
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A series of lanthanide glutarates: lanthanide contraction effect on crystal frameworks of lanthanide glutarates†
Xiao-Feng Tana,
Jian Zhou *a,
Hua-Hong Zou*b,
Lianshe Fu*c,
Qiuling Tanga and
Peng Wanga
*a,
Hua-Hong Zou*b,
Lianshe Fu*c,
Qiuling Tanga and
Peng Wanga
aCollege of Chemistry, Chongqing Normal University, Chongqing, 401331, P. R. China. E-mail: jianzhou888888@163.com
bState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry & Pharmacy of Guangxi Normal University, Guilin, 541004, P. R. China
cDepartment of Physics and CICECO-Aveiro Institute of Materials, University of Aveiro, 3810-193, Aveiro, Portugal
First published on 24th March 2017
Abstract
A series of lanthanide glutarates [Ln(phen)(glu)Cl]n {Ln = Y (1a), Tm (1b); phen = 1,10-phenanthroline; glu = glutarate}, [Ln2(phen)2(glu)3]n {Ln = Ce (2a), Tb (2b), Ho (2c)} and [La2(glu)3(H2O)3]n·5nH2O (3) have been hydrothermally synthesized and characterized structurally. Compounds 1a–b are isostructural and consist of 1-D neutral [Ln(phen)(glu)Cl]n chains, which are built up from the linkages of [Ln(phen)Cl]3+ ions and glutarate ligands. Compounds 2a–c are isostructural and contain 2-D [Ln2(phen)2(glu)3]n layers, where Ln3+ ions are connected by three kinds of glutarate ligands. The 3-D framework of compound 3 is constructed by the linkages of La3+ ions and glutarate ligands. Although some 3-D lanthanide glutarates have been reported, they exhibit a very robust structural type, whose structure is not changed by different Ln3+ ions, but compound 3 shows a new structural type. A systematic investigation of six lanthanide glutarates and some reported compounds revealed that the well-known lanthanide contraction has a significant influence on the formation of lanthanide glutarates. The photoluminescent properties of 1b and 2b, and magnetic properties of 1b, 2b and 2c have been studied.
Introduction
Lanthanide coordination polymers are of great interest due to their fantastic topological structures, and potentially applicable magnetic and photoluminescence properties.1 The magnetic properties could arise from the intrinsic large spin ground state and the strong uniaxial magnetic anisotropy of some trivalent lanthanide ions (Ln3+),2 while the luminescence of Ln3+ ions is possible from intra-configurational f–f transitions, which show fascinating luminescence in the visible or near-infrared spectral regions upon irradiation with ultraviolet radiation.3 Due to the low absorption of the forbidden f–f transitions, the emissions of Ln3+ ions are weak. In order to enhance the emissive efficiency and decrease the internal non-radiative decay processes, two strategies may be generally utilized. One is to prevent water molecules from binding to Ln3+ ions, as well as high-vibrational O–H, N–H and C–H oscillators that tend to quench photoluminescence,4 the other is to promote the antenna effect, which pumps up Ln3+ emission by using π-conjugated organic aromatic chromophores directly coordinated to Ln3+ ions.5 Bidentate neutral 1,10-phenanthroline (phen) is one of the widest used chromophoric ligand in the design of photoluminescent lanthanide complexes, because it can chelate to Ln3+ ion, which may not only encapsulate and protect the Ln3+ ion from the water molecules, but also absorb and efficiently transfer energy onto the Ln3+ excited states. As expected, a large number of photoluminescent Ln-complexes containing neutral phen and its derivatives have been synthesized,6 but their solids are difficult to directly use as photoluminescent sources, mainly because phen or its derivatives acting as terminating group prevent further connections, resulting in molecules that show poor thermal stability, weak moisture stability and feeble mechanical strength.Glutaric acid is one of excellent long-chain aliphatic dicarboxylic acids with flexible bridging capability for the construction of a diversity of lanthanide coordination polymers.7 But water molecule is easily bound to Ln3+ ion in the absence of chelating chromophoric ligand, which could lead to efficient nonradiative deactivation of their excited states. It is expected that phen or its derivatives in conjunction with glutaric acid may generate a new class of lanthanide coordination polymers with enhanced thermal stability, high luminescence quantum yield and relatively long-lived emission. However, a few lanthanide glutarates containing phen or its derivatives were prepared under certain synthetic conditions,8 where the significant influence of lanthanide contraction on their structures and the relationship between the structures and photoluminescent properties have been rarely reported to the best of our knowledge. To further explore the influences on the formation of lanthanide glutarates, it is necessary to synthesize a series of new lanthanide glutarates for improving their desirable photoluminescent properties. This paper reports the synthesis, crystal structures and properties of a series of new lanthanide glutarates [Ln(phen)(glu)Cl]n {Ln = Y (1a), Tm (1b)}, [Ln2(phen)2(glu)3]n {Ln = Ce (2a), Tb (2b), Ho (2c)} and [La2(glu)3(H2O)3]n·5nH2O (3). A systematic investigation of six lanthanide glutarates and some reported compounds exhibited that the well-known lanthanide contraction has a significant influence on the formation of lanthanide glutarates.
