
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DFT study of single water molecule adsorption on the (100) and (101) surfaces of KH2PO4
Lei Zhang,
Yulin Wu,
Yao Liu * and
Hui Li
* and
Hui Li
Key Laboratory for Liquid-Solid Structural Evolution and Processing of Materials, Ministry of Education, Shandong University, Jinan 250061, China
First published on 16th May 2017
Abstract
Potassium dihydrogen phosphate (KH2PO4; KDP) is a widely used non-linear optical material. A critical issue associated with KDP crystals is that the water molecules in the environment inevitably adhere to the surface and affect the surface electronic and physical properties. In order to understand how water interacts with the surface, the adsorption of a single water molecule on the external surfaces of a KDP crystal, including (100) and (101) surfaces, was theoretically investigated based on density functional theory (DFT) method. The most favorable adsorption sites on both surfaces were considered based on the adsorption energy. The electron density difference and partial density of states were calculated to analyze the bond formation during the adsorption process. It was found that the water molecule can be adsorbed in two different ways—forming two hydrogen bonds with the O and H atoms on the surface or forming a hydrogen bond and an O–K chemical bond with the O and K atoms on the surface. We also found that the surface atoms and the pulling effect between two high potential states on the surface play a dominating role in the adsorption process.
1. Introduction
Potassium dihydrogen phosphate (KH2PO4; KDP) is a widely used non-linear optical material with large electro-optical and nonlinear optical coefficients and a high laser damage threshold.1–5 It has wide practical applications in laser frequency converters, electro-optic modulators, optical switches and other high technology fields owing to its unique properties.6,7Large-scale KDP crystals that meet the need of high power laser devices for large size frequency doubling crystals are mainly grown from its saturated aqueous solution.8–10 The growth of crystals takes place at the interface between the crystal and the solution, and the atomic structure at this interface plays a primary role in the structure and morphology of the crystal. Thus, many people have studied the structure of the interface. For example, Vries11 found that the {101} faces of KDP crystal in solution are terminated by a layer of K+ ions and not by H2PO4− groups. Reedijk et al. studied interface-induced ordering in the first four layers of water molecules through X-ray diffraction. They found that the first two layers behave like ice and are strongly bound to the surface.12 KDP crystal is also fragile, thus it is extremely difficult to machine via traditional machining methods. Owing to its water-soluble property, micro water dissolution machining method and micro-deliquescence polishing method have been applied to machine KDP crystals.13,14 The dissolution and deliquescence taking place at the surface of the KDP crystals improved the machining quality of KDP optical crystals. Besides the producing and machining processes, the application and storage of KDP are also strongly affected by water. To minimize the Fresnel losses from the optical surface, the KDP surface is usually covered with a sol–gel coating.15 However, people found that etch pits can develop at the KDP surface beneath the coating after exposure to ambient humidity.16 Water adsorbed from the environment into the coating contacts the KDP crystal surface, causing etch pits nucleation at high undersaturation.17
As we can see, during the producing, machining and application processes, the adsorption of water can have a significant influence on the surface electronic and physical properties of KDP crystals. Therefore, to control the influences of water adsorption, it is essential to understand the interaction between water and KDP crystal surfaces. So far, researchers have mainly focused on two aspects of this issue. One is the structure of the KDP surface in aqueous solution, where they found the K+ ions termination at the {101} faces11 and the growth morphology of the {100} faces.18 The other one is the structure of the liquid part of the interface where they found the liquid order at the interface12 and a strongly hydrogen bound water layer close to the KDP surface.19,20 However there are very few theoretical studies of the adsorption of water molecules on KDP surfaces, while this kind of study has been a research hotspot in other optical materials. For example, water adsorption on TiO2 was investigated by ab initio study, showing that molecular adsorption is more favorable than dissociative chemisorption.21 The adsorption of water on the potassium sodium tantalite niobate (KNTN) (001) surface was studied by Wang, who revealed the energetically favorable configurations for water monomer adsorption.22 Giordano et al. studied water molecules on the MgO (100) surface, finding the first theoretical evidence of water dissociation on the perfect MgO (100) surface.23
Overall, there are urgent realistic and theoretical needs for the study of the interaction between water molecules and KDP surfaces. In this paper, we are trying to study where a water molecule can be adsorbed more stably on the (100) and (101) surfaces and the differences in how the water molecule is adsorbed on the two surfaces of a KDP crystal. This would help us understand the nature of the interaction between a water molecule and the surfaces of a KDP crystal, and guide us to produce, machine and protect KDP crystal more effectively.
2. Computational details
KDP has a I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d space group and crystal lattice parameters of a = b = 7.453 Å, c = 6.973 Å.24 First, a unit cell of bulk KDP crystal was generated and then optimized. When growing from aqueous solution, two different surfaces are observed on the morphology of the KDP crystal—the (100) and (101) surfaces. Thus, these two surfaces were chosen to be calculated in our work (see Fig. 1(c)). When cleaving the (101) surface, there are two possible ways: one produces a cation (K+)-terminated surface and the other produces a dihydrogen phosphate (H2PO4−)-terminated surface. According to the work by Vries, which indicated K+ termination at the (101) surface of KDP,11 only the K+-terminated surface was considered in our calculations. The KDP (100) and (101) surfaces were modeled by a slab of three layers (1 × 1 unit cell along the x and y directions) in the z-direction. The thickness of a single layer equals the interplanar distance of the (100) and (101) surfaces. A vacuum spacing in the z-direction of 30 Å was also used to avoid the influence of periodic boundary conditions (x, y and z direction).
2d space group and crystal lattice parameters of a = b = 7.453 Å, c = 6.973 Å.24 First, a unit cell of bulk KDP crystal was generated and then optimized. When growing from aqueous solution, two different surfaces are observed on the morphology of the KDP crystal—the (100) and (101) surfaces. Thus, these two surfaces were chosen to be calculated in our work (see Fig. 1(c)). When cleaving the (101) surface, there are two possible ways: one produces a cation (K+)-terminated surface and the other produces a dihydrogen phosphate (H2PO4−)-terminated surface. According to the work by Vries, which indicated K+ termination at the (101) surface of KDP,11 only the K+-terminated surface was considered in our calculations. The KDP (100) and (101) surfaces were modeled by a slab of three layers (1 × 1 unit cell along the x and y directions) in the z-direction. The thickness of a single layer equals the interplanar distance of the (100) and (101) surfaces. A vacuum spacing in the z-direction of 30 Å was also used to avoid the influence of periodic boundary conditions (x, y and z direction).
