
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into high temperature hydrothermal synthesis in the preparation of visible-light active, ordered mesoporous SiO2–TiO2 composited photocatalysts†
Qin Wu,
Chen Liu,
Jinjun Peng and
Fujian Liu *
*
College of Chemistry & Chemical Engineering, Shaoxing University, Shaoxing, Zhejiang 312000, China. E-mail: fjliu@usx.edu.cn
First published on 3rd April 2017
Abstract
Visible-light active, carbon doped and ordered mesoporous SiO2–TiO2 nanocomposites (C-OMS@TiO2-xs) were synthesized from the self-assembly of Si and Ti precursors with block copolymer templates (F127 and P123), hydrothermally treated at 180 °C, and calcination or extraction for removing the templates. XRD patterns and TEM images indicate highly ordered mesopores were formed in C-OMS@TiO2-xs, and TiO2 species with a high crystalline degree were homogeneously dispersed into the samples, which formed a stable “brick-and-mortar” nanostructure. N2 adsorption–desorption isotherms indicate very large BET surface areas (224–553 m2 g−1) and pore volumes (0.45–1.4 cm3 g−1) in these samples. EDX spectra and elemental maps further confirm successful doping and homogeneous dispersion of TiO2 nanocrystals in C-OMS@TiO2-xs. XPS spectra confirmed the doping of carbon atoms into the lattice of TiO2 species under high temperature hydrothermal conditions, which results in C-OMS@TiO2-xs with enhanced visible light response properties, as confirmed by UV-vis diffuse reflectance spectra and photocurrent–time (I–t) curves. Catalytic tests indicate that the synthesized C-OMS@TiO2-xs show excellent activities for degradation of organic pollutants irradiated with visible light. For instance, degradation of highly concentrated rhodamine B (RhB, 100 ppm) can be completely catalyzed in the water within 50 min. This work provides a facile approach to the targeted synthesis of visible active and ordered mesoporous materials.
Introduction
TiO2 and its derived materials have received considerable attention because of their wide applications in the areas of water purification, solar cells, hydrogen production, photoelectric conversion and photocatalysis,1–16 which show characteristics such as good chemical stability, nontoxicity and low cost.1–19 It is well known that the structural characteristics such as crystallinity, morphology, and surface areas strongly affect their functionality.2,20,21 In general, TiO2 based materials with large BET surface areas and good crystallinity are always desired, which could largely enhance the degree of exposure and accessibility of anchored Ti sites to various guest molecules.2,20,21 These structural characteristics could largely enhance their performances in various research fields.One of the most important applications of TiO2 based materials is photocatalysis. However, TiO2 is only sensitive to UV-light due to its large band gap (3.2 eV) in nature, which must be excited by UV-light. However, UV-light accounts for rather limited content of solar radiation energy.22–24 Therefore, extended photocatalytic response of traditional TiO2 materials into visible light region and properly reduce charge-carrier recombination rate would greatly promote their practical applications.22–28 Up to now, several methods have been used to decrease the band gap energy of TiO2, including defects generation by H2 reduction,29–31 doping by transition metal32,33 and nonmetallic ions (S, N, C etc.) into TiO2 lattice.33–35 However, the reported modification processes were usually performed under rather complicated or harsh conditions.27,32–37 Meanwhile, the reported visible light active TiO2 based photocatalysts usually show poor porosity or nanocrystalline morphology, which lead to the drawbacks such as low exposure degree of catalytically active sites, easily sintering and complicated separation processes from the reaction mixtures.2,8,21,31–36
The preparation of mesoporous TiO2 monolith with micrometer sizes could overcome the above problems. However, the mesostructures collapse and lose most of the porosity after being heated up to 300–350 °C because of the intrinsical crystallization of anatase phase.2,21,38–40 An alternative way is to immobilize TiO2 nanocrystals into ordered mesoporous materials, which usually show enhanced stability of mesostructures at high temperatures.2,21,37 Notably, ordered mesoporous materials show unique structural characteristics including controllable morphology, large BET surface areas, versatile frameworks, ordered and uniform mesopores, which strongly enhance the accessibility of catalytically active sites and simplify their regeneration processes.2,21,37,41–43 For instance, Zhao and coworkers successfully immobilized active TiO2 nanocrystals onto the networks of ordered mesoporous TiO2 and carbon, which showed much improved photocatalytic activities for degradation of organic dyes in comparison with commercial P25 irradiated with UV-light.2,21 However, it remains a challenge to synthesize ordered mesoporous TiO2 based photocatalysts with visible-light response.
In this work, we innovatively developed high temperature (180 °C) hydrothermal technology to the synthesis of carbon doped, ordered mesoporous SiO2–TiO2 nanocomposites (C-OMS@TiO2-xs), the ordered mesostructure was formed from self assembly of block copolymer templates with Si, Ti precursors. Interestingly, the unique high temperature (180 °C) hydrothermal technology results in the doping of carbon atom into TiO2 lattice, which makes the synthesized C-OMS@TiO2-xs are sensitive to visible light due to the changes of band gap of TiO2 nanocrystals in these samples. Notably, this phenomenon can not be found in other ordered mesoporous TiO2 based nanocomposited materials.2,21 The usage of hydrothermal technology induced modification of TiO2 materials with heteroatom in order to adjust their band gap have not been reported previously. Combination of the characteristics of large BET surface areas, ordered and uniform mesopores and visible light adsorption property, C-OMS@TiO2-xs show excellent activities and good reusability for degradation of organic pollutants such as rhodamine B (RhB), methyl orange (MO) and p-nitrophenol (PNP) irradiated with visible light.
Results and discussion
The structural ordering of synthesized C-OMS@TiO2-xs was examined by small angle X-ray diffraction (XRD), as shown in Fig. 1. The as synthesized F127–C-OMS@TiO2-4.0 shows three peaks associated with (110), (200), and (211) reflections at the 2θ range of 0.5–2°, indicating the presence of highly ordered body-centered cubic mesostructure (Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) in the sample. After calcination, the diffraction peaks associated with (110) and (200), and (211) reflections become more resolved at the 2θ range of 0.5–2°. The as synthesized F127–C-OMS@TiO2-4.0 and F127–C-OMS@TiO2-4.0 show similar d-spacing values (d110 = 11.13 & 11.40 nm), corresponding to the unit cell parameters of 15.74 & 16.12 nm. Similarly, P123–C-OMS@TiO2-4.0 shows two peaks associated with (100) and (110) reflections at the 2θ range of 0.5–2°, indicating the presence of highly ordered mesopores with 2D hexagonal mesostructures (p6mm), which gives the d100-spacing values at 8.91 nm. The different mesostructures and d100-spacing values of F127–C-OMS@TiO2-4.0 and P123–C-OMS@TiO2-4.0 should be attributed to their different structures of block copolymer templates.
m) in the sample. After calcination, the diffraction peaks associated with (110) and (200), and (211) reflections become more resolved at the 2θ range of 0.5–2°. The as synthesized F127–C-OMS@TiO2-4.0 and F127–C-OMS@TiO2-4.0 show similar d-spacing values (d110 = 11.13 & 11.40 nm), corresponding to the unit cell parameters of 15.74 & 16.12 nm. Similarly, P123–C-OMS@TiO2-4.0 shows two peaks associated with (100) and (110) reflections at the 2θ range of 0.5–2°, indicating the presence of highly ordered mesopores with 2D hexagonal mesostructures (p6mm), which gives the d100-spacing values at 8.91 nm. The different mesostructures and d100-spacing values of F127–C-OMS@TiO2-4.0 and P123–C-OMS@TiO2-4.0 should be attributed to their different structures of block copolymer templates.