Experimental
General remarks
All analytical grade chemicals were obtained commercially and used without further purification. IR spectra were obtained from a powdered sample pelletized with KBr on an ABB Bomen MB 102 series IR spectrophotometer in the range of 400–4000 cm−1. Elemental analyses (C, H, N) were performed on an Elemental Vario EL III analyzer. The photoluminescence spectra were recorded at room temperature with a modular double grating excitation spectrofluorimeter with a TRIAX 320 emission monochromator (Fluorolog-3, Horiba Scientific) coupled to an R928 Hamamatsu photomultiplier. The excitation source was a 450 W Xe arc lamp. The emission spectra were corrected for detection and optical spectral response of the spectrofluorimeter and the excitation spectra were corrected for the spectral distribution of the lamp intensity using a photodiode reference detector. Variable-temperature magnetic susceptibility measurements were carried out in the temperature range of 2–300 K with a Quantum Design MPMS-5 magnetometer. Thermogravimetric analyses (TGA) were performed using a Mettler TGA/SDTA851 thermal analyzer under an air-flow atmosphere with a heating rate of 10 °C min−1 in the temperature region of 25–600 °C.Synthesis of Y(phen)(glu)Cl]n (1a)
A mixture of glutaric acid (0.0264 g, 0.20 mmol), phen (0.0270 g, 0.15 mmol), YCl3 (0.0742 g, 0.38 mmol), and water (2 mL) was stirred for 1 h, and then the pH value of the mixed solution was adjusted to 5–6 by triethylamine. The final mixture was sealed in a 25 mL Teflon lined autoclave and heated at 100 °C for 7 days. After cooling to room temperature slowly, colorless block crystals were isolated. The yield of 1a is 73% based on YCl3. Anal. calcd for 1a, C17H14ClN2O4Y, C 46.98%, H 3.25%, N 6.44%, found: C 47.12%, H 3.28%, N 6.47%. IR (cm−1): 3094(w), 3054(w), 2900(w), 1598(s), 1517(w), 1420(s), 1350(m), 1280(m), 1219(m), 1169(m), 1099(m), 1059(m), 934(vw), 859(s), 817(m), 781(m), 727(s), 650(m), 539(w).Synthesis of Tm(phen)(glu)Cl]n (1b)
The colorless crystals of 1b were prepared similarly from TmCl3 (yield 63% based on TmCl3). Anal. calcd for 1b, C17H14ClN2O4Tm, C 39.67%, H 2.74%, N 5.44%, found: C 39.63%, H 2.76%, N 5.46%. IR (cm−1): 3094(w), 3054(w), 2900(w), 1591(vs), 1517(w), 1467(w), 1413(s), 1350(m), 1280(m), 1212(m), 1156(m), 1106(m), 1052(m), 962(vw), 851(s), 817(m), 781(m), 727(s), 664(m), 540(w).Synthesis of [Ce2(phen)2(glu)3]n (2a)
The yellow crystals of 2a were prepared similarly from CeCl3 (yield 78% based on CeCl3). Anal. calcd for 2a, C39H34Ce2N4O12, C 45.44%, H 3.32%, N 5.43%, found: C 45.46%, H 3.34%, N 5.47%. IR (cm−1): 3081(w), 3040(w), 2923(w), 1598(vs), 1524(s), 1426(s), 1343(m), 1316(s), 1266(m), 1142(m), 1106(m), 1059(m), 857(s), 794(s), 734(s), 677(w), 636(m), 519(m).Synthesis of [Tb2(phen)2(glu)3]n (2b)
The brown crystals of 2b were prepared similarly from TbCl3 (yield 73% based on TbCl3). Anal. calcd for 2b, C39H34N4O12Tb2, C 43.84%, H 3.21%, N 5.24%, found: C 43.86%, H 3.23%, N 5.28%. IR (cm−1): 3013(w), 2894(w), 1544(s), 1454(s), 1413(s), 1350(m), 1280(m), 1219(m), 1183(w), 1065(m), 1009(w), 934(w), 885(m), 824(m), 740(s), 664(m), 520(m).Synthesis of [Ho2(phen)2(glu)3]n (2c)
The red crystals of 2c were prepared similarly from HoCl3 (yield 83% based on HoCl3). Anal. calcd for 2c, C39H34Ho2N4O12, C 43.35%, H 3.17%, N 5.18%, found: C 43.37%, H 3.19%, N 5.21%. IR (cm−1): 3094(w), 3047(w), 2930(w), 1627(s), 1551(s), 1440(s), 1350(m), 1316(m), 1266(m), 1149(m), 1106(m), 1065(m), 851(s), 808(m), 734(s), 670(w), 643(m), 536(m).Synthesis of [La2(glu)3(H2O)3]n·5nH2O (3)
The colorless crystals of 3 were prepared similarly from La2Cl3 (yield 67% based on La2Cl3). Anal. calcd for 3, C15H34La2O20, C 22.18%, H 4.22%, found: C 22.23%, H 4.27%. IR (cm−1): 3435(s), 2950(w), 1551(s), 1440(s), 1323(m), 1259(m), 1169(w), 1072(w), 968(m), 885(m), 801(w), 657(m), 546(m).X-ray crystallography