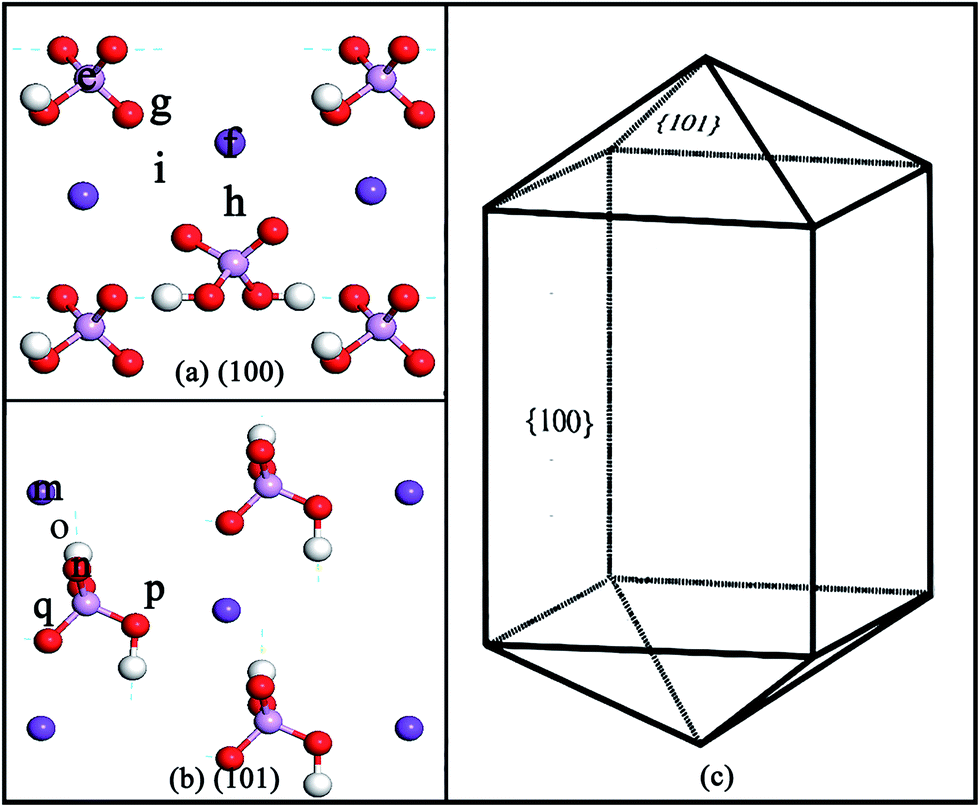 | ||
| Fig. 1 Initial adsorption positions on the KDP (100) surface (a) and (101) surface (b). (c) The idealized KDP crystal with two growth faces, the {101} and the {100}. Dark purple balls represent potassium atoms. Light purple balls represent phosphorus atoms. Red balls represent oxygen atoms. Hydrogen atoms are represented by white balls. | ||
All DFT calculations25 on the KDP surfaces were carried out using the CASTEP code.26 The exchange–correlation potential used in the calculations was the Perdew–Burke–Ernzerhof27 (PBE) form of the generalized gradient approximation (GGA). The electron–ion interactions were described by ultrasoft pseudopotential28 (USP). Geometry optimization was carried out using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) scheme. To describe the dispersion interactions, a semi-empirical correction using the Grimme method29 was adopted. To get converged results and reasonable computation time, a plane-wave energy cutoff of 400 eV was used for geometric optimization, which took the most time of the calculations. To get results of high accuracy, a higher cutoff of 680 eV was used for electronic structure calculations. A 3 × 3 × 1 k-point grid was used in the self-consistent calculations. The convergence tolerances for energy and force were 2 × 10−5 eV per atom and 0.05 eV Å−1 in the self-consistent calculations, respectively.
3. Results and discussion
In this paper, OW and HW represent the oxygen atom and hydrogen atoms in a water molecule, respectively. OP and HP mean the oxygen atom and hydrogen atoms in the related phosphate group, respectively.The initial adsorption positions of the (100) surface and the (101) surface are shown in Fig. 1(a) and (b), respectively. On the (100) surface, positions e and f represent the P top and K top sites, respectively. Positions g and h in at the middle sites of two different P–K bridges. Position i is in the middle site of the K–K bridge. On the (101) surface, positions m and n represent the K top and O top sites, respectively. Positions o and p are at the middle sites of two different O–K bridges (at position o the length of the O–K bridge is 2.60 Å, while at position p the length is 4.36 Å which is significantly longer). Position q is at the middle site of the K–K bridge. Owing to the symmetry of the (100) and (101) surfaces, these initial positions can represent most of the typical adsorption positions on the two surfaces.
We set the adsorption energy as a criterion to determine the stability of the adsorption. It was calculated from the following equation:30,31
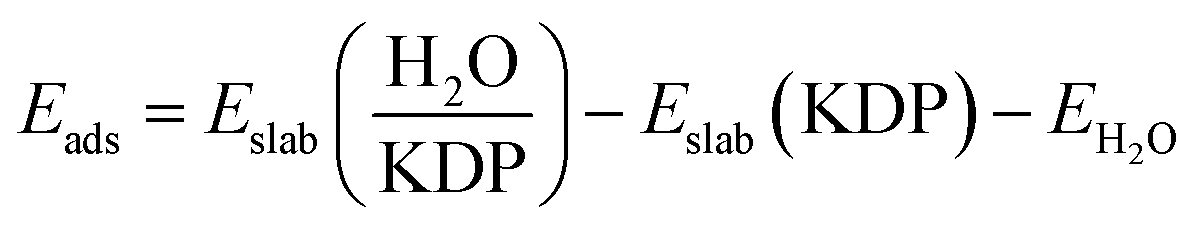
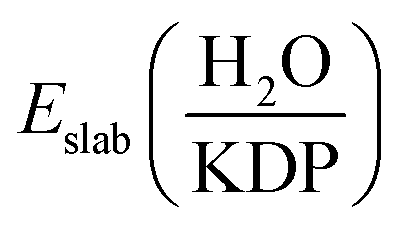 is the total energy of the water-KDP adsorption system. Eslab(KDP) is the energy of a clean KDP surface. EH2O is the energy of an isolated water molecule.
is the total energy of the water-KDP adsorption system. Eslab(KDP) is the energy of a clean KDP surface. EH2O is the energy of an isolated water molecule.
The adsorption energies at given positions on the KDP (100) and (101) surfaces are shown in Table 1. According to the adsorption energy of the water molecule at different positions, we get the two most stable configurations of a single water molecule adsorbed on the (100) surface. They are position e (P top site) and position h (P–K bridge site). On the (101) surface, the two positions where a water molecule can be adsorbed more stably are position o (O–K bridge site) and position p (O–K bridge site). It can be seen that on the (101) surface, the most stable adsorption sites are both O–K bridge positions while on the (100) surface, a single water molecule can be adsorbed more stably not only at the P–K bridge position but also at the P top position.
| Surface | Position | |||||
|---|---|---|---|---|---|---|
| (100) | Eads (eV) | e | f | g | h | i |
| −0.57 | −0.37 | −0.32 | −0.47 | −0.46 | ||
| (101) | Eads (eV) | m | n | o | p | q |
| −0.25 | −0.50 | −0.60 | −0.63 | −0.43 | ||
The adsorption of a water molecule on the surface of materials always results in structure changes within the water molecule. We compared the interatomic distances and angle of an isolated water molecule and an adsorbed one at the most stable adsorption sites in Table 2. It is clear that at all the adsorption sites, the OH bond of the water molecule that is near to the surface is lengthened (by 0.015 Å to 0.027 Å) after the adsorption process while the OH bond that is far from the surface contracts slightly (by 0.001 Å to 0.005 Å). In addition, the adsorption process results in an increase in the HW–OW–HW bond angle of the water molecule. It could be expected that some electron density may be transferred between the water molecule and the KDP (100) and (101) surfaces, as could be validated from our electron density difference analysis.