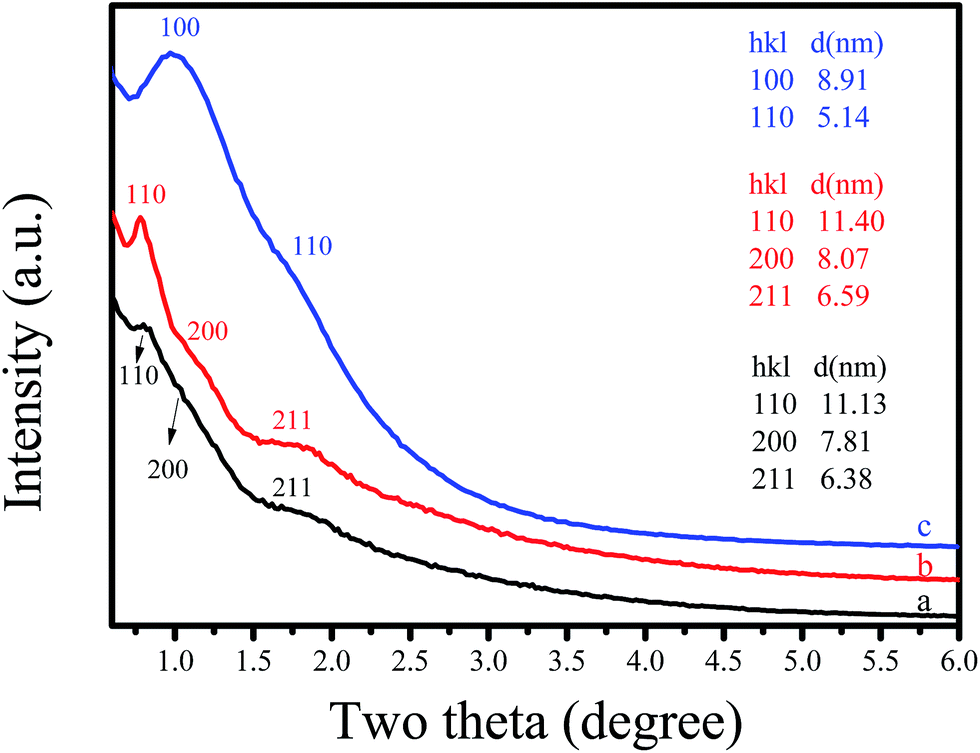 | ||
| Fig. 1 Small-angle XRD pattern of (a) as synthesized F127–C-OMS@TiO2-4.0, (b) F127–C-OMS@TiO2-4.0 and (c) P123–C-OMS@TiO2-4.0. | ||
Fig. 2 shows wide angle XRD patterns of various C-OMS@TiO2-xs and F127–TiO2 synthesized under the same conditions. Notably, as synthesized C-OMS@TiO2-xs exhibit several weak peaks indexed as 101, 004, 200, 105, 211 and 204 reflections, which are assigned to typical anatase phase characteristics. After calcination to remove templates, the characteristic peaks associated with anatase phase become more resolved.4,9,42 Similar result could also be observed in F127–C–TiO2, which suggests the highly crystalline degree of TiO2 active species in both C-OMS@TiO2-xs and F127–C–TiO2. Except for anatase phase, the weak peaks at around 2θ = 27.4, 55.1 and 70.2° associated with rutile phase could also be observed in C-OMS@TiO2-xs and F127–C–TiO2,4,9,42 which exists in the calcinated samples, suggesting polymorphism phenomenon of crystalline TiO2 in these samples.
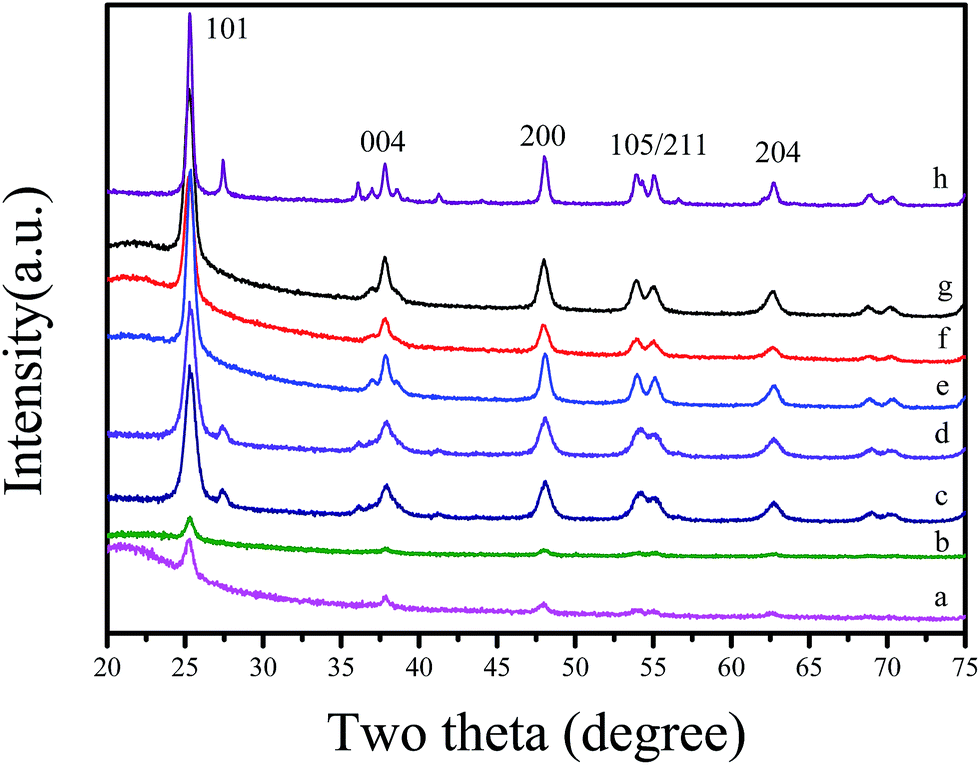 | ||
| Fig. 2 Wide-angle XRD pattern of (a) as synthesized F127–C-OMS@TiO2-4.0, (b) as synthesized P123–C-OMS@TiO2-4.0, (c) F127–C-OMS@TiO2-2.0, (d) F127–C-OMS@TiO2-4.0, (e) F127–C-OMS@TiO2-6.0, (f) F127–C-OMS@TiO2-8.0, (g) P123–C-OMS@TiO2-4.0 and (h) F127–C–TiO2. | ||
Fig. 3 shows the N2 adsorption–desorption isotherms at −196 °C and Barrett–Joyner–Halenda (BJH) pore size distributions of synthesized C-OMS@TiO2-xs. The C-OMS@TiO2-xs show typical type-IV isotherms with low N2 uptake at low relative pressure, and high N2 uptake at high relative pressure, suggesting presence abundant mesopores in these samples. The N2 isotherms of C-OMS@TiO2-xs display steep increase at the relative pressure of 0.75–0.99, indicating relatively narrow pore size distributions centered at 11.1–54.2 nm. The calculated textural properties of synthesized C-OMS@TiO2-xs are summarized in Table 1. The BET surface areas and total pore volumes of C-OMS@TiO2-xs are in the range of 224–553 m2 g−1 and 0.45–1.40 cm3 g−1, respectively. It is found that the pore sizes of C-OMS@TiO2-xs increase as the TiO2 amount increases (Fig. 3b), which exhibits linear relationship (Fig. S1†). This phenomenon can be explained by an enlargement of TiO2 nanocrystals onto the network of C-OMS@TiO2-xs, which results in the larger aggregated mesopores in these samples.