Single-crystal X-ray diffraction data for all compounds were collected on a Rigaku Mercury CCD diffractometer using a ω-scan method with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Routine Lorentz polarization and absorption corrections were applied using multi-scan technique. The structures of all compounds were solved by direct methods of SHELXS-97 (ref. 9) and refined by full-matrix least-squares methods on F2 using the SHELXL-97 program package.10 Positions of H atoms from phen and glu ligands were geometrically placed and H atoms were refined isotropically as a riding mode using the default SHELXTL parameters. A summary of crystallographic data is listed in Table 1. 1528897–1528902 contains the supplementary crystallographic data for this paper.†| 1a | 1b | 2a | 2b | 2c | 3 | |
| Formula | C17H14ClN2O4Y | C17H14Cl N2O4Tm | C39H34Ce2N4O12 | C39H34N4O12Tb2 | C39H34Ho2N4O12 | C15H34La2O20 |
| Fw | 434.66 | 514.68 | 1030.94 | 1068.56 | 1080.56 | 812.24 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/n | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
| a, Å | 8.1640(4) | 8.1381(11) | 8.8586(18) | 8.7895(4) | 8.7671(5) | 9.076(2) |
| b, Å | 12.4878(6) | 12.4629(16) | 13.565(3) | 13.4391(7) | 13.4002(7) | 9.867(3) |
| c, Å | 16.1848(8) | 16.161(3) | 16.392(3) | 16.4098(9) | 16.4124(11) | 16.303(4) |
| α, deg | 90 | 90 | 84.12(3) | 83.909(2) | 83.911(2) | 103.349(16) |
| β, deg | 94.013(2) | 94.034(5) | 83.86(3) | 84.340(2) | 84.424(2) | 104.110(9) |
| γ, deg | 90 | 90 | 75.14(3) | 74.984(2) | 75.048(2) | 98.526(10) |
| V, Å3 | 1646.00(14) | 1635.1(4) | 1887.2(7) | 1856.50(16) | 1847.46(19) | 1345.0(6) |
| Z | 4 | 4 | 2 | 2 | 2 | 2 |
| T, K | 293(2) | 297(2) | 293(2) | 300(2) | 297(2) | 293(2) |
| Calcd density, Mg m−3 | 1.754 | 2.092 | 1.814 | 1.912 | 1.942 | 2.006 |
| F(000) | 872 | 992 | 1016 | 1044 | 1052 | 796 |
| 2θ (max), deg | 50.20 | 50.80 | 36.06 | 50.20 | 50.20 | 50.20 |
| Total reflns collected | 134![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 409 409 |
9275 | 14486 |
18336 |
17479 |
49759 |
| Unique reflns | 2927 | 2967 | 2610 | 6573 | 6519 | 4781 |
| R1 [I > 2σ(I)] | 0.0402 | 0.0429 | 0.0242 | 0.0334 | 0.0342 | 0.0281 |
| wR2 (all data) | 0.1311 | 0.0719 | 0.0582 | 0.0561 | 0.0532 | 0.0791 |
Results and discussion
Crystal structure
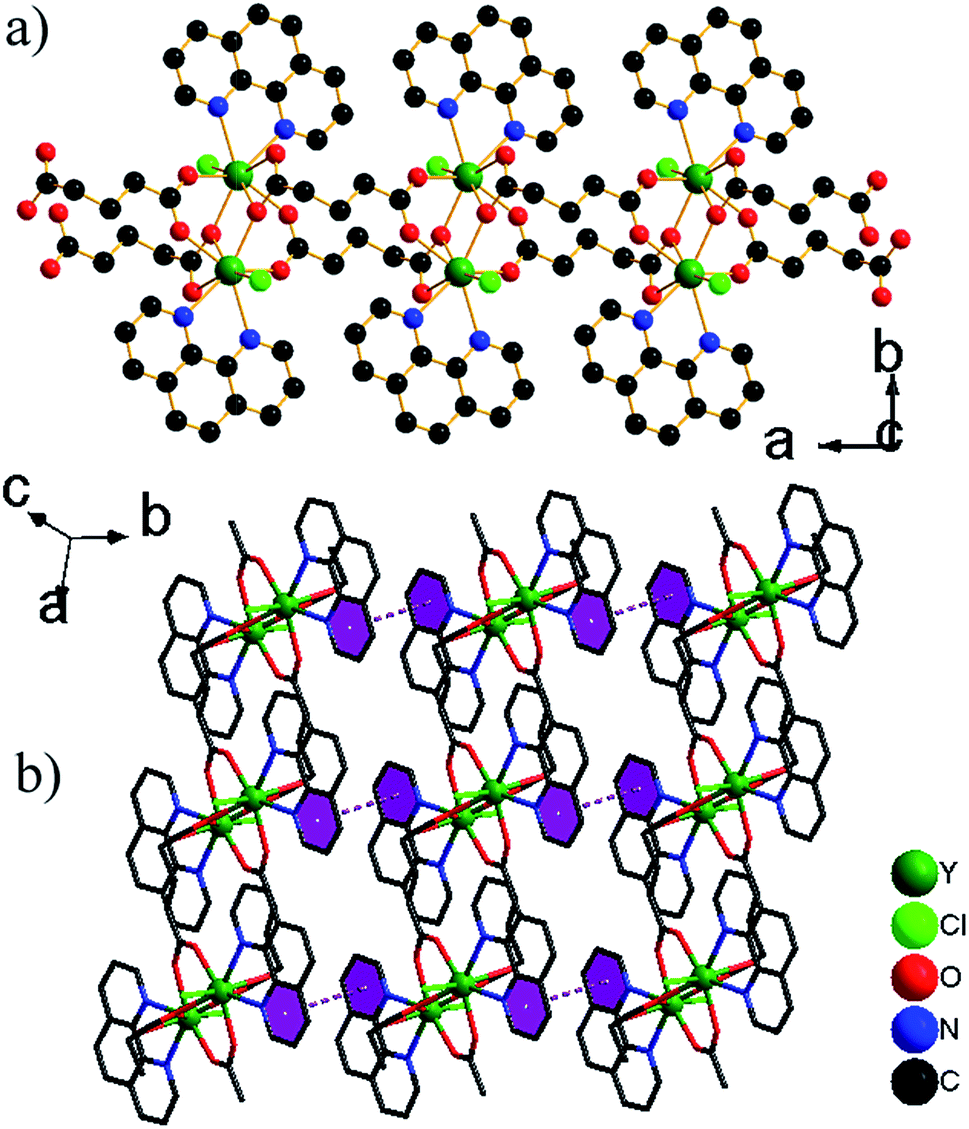 | ||
| Fig. 1 (a) 1-D chain in 1a. (b) Part of the crystal structure of 1a, showing the formation of a layer constructed by π–π stacking interactions. H atoms bonded to C/N atoms have been omitted for clarity. | ||
. The asymmetric unit contains two Ce3+ ions, three glu ligands and two phen ligands (Fig. S3†). The coordination geometry of each Ce3+ ion can be described as a distorted capped trigonal antiprism comprised of seven O atoms of five glu ligands and two N atoms of phen ligand. The Ce–O/N bond lengths are 2.4208(12)–2.7425(15) Å, which are compared with those in other Ce3+ complexes containing carboxylate groups and phen ligands.12 There are three crystallographically distinct glu ligands, which exhibit two different conformations, namely anti–anti and anti–gauche. One adopts anti–anti conformation, illustrated by the C6–C7–C8–C9 [178.22(4)°] and C7–C8–C9–C10 [178.84(4)°] torsion angles, the other adopt the distorted anti–gauche conformation demonstrated by the C1–C2–C3–C4 [63.82(6)°] or C11–C12–C13–C14 [62.48(5)°] torsion angles, and C2–C3–C4–C5 [172.85(4)°] or C12–C13–C14–C15 [176.58(4)°] torsion angles, respectively. The Ce3+ ions are grouped in pairs by bridging carboxylate O atoms. Each pair is connected to four neighbouring pairs via glu ligands, resulting in network of (4,4) topology with the total Schläfli symbol of 44·62 and the long vertex symbol of 4·4·4·4 (Fig. 2a). A 3-D network structure is formed by π–π stacking interactions between phen ligands in the adjacent layers with mean centroid-to-centroid distances of 3.726 Å (Fig. 2b–c).