| Structure | Isolated | Position e | Position h | Position o | Position p |
|---|---|---|---|---|---|
| dOH(1) (Å) | 0.978 | 0.993 | 0.993 | 1.005 | 1.002 |
| dOH(2) (Å) | 0.978 | 0.977 | 0.976 | 0.973 | 0.975 |
| ∠HW–OW–HW (degree) | 104.503 | 105.736 | 105.819 | 107.278 | 105.936 |
3.1. Adsorption of an isolated water molecule on the (100) surface
The geometries of water molecule adsorption at positions e and h on the KDP (100) surface are shown in Fig. 2(a) and (b), respectively. The details of the bond lengths, bond angles and adsorption energy are shown in Table 3. It can be observed that, at position e, one H atom of the water molecule is pulled toward the O atom of the phosphate group (dHW–OP = 1.91 Å and ∠OW–HW⋯OP (shown as ∠1 in Fig. 2) = 147.0°) and the O atom of the water molecule is drawn near to the H atom of the phosphate group (dHP–OW = 1.87 Å and ∠OP–HP⋯OW (shown as ∠2 in Fig. 2) = 162.2°). The geometry of the water molecule adsorption at position e suggests that two hydrogen bonds (OW–HW⋯OP and OP–HP⋯OW) may be formed during the adsorption process.32 At position h, one H atom of the water molecule is attracted to the O atom of the phosphate group (dHW–OP = 1.84 Å and ∠OW–HW⋯OP (shown as ∠3 in Fig. 2) = 154.1°) and the O atom of the water molecule is drawn near to the K atom on the surface (dOW–K = 3.01 Å), indicating a hydrogen bond (OW–HW⋯OP) and an OW–K chemical bond formed during water molecule adsorption at position h.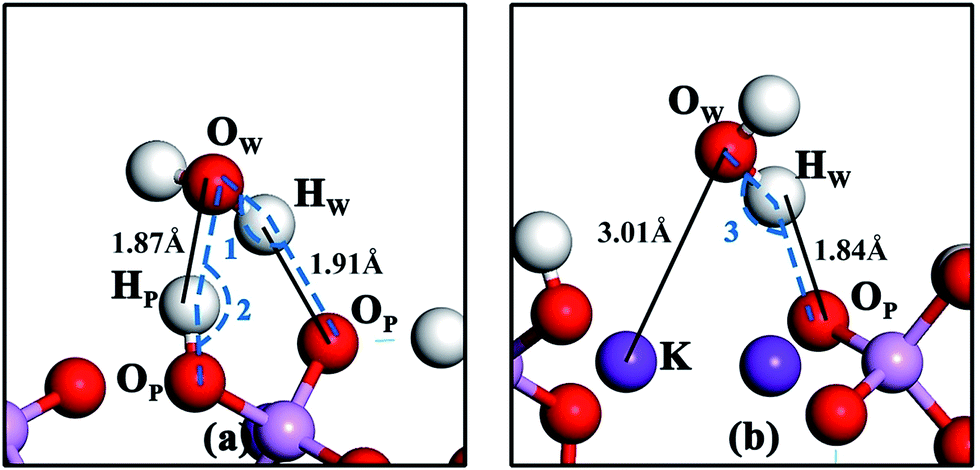 | ||
| Fig. 2 Geometries of water molecule adsorption at positions e (a) and h (b) on the KDP (100) surface. ∠1 is the angle from OW via HW to OP. ∠2 is the angle from OP via HP to OW. ∠3 is the angle from OW via HW to OP. | ||
| Structure | Eads (eV) | ∠OW–HW⋯OP (degree) | ∠OP–HP⋯OW (degree) | dOW–K (Å) | dHW–OP (Å) | dHP–OW (Å) |
|---|---|---|---|---|---|---|
| Position e | −0.57 | 147.0 | 162.2 | — | 1.91 | 1.87 |
| Position h | −0.47 | 154.1 | — | 3.01 | 1.84 | — |
Electron density difference maps have been used to provide a deeper understanding of the electron density transfer while the water molecule is adsorbed on KDP surface. The electron density difference Δρ is defined by the following equation:
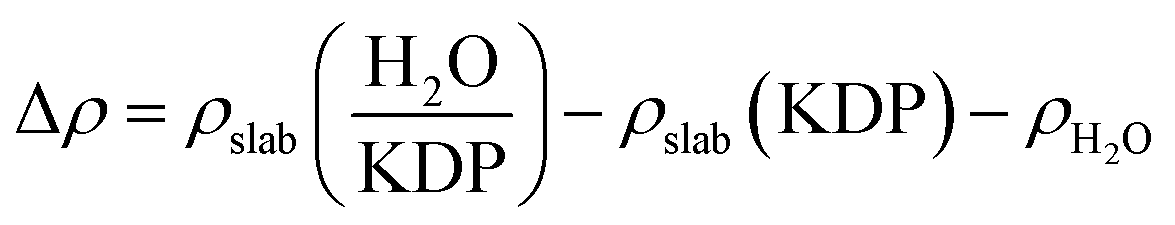
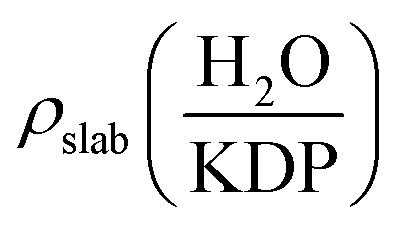 , ρslab(KDP) and ρH2O are the electron densities of the water–KDP adsorption system after adsorption, the clean KDP surface and the free water molecule before adsorption, respectively. To get a clear picture of the difference in electron density, slices are made through related atoms (at position e shown in Fig. 3(a): slice 1 through HP and OW and slice 2 through HW and OP; at position h shown in Fig. 3(b): slice 3 through HW and OP and slice 4 through OW and K). It clearly exhibits an increase of electron density between HW and OP and between OW and HP (see Fig. 3(a)). Such charge distribution illustrates that two hydrogen bonds are formed between the water molecule and the neighbouring phosphate group. Fig. 3(b) shows an increase of electron density between HW and OP, which indicates that a hydrogen bond formed at position h. Meanwhile, the electrons of the K atom transfer to the neighbouring O atoms of the water and phosphate group, which indicates an O–K chemical bond formed during the adsorption process. The electron transference between the water molecule and the KDP (100) surface indicates that H and O atoms of the phosphate group and K atoms exposed on the surface are the functional atoms that dominate the adsorption process.