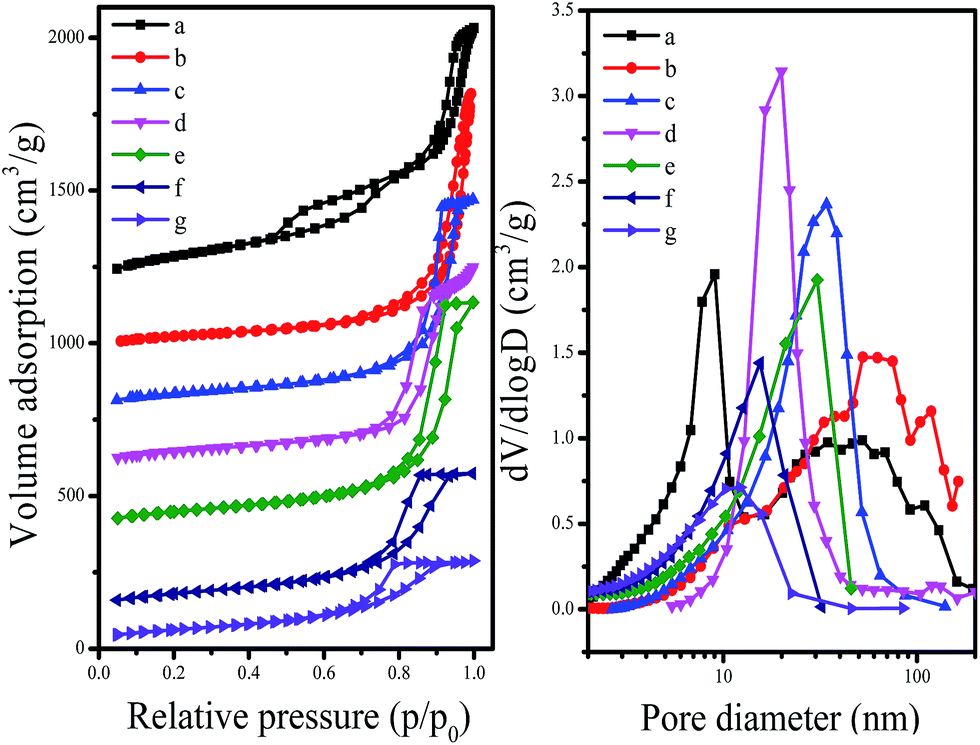 | ||
| Fig. 3 N2 isotherms at −196 °C and BJH pore size distributions of (a) F127–C-OMS@TiO2-9.0, (b) F127–C-OMS@TiO2-2.0, (c) F127–C-OMS@TiO2-4.0, (d) P123–C-OMS@TiO2-2.0, (e) F127–C-OMS@TiO2-6.0, (f) F127–C-OMS@TiO2-8.0 and (g) P123–C-OMS@TiO2-4.0. The isotherms of (a)–(f) are offset 1000, 900, 700, 500, 300 and 100 cm3 g−1, respectively. | ||
| Run | Samples | SBETa (m2 g−1) | Vta (cm3 g−1) | Dpb (nm) |
|---|---|---|---|---|
| a BET surface areas and pore volumes were estimated from N2 adsorption isotherms.b Pore size distribution was estimated from BJH model. | ||||
| 1 | F127–C-OMS@TiO2-2.0 | 230 | 1.28 | 54.2 |
| 2 | F127–C-OMS@TiO2-4.0 | 260 | 1.12 | 34.5 |
| 3 | F127–C-OMS@TiO2-6.0 | 303 | 1.19 | 29.1 |
| 4 | F127–C-OMS@TiO2-8.0 | 288 | 0.73 | 15.2 |
| 5 | F127–C-OMS@TiO2-9.0 | 553 | 1.40 | 8.6 |
| 6 | P123–C-OMS@TiO2-2.0 | 279 | 1.06 | 18.9 |
| 7 | P123–C-OMS@TiO2-4.0 | 224 | 0.45 | 11.1 |
Fig. 4 shows TEM and HR-TEM images of P123–C-OMS@TiO2-4.0 and F127–C-OMS@TiO2-4.0. P123–C-OMS@TiO2-4.0 shows highly ordered mesopores with hexagonal symmetry (Fig. 4a and b), and TiO2 nanocrystals species with uniform sizes were homogeneously dispersed onto the network of P123–C-OMS@TiO2-4.0 (Fig. 4b), which formed the ‘brick-and-mortar’ nanostructures. This is in agreement with small angle XRD results. Similarly, highly ordered mesopores with body centered structure (Imm) could also be found in F127–C-OMS@TiO2-4.0 (Fig. 4d). The high resolution TEM images show that the TiO2 species confined into nanopores of F127–C-OMS@TiO2-4.0 and P123–C-OMS@TiO2-4.0, exhibit several crystal lattice (Fig. 4c, e and f), which is in consistent with the wide angle XRD results. Furthermore, abundant mesopores of F127–C-OMS@TiO2-4.0 could also be confirmed by STEM image (Fig. 5a), which exhibits the signals of Ti, Si, C and O (Fig. 5b). In addition, the elemental maps indicate the signals of Ti, Si, C and O formed a “brick-and-mortar” nanostructure, which were homogeneously dispersed into F127–C-OMS@TiO2-4.0 (Fig. 5c–f).
 | ||
| Fig. 4 TEM images of calcinated P123–C-OMS@TiO2-4.0 (a–c) and F127–C-OMS@TiO2-4.0 (d–f). | ||
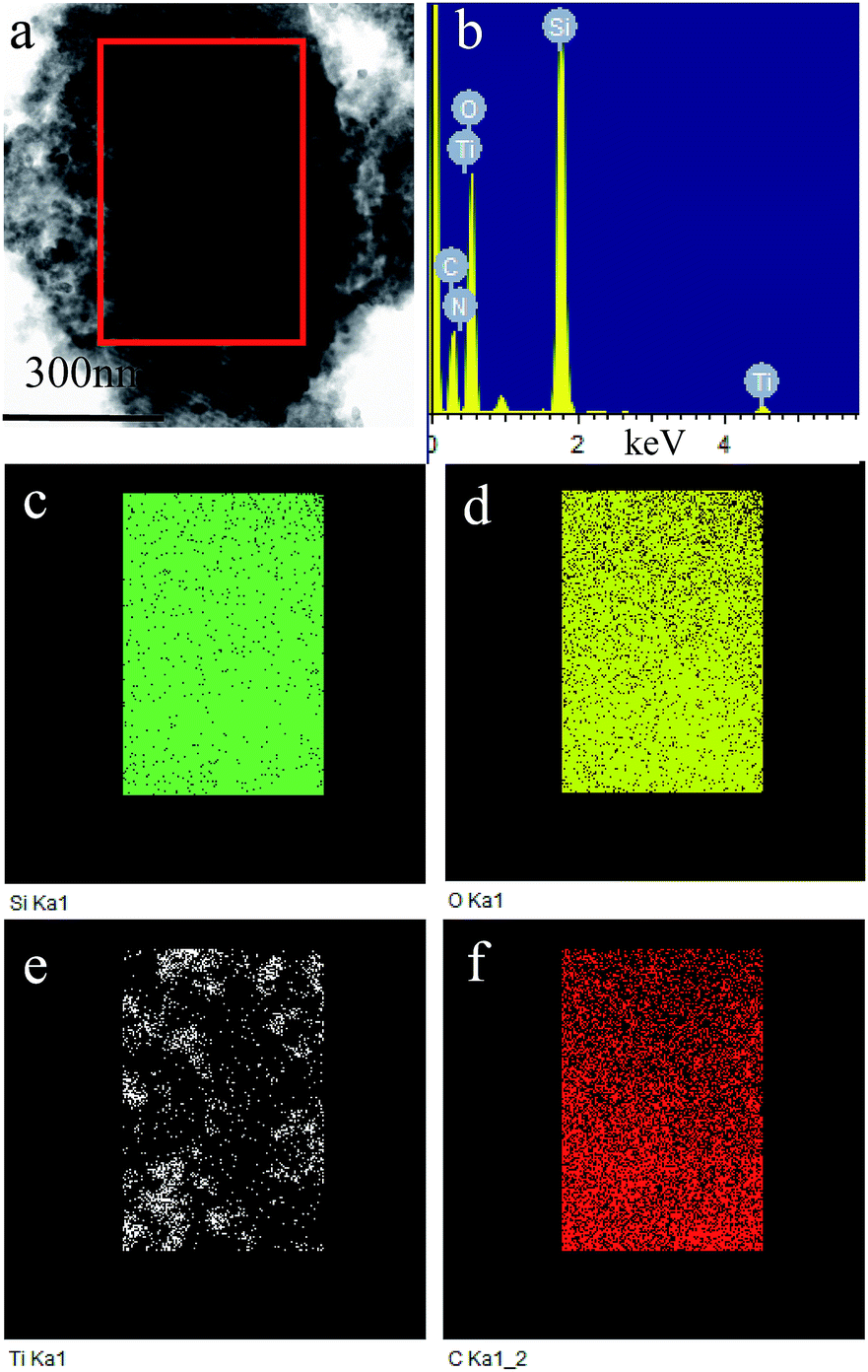 | ||
| Fig. 5 (a) STEM image, (b) EDX spectrum and (c–f) elemental maps of F127–C-OMS@TiO2-4.0. | ||
Fig. 6 shows XPS spectra of F127–C-OMS@TiO2-4.0 and as synthesized F127–C-OMS@TiO2-4.0, which exhibit the signals of C 1s, O 1s, Ti 2p and Si 2p. The peaks associated with Ti 2p are centered at around 458.5 and 464.3 eV, which are assigned to Ti4+ in TiO2 lattices. The peak associated with Si 2p is centered at around 103.8 eV, which is assigned to Si4+ in SiO2 species. The peaks associated with O 1s are centered at around 530.0 and 533.3 eV, which are assigned to the signals of Ti–O and Si–O, respectively.44 The peaks associated with C 1s can be fitted and deconvoluted into two peaks centered at around 284.8 and 286.7 eV, which are attributed to the signals of C–C and C–O bonds respectively. The formation of C–O bond should be attributed to the doping of C atom in the lattice of TiO2 under unique high temperature hydrothermal condition, which changes the band gap of TiO2 species in C-OMS@TiO2-xs. In addition, the C content of F127–C-OMS@TiO2-4.0 was also analyzed by elemental analysis, which shows C content of 0.8 wt%, further confirming the doping of C atom in OMS@TiO2-xs. The above results confirm the successful preparation of carbon doped C-OMS@TiO2-xs nanocomposited photocatalysts, in good agreement with EDX and elemental maps results.