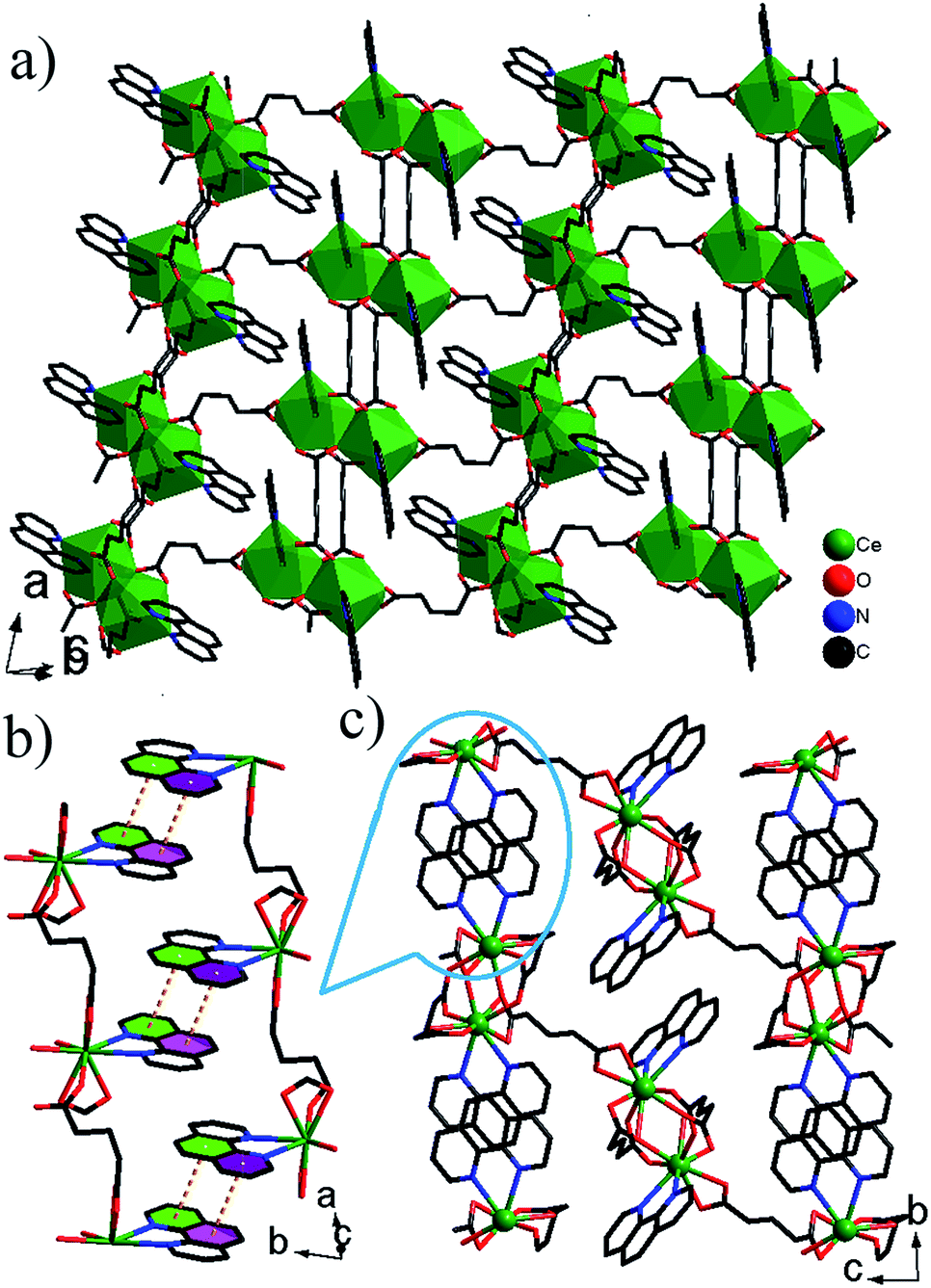 | ||
| Fig. 2 (a) 2-D layer in 2a. (b) 3-D network structure in 2a. (c) π–π stacking interactions between phen ligands. H atoms bonded to C/N atoms have been omitted for clarity. | ||
. The asymmetric unit contains two La3+ ions, three glu ligands, three coordinated H2O molecules and five free H2O molecules (Fig. 3a). The La(1)3+ ion adopts a tetracapped trigonal prism with eight O atoms of four glu ligands and two H2O molecules (Fig. 3b), while the La(2)3+ ion adopts a monocapped dodecahedron with eight O atoms of six glu ligands and one H2O molecules (Fig. 3c). The La–O bond distances are in the range from 2.463(3) Å to 2.897(3) Å. There are three kinds of coordination modes of glu ligands with the anti–gauche conformation in 3 (Fig. 3d–f). The first type (L1) is one chelating–anti and one anti–syn COO ends. The second type (L2) is one chelating and one chelating–anti COO ends. The third type (L3) is one chelating–anti and one chelating–anti COO ends. La(1)3+ and La3+ ions are bridged by two –COO groups in the anti–syn mode from two glu ligands and one –COO group in the chelating–anti mode from one glu ligand to La2 dimer with a La⋯La distance of 4.301(1) Å. The La2 dimer units are interconnected via chelating–anti –COO groups to produce a 1-D infinite chain, which is further connected by ligands L2 and L3 into a layer parallel to the (010) plane (Fig. 3g). These layers are pillared by ligand L1 with its chelating–anti –COO group connecting one layer, and its anti–syn –COO group connecting the adjacent layer, leading to a novel 3-D network structure with the 1-D channels occupied by free water molecules (Fig. 3h–i). There are a lot of O–H⋯O H-bonds between the coordinated H2O molecules and O atoms of –COO– groups, which play an important role in stabilizing 3 in the solid state. Moreover, the coordinated H2O and free H2O molecules interact also by O–H⋯O H-bonds, resulting in the formation of chain-like (H2O)3 unit and (H2O)4 ring (Fig. S4†), which are retained within the 1-D channels. The O⋯O distances are in the range of 2.746(5)–2.946(5) Å and the O–H⋯O angles vary from 143.7 to 177.6°, and are consistent with the values reported in the literature.7
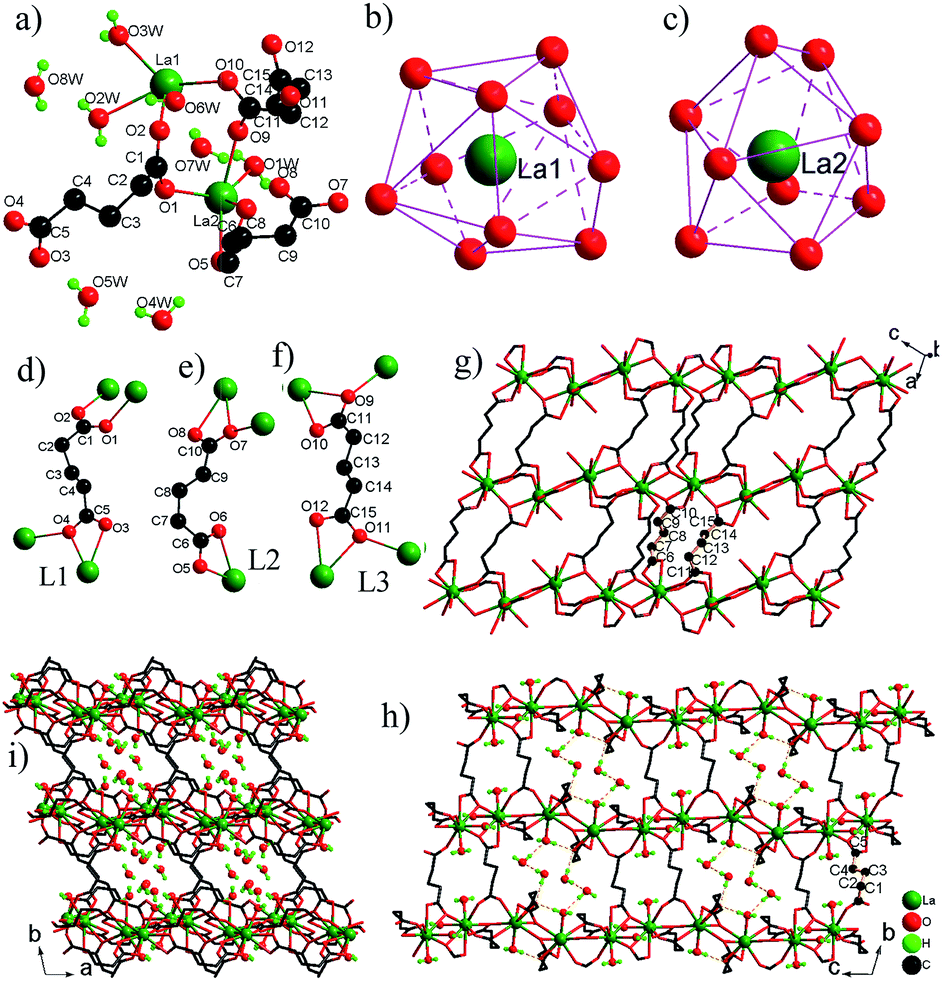 | ||
| Fig. 3 (a) The asymmetric unit of 3. (b and c) The coordination environments of La3+ ions. (d–f) Coordination modes of glu ligand in 3. (g) 2-D layer. View of the 3-D framework in 3 along the [100] (h) and [001] (i) directions. H atoms bonded to C atoms have been omitted for clarity. | ||