, ρslab(KDP) and ρH2O are the electron densities of the water–KDP adsorption system after adsorption, the clean KDP surface and the free water molecule before adsorption, respectively. To get a clear picture of the difference in electron density, slices are made through related atoms (at position e shown in Fig. 3(a): slice 1 through HP and OW and slice 2 through HW and OP; at position h shown in Fig. 3(b): slice 3 through HW and OP and slice 4 through OW and K). It clearly exhibits an increase of electron density between HW and OP and between OW and HP (see Fig. 3(a)). Such charge distribution illustrates that two hydrogen bonds are formed between the water molecule and the neighbouring phosphate group. Fig. 3(b) shows an increase of electron density between HW and OP, which indicates that a hydrogen bond formed at position h. Meanwhile, the electrons of the K atom transfer to the neighbouring O atoms of the water and phosphate group, which indicates an O–K chemical bond formed during the adsorption process. The electron transference between the water molecule and the KDP (100) surface indicates that H and O atoms of the phosphate group and K atoms exposed on the surface are the functional atoms that dominate the adsorption process.
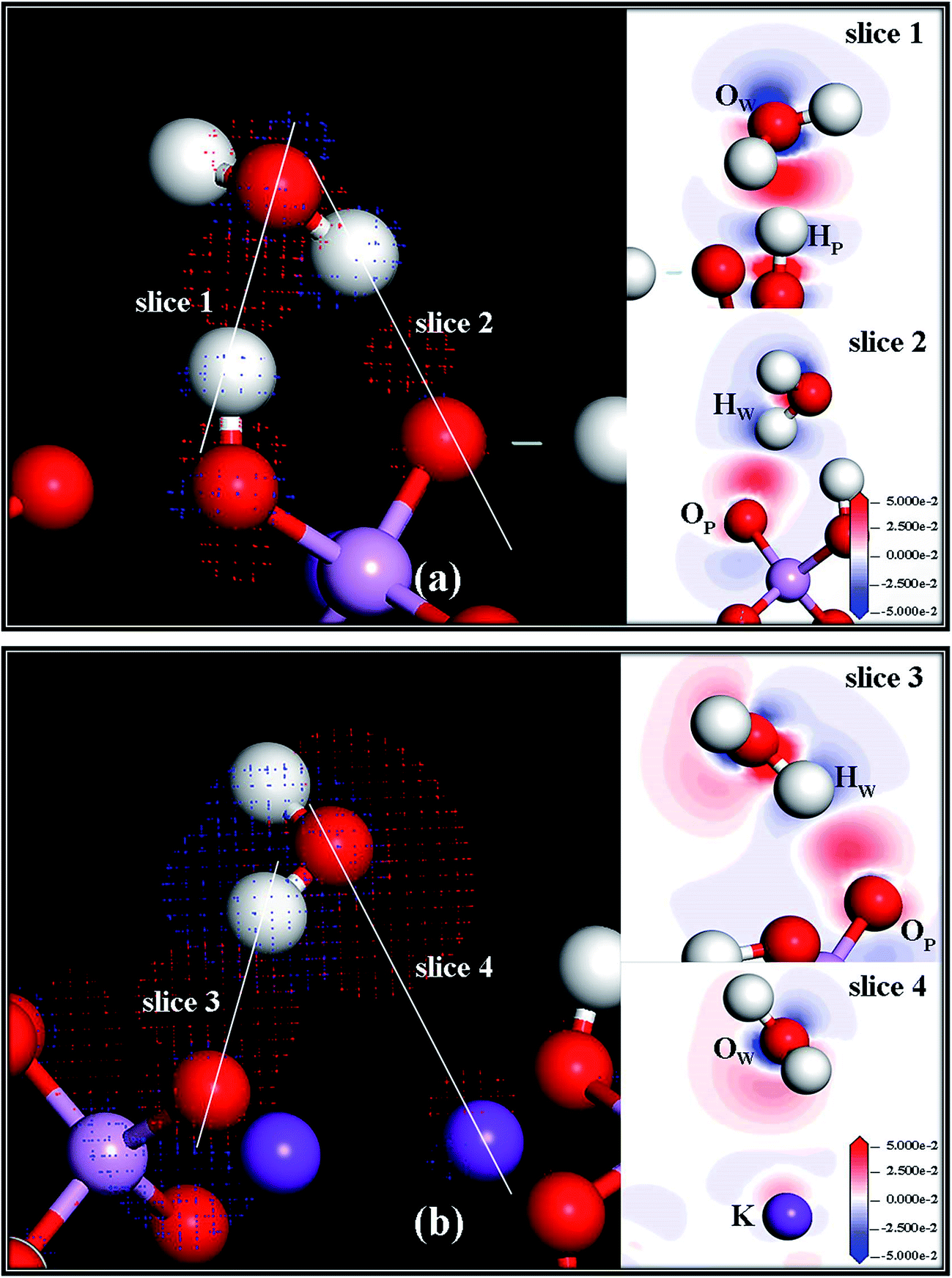 | ||
| Fig. 3 Electron density difference maps between the water molecule and the KDP (100) surface at position e (a) and position h (b). Blue and red colors represent depletion and accumulation of electron density, respectively. Positive values indicate accumulation of electron density. Negative values correspond to depletion of electron density. | ||
The atomic partial densities of states (PDOSs) of selected atoms of the water molecule and the surface were calculated to study the interaction between the functional atoms on the surface and the water molecule, as shown in Fig. 4. At position e, the resonance peaks between HW 1s states and OP 1s and 2p states are at energies of −20.2 eV, −6.5 eV and −2.8 eV, respectively. The resonance peaks between the OW 2p states and the HP 1s states are in a wide range from −8.9 eV to −2.0 eV. These resonance peaks can be attributed to the formation of hydrogen bonds. As mentioned above, at position e, dHP–OW = 1.87 Å is shorter than dHW–OP = 1.91 Å and we noticed that the two states between HP and OW have more similar shapes than that between HW and OP, which may indicate that the hydrogen bond between HP and OW is stronger.