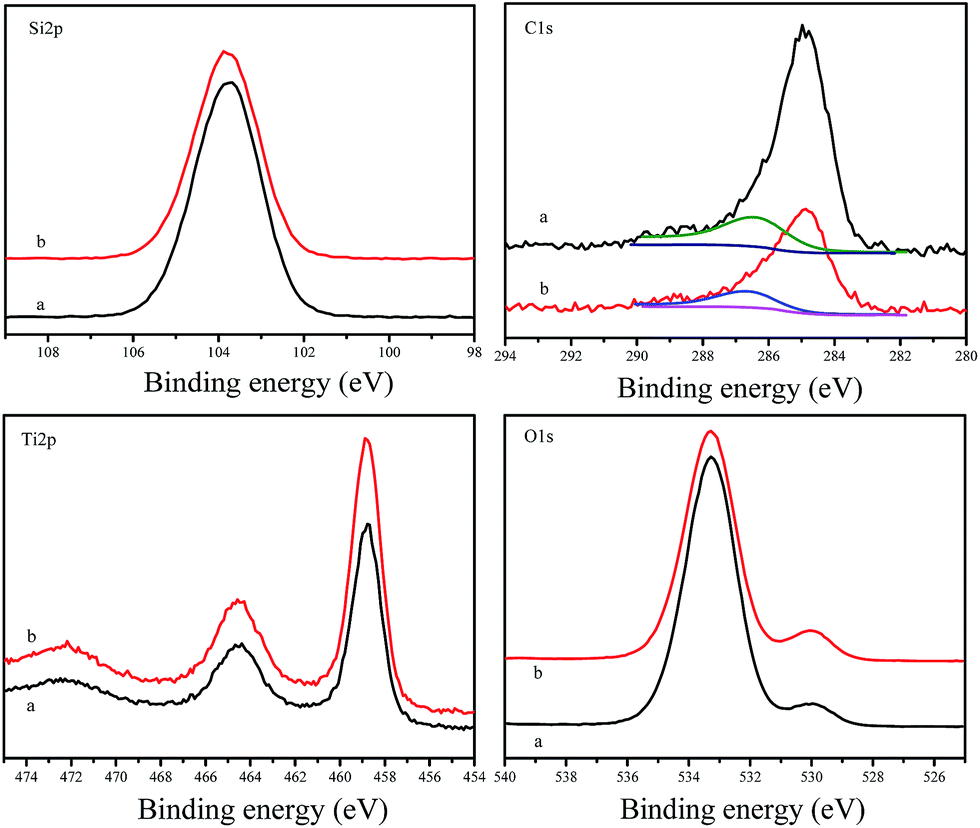 | ||
| Fig. 6 XPS spectra of (a) as synthesized F127–C-OMS@TiO2-4.0 and (b) calcinated F127–C-OMS@TiO2-4.0. | ||
Fig. 7 shows UV-vis diffuse reflectance spectra of as synthesized F127–C-OMS@TiO2-4.0-as, F127–C-OMS@TiO2-4.0, P123–C-OMS@TiO2-4.0, F127–C–TiO2, F127-OMS and commercial P25. Notably, F127-OMS and commercial P25 show the adsorption edges start from 400 nm and nearly no adsorption intensity in the visible-light region. In contrast, as synthesized F127–C-OMS@TiO2-4.0, F127–C-OMS@TiO2-4.0, P123–C-OMS@TiO2-4.0 and F127–C–TiO2 show relatively weak adsorption steps in visible-light region and strong adsorption peaks in UV-light region, which start from 650 nm. The visible light response property should be attributed to their unique high temperature hydrothermal synthesis technology, which results in the doping of carbon heteroatom in the TiO2 lattices. The adjusting of band gap of TiO2 nanocrystals in porous polymers via facile high temperature solvothermal technology has been reported by us previously.37 Visible-light adsorption property of C-OMS@TiO2-xs and F127–C–TiO2 plays the key factor for their photocatalytic applications irradiated with visible light.
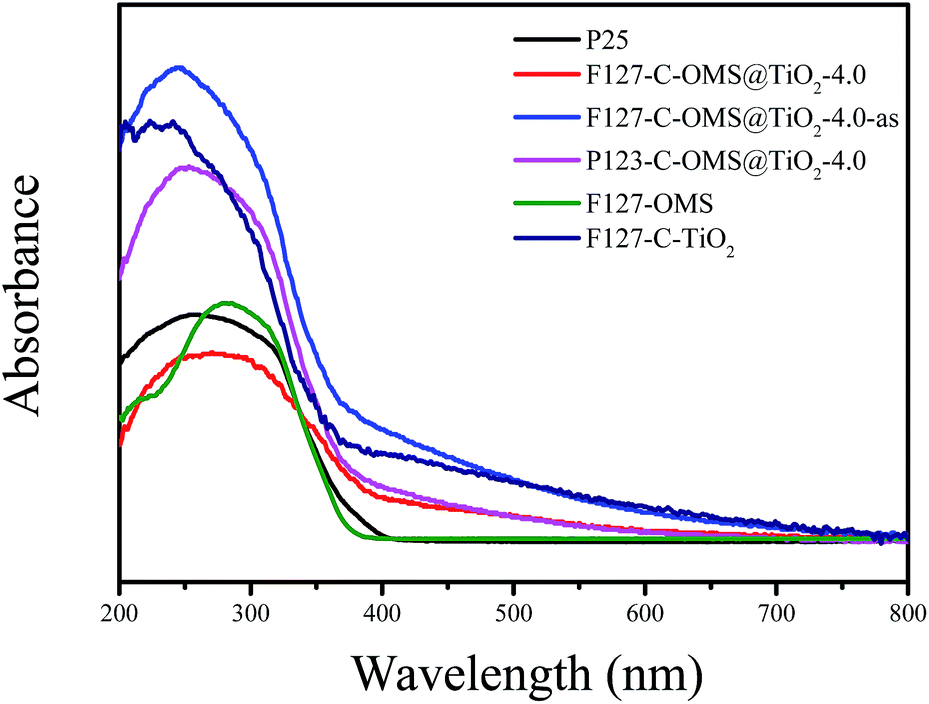 | ||
| Fig. 7 UV-vis diffuse reflectance spectra of various samples. | ||
Fig. 8 shows photocurrent–time (I–t) curves of as synthesized P123–C-OMS@TiO2-2.0-as, as synthesized F127–C-OMS@TiO2-2.0-as, and calcinated F127–C-OMS@TiO2-2.0 under visible light irradiation (λ > 400 nm). It is well known that photoelectron chemistry test was thought to be a powerful tool to monitor the light response, generation and transportation of eCB− of various photocatalysts. In this work, the photoelectrochemical properties of C-OMS@TiO2-xs materials are studied by using the P123–C-OMS@TiO2-2.0-as, F127–C-OMS@TiO2-2.0-as and F127–C-OMS@TiO2-2.0 as the working electrode, where Pt wire and Ag/AgCl (saturated KCl) are used as the counter and reference electrodes, respectively. Under the irradiation with visible light, the photocurrent is produced and recorded. Notably, the photocurrent with different densities formed by employing the synthesized C-OMS@TiO2-xs as the working electrodes, which confirm their visible photoresponse, in agreement with the result of UV-vis diffuse reflectance spectra. Meanwhile, the generated photocurrents are reproducible and stable during five on-off intermittent irradiation cycles, which show photocurrent intensities ranged from 1.68–1.83 μA. The presence of photocurrent is mainly resulted from the fast separation and transportation of the photogenerated eCB− on the surface of the working electrodes. The higher visible photocurrent intensity indicates the higher density of photogenerated carriers, which results in the slower eCB−–hVB+ recombination rate. Compared with F127–C-OMS@TiO2-2.0, relatively higher visible photocurrent intensity of F127–C-OMS@TiO2-2.0-as should be attributed to its higher content of doped carbon.45 The above characteristics were favorable for the enhancement of their visible light induced photocatalytic activities.
