
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Urolithin C, a gut metabolite of ellagic acid, induces apoptosis in PC12 cells through a mitochondria-mediated pathway†
Peipei Yina,
Jianwei Zhanga,
Linlin Yana,
Lingguang Yanga,
Liwei Suna,
Lingling Shia,
Chao Ma*ab and
Yujun Liu *a
*a
aNational Engineering Laboratory for Tree Breeding, College of Biological Sciences and Biotechnology, Beijing Forestry University, Beijing 100083, China. E-mail: happy62889@126.com; jwzhang@iccas.ac.cn; yanlinlin155@163.com; yanglingguangxdjqz@163.com; lsun2013@bjfu.edu.cn; linglingshi2005@163.com; yjliubio@bjfu.edu.cn
bBeijing Key Laboratory of Forest Food Processing and Safety, College of Biological Sciences and Biotechnology, Beijing Forestry University, Beijing 100083, China. E-mail: machao@bjfu.edu.cn
First published on 20th March 2017
Abstract
Urolithins (Uros), metabolites of ellagitannins (ET) and ellagic acid (EA) produced by gut microbiota, showed better bioavailability and extensive bioactivity, and were considered as the active compounds responsible for the health benefits exerted by ET-containing foodstuffs. Here, we chemically synthesized three Uros including Uros A, B, and C and compared their anti-proliferative activities with that of EA in PC12 cells. MTT assay showed that EA significantly promoted, while Uros significantly inhibited the proliferation of PC12 cells, among which UroC showed the strongest anti-proliferation. UroC treatment actively increased the lactate dehydrogenase (LDH) release and lipid peroxidation malondialdehyde (MDA), stimulated reactive oxygen species (ROS) formation and mitochondrial membrane depolarization, and caused calcium dyshomeostasis. Furthermore, flow cytometry analysis showed that UroC treatment induced apoptosis and S phase cell cycle arrest with increasing UroC concentrations. Consequently, UroC also induced imbalance in the Bcl-2/Bax ratio, which triggered the caspase cascade, thereby shifting the balance in favor of apoptosis, as evidenced by western blotting and real-time PCR. These observations indicated that UroC possessed significantly different anti-proliferation activities from EA, and actively induced cell apoptosis through a mitochondria-mediated pathway.
Introduction
Health effects of many fruits and nuts, such as pomegranate, strawberry, raspberry, blackberry, grapes, walnuts and chestnuts, have been associated with a high level of antioxidant polyphenols, particularly ellagitannins (ET),1,2 which are partially converted into ellagic acid (EA) in the upper gastrointestinal tract.3 Both ET and EA show potential preventive effects against chronic diseases such as cancer, diabetes, cardiovascular diseases, and neurodegenerative disease in animal and human studies.1 However, it is well established that absorptions of ET and EA by the gastrointestinal tract are extremely poor, and the unabsorbed compounds are extensively metabolized by the gut microbiota in the colon.4–8 The poor bioavailabilities of ET and EA as well as their extensive metabolism in the gastrointestinal tract have raised the question whether these parent molecules, found as such in the ellagitannin-containing foodstuffs, were the real active compounds responsible for the health benefits.1Metabolism studies revealed that ET and EA were metabolized into urolithins (Uros), dibenzopyran-6-one derivatives with different hydroxyl substitutions, by gut microbiota through losing one of the two lactones present in EA and by successive removals of hydroxyls through dehydroxylase.5 There are a number of Uros produced by gut microbiota, and the main metabolites detected in the plasma and urine of rats were urolithin A (UroA), urolithin B (UroB) and urolithin C (UroC) (see Fig. 1) after oral administration of pomegranate ET4 and other ET-containing foodstuffs such as walnuts, strawberries, and raspberries.6 Once produced, the Uros could be absorbed, circulate in plasma and accumulate in urine as glucuronide and sulphate conjugates or can be directly excreted in feces as aglycones.9 The main metabolites detected in the plasma of humans after the intake of ET-containing foods were the glucuronides, sulphates and methylates of UroA, UroB and UroC with the total concentration of these metabolites ranging from ∼0.2 μM up to ∼18.6 μM,3,10 and these values show a large variability between individuals and between days.5,11 In addition, it had been reported that Uros could also undergo extensive biotransformations in cardiomyocytes and fibroblasts cells in vitro, and a relevant phase II metabolism characterized by (de)glucuronidation, methylation, and sulphation reactions could be observed in the cell cultures.12 Formations of Uros A, B, and C have been recently confirmed by Piwowarski J. P.,13 who determined Uros A, B, and C by ex vivo incubation of human fecal samples after ingestion of aqueous extracts from several ET-containing plant materials. Uros A, B, and C are bioavailable and could reach micromolar concentrations in the blood stream in vivo.4,8 Therefore, ET and EA may exert local health benefits on the gastrointestinal tract, but systemic health benefits result more likely from Uros, such as Uros A, B and/or C, rather than ET and EA themselves, as a consequence of the consumption of ET-containing foodstuffs.3 In this regard, there is an urgent need to assess the direct biological activities of Uros, which could be the missing link to explain the health benefits associated with the consumption of ET-containing foodstuffs.1
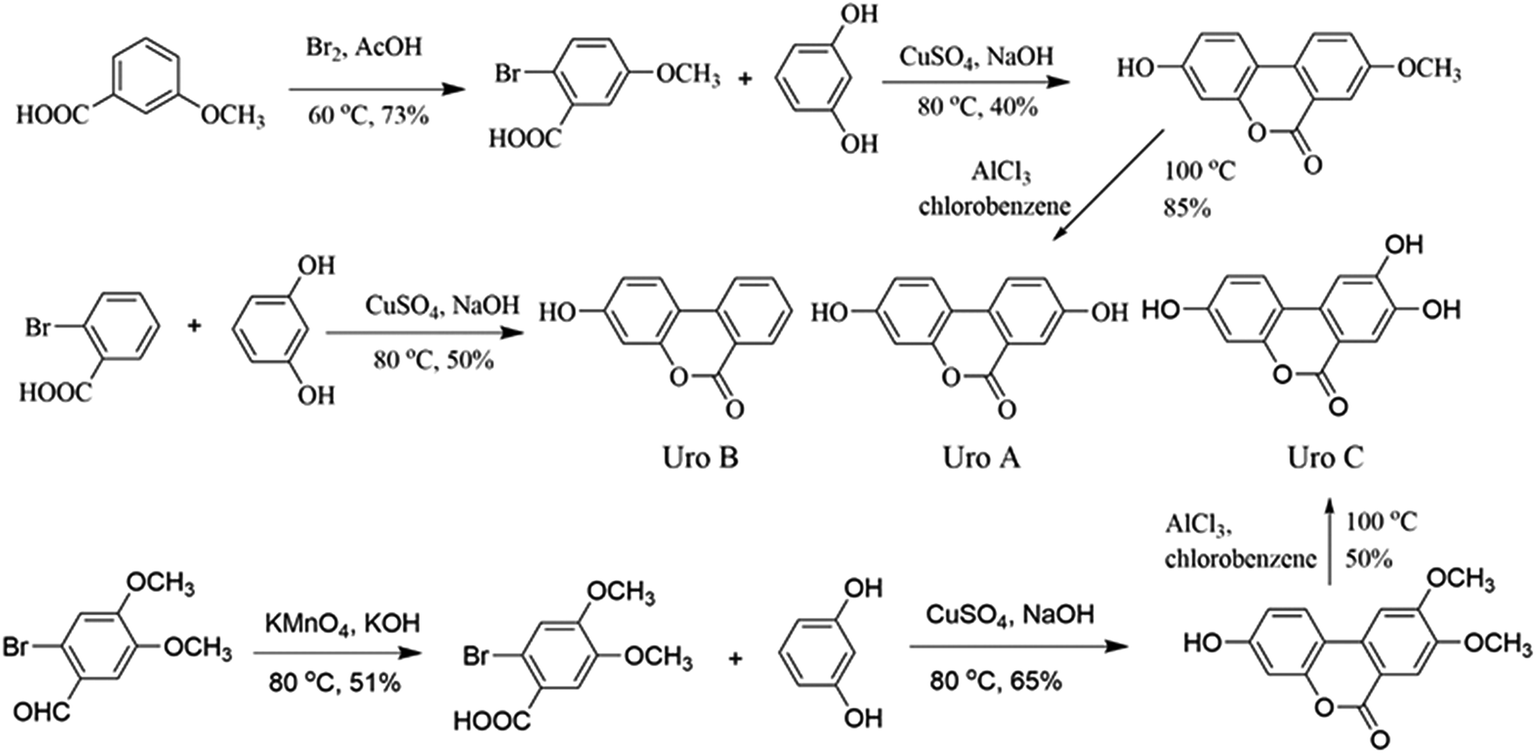 | ||
| Fig. 1 Organic synthesis of Uros A, B and C. | ||
Recently, several studies have described the antioxidant,14 anti-inflammation,10,15 anticancer16–20 and anti-glycation21 activities of Uros. Moreover, Ryu et al.22 reported that UroA could induce mitophagy and prolong lifespan in C. elegans and increase muscle function in rodents. However, few studies have been carried out on the neuroprotective/neurotoxicity activities of Uros,23 although ET-containing foodstuffs showed significant neuroprotective activities in many studies.24–26
In recent years, there are increasing researches about the biological effects of urolithins assayed on human cell lines, such as Caco-2 (human colon carcinoma cell line), CCD18-Co (human colon fibroblast cell line), HEK T293 (human embryonic kidney cell line), HL-60 (promyelocytic leukaemia cells) and MCF-7 (human breast cancer cell line).1 However, relatively little is known about the effects of urolithins on nerve cells and the underlying molecular mechanisms of apoptosis by urolithins in never cells have not been fully determined. PC12 cells derived from pheochromocytoma of rat adrenal medulla, has an embryonic origin from the neural crest and thus shows a variety of neuronal properties, including neurotransmitter release, neurite growth, and response to nerve growth factor, and it has been used in thousands of studies to evaluate neurophysiology, neuropathology and neuropharmacology.27,28 In this study, we establish the synthesis methods of Uros A, B and C. The aim of the present study was to evaluate the cytotoxicity of EA and Uros A, B and C on rat pheochromocytoma PC12 cells, as well as to elucidate the underlying mechanism of UroC.
Materials and methods
Reagents for organic synthesis and cellular determination
3-Methoxybenzoic, Br2, acetic acid, resorcinol, NaOH, CuSO4, AlCl3, KOH, chlorobenzene, and potassium permanganate were purchased from Sinopharm Chemical Reagent Beijing Co., Ltd (China) for the synthesis of Uros A, B and C. RPMI-1640 medium, trypsin, penicillin/streptomycin, and sodium pyruvate were obtained from Gibco Life Technologies (NY, USA). Equine serum, fetal bovine serum (FBS), and PBS were purchased from Hyclone (Utah, USA). Bicinchoninic acid (BCA) protein, lipid peroxidation malondialdehyde (MDA), and lactate dehydrogenase (LDH) cytotoxicity detection kits were obtained from Nanjing Jiancheng Bioengineering Institute (China). 2′,7′-Dichlorofluorescin diacetate (DCFH-DA), diaminofluorescein-FM diacetate (DAF-FM DA), Annexin V-FITC/PI cell apoptosis, cell cycle detection kits, and Trizol Reagent were purchased from Invitrogen (CA, USA). Sodium nitroprusside (SNP) and EA were obtained from Sigma-Aldrich (MO, USA). FastQuant RT Kit (with gDNase) and SuperReal PreMix Plus (SYBR Green) kit were bought from Qiagen (Düsseldorf, Germany).Cell culture and treatments
PC12 cells were purchased from Cell Culture Center, China Academy of Medical Sciences & Peking Union Medical College and maintained in a RPMI-1640 medium supplemented with 5% FBS, 10% equine serum, 1% sodium pyruvate, 100 units per ml penicillin and 100 μg ml−1 streptomycin at 37 °C with 5% CO2 in a humidified atmosphere. They were plated in 96-, 12-, or 6-well plates at appropriate fusion rate prior to administration of UroA, UroB, UroC, and EA dissolved in DMSO. The final concentration of DMSO in each well was less than 0.15% and there was no observable toxicity in our studies.Cell viability and cytotoxicity assays
Cell viability was assessed using the MTT assay, which evaluated mitochondrial activity by measuring the conversion of yellow tetrazolium salt to purple formazan crystals by mitochondrial dehydrogenase. Briefly, the monolayer PC12 cells were trypsinized and cell count was adjusted to 1.0 × 105 cells per ml using the RPMI-1640 medium containing 5% FBS and 10% equine serum. To each well of the 96-well cell culture plate, 100 μl diluted cell suspension was added. After 12 h incubation when a partial monolayer was formed, the supernatant was flicked off and 100 μl serum-free RPMI-1640 medium containing different concentrations of EA, UroA, UroB, or UroC was added. After another 12 h incubation, 20 μl MTT in PBS (5 mg ml−1) was added to each well. The plate was incubated for 4 h at 37 °C in 5% CO2 atmosphere. The supernatant was removed, 150 μl DMSO was added, and the plate was gently shaken to solubilize the formed formazan. Absorbance was measured at 570 nm using a microplate reader (Tecan infinite 200, Swiss) and inhibitory effects of drugs on cell growth were assessed as percent of cell viability. Control cells treated with the vehicle were considered as 100% viable.Cell cytotoxicity was evaluated by measuring the LDH levels in the cell culture supernatant using LDH kit according to the manufacture's protocol. Briefly, after treatment with different concentrations of UroC, cell-culture media were collected and added to the microplate pre-coated with an antibody specific to LDH, then avidin conjugated to Horseradish Peroxidase (HRP) was added to each well and incubated. Next, a TMB (3,3′,5,5′-tetramethyl-benzidine) substrate solution was added to each well and incubated. The enzyme–substrate reaction was terminated by the addition of a sulphuric acid solution and the color change was measured spectrophotometrically at 450 nm using the microplate reader. Protein concentrations were measured with a BCA kit, and the LDH levels were then calculated and expressed as IU LDH per g protein (IU per g Prot).
Flow-cytometric analyses of cell apoptosis and cell cycle arrest
UroC-induced apoptosis was measured using an Annexin V-FITC/PI apoptosis detection kit according to the manufacture's protocol. Briefly, PC12 cells were seeded in 6-well plates at a 70–80% fusion rate. After treatment with UroC, cells were collected and centrifuged at 3000 rpm for 1.5 min, and washed with cold PBS. Approximately 1 × 106 cells per ml were resuspended in 300 μl binding buffer and the cells were incubated with 5 μl Annexin V-FITC for 15 min. After that, 5 μl PI and 200 μl binding buffer were added before detecting with a flow cytometer (FACS Calibur, BD Biosciences).The cell cycle detection kit was used to test cell cycle arrest. In brief, PC12 cells were seeded at a density of 2 × 106 cells per well until the logarithmic phase and treated with various concentrations of UroC for 5 h. Treated cells were then collected by trypsinization, washed twice with cold PBS, and fixed with 75% ice-cold ethanol at 4 °C for 20 h. Next, the fixed cells were centrifuged, re-suspended in PBS and maintained at room temperature for 15 min. Permeabilization and staining of the cells were conducted after incubation for 30 min at room temperature in the dark with PBS containing 5 μg ml−1 PI, 0.1% Triton X-100, and 50 μg ml−1 RNase A. Measurements were performed with the flow cytometer. The PC12 cells were excited at 488 nm with an argon laser source and fluorescence was detected through a 563–607 nm band pass filter (FL-2 channel).
Measurements of intracellular ROS, NO, MDA and Ca2+ levels, and mitochondrial membrane potential (ΔΨm)
Intracellular changes in ROS generation after UroC treatment were measured by staining the cells with DCFH-DA, which could be converted into the cell membrane impermeable nonfluorescent compound DCFH by intracellular esterases. DCFH was then oxidized by ROS to produce a highly fluorescent DCF, which was proportional to the amount of ROS.19 The ability of UroC to generate ROS in PC12 cells was estimated as described earlier19 with some modifications. PC12 cells were treated with 10 and 100 μg ml−1 UroC for 2 h. Afterwards, the cells were washed 3 times with PBS, and further incubated with 2.5 μM DCFH-DA in the Kreb's ringer solution at 37 °C for 20 min. Finally, the cells were washed 3 times with PBS, and the fluorescence intensity was determined by a fluorescence microscopy (Leica DMI4000B, Germany). H2O2 of 100 μM was used as the positive control.Cellular concentration of NO was an important oxidative damage indicator. NO levels after UroC treatment were measured with DAF-FM DA staining described by Sheng et al.29 In brief, PC12 cells were treated with 10 and 100 μg ml−1 UroC for 2 h and the cells were washed twice with PBS. After that, the cells were further incubated with 5 μM DAF-FM DA at 37 °C for 35 min. Finally, the cells were washed 3 times with PBS, and the fluorescence intensity was determined using the fluorescence microscopy. SNP at 200 μg ml−1 was used as the positive control.
MDA was a lipid peroxidation product by ROS and the content of MDA can reflect the degree of lipid peroxidation of cells. MDA levels after UroC treatment were measured by using the method of Sheng et al.29 After UroC treatment, PC12 cells were washed 3 times with PBS, collected, and then centrifuged at 4000 rpm min−1 for 3 min. The cells were ultrasonically broken down in an ice bath and the MDA levels were measured using the MDA assay kit according to the manufacturer's protocol. MDA levels were expressed as nmol mg−1 protein (nmol mg−1 Prot).
Fluo-3AM was used for the determination of intracellular Ca2+.30 In brief, PC12 cells were treated with different concentrations of UroC for 2 h. After which, the cells were washed twice with HBSS and incubated with 5 μM Fluo-3 AM for at least 30 min at 37 °C. The cells were then washed with HBSS to remove the excess probe and incubated for another 30 min at 37 °C. Fluorescence was determined by the fluorescence microscopy.
Mitochondrial membrane potential was assessed using rhodamine 123 as described by Xue et al.31 with some modifications. Briefly, PC12 cells were seeded in confocal dishes at 2 × 105 cells per dish. After treatment with UroC, the cells were washed twice with PBS and incubated with Kreb's ringer buffer containing 2 μM rhodamine 123 at 37 °C for 30 min. The cells were then washed again with PBS to remove the excess stain, and analyzed for changes in ΔΨm using the fluorescence microscopy.
Western blot analysis of apoptosis-associated proteins
Western-blot analysis was performed as described previously.32 Briefly, UroC-treated cells were washed 3 times in precooling PBS and lysed on ice in 300 μl lysis buffer containing 1% PMSF. Thirty min later, the cell lysates were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 4 °C for 5 min and the protein concentrations in the supernatant were determined using the BCA method. The rest of supernatant was boiled at 100 °C for 6 min, and the supernatants were stored at −80 °C until further use. Equal amounts of proteins were separated on a 12% sodium dodecyl sulfate (SDS)-PAGE and transferred to nitrocellulose membrane by electroblotting. The membranes were blocked in Tris-buffered saline-Tween solution (TBST) containing 5% (w/v) non-fat milk for 2 h at 4 °C, followed by overnight incubation at 4 °C with primary antibodies. After washing with TBST buffer for 3 times, the membranes were incubated with anti-secondary antibody for 4 h at 4 °C. Detection was achieved by chemical fluorescence following an enhanced chemiluminescence western blotting protocol. To confirm equal loading, membranes were reprobed with GADPH antibody.
000 rpm and 4 °C for 5 min and the protein concentrations in the supernatant were determined using the BCA method. The rest of supernatant was boiled at 100 °C for 6 min, and the supernatants were stored at −80 °C until further use. Equal amounts of proteins were separated on a 12% sodium dodecyl sulfate (SDS)-PAGE and transferred to nitrocellulose membrane by electroblotting. The membranes were blocked in Tris-buffered saline-Tween solution (TBST) containing 5% (w/v) non-fat milk for 2 h at 4 °C, followed by overnight incubation at 4 °C with primary antibodies. After washing with TBST buffer for 3 times, the membranes were incubated with anti-secondary antibody for 4 h at 4 °C. Detection was achieved by chemical fluorescence following an enhanced chemiluminescence western blotting protocol. To confirm equal loading, membranes were reprobed with GADPH antibody.
Real-time PCR analysis of apoptosis-associated mRNAs
For mRNA analysis, the real-time (RT) reaction was performed according to Sobenin et al.33 with some modifications. Briefly, PC12 cells were plated at 2 × 105 cells per ml in 6-well plates for 12 h, then incubated with different concentrations (0, 5, 10 and 50 μg ml−1) of UroC for 24 h. Total RNA from PC12 cells was obtained using the Trizol Reagent following the manufacturer's instructions. Four microliter of total RNA was reverse-transcribed to synthesize cDNA using the FastQuant RT Kit according to the manufacturer's protocol. Real-time PCR was performed using the SuperReal PreMix Plus kit. Reactions were prepared in a total volume of 20 μl, containing 10 μl of 2× SuperReal PreMix Plus, 0.6 μl of 10 μM Forward Primer, 0.6 μl of 10 μM Reverse Primer, 1.8 μl cDNA template, and 7 μl ddH2O. The cycling parameters were performed as following: 95 °C for 15 min (initial denaturation), 40 cycles at 95 °C for 10 s (denaturation), 55 °C for 20 s (annealing), and 72 °C for 20 s (extension). Dissociation curves for each amplicon were analyzed by keeping at 65 °C for 31 min, then heating to 95 °C and reading at each 0.5 °C to verify the specificity of each amplification reaction. For Bax, Bcl-2, caspase-3, caspase-9, and β-actin with sizes of 204, 232, 210, 187, and 218 bp, the primers were 5′-GTGGTTGCCCTCTTCTACTTTG-3′ (F) and 5′-CACAAAGATGGTC ACTGTCTGC-3′ (R), 5′-ACGAGTGGGATACTGGAGATG-3′ (F) and 5′-TAGCGACGAGAGAAGTCATCC-3′ (R), 5′-TGGCCCTGAAATACGAAGTC-3′ (F) and 5′-GGCAGTAGTCGCCTCTGAAG-3′ (R), 5′-TGTTTTGCAGGT CGCCAATG-3′ (F) and 5′-GCTTCACGGGACAGTTCTGA-3′ (R), and 5′-CACCCGCGAGTACAACCTTC-3′ (F) and 5′-CCCATACCCACCATCACACC-3′ (R), respectively.Statistical analysis
All experiments were carried out at least in triplicates and data were expressed as mean ± standard deviation (SD). The statistical significance (t-test: two-sample equal variance, using two-tailed distribution) was determined by using Microsoft Excel statistical software (Microsoft Office Excel 2016, Microsoft Corp. Redmond, WA, USA). p < 0.05 was set to be significant.Results
Organic synthesis and structure identification of Uros A, B and C
Uros were synthesized according to reported protocols34,35 with modification (Fig. 1), and identifications were conducted based on their 1H NMR and 13C NMR spectra.:AcOH = 19:100) was dropped into 50 ml 3-methoxybenzoic solution (0.40 g ml−1), and after reacting in an ice water bath overnight, the mixture was added to ice to yield 2-bromo-5-methoxybenzoic acid (A1, 21.23 g, yield 73%). Then, 20.0 g A1, 19.9 g resorcinol, and 7.0 g NaOH in 35 ml water were refluxed at 80 °C for 30 min. After the addition of 20 ml aqueous CuSO4 (10%), the mixture was refluxed for a further 60 min and 3-hydro-8-methoxy-6H-dibenzo-[b,d]pyran-6-one (A2, 8.76 g, 40%) precipitated as light pink powders. Finally, A2 (8.0 g) and AlCl3 (8.83 g) in chlorobenzene (60 ml) was refluxed overnight, after which the mixture was added to ice and UroA precipitated as white powders (6.41 g, 85%). The residual solid was purified by column chromatography on silica (10% ethyl acetate/hexane) and the final purity of UroA was 98%.Proton and carbon signals in 1H NMR (see Fig. S1†) and 13C NMR (Fig. S2†) of UroA were allocated as following. 1H NMR (300 MHz, d6-DMSO, δ): 6.73 (d, J = 1.8 Hz, ArH, 1H), 6.81 (dd, J1 = 2.4 Hz, J2 = 8.5 Hz, ArH, 1H), 7.33 (dd, J1 = 2.4 Hz, J2 = 8.5 Hz, ArH, 1H), 7.52 (d, J = 2.7 Hz, ArH, 1H), 8.03 (d, J = 8.8 Hz, ArH, 1H), 8.11 (d, J = 8.8 Hz, ArH, 1H), 10.2 (s, 1H, ArH), 10.2 (s, 1H, ArH); 13C NMR (100 MHz, d6-DMSO, δ): 161.1, 159.0, 157.4, 151.4, 127.4, 124.6, 124.2, 124.0, 120.6, 14.0, 113.5, 110.3, 103.3.
Proton and carbon signals in 1H NMR (Fig. S3†) and 13C NMR (Fig. S4†) of UroB were allocated as following. 1H NMR (300 MHz, d6-DMSO, δ): 6.74 (d, J = 2.4 Hz, ArH, 1H), 6.83 (dd, J1 = 2.4 Hz, J2 = 8.7 Hz, ArH, 1H), 7.51–7.56 (m, 1H, ArH), 7.83–7.88 (m, 1H, ArH), 8.11 (s, 1H, ArH), 8.14–8.24 (m, ArH, 2H), 10.4 (brs, 1H, ArOH); 13C NMR (100 MHz, d6-DMSO, δ): 161.1, 160.4, 152.6, 135.7, 135.6, 130.1, 128.1, 125.3, 122.1, 119.4, 113.6, 109.8, 103.4.
Proton and carbon signals in 1H NMR (Fig. S5†) and 13C NMR (Fig. S6†) of UroC were allocated as following. 1H NMR (300 MHz, d6-DMSO, δ): 6.69 (s, 1H, ArH), 6.78 (d, J = 8.2 Hz, ArH, 1H), 7.45 (s, 1H, ArH), 7.49 (s, 1H, ArH), 7.82 (d, J = 8.4 Hz, ArH, 1H), 10.0 (brs, 1H, ArOH), 10.1 (brs, 1H, ArOH), 10.4 (brs, 1H, ArOH); 13C NMR (100 MHz, d6-DMSO, δ): 160.8, 159.1, 153.9, 151.9, 146.6, 129.6, 124.2, 114.7, 113.3, 111.4, 110.3, 107.3, 101.3.
Cytotoxicity of EA and Uros in PC12 cells
EA and Uros were examined for their ability to inhibit proliferation of PC12 cells using MTT assay, which was usually used as an index of cell viability by measuring mitochondrial dehydrogenase activity based on its ability of cleaving tetrazolium ring to produce formazan. As shown in Fig. 2A, EA could increase to certain extent at 10 and 100 μg ml−1, while significantly promoted proliferation of PC12 cells at 300 μg ml−1. In contrast, Uros significantly inhibited the proliferation of PC12 cells at high concentrations, among which UroC showed the most significant cytotoxicity. These observations indicated that Uros, produced by the gut microbiota probably from EA, possessed significantly different bioactivities to those of EA. Furthermore, the cytotoxicity of UroC was confirmed with LDH assay, which was usually used to quantitatively measure LDH released into the medium from damaged cells as a biomarker for cellular cytotoxicity and cytolysis. As shown in Fig. 2B, LDH levels in medium increased significantly with increasing UroC concentrations, indicating that PC12 cells were severely damaged. In view of its potent cytotoxicity, UroC was chosen for further research to clarify the cytotoxic mechanism in PC12 cells.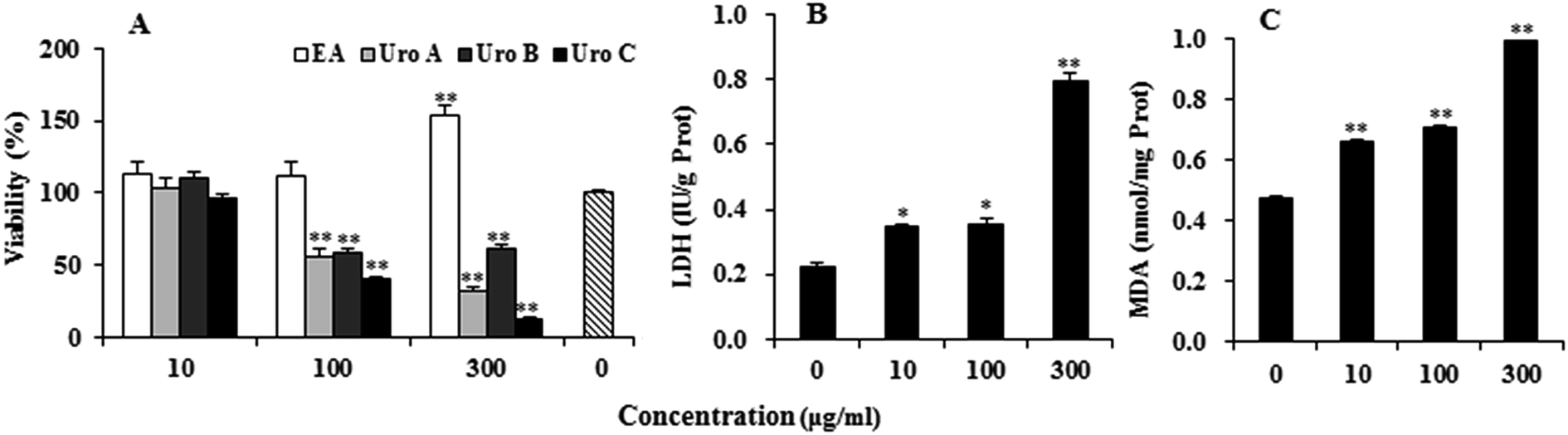 | ||
| Fig. 2 Effects of EA and Uros A, B and C on PC12 cells proliferation measured by MTT (A), and release of LDH (B) and formation of MDA (C) treated with different concentrations of UroC. For each treatment, cells were treated with drugs for 12 h at 37 °C in 5% CO2 atmosphere. Single (*) and double (**) asterisks indicate significant difference at p < 0.05 and extremely significant difference at p < 0.01 compared to the control, respectively. | ||
UroC-induced cell apoptosis and S phase arrest in PC12 cells
To examine the effect of UroC on apoptosis in PC12 cells, Annexin V-FITC/PI double staining was used to distinguish apoptotic cells from damaged cells. Total apoptotic rate was calculated as apoptotic rate of cells in the upper right quadrant plus the lower right quadrant (Fig. 3A–E). The percentage of apoptotic cells after UroC treatment increased with increasing concentrations. The percentage of apoptotic cells was 0.80% in control group (0 μg ml−1) and there was no significant difference between 0 (Fig. 3A) and 100 μg ml−1 (Fig. 3B) UroC. However, cells treated with 200 (Fig. 3C) and 300 μg ml−1 (Fig. 3D) UroC exhibited extremely significant differences compared to the control, with the percentage of apoptotic cells being 43.77% and 67.42%, respectively. These results show that UroC could inhibit the proliferation of PC12 cells by inducing apoptosis.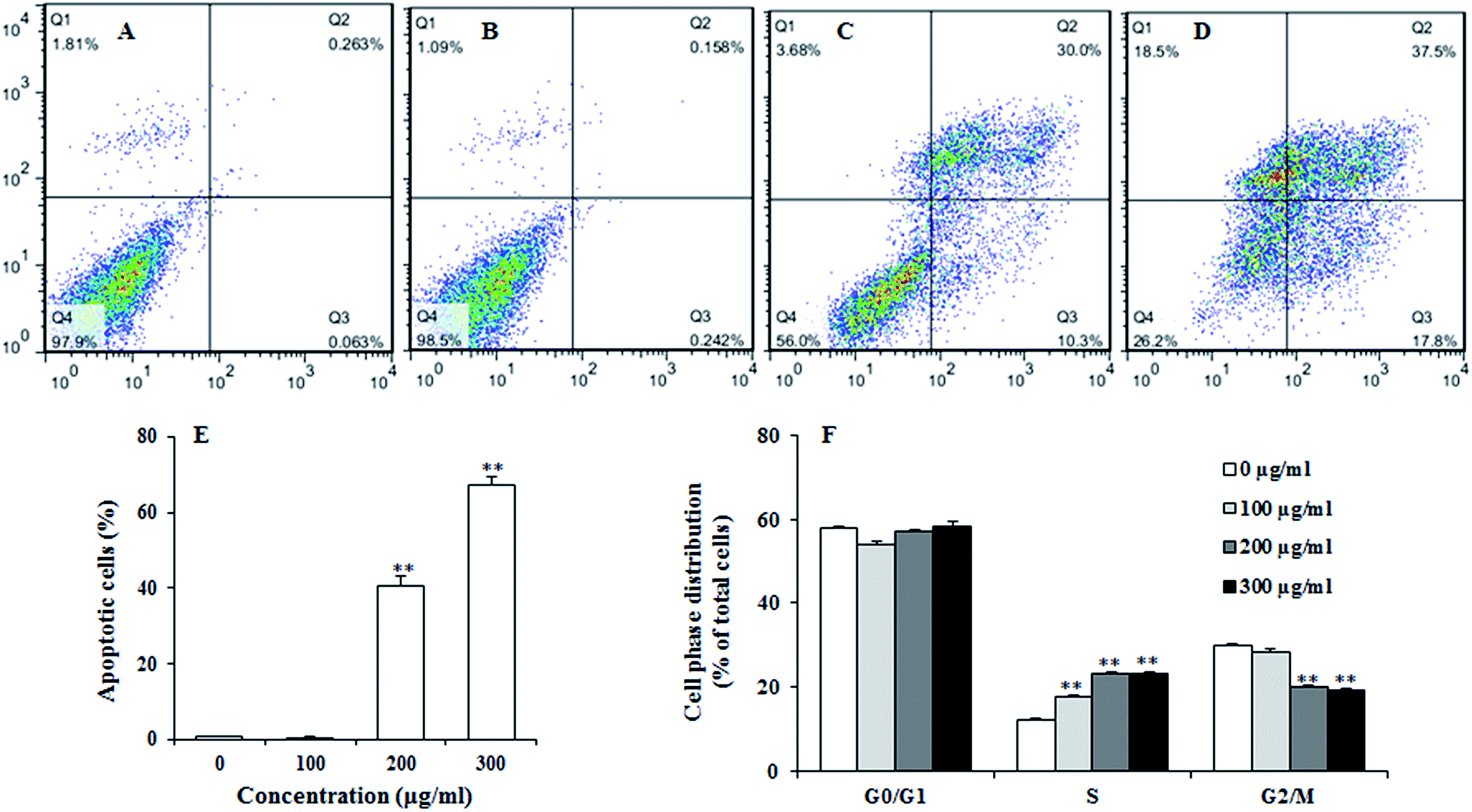 | ||
| Fig. 3 Annexin V-FITC/PI flow cytometric detection of apoptosis in PC12 cells treated with 0 (A), 100 (B), 200 (C) and 300 (D) μg ml−1 UroC for 5 h at 37 °C in 5% CO2 atmosphere before determination. (E) Percentage of apoptotic cells after treatments with different concentrations of UroC. (F) Cell cycle alteration was determined with PI after treatments with different concentrations of UroC for 5 h. Data are calculated as percentages of cells in different phases and expressed as mean ± SD (n = 3). Double (**) asterisks indicate extremely significant difference at p < 0.01 compared to the control. | ||
To assess whether UroC-induced cell apoptosis was due to arrest at a specific point of the cell cycle, PC12 cells treated with different concentrations of UroC were detected using flow cytometry. As shown in Fig. 3F, cell cycle tended to arrest at S phase after UroC-treatment. The percentage of S phase in control group was about 12.18%, while it significantly increased to 17.63%, 23.08% and 23.29% in 100, 200 and 300 μg ml−1 treated groups, respectively. This increase in S cell population was mostly at the expense of G2/M phase cell populations. Therefore, anti-proliferation activity of UroC could attribute to apoptotic inducement and S phase cell cycle arrest.
UroC-induced changes in cellular oxidative status
Reactive oxygen species (ROS) such as superoxide and hydrogen peroxide are continually produced during cellular metabolic processes. ROS generation is normally counterbalanced by the action of antioxidant enzymes and other redox molecules. However, excess ROS can lead to cellular injury in forms of damaged DNA, lipids, and proteins. MDA is one of the most prevalent byproducts of lipid peroxidation during oxidative stress. In addition, NO can stimulate formation of a variety of free radicals and other oxidant radicals via mitochondria, leading to activation of caspase.36,37As shown in Fig. 4A, UroC dramatically increased ROS levels with increasing concentrations in PC12 cells, while no significant changes were found in NO levels after UroC treatment (Fig. 4B). In addition, MDA levels in PC12 cells were also significantly enhanced after UroC treatment in a dose-dependent manner (Fig. 2C). These data indicated that UroC could induce severe oxidative stress in PC12 cells with increased ROS and MDA levels, while not affecting NO levels.
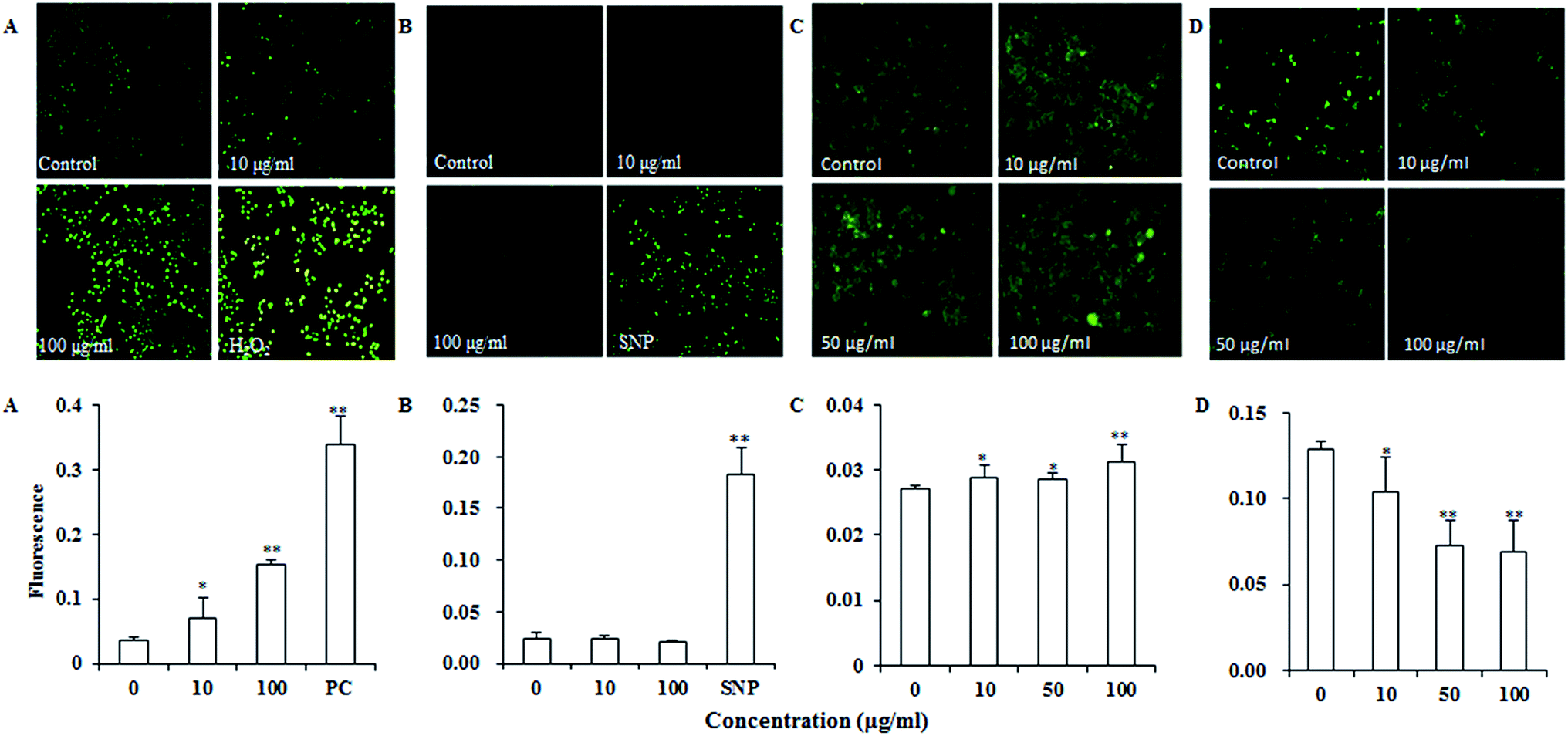 | ||
| Fig. 4 Effects of UroC on intracellular ROS (A), NO (B) and Ca2+ (C) levels and ΔΨm (D) in PC12 cells treated with different concentrations for 2 h before determination and detected with fluorescence microscope. (A) ROS level assessed via DCFH-DA staining. PC, 100 μM H2O2 as positive control. (B) NO level assessed via DAF-FM DA staining. SNP, 200 μg ml−1 sodium nitroprusside as positive control. (C) Ca2+ level assessed via Fluo-3AM staining. (D) ΔΨm assessed via Rhodamine 123 staining. Single (*) and double (**) asterisks indicate significant difference at p < 0.05 and extremely significant difference at p < 0.01 compared to the control, respectively. | ||
UroC-induced dysfunction of mitochondrial membrane potential and dyshomeostasis of Ca2+ in PC12 cells
Calcium dyshomeostasis will cause cell dysfunction and metabolic disorders, even apoptosis. Fig. 4C shows that Ca2+ levels are significantly higher than that of the control, indicating that UroC treatment caused significant calcium dyshomeostasis in PC12 cells. Mitochondrial dysfunction is another important trait of apoptosis since mitochondria are the critical mediators of apoptosis in the intrinsic pathway. In this experiment, rhodamine 123 was used to analyze the ΔΨm of PC12 cells. As shown in Fig. 4D, after UroC treatment, a significant decrease in mean fluorescence intensity was observed compared to that of the control. Moreover, the effect was gradually enhanced with increasing concentrations. These data indicated that UroC-induced apoptosis was accompanied by a collapse in the ΔΨm.UroC-induced decrease of Bcl-2/Bax protein ratio
Bcl-2 family proteins have been shown to play an important role in the regulation of mitochondria-mediated apoptosis, and are the upstream regulator of the ΔΨm. To detect whether apoptosis-associated proteins Bax and Bcl-2 were involved in UroC-induced apoptosis, western blotting was used to determine Bcl-2/Bax ratio in PC12 cells. As shown in Fig. 5A and B, the ratio was down-regulated in PC12 cells after UroC treatment. That is, UroC-induced apoptosis was accompanied by more imbalance of Bcl-2/Bax.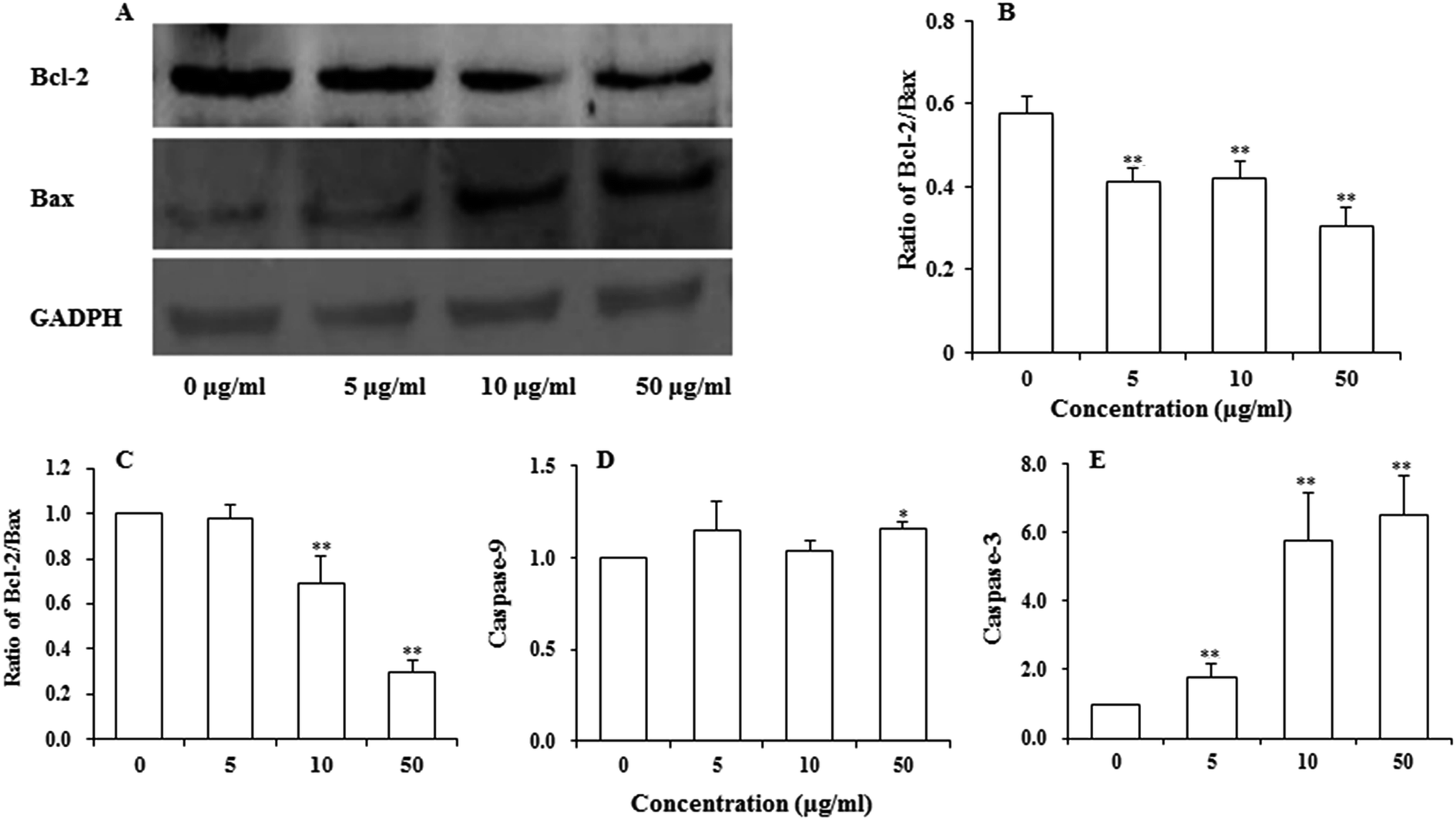 | ||
| Fig. 5 Changes in apoptosis-associated regulatory proteins (A and B) and expressions of mRNAs (C–E) analyzed by western blot and real-time PCR, respectively, in PC12 cells treated with different concentrations of UroC (0, 5, 10, and 50 μg ml−1) for 24 h. (A) Western blotting of Bcl-2 and Bax proteins; (B) Bcl-2/Bax ratios quantified by Image J software; (C) expression ratio of Bcl-2/Bax mRNA; (D) expression of caspase-9 mRNA; (E) expression of caspase-3 mRNA. Data in (B–E) are expressed as mean ± SD (n = 3). Single (*) and double (**) asterisks indicate significant difference at p < 0.05 and double (**) asterisks indicate extremely significant difference at p < 0.01 compared to the control. | ||
UroC-induced change in apoptosis-associated mRNAs
Major gene groups that regulate apoptosis are those in the Bcl-2 family. It had been reported that Bcl-2 members (e.g., Bcl-2 and p53) protect against multiple signals that lead to cell death whereas Bax members (e.g., Bax, Bad) induce apoptosis.38 Fig. 5C shows that mRNA ratio of Bcl-2/Bax decreased with increasing UroC concentrations. Compared to the control (0 μg ml−1), there was no significant difference at 5 μg ml−1, while extremely significant differences were found at 10 and 50 μg ml−1. These results suggest that UroC might induce PC12 cells apoptosis through regulation of Bcl-2/Bax ratio at the mRNA level.As to the caspase-9 expression (Fig. 5D), no significant difference was observed between control and two groups treated with lower UroC (5 and 10 μg ml−1), while an extremely significant increase was found in the 50 μg ml−1 group, indicating that caspase-9 might also be involved in UroC-induced cell apoptosis, but with a less extent. Caspase-3, a downstream effector in the caspase cascade, is considered as an essential executor for mitochondria-dependent apoptotic pathway.31 As shown in Fig. 5E, UroC increased the level of relative caspase-3 mRNA in a concentration-dependent manner, and there existed extremely significant differences after UroC treatments compared to the control (0 μg ml−1). These results suggest that the mitochondrial pathway is potentially involved in UroC-induced cell apoptosis.
Discussion
Uros A, B, and C were gut microbiota metabolites derived from EA and ET. It is conceived that the health effects of ET-containing products could be associated with these gut-produced Uros and their conjugates. Uros are not common molecules in nature and they could be detected in plasma, urine or feces of mammals at concentrations ranging between high nM and low μM. In view of the small quantity of Uros, it is significant to establish the chemosynthetic method of Uros. In this study, we successfully synthesized three Uros A, B and C with purify of 98%, 96% and 95%, respectively. To clarify potential bioactive compounds after consumption of ET-containing foodstuffs, cytotoxicity activities of EA and Uros A, B, and C were tested. It was found that EA promoted in a dose-dependent manner, however, Uros A, B, and C significantly inhibited proliferation of PC12 cells, among which UroC showed the strongest inhibitory effects. And this was consistent with result of Stolarczyk et al., 39 who found that UroC displayed the strongest activity in the inhibition of prostate cells proliferation. After treatment with 300 μg ml−1 UroC, nearly 90% of PC12 cells were inhibited (Fig. 2A), in contrary to only about 55% of HepG2 cells were inhibited (data not shown). These observations indicated that PC12 cells were more sensitive than HepG2 to cytotoxic effects of UroC. Variation between the two types of cells could be due to differences in membrane composition, proliferation ability, endogenous expression of cellular proteins,19 etc. LDH, existing in cells, could be released into the medium if cells were damaged, thus LDH level in the medium reflected membrane integrity. In the present study, LDH level in the medium increased significantly after UroC treatment (Fig. 2B), indicating that UroC led to loss of membrane integrity and increased membrane penetrability. The significant cytotoxicity of UroC might be elucidated by the apoptosis-induced mechanism.Apoptosis and cell cycle arrest are two major causes of cell growth inhibition.40 Dysregulation of cell cycle is also associated with tumorigenesis.41 In our study, we found that UroC induced apoptosis in PC12 cells in a dose-dependent manner, and that it arrested cell cycle at S phase (Fig. 3). A previous study has shown that Uros A and B have inhibitory activity against three human colon cancer cell lines (Caco-2, SW480 and HT-29) and that they arrested the cells at S and/or G2/M phases, while UroC arrested the colon cells (Caco-2, SW480 and HT-29) at S phase,42 which was in accordance with our study. These results indicated that UroC potentially exerts inhibitory activity against cell proliferation through both apoptotic induction and cell cycle arrest.
Apoptosis could be promoted by generation of free radicals during oxidative stress, involving a variety of signaling events such as lipid peroxidation, intracellular Ca2+ dyshomeostasis, mitochondrial membrane depolarization, and changes in levels of apoptotic effectors such as Bcl-2, Bax, and caspase-3.41,43,44 It has been previously reported that increased generation of intracellular ROS could also induce cell cycle arrest at S phase.40,45,46 By ROS stimulation, a variety of unsaturated fatty acids in biological membrane are degraded to MDA via lipid peroxidation.47 ROS can also inhibit endoplasmic reticulum calcium pumps and facilitates the dumping of stored calcium from endoplasmic reticulum to cytosol. Mitochondria take up calcium and initiate apoptosis through opening of their permeability transition pores.48 Furthermore, ROS can disrupt mitochondria membrane potential, which is regarded as an indicator of mitochondrial damage and an early stage of apoptosis. In the present study, UroC-induced apoptosis was accompanied by increased levels of ROS and MDA, Ca2+ dyshomeostasis, and mitochondrial membrane depolarization (Fig. 2C and 4A, C and D). It has been reported that NO stimulates formation of a variety of free radicals and other oxidant radicals in mitochondria, leading to activation of caspase cascade.36,37 However, our study indicated that NO was not involved in UroC-induced apoptosis (Fig. 4B).
Bcl-2 family proteins have been shown to play an important role in regulation of mitochondria-mediated apoptosis, and are upstream regulators of ΔΨm.31,49 To investigate whether UroC can trigger an intrinsic apoptotic cascade in PC12 cells, real-time PCR and western blotting analyses were performed to quantify expression ratios of Bcl-2/Bax genes and proteins. The results demonstrated that ratios of Bcl-2/Bax genes and proteins both decreased after UroC treatment (Fig. 5A–C), suggesting that UroC might induce apoptosis through intrinsic pathway. Furthermore, caspase-9 mRNA exhibited also an increase compared to control group after UroC treatment, though to a less extent (Fig. 5D). Caspase-3, the most downstream effector in caspase cascade, is considered as an essential executor for mitochondria-dependent apoptotic pathway.31,50 An increase of caspase-3 genes was observed in UroC-treated cells (Fig. 5E), suggesting that Uro C induced apoptosis in PC12 cells through caspase-3 activation.
Uros constitute a family of metabolites produced from EA and ET. It was found that the glucuronide and sulphate conjugates are mainly found in plasma and urine while several urolithin aglycones could be present in fecal samples.51,52 And there were several reports about the biological activity of the glucuronides and methylates of Uros. Giménez-Bastida et al.10 found that UroA and UroB glucuronides exhibited beneficial effects against cardiovascular diseases, which attributed to the consumption of ET-containing foods. And it reported that UroB and its glucuronide succeeded in preventing inflammatory responses in cardiomyocytes.12 In addition, Dellafiora et al.53 evaluated the effect of phase II metabolism (glucuronidation, sulphation, and methylation) on the estrogenicity of urolithins on α- and β-oestrogen receptor isoforms by using an in silico approach. The methylated UroA could suppresses cell viability of DU145 human prostate cancer cells.54 Therefore, it is necessary to do further research on the glucuronides, sulfates and methylates conjugates of Uros in the future.
A limitation of this work was that the different experimental concentrations of Uros were 5–300 μg ml−1, which were beyond the physiological concentration range, but could have maximized the impact of these results. And the concentrations in our study were in accordance with Sharma et al.,55 who reported that UroA could induce Wnt pathway inhibition in HEK T293 cells at 0.2–200 μg ml−1.
Conclusion
In conclusion, UroC, as one of the major gut microbiota metabolites derived from EA and ET, showed potent cytotoxicity in PC12 cells compared to EA. UroC treatment damaged membrane integrity, increased LDH release and membrane lipid peroxidation, and stimulated ROS generation. Furthermore, it induced cell apoptosis and cell cycle arrest at S phase through a mitochondria-mediated pathway, involving mitochondrial membrane depolarization and calcium dyshomeostasis. Consequently, UroC induced imbalance of Bcl-2/Bax ratio and triggered the caspase cascade, thereby shifting the balance in favor of apoptosis (Fig. 6). These findings would not only broaden our knowledge on bioactivities of gut microbiota metabolites derived from EA and ET, but also facilitate attempts on developing UroC as a novel antitumor agent in the future.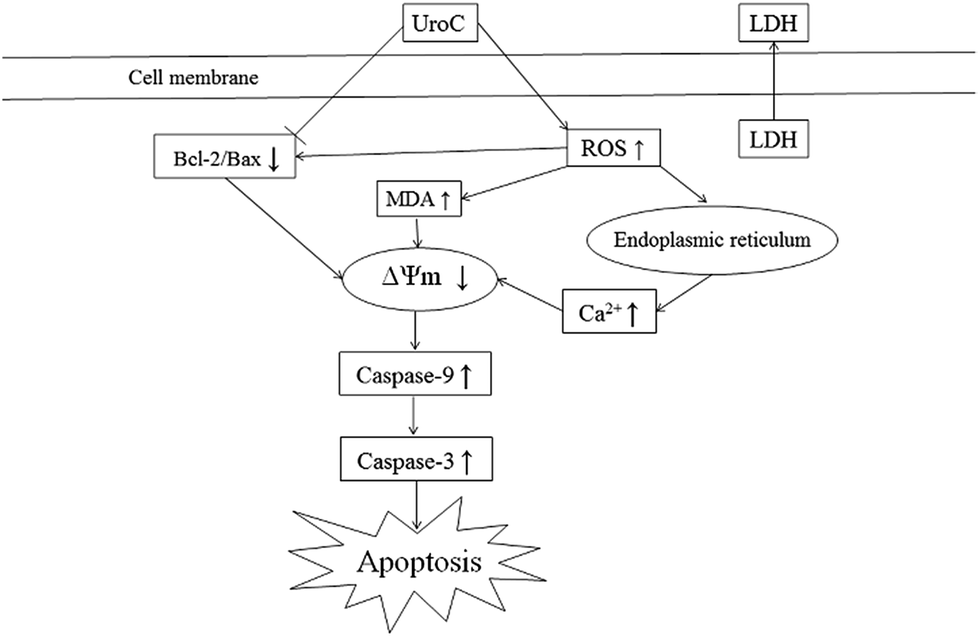 | ||
| Fig. 6 Proposed mechanism of UroC-induced apoptosis in PC12 cells. | ||
Acknowledgements
The study was financially supported by the Fundamental Research Funds for the Central Universities (No. YX2015-13 and BLYJ201412), Special Fund for Beijing Common Construction Project (No. 2014GJ01) and Beijing Higher Education Young Elite Teacher Project (No. YETP0757). We would like to thank Cesar from School of Medicine, Yale University for English editing.References
- J. C. Espín, M. Larrosa, M. T. García-Conesa and F. Tomás-Barberán, Evid. Based Complement. Alternat. Med., 2013, 2013, 270418 CrossRef PubMed.
- M. I. Gil, F. A. Tomas-Barberan, B. Hess-Pierce, D. M. Holcroft and A. A. Kader, J. Agric. Food Chem., 2000, 48, 4581–4589 CrossRef CAS PubMed.
- C. Garcia-Muñoz and F. Vaillant, Crit. Rev. Food Sci. Nutr., 2014, 54, 1584–1598 CrossRef PubMed.
- B. Cerdá, R. Llorach, J. J. Cerón, J. C. Espín and F. A. Tomás-Barberán, Eur. J. Nutr., 2003, 42, 18–28 CrossRef PubMed.
- B. Cerdá, J. C. Espín, S. Parra, P. Martínez and F. A. Tomás-Barberán, Eur. J. Nutr., 2004, 43, 205–220 CrossRef PubMed.
- B. Cerdá, F. A. Tomás-Barberán and J. C. Espín, J. Agric. Food Chem., 2005, 53, 227–235 CrossRef PubMed.
- S. U. Mertens-Talcott, P. Jilma-Stohlawetz, J. Rios, L. Hingorani and H. Derendorf, J. Agric. Food Chem., 2006, 54, 8956–8961 CrossRef CAS PubMed.
- N. P. Seeram, S. M. Henning, Y. Zhang, M. Suchard, Z. Li and D. Heber, J. Nutr., 2006, 136, 2481–2485 CAS.
- R. García-Villalba, J. C. Espín and F. A. Tomás-Barberán, J. Chromatogr. A, 2016, 1428, 162–175 CrossRef PubMed.
- J. A. Giménez-Bastida, A. González-Sarrías, M. Larrosa, F. Tomás-Barberán, J. C. Espín and M. T. García-Conesa, Mol. Nutr. Food Res., 2012, 56, 784–796 Search PubMed.
- A. González-Sarrías, J. A. Giménez-Bastida, M. T. García-Conesa, M. B. Gómez-Sánchez, N. V. García-Talavera, A. Gil-Lzquierdo, C. Sánchez-Álvarez, L. O. Fontana-Compiano, J. P. Morga-Egea, F. A. Pastor-Quirante, F. Martínez-Díaz, F. A. Tomás-Barberán and J. C. Espín, Mol. Nutr. Food Res., 2010, 54, 311–322 Search PubMed.
- R. Sala, P. Mena, M. Savi, F. Brighenti, A. Crozier, M. Miragoli, D. Stilli and D. Del Rio, J. Funct. Foods, 2015, 15, 97–105 CrossRef CAS.
- J. P. Piwowarski, S. Granica, M. Zwierzynska, J. Stefanska, P. Schopohl, M. F. Melzig and A. K. Kiss, J. Ethnopharmacol., 2014, 155, 801–809 CrossRef CAS PubMed.
- T. Kallio, J. Kallio, M. Jaakkola, M. Maki, P. Kilpelainen and V. Virtanen, J. Agric. Food Chem., 2013, 61, 10720–10729 CrossRef CAS PubMed.
- J. A. Giménez-Bastida, M. Larrosa, A. González-Sarrías, F. Tomás-Barberán, J. C. Espîn and M. T. García-Conesa, J. Agric. Food Chem., 2012, 60, 8866–8876 CrossRef PubMed.
- A. González-Sarrías, J. C. Espín, F. A. Tomás-Barberán and M. T. García-Conesa, Mol. Nutr. Food Res., 2009, 53, 686–698 Search PubMed.
- S. G. Kasimsetty, D. Bialonska, M. K. Reddy, G. Ma, S. I. Khan and D. Ferreira, J. Agric. Food Chem., 2010, 58, 2180–2187 CrossRef CAS PubMed.
- S. G. Kasimsetty, D. Bialonska, M. K. Reddy, C. Thornton, K. L. Willett and D. Ferreira, J. Agric. Food Chem., 2009, 57, 10636–10644 CrossRef CAS PubMed.
- G. Madhubabu and S. Yenugu, Toxicol. in Vitro, 2014, 28, 1386–1395 CrossRef CAS PubMed.
- R. Vicinanza, Y. Zhang, S. M. Henning and D. Heber, Evid. Based Complement. Alternat. Med., 2013, 2013, 247504 Search PubMed.
- W. Liu, H. Ma, L. Frost, T. Yuan, J. A. Dain and N. P. Seeram, Food Funct., 2014, 5, 2996–3004 CAS.
- D. Ryu, L. Mouchiroud, P. A. Andreux, E. Katsyuba, N. Moullan, A. A. Nicolet-dit-Félix, E. G. Williams, P. Jha, G. Lo Sasso, D. Huzard, P. Aebischer, C. Sandi, C. Rinsch and J. Auwerx, Nat. Med., 2016, 22, 879–888 CrossRef CAS PubMed.
- E. Verzelloni, C. Pellacani, D. Tagliazucchi, S. Tagliaferri, L. Calani, L. G. Costa, F. Brighenti, G. Borges, A. Crozier, A. Conte and D. Del Rio, Mol. Nutr. Food Res., 2011, 55(Suppl. 1), S35–S43 CAS.
- S. J. Choi, J. H. Lee, H. J. Heo, H. Y. Cho, H. K. Kim, C. J. Kim, M. O. Kim, S. H. Suh and D. H. Shin, J. Med. Food, 2011, 14, 695–701 CrossRef CAS PubMed.
- F. Forouzanfar, A. Afkhami Goli, E. Asadpour, A. Ghorbani and H. R. Sadeghnia, Evid. Based Complement. Alternat. Med., 2013, 2013, 716730 Search PubMed.
- S. Subash, M. M. Essa, A. Al-Asmi, S. Al-Adawi, R. Vaishnav, N. Braidy, T. Manivasagam and G. J. Guillemin, J. Tradit. Complement Med., 2014, 4, 232–238 CrossRef PubMed.
- C. Zhang, C. Li, S. Chen, Z. Li, X. Jia, K. Wang, J. Bao, Y. Liang, X. Wang, M. Chen, P. Li, H. Su, J. B. Wan, S. M. Lee, K. Liu and C. He, Redox Biol., 2017, 11, 1–11 CrossRef CAS PubMed.
- X. Wang, W. Xu, M. Fan, T. Meng, X. Chen, Y. Jiang, D. Zhu, W. Hu, J. Gong, S. Feng, J. Wu and Y. Li, Environ. Toxicol. Pharmacol., 2016, 43, 193–202 CrossRef CAS PubMed.
- B. Sheng, K. Gong, Y. Niu, L. Liu, Y. Yan, G. Lu, L. Zhang, M. Hu, N. Zhao, X. Zhang, P. Tang and Y. Gong, Free Radical Biol. Med., 2009, 46, 1362–1375 CrossRef CAS PubMed.
- Q. Q. Huang, M. Fang, H. Q. Zhang and S. B. Xue, Cell Res., 1996, 6, 23–30 CrossRef.
- L. Xue, M. Li, T. Chen, H. Sun, J. Zhu, X. Li, F. Wu, B. Wang, J. Li and Y. Chen, Int. J. Mol. Med., 2014, 34, 119–129 CAS.
- M. Singh, H. Sharma and N. Singh, Mitochondrion, 2007, 7, 367–373 CrossRef CAS PubMed.
- I. A. Sobenin, Y. V. Bobryshev, G. A. Korobov, E. N. Borodachev, A. Y. Postnov and A. N. Orekhov, Exp. Mol. Pathol., 2015, 99, 1–6 CrossRef CAS PubMed.
- J. Pandey, A. K. Jha and K. Hajela, Bioorg. Med. Chem., 2004, 12, 2239–2249 CrossRef CAS PubMed.
- D. Bialonska, S. G. Kasimsetty, S. I. Khan and D. Ferreira, J. Agric. Food Chem., 2009, 57, 10181–10186 CrossRef CAS PubMed.
- J. O. Jin, M. G. Song, Y. N. Kim, J. I. Park and J. Y. Kwak, Mol. Carcinog., 2010, 49, 771–782 CAS.
- J. P. Kolb, Leukemia, 2000, 14, 1685–1694 CrossRef CAS PubMed.
- R. Thangam, M. Sathuvan, A. Poongodi, V. Suresh, K. Pazhanichamy, S. Sivasubramanian, N. Kanipandian, N. Ganesan, R. Rengasamy, R. Thirumurugan and S. Kannan, Carbohydr. Polym., 2014, 107, 138–150 CrossRef CAS PubMed.
- M. Stolarczyk, J. P. Piwowarski, S. Granica, J. Stefańska, M. Naruszewicz and A. K. Kiss, Phytother Res., 2013, 27, 1842–1848 CrossRef CAS PubMed.
- M. Khan, C. Ding, A. Rasul, F. Yi, T. Li, H. Gao, R. Gao, L. Zhong, K. Zhang, X. Fang and T. Ma, Int. J. Biol. Sci., 2012, 8, 533–547 CrossRef CAS PubMed.
- S. H. Wu, L. W. Hang, J. S. Yang, H. Y. Chen, H. Y. Lin, J. H. Chiang, C. C. Lu, J. L. Yang, T. Y. Lai, Y. C. Ko and J. G. Chung, Anticancer Res., 2010, 30, 2125–2133 CAS.
- A. Gonzalez-Sarrias, J. A. Gimenez-Bastida, M. A. Nunez-Sanchez, M. Larrosa, M. T. Garcia-Conesa, F. A. Tomas-Barberan and J. C. Espin, Eur. J. Nutr., 2014, 53, 853–864 CrossRef CAS PubMed.
- L. A. Booth, S. Tavallai, H. A. Hamed, N. Cruickshanks and P. Dent, Cell. Signalling, 2014, 26, 549–555 CrossRef CAS PubMed.
- Z. Qiu, B. Zhou, L. Jin, H. Yu, L. Liu, Y. Liu, C. Qin, S. Xie and F. Zhu, Food Chem. Toxicol., 2013, 59, 428–437 CrossRef CAS PubMed.
- S. C. Hsu, C. L. Kuo, J. P. Lin, J. H. Lee, C. C. Lin, C. C. Su, H. J. Lin and J. G. Chung, Anticancer Res., 2007, 27, 2415–2425 CAS.
- T. H. Chiu, W. W. Lai, T. C. Hsia, J. S. Yang, T. Y. Lai, P. P. Wu, C. Y. Ma, C. C. Yeh, C. C. Ho, H. F. Lu, W. G. Wood and J. G. Chung, Anticancer Res., 2009, 29, 4503–4511 CAS.
- T. Finkel, Curr. Opin. Cell Biol., 2003, 15, 247–254 CrossRef CAS PubMed.
- G. Hajnóczky, G. Csordás, S. Das, C. Garcia-Perez, M. Saotome, S. Sinha Roy and M. Yi, Cell Calcium, 2006, 40, 553–560 CrossRef PubMed.
- Y. H. Ling, R. Lin and R. Perezsoler, Mol. Pharmacol., 2008, 74, 793–806 CrossRef CAS PubMed.
- M. O. Hengartner, Nature, 2000, 407, 770–776 CrossRef CAS PubMed.
- J. C. Espín, R. Gonzálezbarrio, B. Cerdá, C. Lópezbote, A. I. Rey and F. A. Tomásbarberán, J. Agric. Food Chem., 2007, 55, 10476 CrossRef PubMed.
- B. Pfundstein, R. Haubner, G. Würtele, N. Gehres, C. M. Ulrich and R. W. Owen, J. Agric. Food Chem., 2014, 62, 10264–10273 CrossRef CAS PubMed.
- L. Dellafiora, P. Mena, P. Cozzini, F. Brighenti and D. Del Rio, Food Funct., 2013, 4, 1442–1451 CAS.
- B. Zhou, J. Wang, G. Zheng and Z. Qiu, Food Chem. Toxicol., 2016, 97, 375–384 CrossRef CAS PubMed.
- M. Sharma, L. Li, J. Celver, C. Killian, A. Kovoor and N. P. Seeram, J. Agric. Food Chem., 2010, 58, 3965–3969 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01548h |
| This journal is © The Royal Society of Chemistry 2017 |