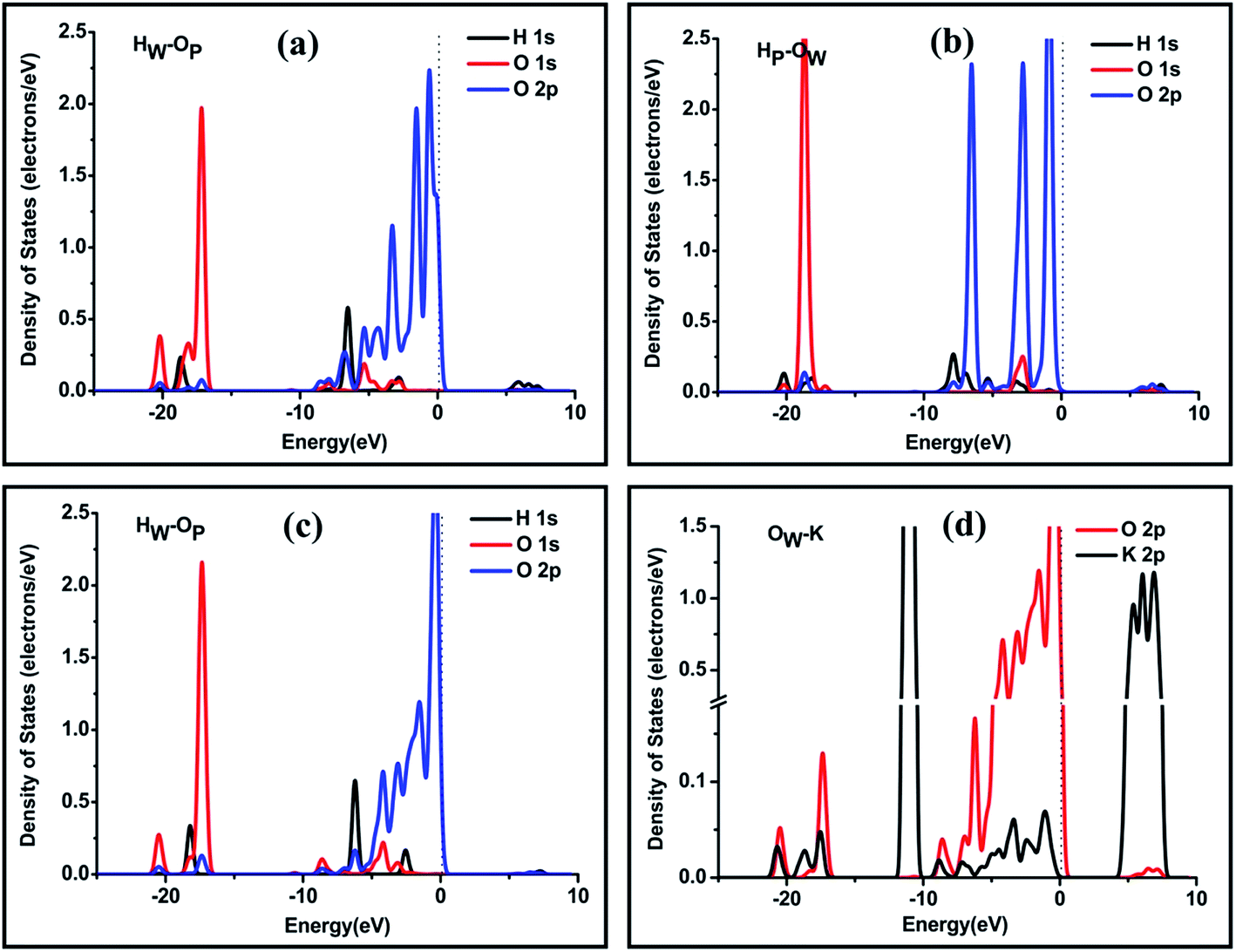 | ||
| Fig. 4 PDOSs of selected atoms of the water molecule and the (100) surface at position e: (a) atomic orbitals of the H atom of the water molecule and the O atom of the phosphate group and (b) atomic orbitals of the H atom of phosphate group and the O atom of the water molecule. PDOS of selected atoms of water molecule and the (100) surface at position h: (c) atomic orbitals of the H atom of the water molecule and the O atom of the phosphate group and (d) atomic orbitals of the O atom of the water molecule and the K atom of the surface. The zero energy is the Fermi level. | ||
Fig. 4(c) shows that, at position h, the resonance peaks between the HW 1s state and OP 1 and 2p states are at energies of −18.2 eV, −6.2 eV and −2.5 eV, respectively. Like the two states between HW and OP at position e, their shapes are not completely similar. One reason may be that the hydrogen bond between HW and OP is relatively weak. The other reason may be that the OP is bonded tightly to other atoms on the KDP surface, thus the peaks in the PDOS of OP are not only reflecting the bonding between HW and OP but also reflecting the bonding with other atoms on the surface. There are two main resonance peaks at energies of −17.5 eV and a wide range from −9.5 eV to 0 eV between the OW 2p states and the K 2p states, see Fig. 4(d), which reveals that the interaction between the OW and the surface K atom is relatively strong.
3.2. Adsorption of an isolated water molecule on the (101) surface
Geometries of water molecule adsorption at positions o and p on the KDP (101) surface are shown in Fig. 5(a) and (b) respectively. The details of bond lengths, bond angles and adsorption energy are shown in Table 4. As shown in Fig. 5, the geometries of water molecule adsorption at positions o and p on the KDP (101) surface are quite similar. Taking the geometry at position o as an example, one H atom of the water molecule is pulled toward the O atom of the phosphate group, indicating a strong interaction between HW and OP. The short HW–OP distance and the large OW–HW⋯OP angle (shown in Table 4) imply that this interaction is the hydrogen bond (OW–HW⋯OP) formed during the adsorption process. Meanwhile, the O atom of the water molecule is attracted to the K atom of the surface, which suggests that an OW–K chemical bond is formed during the adsorption process at position o. The similar geometry at position p leads to the same conclusion that a hydrogen bond (OW–HW⋯OP) and an OW–K chemical bond are formed when a single water molecule is adsorbed at position p.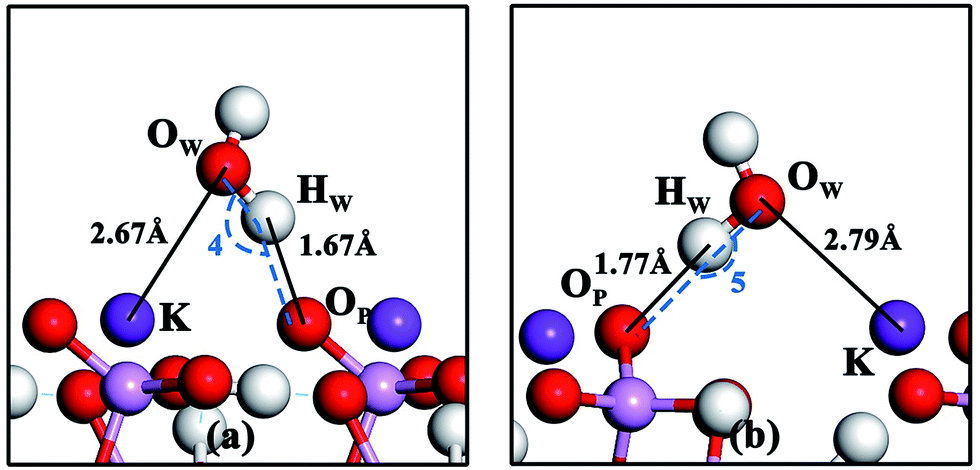 | ||
| Fig. 5 Geometries of water molecule adsorption at positions o (a) and p (b) on the KDP (101) surface. ∠4 and ∠5 are the angles from OW via HW to OP. | ||
| Structure | Eads (eV) | ∠OW–HW⋯OP (degree) | ∠OP–HP⋯OW (degree) | dOW–K (Å) | dHW–OP (Å) | dHP–OW (Å) |
|---|---|---|---|---|---|---|
| Position o | −0.60 | 156.1 | — | 2.67 | 1.67 | — |
| Position p | −0.63 | 173.3 | — | 2.79 | 1.77 | — |
The electron density difference maps between the water molecule and the KDP (101) surface at positions o and p are shown in Fig. 6(a) and (b), respectively. In addition, slices are made through related atoms (at position o shown in Fig. 6(a): slice 5 through HW and OP and slice 6 through OW and K; at position p shown in Fig. 6(b): slice 7 through HW and OP, slice 8 through OW and K) to show the difference in the electron density clearly. The electron density transfer of these two positions is similar too. The electrons of HW transfer to the neighbouring OP, which indicates that a hydrogen bond is formed. Meanwhile, the electrons of the K atom transfer to the neighbouring OW, which indicates that an O–K chemical bond is formed during the adsorption process.