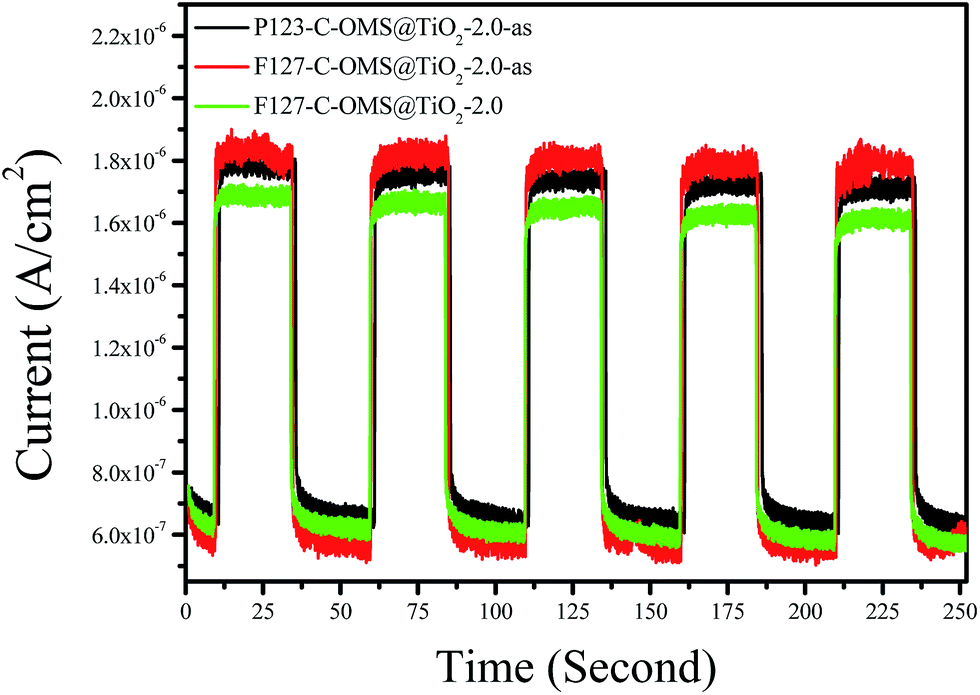 | ||
| Fig. 8 Photocurrent responses of various C-OMS@TiO2-xs electrodes in 0.01 mol L−1 Na2SO4 electrolyte solution under visible light irradiation (λ > 400 nm). The working electrode potential is constant at +1.0 V. | ||
Fig. 9 shows TG curves of F127–C-OMS@TiO2-4.0 and P123–C-OMS@TiO2-4.0 before and after calcination. The as synthesized samples show two weight loss steps ranged from 50 to 150 and 230 to 500 °C, which were assigned to the desorption of water and decomposition of copolymer templates in the samples. After removing the templates by calcination, the samples show only one weight loss steps ranged from 50 to 150 °C due to the desorption of water. Notably, these samples show nearly no obvious weight loss or exothermic signals in the temperatures range from 600–1000 °C, which indicates their ultrastable frameworks.
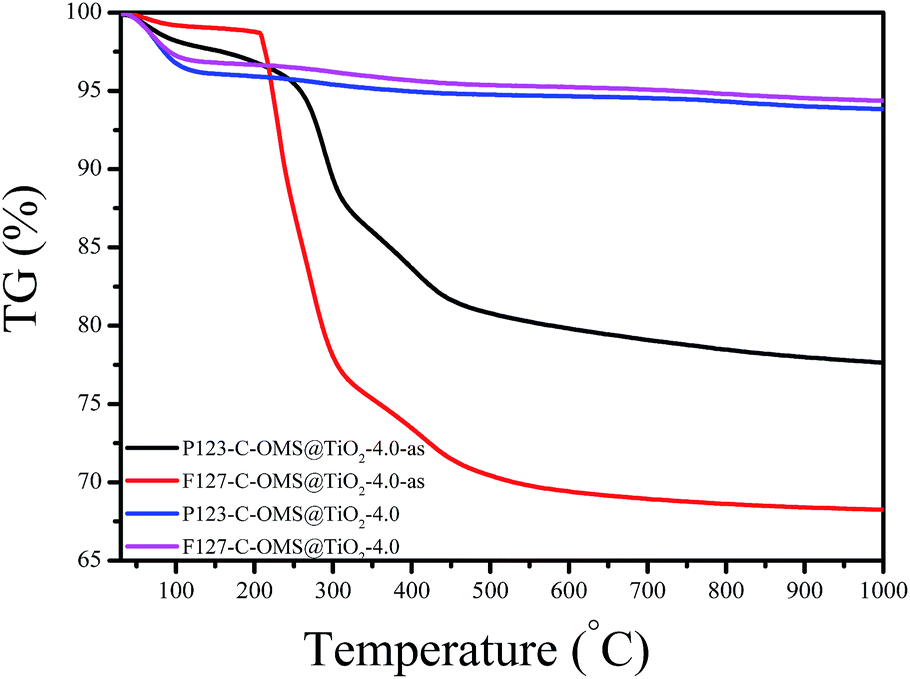 | ||
| Fig. 9 TG curves of various C-OMS@TiO2-xs. | ||
Fig. 10 shows the concentrations versus reaction time for degradation of RhB over C-OMS@TiO2-xs, F127–C–TiO2 and commercial P25 irradiated with visible light. Before irradiation, the reaction mixture was stirred in darkness for 30 min to reach adsorption equilibrium, and all the samples show limited adsorption capacities for RhB. When the reaction mixture was exposed to visible light, the concentrations of RhB quickly decreased catalyzed by C-OMS@TiO2-xs and F127–C–TiO2, which is much better than that of commercial P25. For instance, the concentrations of RhB decrease from 97.4 to 74.2 ppm only after 10 min reaction in presence of F127–C–TiO2. Further prolong the reaction time up to 30 min, the concentration of RhB was decreased up to 9.7 ppm, which can be fully decomposed after 40 min irradiation. Similarly, the concentrations of RhB change from 86.5–98.5 to 61.7–78.3 ppm after 10 min reaction in presence of C-OMS@TiO2-xs catalysts; further prolong the reaction time to 50 min, nearly all of RhB has been catalyzed decomposition. In contrast, the concentration of RhB was as high as 92.1 ppm after 30 min reaction catalyzed by commercial P25. Even prolong the reaction time to 80 min, the concentration of RhB was still up to 81.5 ppm catalyzed by commercial P25. The excellent activities of C-OMS@TiO2-xs and F127–C–TiO2 in visible light region were attributed to their enhanced visible light adsorption properties. In fact, SiO2 and TiO2 act as the role of mortar and brick in C-OMS@TiO2-xs, which formed unique “brick-and-mortar” nanostructure. The “brick-and-mortar” structure was favorable for formation of ordered mesopores in C-OMS@TiO2-xs, which strongly enhance mass transfer and exposure degree of TiO2 active sites in process of photocatalysis. Therefore, although F127–C-OMS@TiO2-2.0-as show much lower TiO2 active site in comparison with F127–C–TiO2, the degradation activity of F127–C-OMS@TiO2-2.0-as is still as comparable as that of F127–C–TiO2.
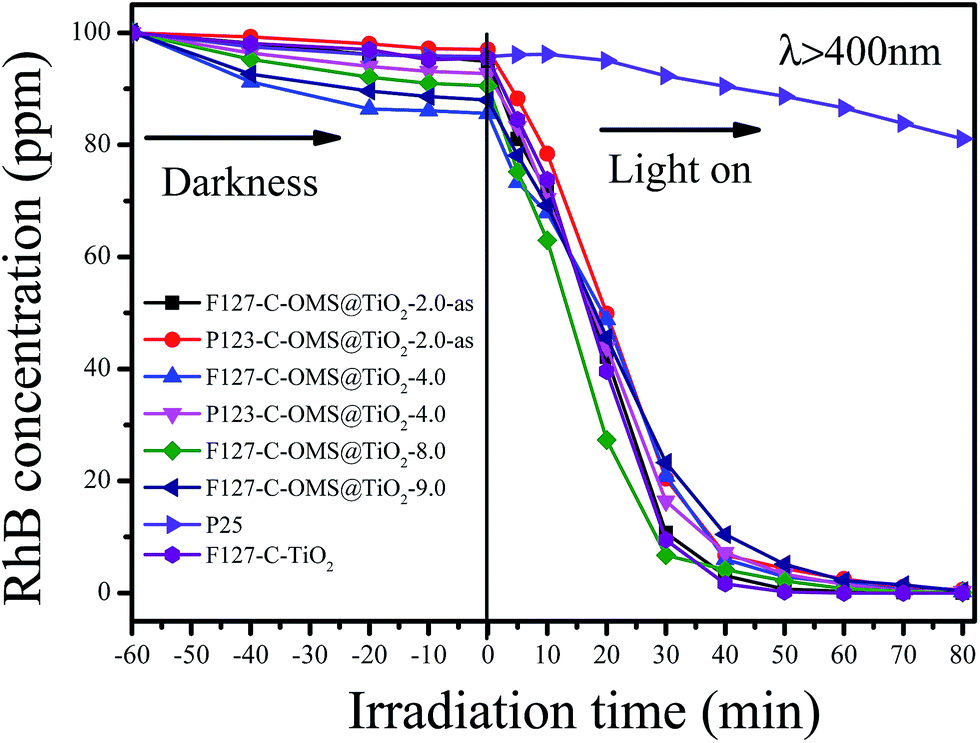 | ||
| Fig. 10 Adsorption property and photocatalytic activity of various samples towards the degradation of RhB irradiated with visible light (400 < λ < 680 nm). | ||
The degradation processes of RhB were also monitored by UV-vis adsorption spectra (Fig. S2†). The adsorption peaks associated with RhB decreases with increase in reaction time catalyzed by F127–C-OMS@TiO2-4.0 photocatalyst. The peaks associated with RhB nearly disappeared when the reaction mixture was irradiated with visible light for 80 min, and no other additional peaks could be observed, which further confirm completely decomposition of RhB in the water. Meanwhile, the TOC values of reaction mixture catalyzed by F127–C-OMS@TiO2-4.0 and F127–C–TiO2 were also investigated (as shown in Fig. S3†), which decrease with the reaction time increase. When the irradiation time was up to 150 min, nearly 90% of organic carbon was removed from the reaction mixture irradiated with visible light, which basically avoids the secondary pollution of the water. On the other hand, C-OMS@TiO2-xs show very good reusability. For instance, the decreasing of activity of P123–C-OMS@TiO2-2.0 can not be observed after 5 cycles, suggesting the long-term stabilities of C-OMS@TiO2-xs in visible light induced photocatalysis (Fig. 11). Good reusability should be attributed to their very stable framework, abundant and ordered mesopores, which strongly enhance their anti-choking and anti-sintering abilities.
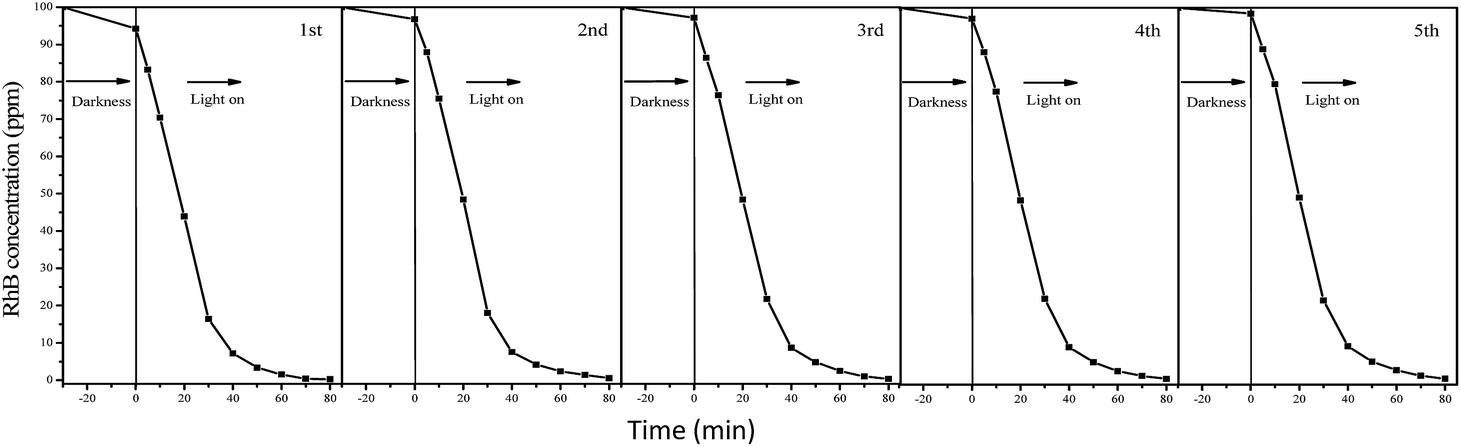 | ||
| Fig. 11 Cycling of P123–C-OMS@TiO2-2.0 in degradation of RhB in the water irradiated with visible light (λ > 400 nm). | ||
Except for RhB, C-OMS@TiO2-xs were also very active for catalyzing degradation of MO and PNP in the water. Fig. 12 shows kinetic curves for degradation of MO over various samples irradiated with visible light. Notably, all the samples show limited adsorption contents for MO under darkness. When the reaction mixture was exposed to visible light, the concentrations of MO quickly decreased catalyzed by as made F127–C-OMS@TiO2-2.0, as made P123–C-OMS@TiO2-2.0 and F127–TiO2, nearly all of MO was decomposed (from 29.3 to 0.3 ppm) within 180 min of irradiation. In contrast, the concentration of MO was still up to 26.3 ppm after 180 min of reaction in presence of commercial P25. Similar results could also be observed for degradation of PNP, after 140 min irradiation with visible light, nearly all of PNP in the water has been catalyzed decomposition in presence of F127–C-OMS@TiO2-2.0 and P123–C-OMS@TiO2-2.0 photocatalysts (Fig. 13). The above results confirmed that C-OMS@TiO2-xs and F127–C–TiO2 could be used as visible light active, highly efficient and reusable photocatalysts for degradation of organic pollutants in the water.