Although 3-D frameworks of 3 and reported lanthanide glutarates [Ln(glu)3(H2O)2]n·4nH2O [Ln = Pr (4a), Nd (4b), Sm (4c), Eu (4d), Gd (4e), Dy (4f), Ho (4g) and Y (4h)]7a–c are built from Ln3+ ions and glu ligands, the structures are completely different: (a) different 1-D inorganic chains: 1-D chains of 4a–h is constructed by only type of [LnO8(H2O)] polyhedra sharing edge via two –COO group bridges of glu ligands, while 1-D chain of 3 is based on two types of [LaO8(H2O)x] (x = 1, 2) polyhedra. (b) Different bridging modes of glu ligands: the structures of 4a–h possess two types of bridging modes, namely one has two chelating –COO groups, the other has two chelating–anti –COO groups, whereas the structure of 3 has three kinds of bridging modes, where two –COO groups of each glu ligand adopt different coordination modes. (c) Different space groups and sizes of channels: 3 crystallizes in space group P and has large rectangular channels that are filled by free water molecules, but 4a–h crystallizes in space group C2/c and has small circular channels, where free water molecules are incorporated.
IR spectra and photoluminescent properties
The IR spectra of all compounds show the characteristic bands of the –COO− groups in the range of 1627–1517 cm−1 for vC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–O asymmetric stretching and 1467–1343 cm−1 for vCO/C–O symmetric stretching. The weak bands at 3094–3010 cm−1 in 1a–b and 2a–c belong to the vC–H stretching of phen ligand, but no similar peaks in 3 appear, which further confirms the absence of phen ligand. The broad band at 3435 cm−1 can be attributed to vO–H stretching of water molecules in 3. Upon excitation at 345 nm, 1b shows a broad band centered at 388 nm (Fig. S5†). This band is attributed to the ligand emission due to its short lifetime (∼15 μs obtained from its decay curve). The weak shoulder at 475 nm is assigned to the transition from the 1G4 excited state to the 3H6 ground state.
O/C–O asymmetric stretching and 1467–1343 cm−1 for vCO/C–O symmetric stretching. The weak bands at 3094–3010 cm−1 in 1a–b and 2a–c belong to the vC–H stretching of phen ligand, but no similar peaks in 3 appear, which further confirms the absence of phen ligand. The broad band at 3435 cm−1 can be attributed to vO–H stretching of water molecules in 3. Upon excitation at 345 nm, 1b shows a broad band centered at 388 nm (Fig. S5†). This band is attributed to the ligand emission due to its short lifetime (∼15 μs obtained from its decay curve). The weak shoulder at 475 nm is assigned to the transition from the 1G4 excited state to the 3H6 ground state.
For 2b, the excitation spectrum was obtained by monitoring the most intense emission wavelength of Tb3+ at 545 nm (Fig. 4a). It shows a broad band ranging from 240 to about 400 nm attributed to electronic transitions from the ground state level (π) S0 to the excited level (π*) S1 of the organic ligand. The excitation spectrum also displays some weak sharp peaks at 326, 369, 378 and 487 nm assigned to Tb3+ ion intra f–f transitions 7F6 → 5D1, 7F6 → 5L10, 7F6 → 5G6 and 7F6 → 5D4, respectively. The dominate excitation bands are from the ligand, indicating that a sensitization of Tb3+ luminescence is mainly through an indirect energy transfer process from ligand to Tb3+ ions. Upon excitation at the most intensive wavelength 280 nm (Fig. 4b), the emission spectrum exhibits four strong emission bands at 489, 545, 586, and 622 nm corresponding to 5D4 → 7FJ (J = 6, 5, 4, 3), and three weak bands located at 645, 668 and 679 nm arising from the 5D4 → 7FJ (J = 2, 1, 0), respectively, with the 5D4 → 7F5 emission as the dominant band. Since no emission bands from the ligand were observed, it is further demonstrated that there is an intramolecular energy transfer from the ligand to Tb3+ ions. The intensities of characteristic Tb3+ emissions are obviously stronger than those of [Tb(H2O)]2(glu)3·4H2O,7c which contain the coordinated H2O molecules. This result demonstrated that the introduction of chromophoric phen ligand into the frameworks of lanthanide glutarates can enhance the intensity of Ln3+ emissions. The luminescence decay curve of Tb3+ related to the 5D4 → 7F5 emission (545 nm) was shown in Fig. 4c. The decay curve is singly exponential, confirming that all Tb3+ ions lie in the same average environment. The luminescence lifetime was determined to be 0.414 ms. The CIE chromaticity coordinate was calculated to be (0.35, 0.59), as shown in Fig. 4d.
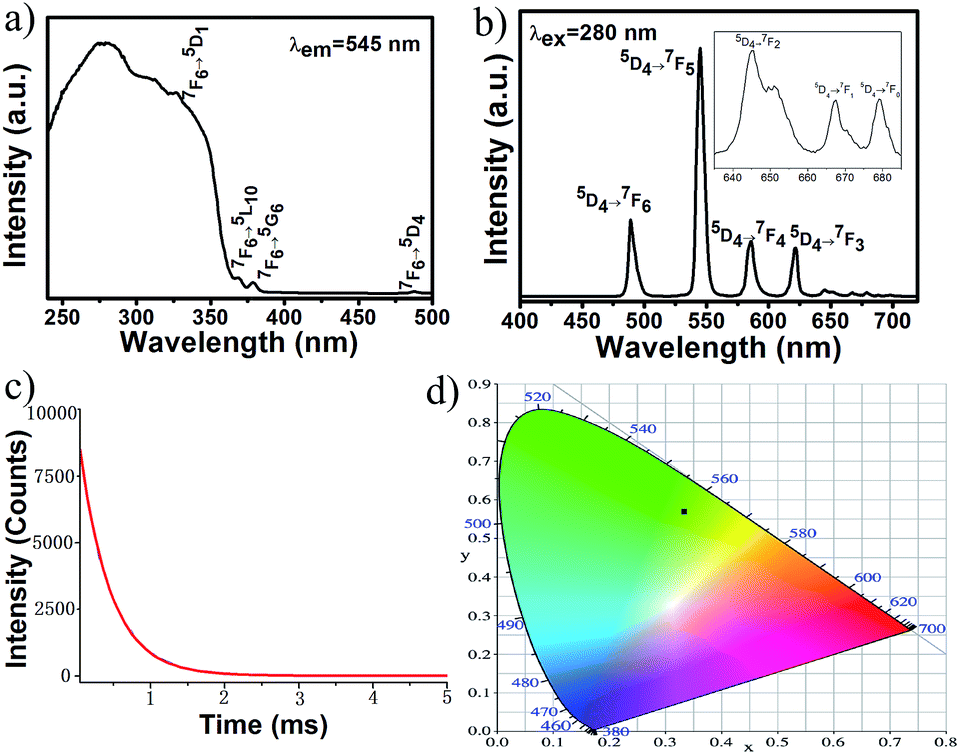 | ||
| Fig. 4 (a) Excitation spectrum for 2b (λem = 545 nm). (b) Emission spectrum for 2b (λex = 280 nm, the enlarged part shows the emission bands between 635 to 685 nm). (c) Decay curve for 2b. (d) CIE chromaticity diagram for 2b at λex = 280 nm. | ||
Magnetic properties
The magnetic susceptibilities of compounds 1b, 2b and 2c were measured in the temperature range 2–300 K under an applied magnetic field of 1 kOe. For 1b, the χMT value (7.37 cm3 K mol−1) at room temperature is close to the theoretical value for one non-interacting Tm3+ ion (7.08 cm3 K mol−1 for Tm3+, 3H6, S = 1, L = 5, J = 6, g = 7/6). With lowering temperature, χMT value shows a slight decrease for 1b (Fig. 5a), as a result of as a consequence of the depopulation of sublevels of the ground J multiplet split by the crystal field13 and possible antiferromagnetic interactions.14 Antiferromagnetic interactions between Tm3+ ions can be also confirmed by the smaller Tm–O–Tm angle value of 106.97°, because the rule is that Ln–O–Ln angles below 113.50° are assumed to cause an antiferromagnetic exchange in the literature.15 For 2b, the χMT value at room temperature is 23.61 cm3 K mol−1 (Fig. 5b), which is in good agreement with the expected theoretical values for two uncoupled Tb3+ ions (23.64 cm3 K mol−1 for Tb3+, 7F6, S = 3, L = 3, J = 6, g = 3/2). Upon cooling, χMT increases to a maximum of 26.33 cm3 K mol−1 at 54 K. This magnetic behavior typifies the ferromagnetic coupling interactions between adjacent Tb3+ centers. A sudden decrease of the χMT value below 54 K suggests that is a consequence of the depopulation of sublevels of the ground J multiplet split by the crystal field and antiferromagnetic interactions. The ferromagnetic interactions provided by chelating–anti –COO bridges2a in the dimer could also be concomitant with significant antiferromagnetic interactions, due to the Tb–O–Tb angle values in the range of 104.54–107.54° below 113.50°. The classical syn–anti –COO bridges within the dimeric entity appear as the most likely pathways for the antiferromagnetic exchange.2a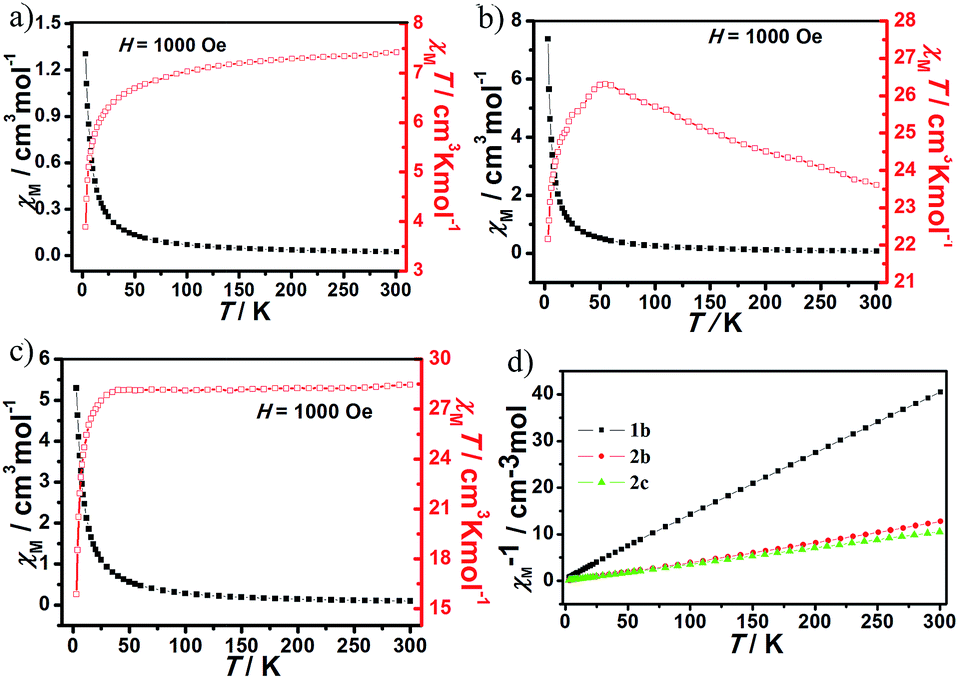 | ||
| Fig. 5 The plot of χMT versus T for 1b (a), 2b (b) and 2c (c). (d) Plot of χM−1 versus T for 1b, 2b and 2c. | ||
For 2c, the χMT value (28.46 cm3 K mol−1) at room temperature (Fig. 5c) is as expected for two magnetically isolated Ho3+ ion (28.14 cm3 K mol−1 for Ho3+, 5I8, S = 2, L = 6, J = 8, g = 5/4). As the temperature is lowered, this value remains practically constant until 28 K and it decreases further to reach a value of 2.46 cm3 K mol−1 at 3 K. This plot can be indicative of the existence of an antiferromagnetic coupling between the Ho3+ ions, which is further confirmed by the smaller Ho–O–Ho angle values of 103.81–107.58° below 113.50°.15 The χM−1 versus T over the entire temperature range for compounds 1b, 2b and 2c can be fitted to the Curie–Weiss law, χM = C/(T − θ) with the Curie constant C = 7.47, 23.95 and 28.47 cm3 K mol−1, and the Weiss constant θ = −4.53, 2.65 and −1.29, respectively (Fig. 5d).
Conclusions
A series of lanthanide glutarates were isolated as single crystals under mild hydrothermal conditions in the presence of phen. The lighter Ln3+ ions form the 2-D layers, while the heavier ones form the 1-D chains, where the change in their crystal architectures is obviously related to the well-known lanthanide contraction. The chromophoric phen ligand was incorporated successfully into the frameworks of lanthanide glutarates and prevents water molecules from binding to Ln3+ ions, which could lead to the enhancement of characteristic Ln3+ emissions, for instance, 2b can emit stronger green luminescence at 545 nm with long luminescent lifetime, making 2b a good candidate for potential green materials. Moreover, the interconnections of Ln3+ ions and glu ligands lead to very robust structural type [Ln(glu)3(H2O)2]n·4nH2O, whose 3-D networks containing 1-D inorganic chains of edge-sharing [LnO8(H2O)] polyhedra are not changed by different Ln3+ ions, but 3-D framework of 3 contains 1-D inorganic chains based on two types of [LaO8(H2O)x] (x = 1, 2) polyhedra, which exhibits a new structural type. The successful synthesis of lanthanide glutarates containing chromophoric phen ligand not only enriches the existing field of lanthanide coordination polymers but also opens possibilities for synthesizing other novel photoluminescent lanthanide glutarates using chromophoric phen derivatives units under desirable conditions.Acknowledgements
This work was supported by the NNSF of china (No. 21671029 and 21601038), the NSF of Chongqing municipality (No. cstc2014jcyjA50002, cstc2015jcyjBX0117), Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER through Programa Operacional Factores de Competitividade (COMPETE), and CICECO-Aveiro Institute of Materials, POCI-01-0145-FEDER-007679 (FCT Ref. UID/CTM/50011/2013), financed by national funds through the FCT/MEC and when appropriate co-financed by FEDER under the PT2020 Partnership Agreement, and Program for Excellent Talents in Chongqing Higher Education Institutions. The authors are also grateful to Chongqing Normal University for financial support (14CSLJ02), graduate innovative research projects of Chongqing municipality (CYS16147) and the innovative training program of university student (201610637008).Notes and references
- (a) N. Ishikawa, T. Iino and Y. Kaizu, J. Am. Chem. Soc., 2002, 124, 11440 CrossRef CAS PubMed; (b) G. Novitchi, W. Wernsdorfer, L. F. Chibotaru, J. Costes, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2009, 48, 1614 CrossRef CAS PubMed; (c) C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS PubMed; (d) P. Lin, T. J. Burchell, R. Clérac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848 CrossRef CAS PubMed; (e) J. Peng, X. Kong, Q. Zhang, M. Orendac, J. Prokleska, Y. Ren, L. Long, Z. Zheng and L. Zheng, J. Am. Chem. Soc., 2014, 136, 17938 CrossRef CAS PubMed; (f) D. D'Alessio, A. N. Sobolev, B. W. Skelton, R. O. Fuller, R. C. Woodward, N. A. Lengkeek, B. H. Fraser, M. Massi and M. I. Ogden, J. Am. Chem. Soc., 2014, 136, 15122 CrossRef PubMed; (g) J. M. Clemente-Juan, E. Coronado and A. Gaita-Arino, Chem. Soc. Rev., 2012, 41, 7464 RSC.
- (a) Y. Zheng, Z. Zheng and X. Chen, Coord. Chem. Rev., 2014, 258–259, 1 CrossRef CAS; (b) P. Zhang, L. Zhang, C. Wang, S. Xue, S. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484 CrossRef CAS PubMed; (c) N. Ishikawa, M. Sugita and W. Wernsdorfer, J. Am. Chem. Soc., 2005, 127, 3650 CrossRef CAS PubMed; (d) N. Ishikawa, T. Iino and Y. Kaizu, J. Am. Chem. Soc., 2002, 124, 11440 CrossRef CAS PubMed; (e) A. B. Canaj, D. I. Tzimopoulos, M. Siczek, T. Lis, R. Inglis and C. J. Milios, Inorg. Chem., 2015, 54, 7089 CrossRef CAS PubMed; (f) X. Tan, J. Zhou, L. Fu, H. Xiao, H. Zou and Q. Tang, Dalton Trans., 2016, 45, 5253 RSC.
- (a) Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76 CrossRef CAS; (b) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed; (c) X. Yang, D. Schipper, R. A. Jones, B. J. Holliday, S. Huang and L. A. Lytwak, J. Am. Chem. Soc., 2013, 135, 8468 CrossRef CAS PubMed; (d) J. Rausch, V. Lorenz, C. G. Hrib, V. Frettlöh, M. Adlung, C. Wickleder, L. Hilfert, P. G. Jones and F. T. Edelmann, Inorg. Chem., 2014, 53, 11662 CrossRef CAS PubMed; (e) X. Feng, Y. Feng, J. J. Chen, S. Ng, L. Wang and J. Guo, Dalton Trans., 2015, 44, 804 RSC.
- (a) G. Stein and E. Wurzberg, J. Chem. Phys., 1975, 62, 208 CrossRef CAS; (b) Y. Haas and G. Stein, J. Phys. Chem., 1971, 75, 3677 CrossRef CAS.
- (a) L. N. Puntus, K. A. Lyssenko, I. S. Pekareva and J. G. Bünzli, J. Phys. Chem. B, 2009, 113, 9265 CrossRef CAS PubMed; (b) R. Martínez-Manez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed; (c) X. P. Yang, R. A. Jones, M. M. Oye, A. L. Holmes and W.-K. Wong, Cryst. Growth Des., 2006, 6, 2122 CrossRef CAS; (d) J.-C. G. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897 CrossRef.
- (a) X. Huang, Q. Wang, X. Yan, J. Xu, W. Liu, Q. Wang and Y. Tang, J. Phys. Chem. C, 2011, 115, 2332 CrossRef CAS; (b) Y. Hasegawa, S. Tsuruoka, T. Yoshida, H. Kawai and T. Kawai, J. Phys. Chem. A, 2008, 112, 803 CrossRef CAS PubMed; (c) P. Lenaerts, A. Storms, J. Mullens, J. D'Haen, C. Görller-Walrand, K. Binnemans and K. Driesen, Chem. Mater., 2005, 17, 5194 CrossRef CAS; (d) X. Ma, X. Li, Y. Cha and L. Jin, Cryst. Growth Des., 2012, 12, 5227 CrossRef CAS; (e) L. N. Puntus, K. A. Lyssenko, M. Y. Antipin and J. G. Bünzli, Inorg. Chem., 2008, 47, 11095 CrossRef CAS PubMed.
- (a) E. Antic-Fidancev, F. Serpaggi and G. Férey, J. Alloys Compd., 2002, 340, 88 CrossRef CAS; (b) F. Serpaggi and G. Férey, J. Mater. Chem., 1998, 8, 2737 RSC; (c) B. Yu, C. Xie, X. Wang, R. Wang, G. Shen and D. Shen, J. Coord. Chem., 2007, 60, 1817 CrossRef CAS; (d) C. Wang, Y. Xing, Z. Li, J. Li, X. Zeng, M. Ge and S. Niu, J. Mol. Struct., 2009, 931, 76 CrossRef CAS; (e) P. I. Girginova, F. A. Almeida Paz, P. C. R. Soares-Santos, R. A. Sá Ferreira, L. D. Carlos, V. S. Amaral, J. Klinowski, H. I. S. Nogueira and T. Trindade, Eur. J. Inorg. Chem., 2007, 4238 CrossRef CAS; (f) Y. Kim, Y. Park and D. Jung, Dalton Trans., 2005, 2603 RSC; (g) A. Dimos, D. Tsaousis, A. Michaelides, S. Skoulika, S. Golhen, L. Ouahab, C. Didierjean and A. Aubry, Chem. Mater., 2002, 14, 2616 CrossRef CAS.
- (a) D. Wei, H. Xie, S. Huang, J. Zhou and G. Lu, J. Coord. Chem., 2011, 64, 424 CrossRef CAS; (b) L. Zhang, Y. Wan and L. Jin, J. Mol. Struct., 2003, 646, 169 CrossRef CAS.
- (a) G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- (a) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) K. P. Carter, S. J. A. Pope and C. L. Cahil, CrystEngComm, 2014, 16, 1873 RSC; (b) C. Wang, Z. Wang, F. Gu and G. Guo, J. Mol. Struct., 2010, 979, 92 CrossRef CAS.
- F. Bai, J. Xu, H. Su and X. Gu, Z. Anorg. Allg. Chem., 2012, 638, 2366 Search PubMed.
- (a) Y.-L. Wang, Y. Ma, X. Yang, J.-K. Tang, P. Cheng, Q.-L. Wang, L.-C. Li and D.-Z. Liao, Inorg. Chem., 2013, 52, 7380 CrossRef CAS PubMed; (b) H.-S. Ke, G.-F. Xu, Y.-N. Guo, P. Gamez, C. M. Beavers, S. J. Teat and J.-K. Tang, Chem. Commun., 2010, 46, 6057 RSC.
- (a) G. Abbas, Y. H. Lan, G. E. Kostakis, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2010, 49, 8067 CrossRef CAS PubMed; (b) J. Zhou, R. Zhao, T. Yang, X. Liu, H. Xiao, H. Zou and X. Tan, Dalton Trans., 2015, 44, 7203 RSC; (c) J. Zhou, H. Xiao, H. Xiao, T. Yang, H. Zou, X. Liu, R. Zhao and Q. Tang, Dalton Trans., 2015, 44, 1350 RSC.
- B. Benmerad, K. Aliouane, N. Rahahlia, A. Guehria-Laïdoudi, S. Dahaoui and C. Lecomte, J. Rare Earths, 2013, 31, 85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data in CIF format, CIF files, XRD patterns, and some additional figures. CCDC 1528897–1528902. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01552f |
| This journal is © The Royal Society of Chemistry 2017 |