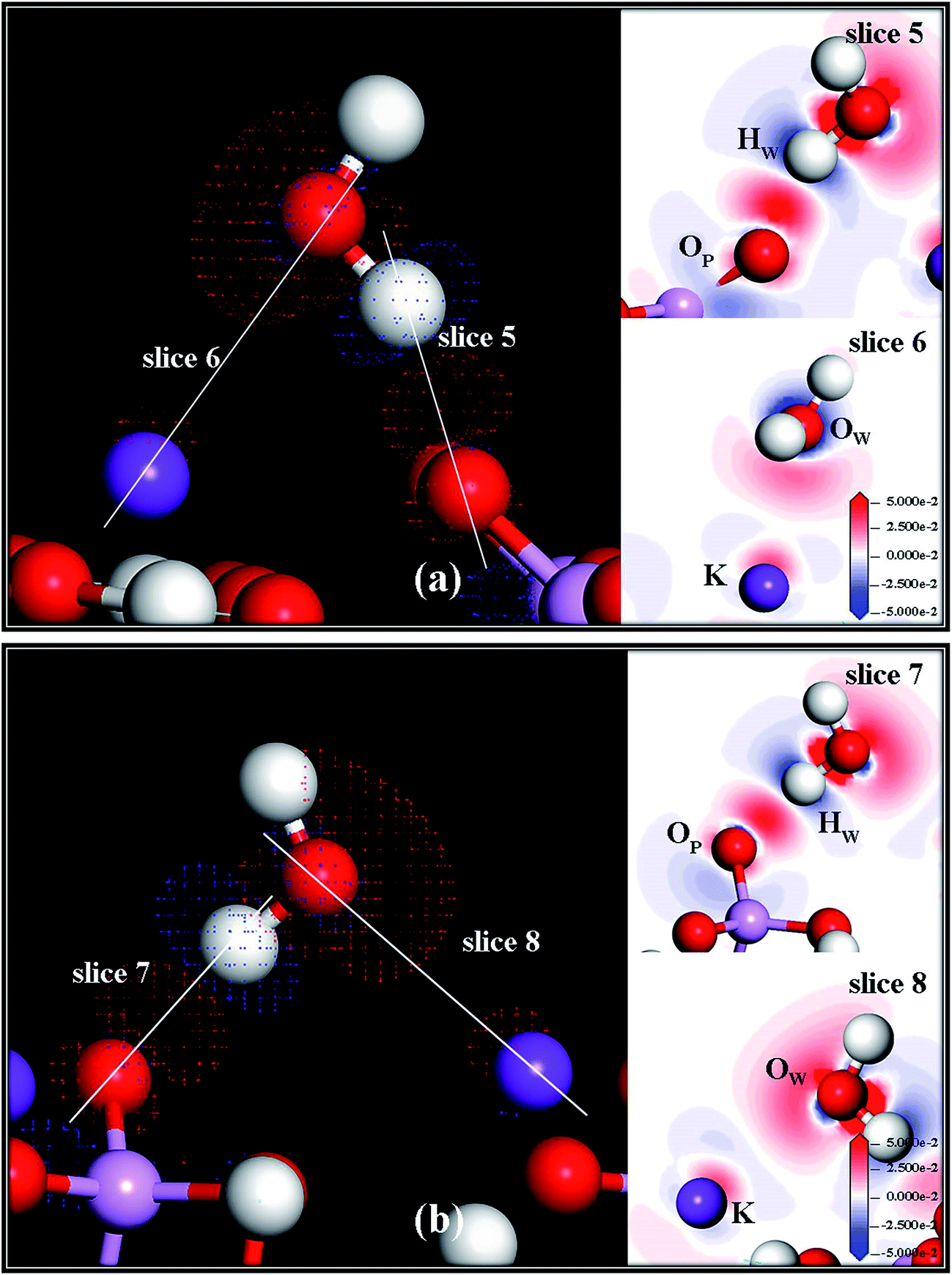 | ||
| Fig. 6 Electron density difference maps between the water molecule and the related phosphate group at position o (a) and position p (b) on the KDP (101) surface. Blue and red colors represent depletion and accumulation of electron density, respectively. Positive values indicate accumulation of electron density. Negative values correspond to depletion of electron density. | ||
We also calculated the PDOSs of the selected atoms, as shown in Fig. 7. At position o, as shown in Fig. 7(a) and (b), the resonance peaks between the HW 1s states and the OP 1s, 2p states are at energies of −20.5 eV and −6.7 eV and range from −3.8 eV to −1.5 eV. These resonance peaks might be attributed to the formation of a hydrogen bond. There are two main resonance peaks at energy of −18.5 eV and a wide range from −8.6 eV to 0 eV between the OW 2p states and the K 2p states, which reveals a strong interaction between OW and K exposed on the surface.