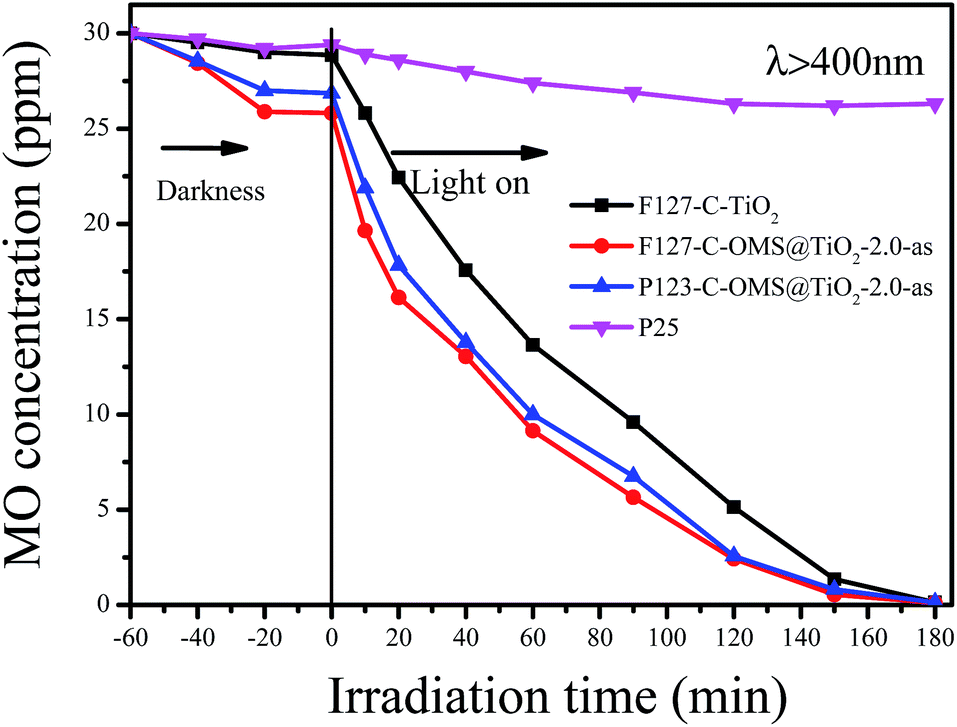 | ||
| Fig. 12 Adsorption property and photocatalytic activity of various samples towards the degradation of MO irradiated with visible light (400 < λ < 680 nm). | ||
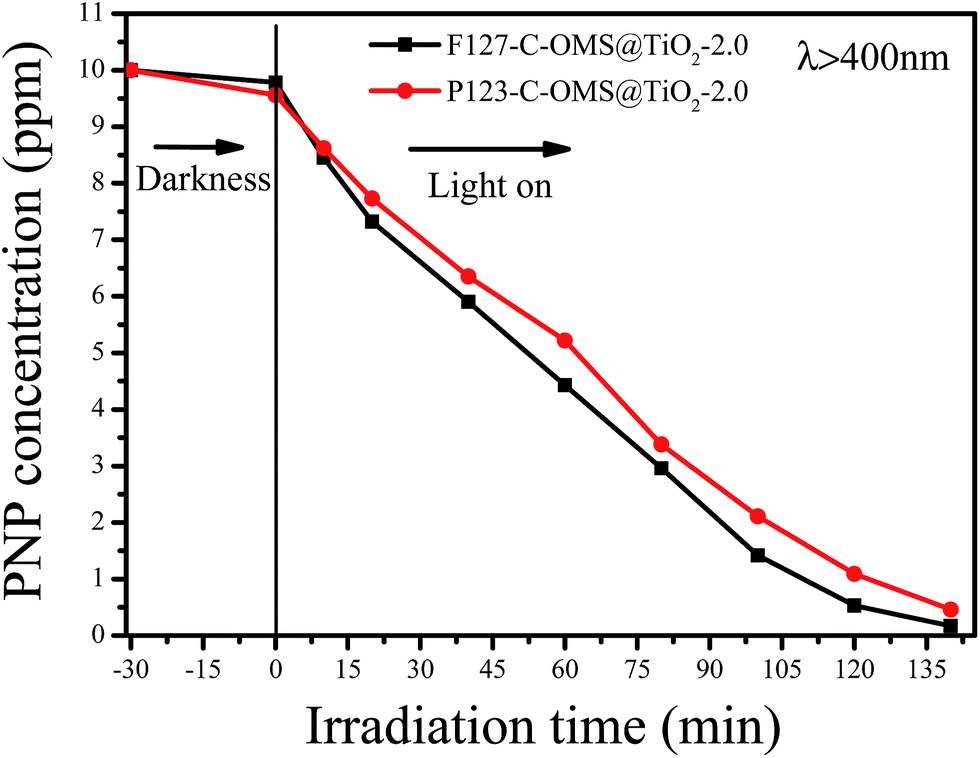 | ||
| Fig. 13 Adsorption property and photocatalytic activity of various samples towards the degradation of PNP irradiated with visible light (400 < λ < 680 nm). | ||
Experimental
Chemicals and reagents
All reagents were of analytical grade and used as purchased without further purification. Tetraethyl orthosilicate (TEOS), nonionic block copolymer template poly(ethyleneoxide)–poly(propyleneoxide)–poly(ethyleneoxide), (Pluronic F127, Mw = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600, P123, Mw = 5800), tetrabutyl titanate (TBT), HCl (36.5 wt%), methyl orange (MO), p-nitrophenol (PNP) and rhodamine-B (RhB) were purchased from Sinopharm Chemical Reagent Co., Ltd. P25 was purchased from Degussa Co. without any treatment.
600, P123, Mw = 5800), tetrabutyl titanate (TBT), HCl (36.5 wt%), methyl orange (MO), p-nitrophenol (PNP) and rhodamine-B (RhB) were purchased from Sinopharm Chemical Reagent Co., Ltd. P25 was purchased from Degussa Co. without any treatment.
Synthesis of C-OMS@TiO2-xs
C-OMS@TiO2-xs were hydrothermally synthesized at the temperatures ranged from 180 to 200 °C from a self-assembly of inorganic precursors and block copolymer surfactants, where x stands for the volume ratio of TEOS to TBT. As a typical run for the synthesis of F127–C-OMS@TiO2-2.0 or P123 – 2.4 g of F127 or P123 was dissolved in 76 mL of water with 3.3 mL of HCl (36.5 wt%), followed by addition of 6.0 mL of TEOS. After stirring at 40 °C for 2–3 h, 3 mL of TBT was then introduced into the mixture. After stirring of the mixture for another 24 h, the reaction mixture was transferred into an autoclave and cured at 180 to 200 °C for 24 h. The product was collected by filtration, drying at 80 °C, and calcination or extraction for removing of templates. Moreover, other OMS@TiO2-xs samples with different volume ratios of TEOS/TBT could also be synthesized as similar procedures.Characterizations
X-ray diffraction (XRD) patterns were obtained with a Rigaku D/Max-2550 using nickel-filtered CuKα radiation. Nitrogen isotherms at the temperature of liquid nitrogen were measured using Micromeritics ASAP 3020 and Tristar system, respectively. The samples were outgassed for 10 h at 150 °C before the measurements. The UV-visible diffuse reflectance spectra were recorded on a Perkin Elmer Lambda 20 UV/vis spectrometer. X-ray photoelectron spectroscopy (XPS) spectra were performed on a Thermo ESCALAB 250 spectrometer with Al Kα radiation, and binding energies were calibrated using the C 1s peak at 284.9 eV. Thermogravimetric analysis (TG) was performed with a PerkinElmer TGA7 analyzer and a Perkin Elmer DTA-1700 furnace tube in flowing air. Transmission electron microscopy (TEM) experiments were performed on a JEM-2100F electron microscope (JEOL, Japan) with an acceleration voltage of 200 kV. The total organic carbon (TOC) of the initial and irradiated samples was recorded on a Shimadzu-5000A analyzer. The concentrations of organic dyes were measured by using a NanoDrop 2000 UV-spectrophotometer based on the standard curves method.Visible light induced photocatalysis
Visible light induced (λ > 400 nm) photocatalytic degradation of RhB was carried out in a cylindrical glass under irradiation by a PLS-SXE300 300 W xenon lamp equipped with a 400 nm cutoff filter. Typically, 10.0 mg of catalyst was added into a cylindrical glass containing 10 mL of RhB aqueous solution with the concentration of 100 ppm (100 mg L−1), which was equipped with a circulation water system to maintain a constant temperature. Before irradiation, the mixture was stirred under darkness for 30 min to achieve sorption equilibrium. At a given time interval of irradiation, small aliquots (0.5 mL) were withdrawn and analyzed by UV spectroscopy at the wavelength of 555 nm. The catalysts could be regenerated by the following procedures: (i) separation of catalysts from reaction mixtures by filtration; (ii) washing the samples with abundant ethanol to remove residual reactants or products; (iii) separation; (iv) drying of the catalyst at 60 °C under vacuum condition. Then, the catalyst was used directly for the next time.Visible light induced (λ > 400 nm) photocatalytic degradation of MO and PNP was also carried out in a cylindrical glass under irradiation by a PLS-SXE300 300 W xenon lamp equipped with a 400 nm cutoff filter. Typically, 5 mg of catalyst was added into a cylindrical glass containing 10 mL of MO (30 ppm) or 10 mL of PNP (10 ppm), which was equipped with a circulation water system to maintain a constant temperature. Before irradiation, the mixture was stirred under darkness for 30 min to achieve the adsorption equilibrium. At a given time interval of irradiation, small aliquots (0.5 mL) were withdrawn and analyzed by UV spectroscopy with the wavelength of 463 (MO) and 317 nm (PNP), respectively.
Conclusions
In summary, we exploited a high temperature hydrothermal technology in this work, which has been successfully used for preparation of ordered mesoporous SiO2@TiO2 nanocomposites with controllable pore sizes and Ti contents. The facile high temperature hydrothermal technology results in the doping of carbon atom into TiO2 lattice in C-OMS@TiO2-xs, which changes the band gap of TiO2 in these samples. The above characteristics make C-OMS@TiO2-xs sensitive to visible light, which show excellent activities for degradation of organic pollutants irradiated with visible light. This work develops a facile and cost effective approach to the synthesis of visible light active, ordered and stable mesoporous TiO2 materials, which is very important for the wide applications of TiO2 and its derived materials in the area of visible light photocatalysis.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21573150, 21203122) and the Natural Science Foundation of Zhejiang Province (LY15B030002). We sincerely thank Prof. Yihang Guo in Northeast Normal University for her kind helps and valuable discussion on this work.Notes and references
- A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- W. Y. Dong, Y. J. Sun, C. W. Lee, W. M. Hua, X. C. Lu, Y. F. Shi, S. C. Zhang, J. M. Chen and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 13894–13904 CrossRef CAS PubMed.
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 283, 37–38 CrossRef.
- Y. J. Lee, J. B. Joo, Y. D. Yin and F. Zaera, ACS Energy Lett., 2016, 1, 52–56 CrossRef CAS.
- S. C. Sun, P. Gao, Y. Yang, P. Yang, Y. Chen and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18126–18131 CAS.
- H. Choi, D. Shin, B. C. Yeo, T. Song, S. S. Han, N. Park and S. Kim, ACS Catal., 2016, 6, 2745–2753 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zhou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- C. P. Sajan, S. Wageh, A. A. Al-Ghamdi, J. G. Yu and S. W. Cao, Nano Res., 2016, 9, 3–27 CrossRef CAS.
- J. M. Meichtry, R. Dillert, D. W. Bahnemann and M. I. Litter, Langmuir, 2015, 31, 6229–6236 CrossRef CAS PubMed.
- Z. G. Xiong and X. S. Zhao, J. Am. Chem. Soc., 2012, 134, 5754–5757 CrossRef CAS PubMed.
- S.-H. Lin, Y.-H. Su, H.-W. Cho, P.-Y. Kung, W.-P. Liao and J.-J. Wu, J. Mater. Chem. A, 2016, 4, 1119–1125 CAS.
- Z. Ren, J. Wang, Z. Pan, K. Zhao, H. Zhang, Y. Li, Y. Zhao, I. Mora-Sero, J. Bisquert and X. Zhong, Chem. Mater., 2015, 27, 8398–8405 CrossRef CAS.
- K. Nakamura, T. Oshikiri, K. Ueno, Y. Wang, Y. Kamata, Y. Kotake and H. Misawa, J. Phys. Chem. Lett., 2016, 7, 1004–1009 CrossRef CAS PubMed.
- E. Guo and L. W. Yin, J. Mater. Chem. A, 2015, 3, 13390–13401 CAS.
- L. Yuan, C. Han, M. Pagliaro and Y.-J. Xu, J. Phys. Chem. C, 2016, 120, 265–273 CAS.
- M. M. Ye, J. Jia, Z. J. Wu, C. X. Qian, R. Chen, P. G. O'Brien, W. Sun, Y. C. Dong and G. A. Ozin, Adv. Energy Mater., 2016, 7, 1601811 CrossRef.
- X. H. Gao, G. R. Li, Y. Y. Xu, Z. L. Hong, C. D. Liang and Z. Lin, Angew. Chem., Int. Ed., 2015, 54, 14331–14335 CrossRef CAS PubMed.
- H. Ren, R. Yu, J. Y. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- T. Kawahara, Y. Konishi, H. Tada, N. Tohge, J. Nishii and S. Ito, Angew. Chem., Int. Ed., 2002, 41, 2811–2813 CrossRef CAS PubMed.
- R. L. Liu, Y. J. Ren, Y. F. Shi, F. Zhang, L. J. Zhang, B. Tu and D. Y. Zhao, Chem. Mater., 2008, 20, 1140–1146 CrossRef CAS.
- C. Wang, L. Yin, L. Zhang, N. Liu, N. Lun and Y. Qi, ACS Appl. Mater. Interfaces, 2010, 2, 3373–3377 CAS.
- T. Pauporté and J. Rathousky, J. Phys. Chem. C, 2007, 111, 7639–7644 Search PubMed.
- W. Dong, Y. Zhu, H. Huang, L. Jiang, H. Zhu, C. Li, B. Chen, Z. Shi and G. Wang, J. Mater. Chem. A, 2013, 1, 10030–10036 CAS.
- Y. Bi, H. Hu, S. Ouyang, Z. Jiao, G. Lu and J. Ye, J. Mater. Chem., 2012, 22, 14847–14850 RSC.
- J. Luo and P. A. Maggard, Adv. Mater., 2006, 18, 514–517 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler Jr, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- M. R. Elahifard, S. Rahimnejad, S. Haghighi and M. R. Gholami, J. Am. Chem. Soc., 2007, 129, 9552–9553 CrossRef CAS PubMed.
- K. Takeuchi, I. Nakamura, O. Matsumoto, S. Sugihara, M. Ando and T. Ihara, Chem. Lett., 2000, 29, 1354–1355 CrossRef.
- T. Ihara, M. Miyoshi, M. Ando, S. Sugihara and Y. Iriyama, J. Mater. Sci., 2001, 36, 4201–4207 CrossRef CAS.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Choi, H. Park and M. R. Hoffmann, J. Phys. Chem. C, 2010, 114, 782–793 Search PubMed.
- B. Xin, Z.-H. Ren, P. Wang, J. Liu, L. Jing and H. Fu, Appl. Surf. Sci., 2007, 253, 4390–4395 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- J.-W. Shi, C. Liu, C. He, J. Li, C. Xie, S.-H. Yang, J.-W. Chen, S. Li and C.-M. Niu, RSC Adv., 2015, 5, 17667–17675 RSC.
- C. Xie, S. Yang, B. Li, H. Wang, J.-W. Shi, G. Li and C. Niu, J. Colloid Interface Sci., 2016, 476, 1–8 CrossRef CAS PubMed.
- F. J. Liu, W. Kong, L. Wang, I. Noshadi, Z. H. Zhang and C. Z. Qi, Nanotechnology, 2015, 26, 085705 CrossRef PubMed.
- D. L. Li, H. S. Zhou and I. Honma, Nat. Mater., 2004, 3, 65–72 CrossRef CAS PubMed.
- S. Y. Choi, M. Mamak, N. Coombs, N. Chopra and G. A. Ozin, Adv. Funct. Mater., 2004, 14, 335–344 CrossRef CAS.
- F. J. Liu, S. F. Zuo, X. D. Xia, J. Sun, Y. C. Zou, L. Wang, C. G. Li and C. Z. Qi, J. Mater. Chem. A, 2013, 1, 4089–4096 CAS.
- P. D. Yang, D. Y. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- Y. J. Li, L. M. Yu, N. Li, W. F. Yan and X. T. Li, J. Colloid Interface Sci., 2015, 450, 246–253 CrossRef CAS PubMed.
- W. J. Ren, Z. H. Ai, F. L. Jia, L. Z. Zhang, X. X. Fan and Z. G. Zou, Appl. Catal., B, 2007, 69, 138–144 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01368j |
| This journal is © The Royal Society of Chemistry 2017 |