ligands with the adjacent molecules. The free CH3CN molecules are located in the channels. | ||
As shown in Fig. 1b, these molecules are close-packed in the solid-state via the offset π–π interactions between the neighboring benzene of the bpy ligands with the shortest inter-ring separation of benzene of 3.45 Å leading to a 3D supramolecular network, which plays a crucial role in stabilizing the solid state structure.
Characterizations
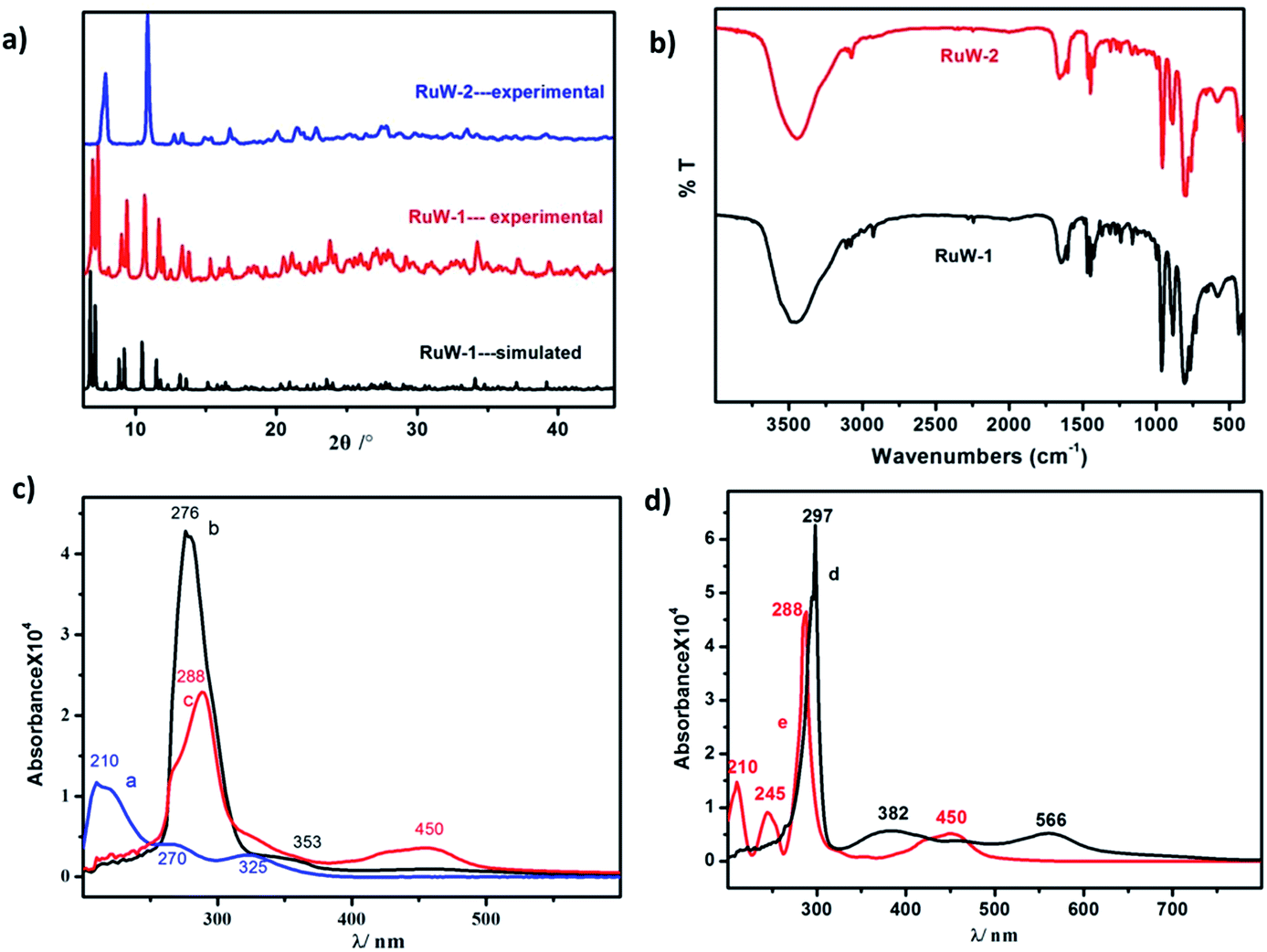 | ||
| Fig. 2 (a) PXRD pattern of RuW-1 and RuW-2, its calculated pattern is based on the single-crystal solution. It is shown that the seven intense peaks ranging from 5 to 11°, (010), (001), (011), (100), (101), (110) and (111) of RuW-1, were nearly identical. (b) IR spectra of RuW-1 and RuW-2. (c) UV-Vis absorption spectra of K4W10O32 (curve a), RuW-1 (curve b) and RuW-2 (curve c). (d) UV-Vis absorption spectra of precursor Ru(bpy)2Cl2 (curve d) and Ru(bpy)3I2 (curve e). | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od (958 cm−1), W–Ob–W (890 cm−1) and W–Oc–W (803 cm−1).19 The vibrational peak of RuW-1 is similar to RuW-2 except for the weak peaks around 2250 cm−1 and 2921 cm−1, which were assigned to the stretching of CN, and the stretching vibration of –CH3, respectively.20NCH3, the absorption band of 353 nm shows a blue shift from 382 nm. Compared with the precursor, for K4W10O32 (Fig. 2c, curve a), Ru(bpy)2Cl2 (Fig. 2d, curve d) and Ru(bpy)3I2 (Fig. 2d, curve e), the bands at 288 nm for RuW-1 and RuW-2 were obviously broadened due to overlapping with the band of the bridging oxygen-to-tungsten charge transfer, which also is the characteristic absorbing peak of K4W10O32.23 The UV-Vis spectra show that the combination of the photosensitizer, Ru(bpy)32+, and K4W10O32 can broaden absorption in the visible region, which will be beneficial to enhance the utilization of sunlight and improve the activity of the photocatalyst, K4W10O32.24NCH3 is an unconjugated ligand, which affects its fluorescence emission ability.
Od (958 cm−1), W–Ob–W (890 cm−1) and W–Oc–W (803 cm−1).19 The vibrational peak of RuW-1 is similar to RuW-2 except for the weak peaks around 2250 cm−1 and 2921 cm−1, which were assigned to the stretching of CN, and the stretching vibration of –CH3, respectively.20NCH3, the absorption band of 353 nm shows a blue shift from 382 nm. Compared with the precursor, for K4W10O32 (Fig. 2c, curve a), Ru(bpy)2Cl2 (Fig. 2d, curve d) and Ru(bpy)3I2 (Fig. 2d, curve e), the bands at 288 nm for RuW-1 and RuW-2 were obviously broadened due to overlapping with the band of the bridging oxygen-to-tungsten charge transfer, which also is the characteristic absorbing peak of K4W10O32.23 The UV-Vis spectra show that the combination of the photosensitizer, Ru(bpy)32+, and K4W10O32 can broaden absorption in the visible region, which will be beneficial to enhance the utilization of sunlight and improve the activity of the photocatalyst, K4W10O32.24NCH3 is an unconjugated ligand, which affects its fluorescence emission ability.
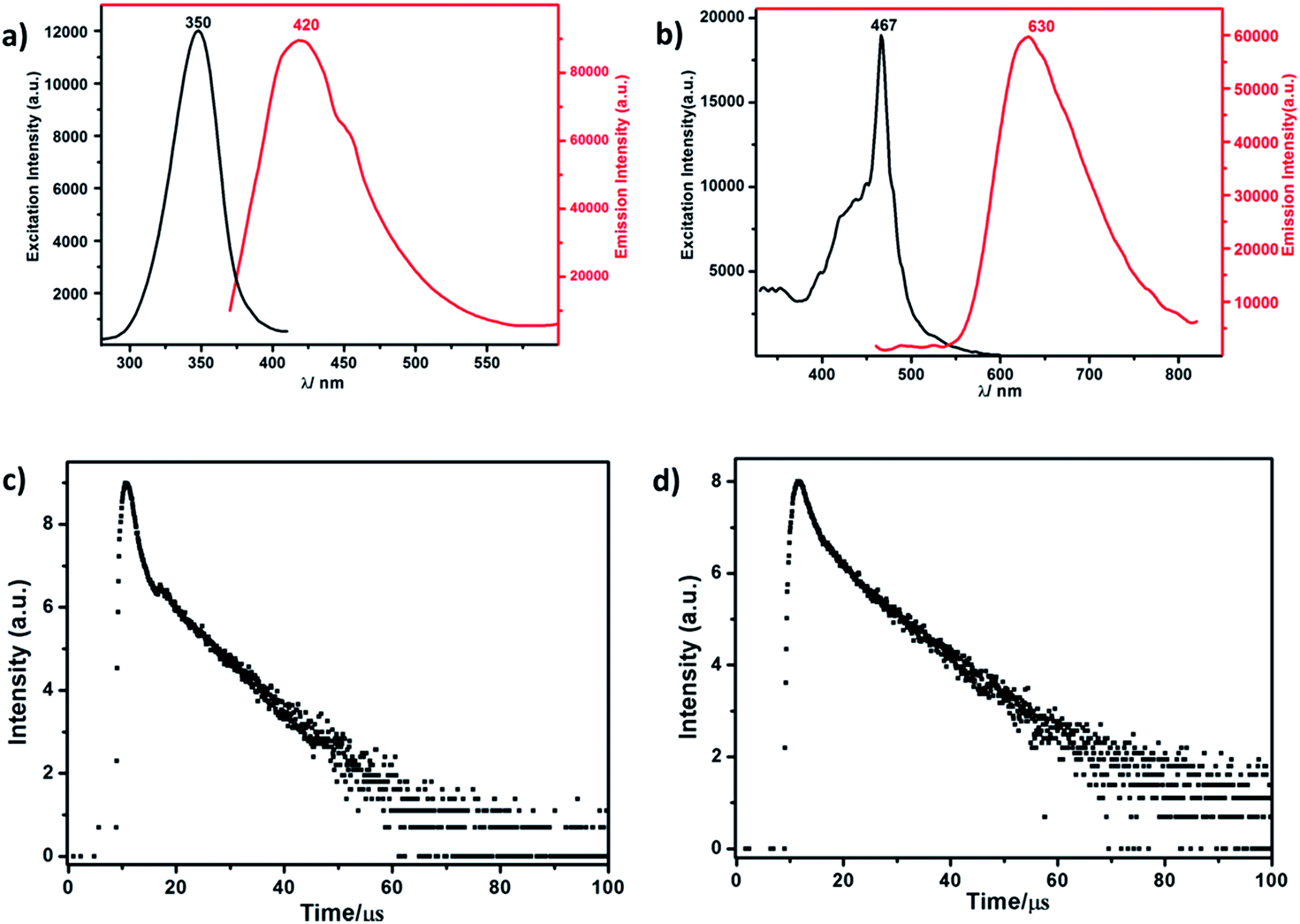 | ||
| Fig. 3 (a) The excitation and emission spectrum of RuW-1. (b) The excitation and emission spectrum of RuW-2. (c) The luminescence decay curve of RuW-1. (d) The luminescence decay curve of RuW-2. | ||
The lifetime curve of RuW-1 can be well fitted to a double exponential function I = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) (where I represents the luminescent intensity, t is the time, τ1 and τ2 are the fast and slow components of the luminescence lifetimes, and A1 and A2 are the pre-exponential factors) affording the luminescence lifetimes τ1 and τ2 as 0.89 μs (37.0%) and 8.06 μs (63.0%), and an agreement factor (χ2) of 1.265 (Fig. 3c). The luminescence lifetime can be calculated by the following equations:
exp(−t/τ1) + A2exp(−t/τ2) (where I represents the luminescent intensity, t is the time, τ1 and τ2 are the fast and slow components of the luminescence lifetimes, and A1 and A2 are the pre-exponential factors) affording the luminescence lifetimes τ1 and τ2 as 0.89 μs (37.0%) and 8.06 μs (63.0%), and an agreement factor (χ2) of 1.265 (Fig. 3c). The luminescence lifetime can be calculated by the following equations:
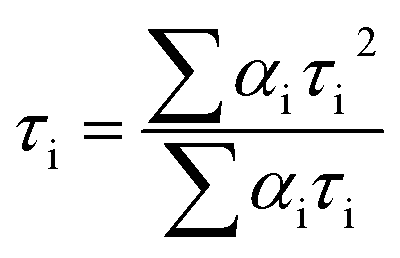
The luminescence of RuW-1 has a lifetime around 7.50 μs at room temperature, which is in agreement with the value 7.80 μs of the precursor Ru(bpy)2Cl2, where τ1 = 0.88 μs (29.4%) and τ2 = 8.11 μs (70.6%). The luminescence decay curve of RuW-2 is also carried out, which can be fitted to a triple exponential function I = A1exp(−t/τ1) + A2exp(−t/τ2) + A3exp(−t/τ3). The fitted luminescence lifetimes τ1, τ2, and τ3 are 1.35 μs (15.9%), 3.75 μs (17.5%) and 10.83 μs (66.6%), respectively, and the agreement factor (χ2) is 1.235 (Fig. 3d). The luminescence has a lifetime of around 10.02 μs at room temperature. Compared to the lifetime of 6.22 μs of the precursor Ru(bpy)3I2, where τ1 = 0.89 μs (62.6%) and τ2 = 7.31 μs (37.4%), the lifetime of RuW-2 is longer.
Nanosecond time-resolved transient difference absorption spectra
The charge-separated state can be detected using a transient absorption spectrum, 20 ns after excitation of a deoxygenated solution of photochromic compounds in CH3CN alone, with a nanosecond laser pulse (Fig. 4a and b). Because the maximum ultraviolet absorption of W10O324− is 325 nm, we selected an excitation wavelength of 350 nm to stimulate the bipyridine–Ru(II) and guarantee W10O324− was free from stimulation. For RuW-2, the difference spectra associated with the species consist of three contributions: the negative bands at 445 nm were assigned as Ru2+ ground state bleaching signals, which indicates the formation of Ru(bpy)33+.26 The other negative bands at 638 nm were assigned as stimulated emission signals, which are well consistent with the fluorescence spectrum of RuW-2.26 The positive bands at 360 nm are very similar to the ground-state absorption of reduced bipyridine, and therefore are assigned to transitions of a reduced bipyridine.26,27 The bands at 320 nm were assigned to the ground state of W10O324−.28 The absorbance at 750 nm may be associated with the formation of the heteropoly blue W10O325− anion. The follow-up spectral changes were characteristic of the formation of a charge-separated state Ru(bpy)33+–W10O325−, indicating a photoinduced intramolecular electron-transfer from the excited state of the *Ru(bpy)32+ moiety to the W10O324− moiety. In comparison, RuW-1 also has excited state transitions around 320 and 750 nm with roughly the same shape, but the bands were assigned to transitions of decatungstate.28 The 530 nm band is associated with the depletion of the Ru(dπ)–bpy(π*) MLCT and the formation of bpy˙− radical states after charge transfer.24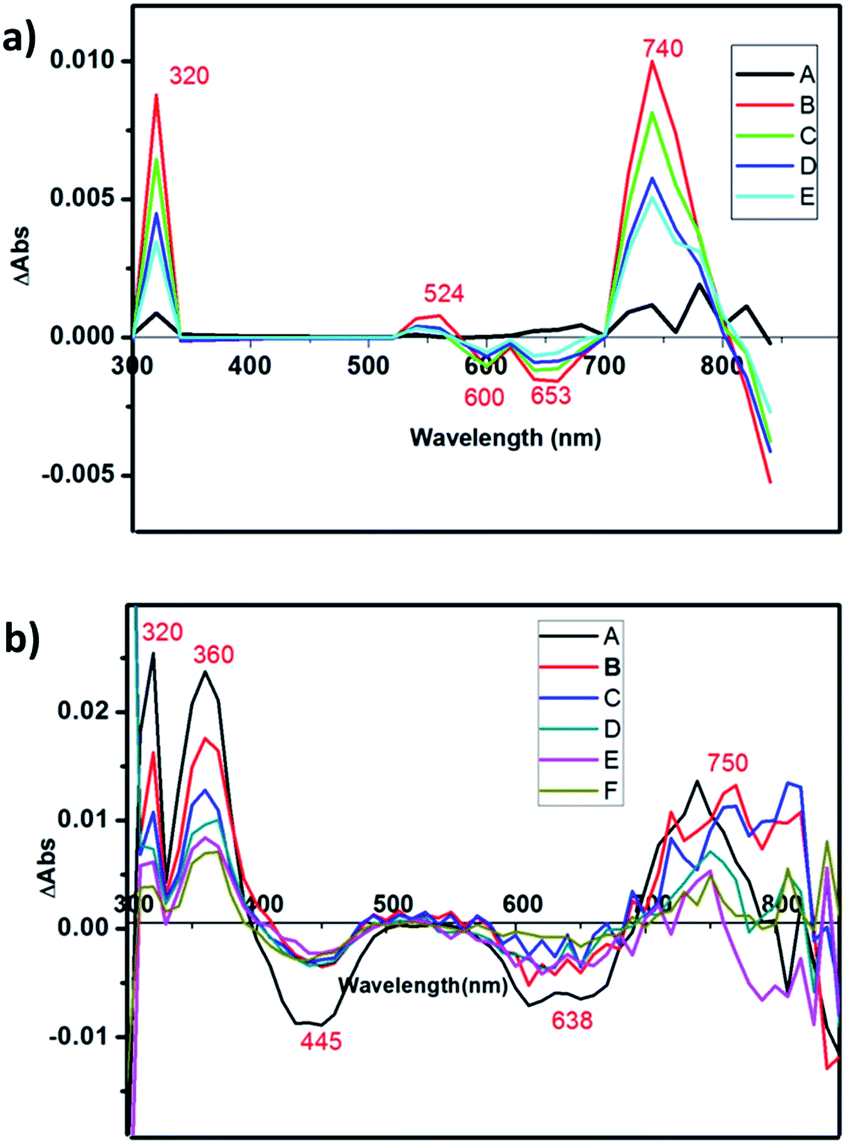 | ||
| Fig. 4 (a) Nanosecond time-resolved transient difference absorption spectra of RuW-1 in CH3CN at room temperature, with data recorded at delays: A-4000 ns; B-4450 ns; C-4675 ns; D-4900 ns; E-5125 ns following excitation by a 350 nm laser pulse. (b) Nanosecond time-resolved transient difference absorption spectra of RuW-2 at room temperature, with data recorded at delays: A-1960 ns, B-2133 ns, C-2306 ns, D-2480 ns, E-2566 ns, F-2783 ns following excitation by a 350 nm laser pulse. | ||
Photocatalytic activity
Because of the strong absorption in the visible-light region and excellent stability under various pH conditions, RhB was chosen as the primary model dye pollutant to examine the photodegradation behaviors of the hybrid RuW-1 and RuW-2 under visible light and UV/visible light. The degradation reactions were carried out after the establishment of the adsorption/desorption equilibrium by magnetic stirring for 30 min in the dark. Under UV/visible light irradiation, the blank experiment showed only weak degradation of RhB after 12 h (Fig. 5a). When in the presence of K4W10O32 as the homogenous catalyst, the degradation efficiency of RhB increased and reached 96.7% after 12 h under UV/visible light (Fig. 5b). It is worth mentioning that when in the presence of RuW-1 (Fig. 5c) and RuW-2 (Fig. 5d), the degradation efficiency was improved significantly. The degradation efficiencies were 91.9% after 10 h and 98.2% after 6 h for RuW-1 and RuW-2, respectively. In the dark, the RuW-2 showed low photocatalytic activity toward RhB (Fig. 5e). However, the visible light irradiation could also lead to rapid degradation of the dye in the presence of RuW-2, which is comparable with the UV/visible light irradiation (Fig. 5f).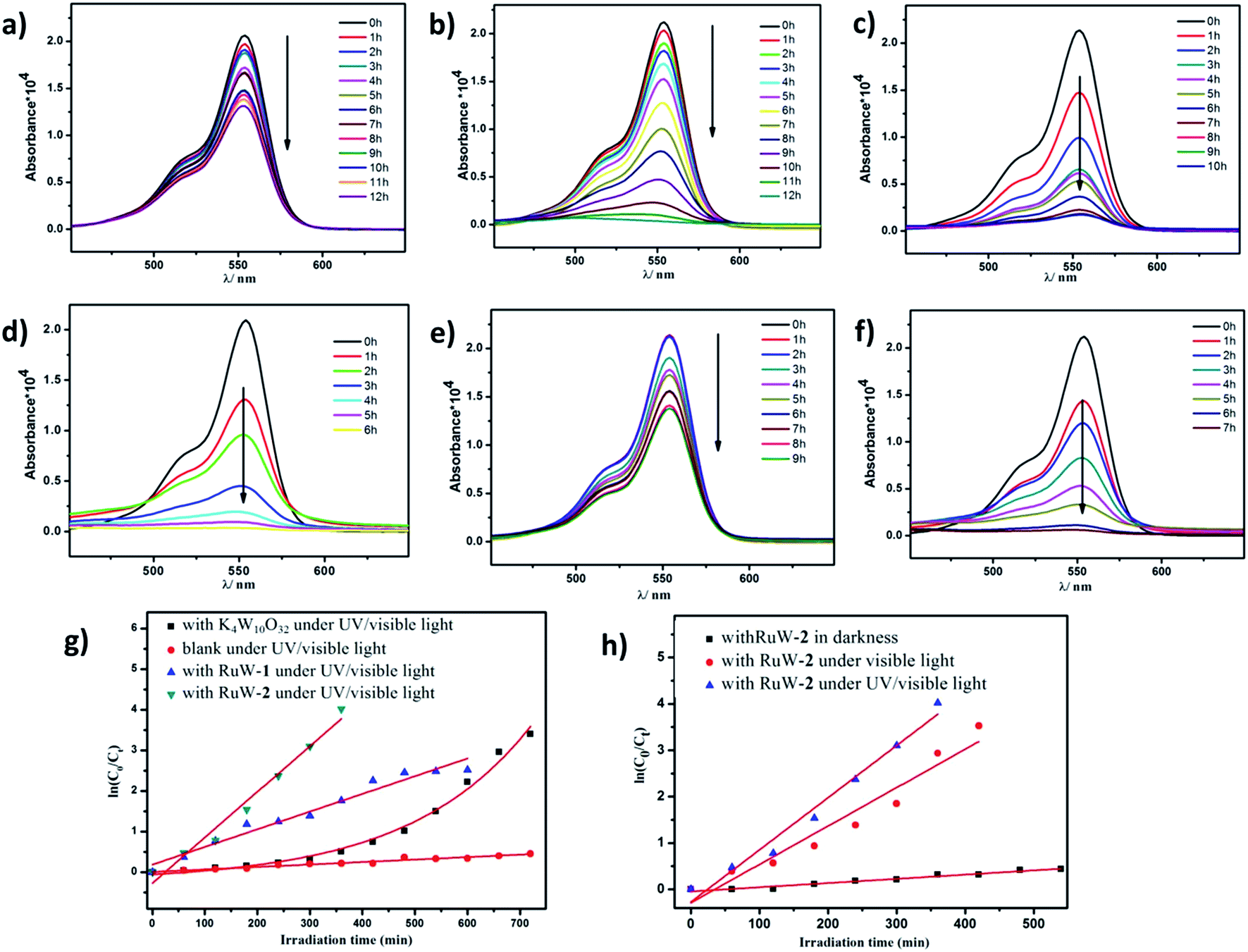 | ||
| Fig. 5 Degradation of RhB in the different reaction systems: (a) blank under UV/visible light; (b) with K4W10O32 under UV/visible light; (c) with RuW-1 under UV/visible light; (d) with RuW-2 under UV/visible light; (e) with RuW-2 in darkness; (f) with RuW-2 under visible light; (g) kinetics equation of RhB degradation by different photocatalyst under UV/visible light; (h) kinetics equation of RhB degradation by RuW-2 irradiated by different light. | ||
The kinetics of RhB photodegradation by the RuW-1 and RuW-2 can be described via the first-order equation (Fig. 5g and h). Fig. 5g shows the linear relationship of ln(C0/Ct) versus the irradiation time for RhB under UV/visible light by different catalysts. All the reaction rate constants are recorded in Table 2. The kinetics constants (kr) of RuW-1 and RuW-2 under the UV/visible light are 4.4 × 10−3 min−1 and 11.3 × 10−3 min−1, respectively. The reaction rate constant of the blank experiment is 0.6 × 10−3 min−1. The experimental result of RhB photodegradation by K4W10O32 shows that the relationship between the RhB concentration and irradiation time is linear, and the reaction order is zero, which may be ascribed to the homogeneous catalytic system. In addition, the kinetics equations of the Rhb photocatalytic degradation by RuW-2 under different light are displayed in Fig. 5h. From the slope of the linear relationship, the reaction rate constants are 8.3 × 10−3 min−1 under visible light and 0.9 × 10−3 min−1 under darkness. The results indicate that not only does the light irradiation play a key role in the degradation, but also polypyridyl–ruthenium as a sensitizer plays an important role in enhancing the photocatalytic activity of K4W10O32 in the visible region. Additionally, the crystals of RuW-1 and RuW-2 were easily isolated from the reaction suspension by filtration alone and reused three times, displaying only a slight decrease in the activity. These results suggest that they are potential heterogenous photocatalysts for reduction of some other organic dyes.
| Catalysts | Irradiation effect | Reaction order | kr(min−1) |
|---|---|---|---|
| Blank | UV/vis | 1 | 0.61 × 10−3 |
| K4W10O32 | UV/vis | 0 | (C0 − Ct)/t |
| RuW-1 | UV/vis | 1 | 4.36 × 10−3 |
| RuW-2 | UV/vis | 1 | 11.25 × 10−3 |
| RuW-2 | Vis | 1 | 8.27 × 10−3 |
| RuW-2 | Darkness | 1 | 0.91 × 10−3 |
Conclusions
In summary, two water-insoluble hybrid photocatalysts, RuW-1 and RuW-2, have been synthesized using decatungstate as an electron acceptor unit and polypyridyl ruthenium coordination compounds as the sensitizers. The photocatalysts exhibit good UV/visible light photocatalytic activities for dye degradation and can be recovered and recycled by a simple procedure. In addition, [Ru(bpy)3]2+ and its derivatives show great potential applications in the fields of photocatalysis and photoinduced water splitting due to their interesting photochemical properties. In the future, we expect to synthesize new solid materials with enhanced photochemical properties, in which the Ru complexes are linked to the POM groups through coordination bonds.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21601048, 21671055 and U1304201), the China Postdoctoral Science Foundation (2015M580626), the Natural Science Foundation Project of Henan province (162102210171, 162300410012), and the State Key Laboratory of Fine Chemicals (KF1602).Notes and references
-
(a) M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS
; (b) I. M. Banat, P. Nigam, D. Singh and R. Marchant, Bioresour. Technol., 1996, 58, 217 CrossRef CAS
-
(a) O. Carp, C. L. Huisman and A. Reller, J. Solid State Chem., 2004, 32, 33 CrossRef CAS
-
(a) A. Mylonas, A. Hiskia, E. Androulaki, D. Dimotikali and E. Papaconstantinou, Phys. Chem. Chem. Phys., 1999, 1, 437 RSC
-
(a) S. Kim, J. Yeo and W. Choi, Appl. Catal., B, 2008, 84, 148 CrossRef CAS
-
(a) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609 RSC
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS
-
(a) A. Sartorel, M. Carraro, F. M. Toma, M. Prato and M. Bonchio, Energy Environ. Sci., 2012, 5, 5592 RSC
-
(a) S. Schönweiz, S. A. Rommel, J. Kübel, M. Micheel, B. Dietzek, S. Rau and C. Streb, Chem.–Eur. J., 2016, 22, 12002 CrossRef PubMed
- Z. B. Han, E. B. Wang, G. Y. Luan, Y. G. Li, C. W. Hu, P. Wang, N. H. Hu and H. Q. Jia, Inorg. Chem. Commun., 2001, 4, 427 CrossRef CAS
- B. Yan, S. A. Hodsdon, Y. F. Li, C. N. Carmichael, Y. Cao and W. P. Pan, J. Solid State Chem., 2011, 184(12), 3179 CrossRef CAS
- J. Song, Z. Luo, H. Zhu, Z. Huang, T. Lian, A. Kaledin, D. G. Musaev, S. Lense, K. I. Hardcastle and C. L. Hill, Inorg. Chim. Acta, 2010, 363(15), 4381 CrossRef CAS
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3335 Search PubMed
- R. A. Palmer and T. S. Piper, Inorg. Chem., 1966, 5, 864 CrossRef CAS
- A. Chemseddine, C. Sanchez, J. Livage, J. P. Launay and M. Fournier, Inorg. Chem., 1984, 23, 2609 CrossRef CAS
- G. M. Sheldrick, SHELXTL97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed
- SMART Data collection software (version 5.629), Bruker AXS Inc., Madison, WI, 2003 Search PubMed
- SAINT, Data reduction software (version 6.45), Bruker AXS Inc., Madison, WI, 2003 Search PubMed
- L. H. Bi, H. Y. Wang, Y. Shen, E. K. Wang and S. J. Dong, Electrochem. Commun., 2003, 5, 913 CrossRef CAS
- M. Bonchioa, M. Carraroa, M. Gardana, G. Scorranoa, E. Driolib and E. Fontananovab, Top. Catal., 2006, 40, 133 CrossRef
- A. A. Eigner, J. A. Rohde, C. C. Knutson and J. A. Phillips, J. Phys. Chem. B, 2007, 111, 1402 CrossRef CAS PubMed
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1982, 21, 3967 CrossRef CAS
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 15089 CAS
- S. C. Termes and M. T. Pope, Inorg. Chem., 1978, 17, 500 CrossRef CAS
- T. E. Keyes, E. Gicquel, L. Guerin, R. J. Forster, V. Hultgren, A. M. Bond and A. G. Wedd, Inorg. Chem., 2003, 42, 7897 CrossRef CAS PubMed
- S. D. Inglez, F. C. A. Lima, A. B. F. Silva, A. R. Simioni, A. C. Tedesco, J. F. S. Daniel, B. S. Lima-Neto and R. M. Carlos, Inorg. Chem., 2007, 46, 5744 CrossRef CAS PubMed
- S. Wallin, J. Davidsson, J. Modin and L. Hammarström, J. Phys. Chem. A, 2005, 109, 4697 CrossRef CAS PubMed
- B. A. McClure and J. J. Rack, Inorg. Chem., 2011, 50, 7586 CrossRef CAS PubMed
- D. C. Duncan and M. A. Fox, J. Phys. Chem. A, 1998, 102, 4559 CrossRef CAS
Footnote |
| † CCDC 1530386. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01342f |
| This journal is © The Royal Society of Chemistry 2017 |