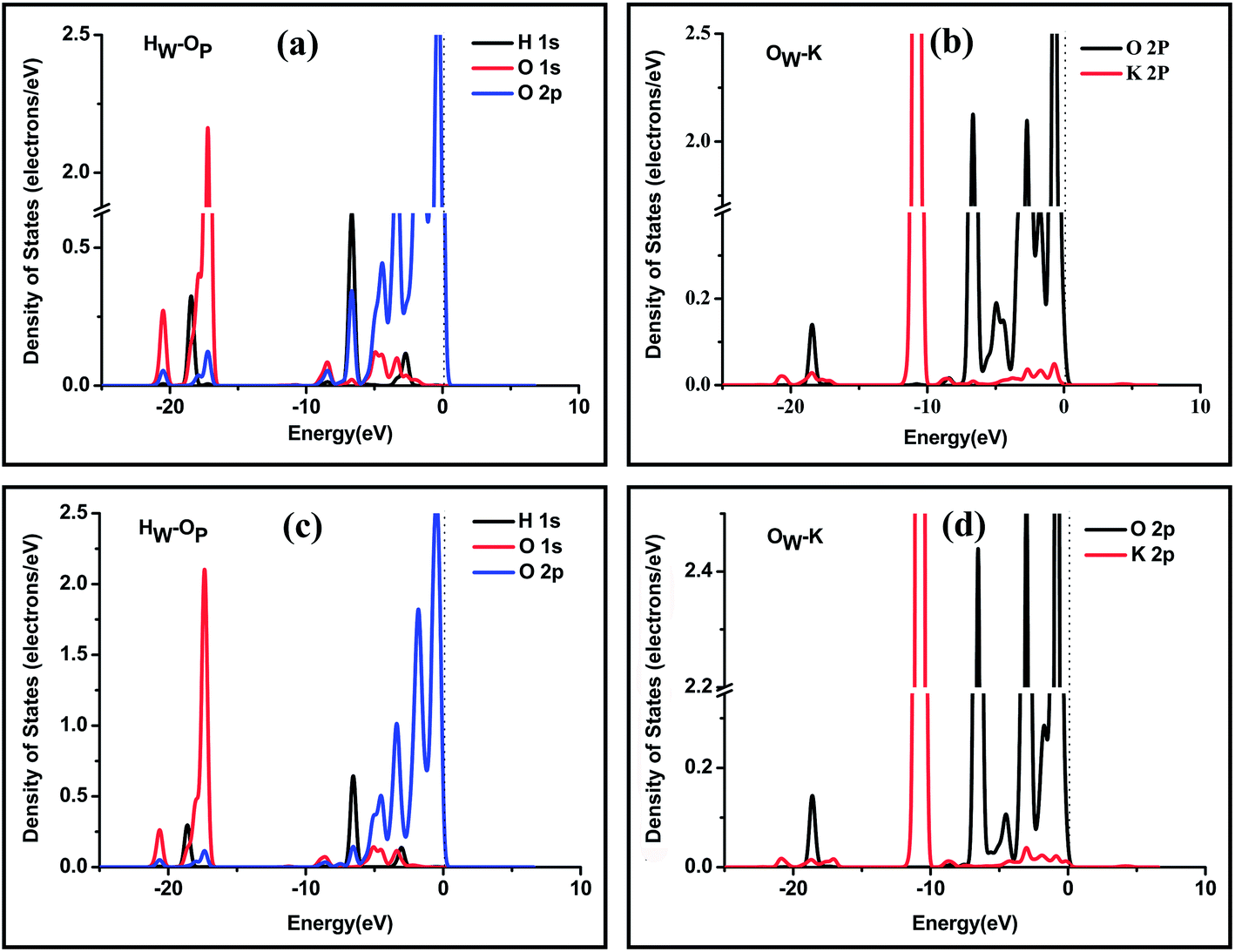 | ||
| Fig. 7 PDOSs of selected atoms of the water molecule and the (101) surface at position o: (a) atomic orbitals of the H atom of the water molecule and the O atom of the phosphate group and (b) atomic orbitals of the O atom of the water molecule and the K atom of the surface. PDOS of selected atoms of the water molecule and the (101) surface at position p: (c) atomic orbitals of the H atom of the water molecule and the O atom of the phosphate group and (d) atomic orbitals of the O atom of the water molecule and the K atom of the surface. The zero energy is the Fermi level. | ||
At position p, shown in Fig. 7(c) and (d), the resonance peaks between the HW 1s states and the OP 1s, 2p states are at energies of −20.5 eV, −6.7 eV and −3.4 eV, respectively. Compared with the PDOSs at position o, the less similar shape of the resonance peaks indicates that the hydrogen bond is weaker than that at position o. We also noticed that dHW–OP is larger at position p (1.77 Å) than that at position o (1.67 Å). The longer bond length also indicates that the hydrogen bond at position p is weaker than that at position o. The resonance peaks between OW 2p states and neighbor K 2p states are at an energy of −19.1 eV and a wide range from −8.7 eV to 0 eV, which proves the formation of O–K chemical bond. Because the distance between OP and K on the (101) surface at position p is longer than that at position o, when a hydrogen bond and an O–K chemical bond are formed, the two bonds have to be stretched to adapt to the longer distance between OP and K, making both bonds weak.
After the comparison and analysis of the adsorption behaviors of a water molecule and two typical surfaces of KDP, we found different interactions between the water molecule and the KDP surfaces. On the KDP (100) surface, the water molecule may form two hydrogen bonds with the neighbor phosphate group (position e) or form an O–K chemical bond with the neighbor K atom and a hydrogen bond with the neighbor phosphate group (position h) during the adsorption process. While on the KDP (101) surface, the water molecule can only be adsorbed by forming an O–K chemical bond and a hydrogen bond. This difference is caused by the different structures of the (100) and (101) surfaces of the KDP crystal. There are three kinds of elements (K, O and H atoms of the phosphate groups) exposed on the (100) surface, making it possible to form two hydrogen bonds (OW–HW⋯OP and OP–HP⋯OW) when a water molecule gets close to a phosphate group. However, on the (101) surface, there are only two kinds of atoms (K and O atoms of the phosphate groups) exposed on the surface. When a water molecule is adsorbed on the KDP (101) surface, it can only form a hydrogen bond and an O–K chemical bond with the surface.
The adsorption style (forming an O–K chemical bond and a hydrogen bond with the surface) at positions o and p on the KDP (101) surface (both positions are O–K bridges) is similar to that at position h on the KDP (100) surface (P–K bridge). Indeed, position e on the KDP (100) surface (P top site) can be treated as a HP–OP bridge, as shown in Fig. 8(a). When we locate the most stable positions on the potential maps of the KDP (100) and (101) surfaces, we discover that all these positions are in the valleys between two peaks of high potential. It seems that the parts of high potential (H, O and K atoms exposed on the surface) on the surface pull water molecules to them (in this paper, the pulling effect is between OW and HP, OW and K and HW and OP). When the attractions of two states with high potential reach a balance, the water molecule is just at the bridge of these two parts. We also noticed that these two elements with high energy must be exposed on the surface and shall be close to each other.
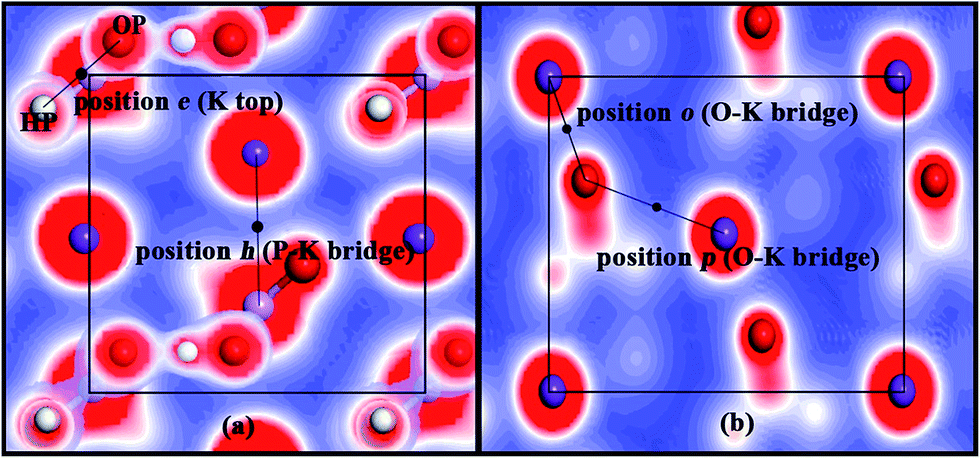 | ||
| Fig. 8 The potential maps of the KDP (100) and (101) surfaces. (a) Represents the KDP (100) surface and (b) represents KDP (101) surface. Red and blue colors mean high potential and low potential, respectively. | ||
We also found that the adsorption energy at the most stable adsorption sites on the KDP (101) surface (−0.60 eV and −0.63 eV) is slightly larger (absolute values) than that on the KDP (100) surface (−0.57 eV and −0.47 eV). This means that a water molecule can be adsorbed more tightly on the KDP (101) surface than on the KDP (100) surface, indicating that the KDP (101) surface can be more easily dissolved than the (100) surface. Furthermore, the pulling effect of the surface mainly affects the O and H atoms of the water molecule, from which we can deduce that the solute, the phosphate group with an O atom and a H atom exposed, can also be more easily adsorbed on the KDP (101) surface. This might be one reason for the KDP (101) surface growing faster than the (100) surface according to experiments.33
4. Conclusion
In this paper, we studied the adsorption of a single water molecule on the (100) and (101) surfaces of a KDP crystal by DFT calculations for the first time, as far as we know. It was found that the most stable adsorption sites on the KDP (100) surface are the P top site and the P–K bridge site while the most stable sites on the KDP (101) surface are both O–K bridge sites. These positions are at the energy valleys between two states of high potential. The stable configurations are the results of the competition and balance of the pulling effect of two parts of high potential on the surface. We also found the O, H and K atoms on the surfaces are the functional atoms that pull the water molecule toward the surfaces, which we believe is the key to controlling the influences of water adsorption. For example, if we were able to make a new kind of coating that could form strong bonds with these functional atoms, the water molecules in the environment would have few available adsorption sites on the surfaces of the KDP crystal and therefore have little effect on the KDP surfaces. The bond formation between a water molecule and the KDP surfaces is also included in our work. The water molecule can be adsorbed in two different ways—forming two hydrogen bonds with the O and H atoms on the surface or forming a hydrogen bond and an O–K bond with the O and K atoms on the surface. Because the atoms on the surface are ordered, water molecules bonding with these atoms would also show an ordered structure corresponding to the arrangement of these surface atoms. This could partly explain the liquid order at the interface from a theoretical point of view.Acknowledgements
We are grateful for the support to one of the authors (Yao Liu) from the National Natural Science Foundation of China [grant No. 51402170] and the Fundamental Research Funds of Shandong University [grant No. 2016WLJH40].References
- R. C. Miller, D. A. Kleinman and A. Savage, Phys. Rev. Lett., 1963, 11, 146–149 CrossRef CAS.
- M. J. Chu and S. S. Lee, J. Appl. Phys., 1985, 57, 2647–2649 CrossRef CAS.
- W. Jamroz and J. Karniewicz, Opt. Quantum Electron., 1979, 11, 23–27 CrossRef CAS.
- M. Izdebski, W. Kucharczyk and R. E. Raab, J. Opt. Soc. Am. A, 2002, 19, 1417–1421 CrossRef CAS PubMed.
- J. E. Swain, S. E. Stokowski, D. Milam and G. C. Kennedy, Appl. Phys. Lett., 1982, 41, 12–14 CrossRef CAS.
- J. J. De Yoreo, A. K. Burnham and P. K. Whitman, Int. Mater. Rev., 2002, 47, 113–152 CrossRef CAS.
- D. Eimerl, Ferroelectrics, 1987, 72, 95–139 CrossRef CAS.
- N. P. Zaitseva, J. J. De Yoreo, M. R. DeHaven, R. L. Vital, K. E. Montgomery, M. Richardson and L. J. Atherton, J. Cryst. Growth, 1997, 180, 255–262 CrossRef CAS.
- X. Zhuang, L. Ye, G. Zheng, G. Su, Y. He, X. Lin and Z. Xu, J. Cryst. Growth, 2011, 318, 700–702 CrossRef CAS.
- M. Nakatsuka, K. Fujioka, T. Kanabe and H. Fujita, J. Cryst. Growth, 1997, 171, 531–537 CrossRef CAS.
- S. A. De Vries, P. Goedtkindt, S. L. Bennett, W. J. Huisman, M. J. Zwanenburg, D. M. Smilgies, J. J. De Yoreo, W. J. P. Van Enckevort, P. Bennema and E. Vlieg, Phys. Rev. Lett., 1998, 80, 2229 CrossRef CAS.
- M. Reedijk, J. Arsic, F. Hollander, S. De Vries and E. Vlieg, Phys. Rev. Lett., 2003, 90, 066103 CrossRef CAS PubMed.
- H. Gao, X. Wang, X. Teng and D. Guo, Sci. China: Technol. Sci., 2015, 58, 1877–1883 CrossRef.
- X. Wang, H. Gao, D. M. Guo, Y. C. Chen and C. P. Song, Adv. Mater. Res., 2013, 797, 423–427 CrossRef.
- I. M. Thomas, Appl. Opt., 1986, 25, 1481–1483 CrossRef CAS PubMed.
- E. K. Wheeler, R. Fluck, B. Woods and P. K. Whitman, Appl. Opt., 2003, 42, 5545–5550 CrossRef CAS PubMed.
- E. K. Wheeler, P. K. Whitman, T. A. Land, J. De Yoreo, C. B. Thorsness, J. H. McWhirter, M. L. Hanna and E. L. Miller, Appl. Phys. A, 2002, 74, 813–823 CrossRef CAS.
- A. G. Stack, J. R. Rustad, J. J. DeYoreo, T. A. Land and W. H. Casey, J. Phys. Chem. B, 2004, 108, 18284–18290 CrossRef CAS.
- D. J. Carter and A. L. Rohl, J. Chem. Theory Comput., 2011, 7, 1604–1609 CrossRef CAS PubMed.
- D. J. Carter, A. L. Rohl and J. D. Gale, J. Chem. Theory Comput., 2006, 2, 797–800 CrossRef CAS PubMed.
- E. V. Stefanovich and T. N. Truong, Chem. Phys. Lett., 1999, 299, 623–629 CrossRef CAS.
- W. Wang, Y. Shen, X. Wang, Z. Zhou and W. Fei, Appl. Surf. Sci., 2014, 298, 102–108 CrossRef CAS.
- L. Giordano, J. Goniakowski and J. Suzanne, Phys. Rev. Lett., 1998, 81, 1271–1273 CrossRef CAS.
- B. Morosin and G. A. Samara, Ferroelectrics, 1972, 3, 49–56 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- D. Y. He, L. J. Qiao, A. A. Volinsky, Y. Bai, M. Wu and W. Y. Chu, Appl. Phys. Lett., 2011, 98, 062905 CrossRef.
- X. Li, B. Wang, T. Y. Zhang and Y. Su, J. Phys. Chem. C, 2014, 118, 15910–15918 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- N. Zaitseva and L. Carman, Prog. Cryst. Growth Charact. Mater., 2001, 43, 1–118 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |