
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly of carbon nanotubes into microparticles with tunable morphologies using droplets in a non-equilibrium state†
Sakurako Tomii,
Masumi Yamada *,
Masahiro Mizuno,
Yasuhiro Yamada,
Takashi Kojima,
Masahito Kushida and
Minoru Seki
*,
Masahiro Mizuno,
Yasuhiro Yamada,
Takashi Kojima,
Masahito Kushida and
Minoru Seki
Department of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: m-yamada@faculty.chiba-u.jp
First published on 23rd March 2017
Abstract
Carbon nanomaterials assembled into micrometer-scale configurations are highly useful because of their unique molecular transport and adsorption properties, improved operability, and versatility in industrial/research applications. Here we propose a facile approach to assemble carbon nanotubes (CNTs) into monodisperse microparticles through rapid condensation and accumulation of CNTs in aqueous droplets in a non-equilibrium state. The droplets were generated by means of a microfluidic process or membrane emulsification, using a water-soluble polar organic solvent as the continuous phase. Because water molecules in the droplets were rapidly dissolved into the continuous phase, the CNTs were dramatically condensed and stable microparticles were finally formed. We prepared microparticles using both multi-walled and single-walled CNTs, and the size was controllable in the range of 10–40 μm simply by changing the initial CNT concentration. Interestingly, the morphologies of the particles were not spherical in many cases, and they were controllable by changing the type of the organic solvent and/or using additives in the dispersed phase. Physicochemical characterization suggested good compatibility of the CNT microparticles when used as supports for catalysts, adsorbents, and electrodes.
Introduction
Carbon nanotubes (CNTs) have attracted growing attention among material scientists because of their unique structural, mechanical, electrical, and thermal properties since their discovery in the 1990s.1 CNTs are being applied to various industrial/research fields,2 as they are potentially useful as new materials for catalyst supports,3,4 electrodes,5,6 adsorbents,7,8 sensors,9–11 and actuators.12,13 In these applications, the high specific surface area and high mechanical strength of CNTs are utilized. In addition, effective molecular interactions in the microenvironments surrounding the CNTs are realized while the physical stability of the material is ensured. For instance, both the high electrical conductivity and high surface area of CNTs are essential for catalyst-loaded electrodes in fuel cells.14In the general process of commercial large-scale CNT production, CNTs are obtained in the form of powder when gas phase reaction schemes, such as the continuous CVD method, are employed.15–17 When CNTs are chemically functionalized, e.g., by depositing catalyst nanoparticles, multistep liquid phase treatments of CNTs that involve medium exchange, removal, condensation, and drying, should be employed. However, in general, dispersed CNTs in a liquid sample are difficult to handle by conventional centrifugation and filtration processes because of their small sizes.18 For instance, it takes several hours to precipitate CNTs in a medium by ultracentrifugation.19 This problem becomes severe when CNTs are used as the supports of catalysts for liquid-phase chemical reactions, because it is difficult to recover the catalyst-loaded CNTs from the reaction solution.18 In addition, once the CNTs are closely packed, it is difficult to control the CNT density because of the relatively high cohesive force, which affects the molecular flux through the accumulated CNTs. If CNTs are assembled into particulates with a size of several micrometers, such materials might potentially be useful because of the good operability via centrifugation/filtration, high property for controlled accumulation, and the high contact efficiency between particles.
In the past decade, researchers have proposed methods to produce microparticles consisting of CNTs, in which mesoporous pores are formed between CNTs. When CNT microparticles are accumulated, macroscale inter-particle spaces are formed at the same time, which will help the smooth flow of molecules for chemical reactions. The techniques for preparing CNT microparticles are roughly classified into 2 types; (1) simultaneous CNT (or carbon nanofiber) growth and particle formation during CVD or pyrolysis processes,20–22 and (2) assembly of pre-synthesized CNTs into particulate forms.23–33 Compared to the former process, where a large-scale particle production is not practical, the latter is advantageous in terms of the high production throughput and controllability in particle size and morphology. One of the most popular approaches is the layer-by-layer assembly or accumulation of CNTs on the core particles (e.g., polymeric particles) as template.23–28 However, this technique requires the removal of the core particles via either etching or calcination, to obtain particles made only of CNTs. This downstream treatment possibly degrades the stability of the CNT layer and causes particle deformation. Water-in-oil (W/O) emulsion systems have also been employed to assemble carbon nanomaterials (CNT and graphite) into particles.29–34 In general, these emulsion-based techniques are capable of precisely controlling the size of the formed particles. However, in most of these studies, microcapsules rather than packed particles were obtained after removing the solvent of the dispersed phase because CNTs were accumulated on the water–oil interface.
In our research group, we have proposed a process to produce microparticles through the rapid shrinkage of droplets in a non-equilibrium state.35–38 Using a polar organic solvent either as the continuous or dispersed phase, droplets of a precursor solution were generated. Because the solvent in the dispersed phase was rapidly extracted to the continuous phase, the precursor substances were instantaneously (typically less than 1 s) concentrated and microparticles were generated. We produced hydrogel,35 polymeric,36 protein,37 and lipid38 microparticles with sizes of several micrometers using this rapid solvent-extraction process. The remarkable advantage of this method is the simplicity in operation with high controllability in particle size and composition. Furthermore, microparticles with various morphologies were obtained by either using additives or controlling the degree of droplet shrinkage.35,36 However, to the best of our knowledge, there have been no reports on the production of non-spherical, closely packed CNT microparticles by using the rapid solvent-extraction process.
In this work, we report a microfluidic process to produce CNT microparticles by assembling CNTs using rapidly dissolving droplets, as shown in Fig. 1(a). Droplets of an aqueous CNT dispersion are formed, which rapidly shrink during flowing through the microchannel. CNTs are dramatically condensed in the droplets and particles are finally formed, and the particle morphology is tunable as it is affected by the dispersion/accumulation behavior of CNTs in the droplets. This method utilizes the adhesion force of CNTs in the W/O droplets to generate particles purely made of CNTs, and hence, there is no need to use any solid templates or cross-linkers. We examined several factors affecting the size and morphology of the formed particles, including the type of the polar organic solvent, CNT concentration, and usage of additives, mainly using multi-walled CNTs (MWCNTs). In addition, the characteristics of the particles in terms of gas adsorption property, electrical conductivity, and microstructures were investigated, using microparticles prepared by membrane emulsification. Furthermore, microparticles made of single-walled CNTs (SWCNTs) were produced.
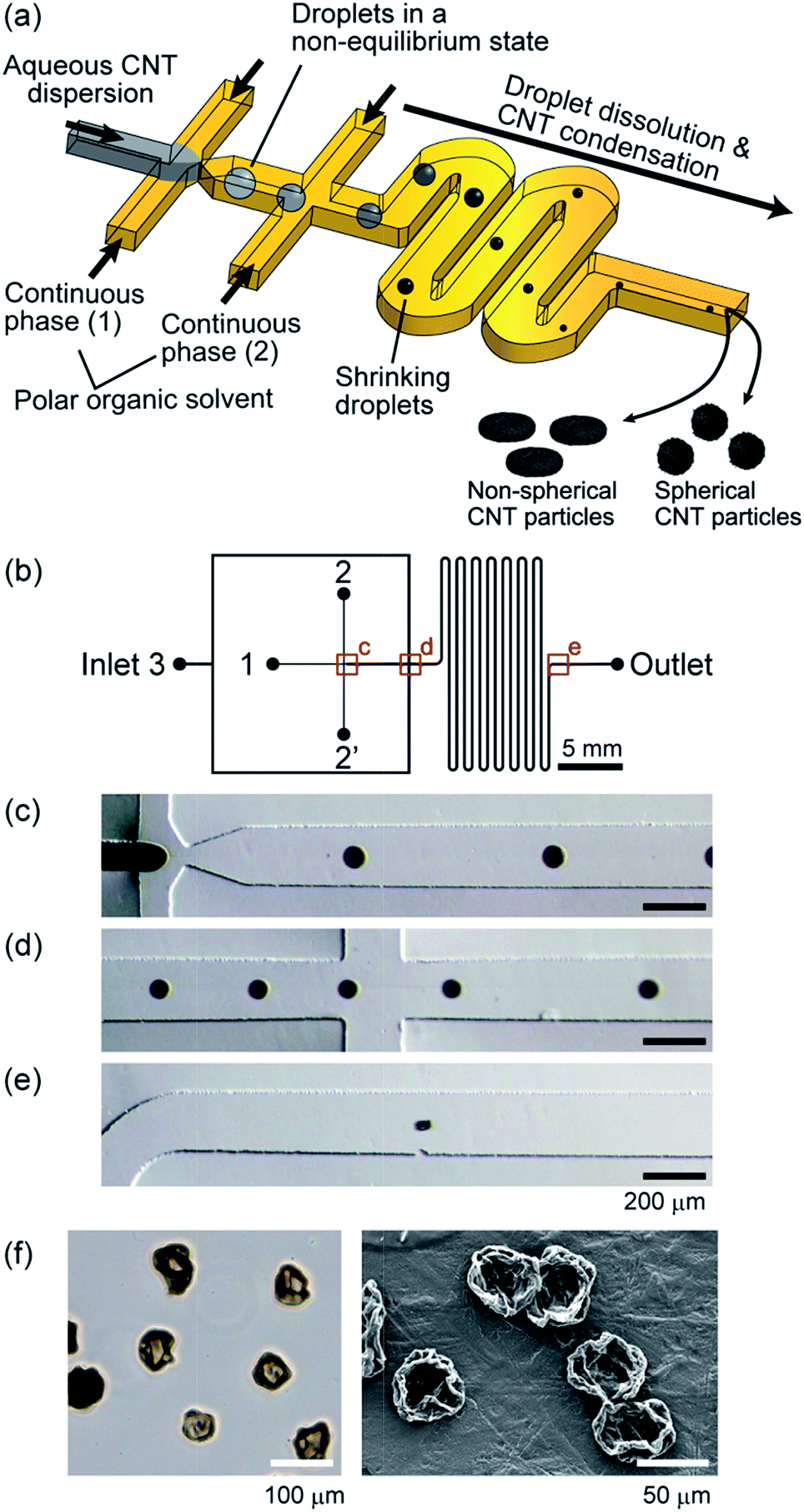 | ||
| Fig. 1 (a) Schematic diagram showing the microfluidic system to produce CNT microparticles. A polar organic solvent was used as the continuous phase, and introduced into the microchannel at two points (1) and (2). (b) Design of the microchannel used to produce CNT microparticles. The main channel width was 200 μm and the depth was ∼100 μm. (c–e) Micrographs showing the droplets/particles flowing in the microchannel. MAc was used as the continuous phase. Each panel corresponds to the position in image (b). (f) Optical and SEM images of the MWCNT particles prepared using MAc as the continuous phase. | ||
Experimental section
Materials
Aqueous suspensions of MWCNTs (2%) and SWCNTs (0.1%), containing an anionic surfactant as a stabilizer, were obtained from Meijo Nano Carbon, Aichi, Japan. Methyl acetate (MAc, >98.0% purity), ethyl acetate (EAc, >99.9% purity), isopropyl acetate (IPAc, >99.0% purity), propyl acetate (PAc, >97.0% purity), sodium alginate (Na-Alg, viscosity of 300–400 mPa s at 1% solution at 20 °C), sodium dodecyl sulfate (SDS), and polyethylene glycol (PEG, MW of 6000) were obtained from Wako Pure Chemical, Osaka, Japan. Triton X-100 was purchased from Sigma-Aldrich, St. Louis, MO, USA. These chemicals were used as received.Fabrication of microfluidic devices
Polydimethylsiloxane (PDMS) microfluidic devices were fabricated using standard soft lithography and replica molding techniques as previously described.39 Initially, SU-8 structures were patterned on a Si wafer, and PDMS prepolymer was cast on this mold. After peeling the PDMS replica off the mold and making inlet/outlet holes, this PDMS plate was bonded with a flat plate via O2 plasma treatment. To ensure the completely hydrophobic surface, the bonded microdevices were incubated in an oven at 150 °C for 3 h.The microchannel design is shown in Fig. 1(b). We employed a flow-focusing microchannel configuration. There were 4 inlets; Inlet 1 for the dispersed phase of the aqueous suspension of CNTs, Inlets 2 and 2′ for the continuous phase, and Inlet 3 for the additional continuous phase. The microchannel depth was uniformly ∼100 μm, and the width of the main channel was 200 μm. Droplets were generated at the orifice with the channel width of 50 μm. The distance between the first confluence (orifice) and the second confluence was 500 μm.
Preparation of CNT particles
To prepare MWCNT microparticles, we diluted the purchased MWCNT suspension with deionized (DI) water, and this suspension was treated by ultrasonication using an ultrasonic cleaner (USK-3R, AS ONE, Osaka, Japan) at room temperature for 30 min. The SWCNT suspension was treated using an ultrasonic homogenizer (US150, Nissei, Aichi, Japan) for 15 min. When the effect of additives on the MWCNT particles was examined, we employed Na-Alg, PEG, Triton X-100, and SDS. Condensed solutions of these additives were prepared, and they were added into the MWCNT suspension, with a final additive concentration of 0.1–0.3% (w/v). As the continuous phase, we used 4 types of polar organic solvents (MAc, EAc, IPAc, and PAc).The MWCNT or SWCNT suspension as a dispersed phase was continuously introduced from Inlet 1, and the polar organic solvent as a continuous phase was introduced from Inlets 2, 2′, and 3 into the microchannel using syringe pumps (KDS 200, KD Scientific, Holliston, MA, USA). The flow rates from Inlets 1, 2, 2′, and 3 were 1, 30, 30, and 400 μL min−1, respectively. The formed CNT particles were continuously collected through a Teflon tube into a centrifuge tube containing the same organic solvent with gentle shaking. When additives were used for the dispersed phase, obtained particles were washed with DI water for several times via gentle centrifugation. The morphologies of the CNT particles in the continuous phase were observed with an optical microscope (IX71, Olympus, Tokyo, Japan) with a CCD camera (DP72, Olympus). In addition, a scanning electron microscope (SEM; VE8800, Keyence, Tokyo, Japan) was used to observe the dried particles. The diameters of the CNT particles and their distributions were analyzed by measuring their area from the captured images using Image J software. On average ∼100 particles were analyzed for each condition.
Preparation of MWCNT particles using membrane emulsification
Porous membranes made of Shirasu porous glass with hydrophobic surface properties and with 30 μm pores (SPG direct connector, SPG technology, Miyazaki, Japan) were employed. As a continuous phase, 300 mL of IPAc was poured into a 500 mL beaker and was vigorously stirred using a magnetic stirrer. A suspension of 0.5% (w/v) MWCNTs was continuously pumped into the continuous phase through the porous membrane, which was dipped in the continuous phase, at a feeding speed of 150 μL min−1 using the syringe pump. After that, the obtained particles were washed with DI water for several times.Characterization of MWCNT particles
To estimate the pore size within the CNT particles, we measured the adsorbed amount of nitrogen gas by a surface area and pore size analyzer (BELSORP-max-N, MicrotracBEL, Osaka, Japan). The CNT particles were washed several times with DI water, dried on an evaporating dish in an oven at 50 °C, and then examined by the analyzer. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method. In addition, the inner structure of the particles was directly observed by transmission electron microscopy (TEM). After embedding CNT particles into epoxy resin, ultra-thin sections with a thickness of 70–80 nm were prepared by ultramicrotomy (ULTTOME V, LKB, Bromma, Sweden). Then the sample was observed by TEM (JEM-2010, JEOL, Tokyo, Japan). Furthermore, we examined the physical characteristics of the particles in a macroscale. We measured the sedimentation rate of CNT particles in DI water. Then the filtering time of water through the film of the accumulated CNT particles was evaluated. As-received CNTs (native CNTs) and graphite (SP270, Nippon Graphite, Shiga, Japan) were also used as control samples for comparison. Finally, the electrical conductivity of the accumulated film of the CNT particles was measured by the four-terminal sensing method using a digital multimeter (VOAC7521A, Iwatsu Electric, Tokyo, Japan). CNT particles prepared by the membrane emulsification technique were employed for these experiments, except for the sedimentation test.Results and discussion
Formation of droplets and MWCNT particles
In the presented scheme for CNT particle generation (Fig. 1(a)), the water molecules in the droplets should be perfectly dissolved into the continuous phase to form condensed particles. In previous reports, W/O droplets in a non-equilibrium condition have been generated to prepare spherical/non-spherical hydrogel particles35 and protein particles.37 In these reports, MAc was used as the continuous phase, which can be dissolved in water at ∼25%. In this study, we first employed MAc, and examined if CNT particles are actually formed. The droplets formed in the microchannel, when the initial MWCNT concentration was 0.5% (w/v), are shown in Fig. 1(c–e). Droplets were continuously generated with an initial average diameter of 83 μm and coefficient of variation (CV) value of 9%, showing a relatively high monodispersity. Because of the introduction of an additional continuous phase from Inlet 3, the inter-droplet distance was increased (Fig. 1(d)), which prevented the droplets from coalescing with each other. The generated droplets rapidly shrank during flowing through the microchannel (retention time of about 700 ms) because of the solvent-extraction into the continuous phase. The droplet shape was deformed near the outlet (Fig. 1(e)), and the length of the major axis was ∼63 μm. We observed the morphology of the particles suspended in the solvent by optical microscopy and those dried on Teflon sheets by SEM. We found that the obtained particles exhibited a non-spherical, deflated beach ball-like morphology with an average diameter of 62 μm (CV of 15%, Fig. 1(f)). We assumed that CNTs were probably condensed at the interface between the continuous/dispersed phase as with the CNT capsule formation using droplets in an equilibrium state,29,30 and a solid membrane (shell) was temporarily formed. The formed shell structure was then deformed into particles with such a unique morphology with the progress of the droplet dissolution.Effect of organic solvent on particle morphology
In addition to MAc, we tested 3 types of acetic esters with different water solubilities as the continuous phase. EAc, IPAc, and PAc were used, which can be dissolved in water at 8, 4, and 2%, respectively. When we used these solvents, droplets with nearly the same average size were produced (80–90 μm). Because of the difference in water solubility, the dissolution speed of the droplets was changed as shown in Fig. 2(a). For example, when IPAc was used as the continuous phase, the droplets shrinkage was suppressed and the droplet dehydration was not completed even at the microchannel exit (the average diameter at this point was ∼65 μm). The particles prepared using different solvents are shown in Fig. 2. The average diameters of the particles were 50 μm (CV of 13%), 31 μm (CV of 7%), and 33 μm (CV of 8%) when EAc, IPAc, and PAc were used, respectively. When EAc was used, we obtained crumpled particles as in the case of using MAc (Fig. 2(b)). On the other hand, when either IPAc or PAc was used, the particle deformation was not so significant, and the particles showed a concave but relatively spherical morphology in terms of increased thickness and compact shape. Interestingly, we found that particles prepared using IPAc and PAc had a number of small pores with diameters of 400–800 nm on the surface (Fig. 2(c and d)). From these results, we confirmed that the morphologies of the particles were significantly affected by the type of the continuous phase.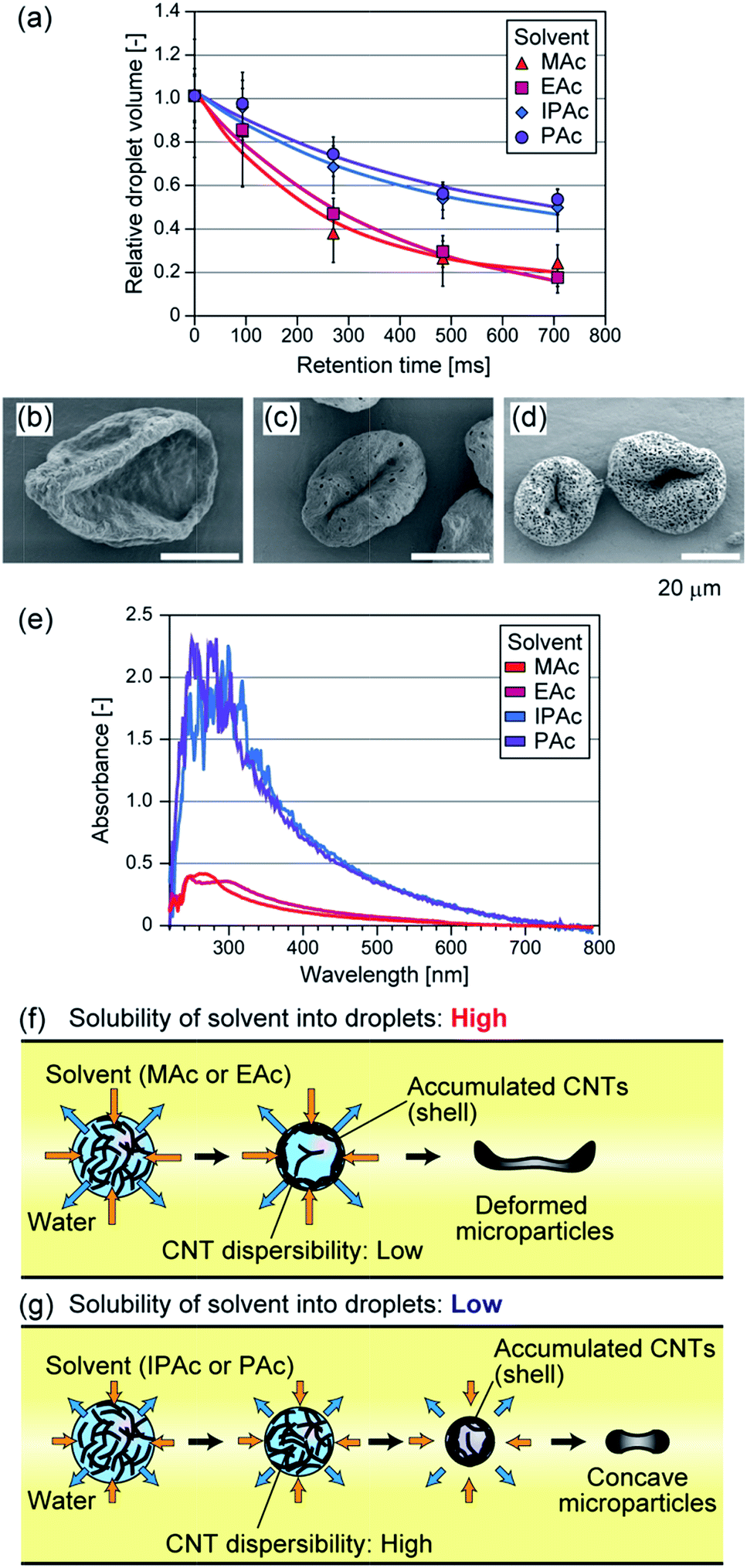 | ||
| Fig. 2 (a) Time course changes in the droplet volumes when 4 types of polar organic solvents were used. (b–d) SEM images of the MWCNT particles prepared using different solvents as the continuous phase: (b) EAc, (c) IPAc, and (d) PAc. (e) The degrees of aggregate formation of CNTs in water samples, equilibrated with a polar organic solvent (MAc, EAc, IPAc, or PAc). Optical/UV absorption spectra of the supernatant of CNT dispersions, when polar organic solvent was added. The initial CNT concentration was 0.001%. (f, g) Schematic images showing the formation behaviors of CNT microparticles, when the solubility of the solvent into droplets is (f) high and (g) low. | ||
We investigated the reason why the particle shape was affected by the type of organic solvent. We suspected that the dispersibility of CNTs in the shrinking droplets was affected by the type of organic solvent, because the solvent molecules were incorporated into the droplets but their concentrations would be different. So we prepared DI water samples equilibrated with MAc, EAc, IPAc, or PAc, and evaluated the degree of aggregate formation of MWCNTs in these samples. MWCNTs were suspended in these samples at a concentration of 0.01 or 0.001% (w/v) by vigorous stirring, and these suspensions were settled at room temperature for 3 days. As a result, CNTs were aggregated and precipitated when DI water was saturated with either MAc or EAc (ESI Fig. S1†). In contrast, CNTs were well dispersed and precipitates were not observed when IPAc or PAc was used, mostly because of the lower solubility of these solvents in water. The absorption spectrum of the supernatant was also measured using a spectrophotometer (Nanodrop 2000C, Thermo Fisher, MA, USA). As shown in Fig. 2(e), strong UV absorption was observed when IPAc or PAc was used, because CNTs were well dispersed and precipitates were not formed. These results clearly demonstrated that the types of polar organic solvents critically affected the dispersibility of CNTs, which would be attributed to the difference in the amount of the dissolved organic solvent in the aqueous droplets. From these observations, we concluded that the dispersibility of CNTs in the shrinking droplets was decreased for MAc and EAc, and the solid shell structure was rapidly formed in the droplet and it was deformed at the earlier stage of droplet dissolution, resulting in the formation of significantly deformed microparticles (Fig. 2(f)). In contrast, the shell formation was not significant when IPAc or PAc was used, resulting in the formation of microparticles with a compact shape (Fig. 2(g)).
Size control of CNT particles
Next, we changed the initial concentration of CNTs for the purpose of controlling the size of the CNT particles. The size distributions of the obtained particles, when the initial CNT concentration was changed from 0.05% (w/v) to 1% (w/v) and when IPAc was used as the continuous phase, are shown in Fig. 3. The average diameters of the particles were 23, 24, 31, and 40 μm, respectively, with the corresponding CV values of 18, 8, 7, and 6%, for the initial CNT concentrations of 0.05, 0.1, 0.5, and 1% (w/v) suspensions, respectively. From this result, we confirmed that the particle diameters can be precisely controlled simply by changing the initial CNT concentration.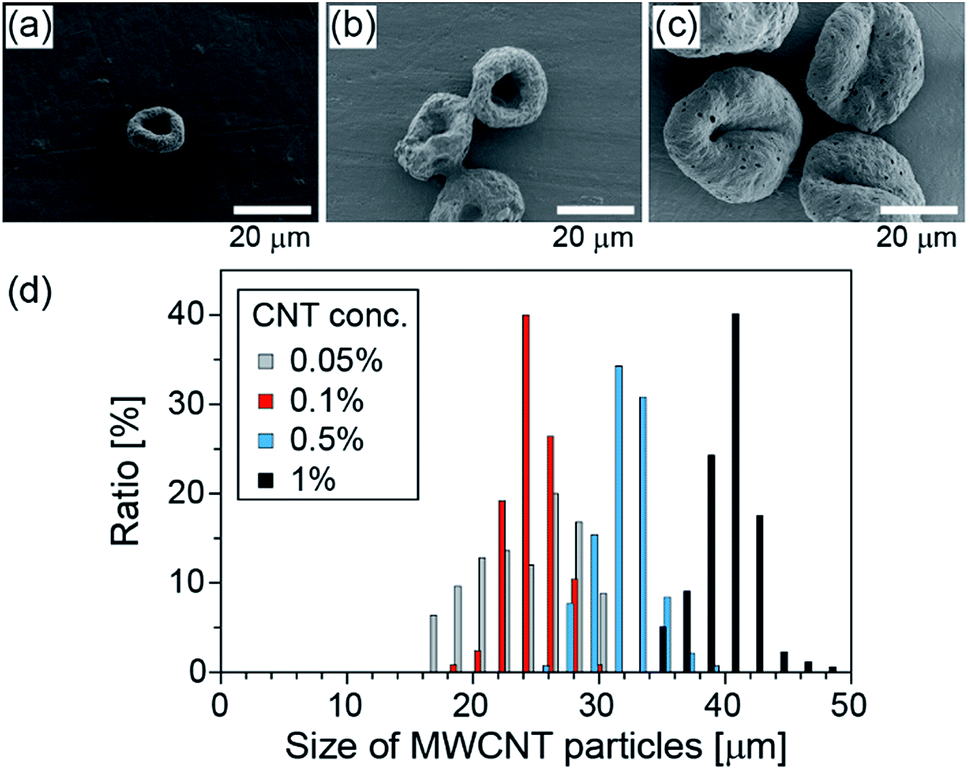 | ||
| Fig. 3 (a–c) SEM images of the MWCNT microparticles prepared under different CNT concentrations: (a) 0.05% (w/v), (b) 0.1% (w/v), and (c) 1% (w/v). (d) Size distributions of the MWCNT particles. IPAc was used as the continuous phase. | ||
Sedimentation behavior of CNT particles
We performed several experiments to characterize the physicochemical properties of the obtained CNT particles. First, we observed the sedimentation behavior of the particles, prepared by using microfluidic devices. When CNTs are chemically functionalized, or when catalyst nanoparticles are loaded on the CNTs, it is necessary to exchange the medium of the CNT dispersion for several times. However, it is not easy to condense CNTs dispersed in a liquid sample and remove the carrier medium.19 We expected that the presented CNT microparticles in a liquid sample would be precipitated much faster than native CNTs, because of their relatively large size. CNT particles with an average diameter of 31 μm were prepared using IPAc as the continuous phase and suspended in DI water. It is notable that the shape of the obtained particles was stable when they were suspended in DI water because a strong van der Waals force is present between closely packed CNTs, and the dispersion stabilizers in the initial CNT sample were removed during the production process. After shaking, we settled the plastic vial and measured the sedimentation speed of the CNT particles. As shown in Fig. 4(a), most CNT particles settled on the bottom of the vial within several minutes, whereas the CNTs remained suspended. Native CNTs did not completely settle even when centrifugation was performed at 6000g for 4 h (Fig. 4(b)). From these results, it was possible to efficiently separate CNT particles from the medium, indicating that the operability was improved, which potentially facilitates the chemical/physical functionalization of the CNTs composing the particles.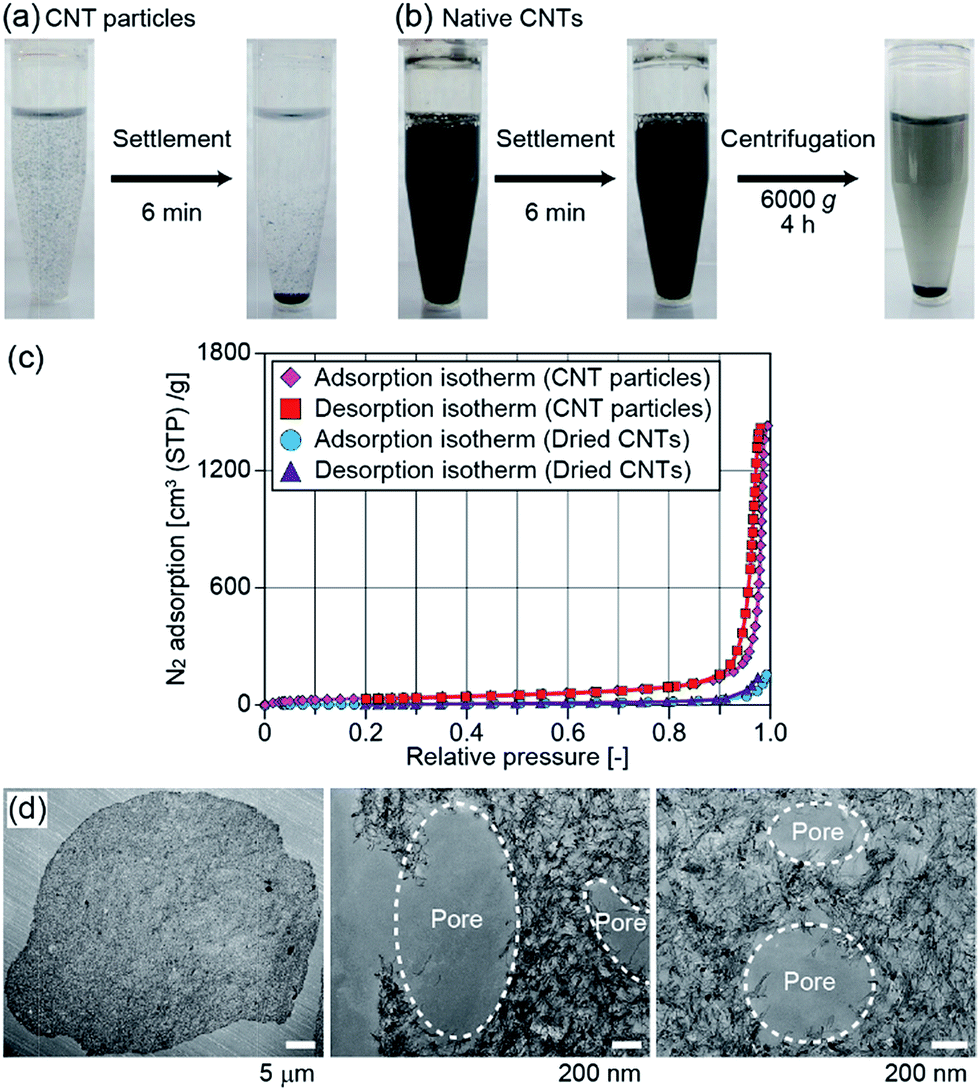 | ||
| Fig. 4 (a, b) Sedimentation of (a) CNT particles and (b) native CNTs in DI water. The CNT concentration was ∼0.15 g mL−1 for each condition. Most CNT particles settled on the bottom of the vial within ∼6 min, whereas a portion of native CNTs remained suspended even after centrifugation at 6000g for 4 h. The total amounts of CNTs were same for these conditions. In (a), CNT particles prepared by using microfluidic devices were used. (c) Adsorption and desorption isotherms of nitrogen of the dried CNT particles and dried CNTs. (d) TEM images of the cross-sections of CNT particles. Pores with sizes of several hundred nanometers were observed on the surface and within the cross section. In (c, d), CNT microparticles prepared by using membrane emulsification were used. | ||
Characterization of CNT particles prepared by membrane emulsification
The production throughput of CNT particles in the presented microfluidic system was not high (∼0.3 mg per particles per h). To further characterize the physicochemical properties of the particles, we employed another process, membrane emulsification, to achieve a larger-scale production. The particles prepared by membrane emulsification using IPAc as the continuous phase are shown in ESI Fig. S2(a).† The average diameter of the particles was 37 μm with a CV value of 17%. Although the size distribution of the obtained particles was larger than that of the microfluidic process (CV of 7%), and the variation of particle shape increased, the particles had a concave morphology as in the case of the microfluidic process (Fig. 2(c)). The production speed was dramatically increased (about 100-fold) compared to the microfluidic system, although the average particle sizes were not significantly different. We used these particles for further characterizations.First, we evaluated the specific surface area of the particles by measuring nitrogen adsorption. Both of the adsorption and desorption isotherms of nitrogen gas for the dried CNT particles are shown in Fig. 4(c). The specific surface area of the particles calculated by the BET method was 137 m2 g−1, and the average pore size was 63 nm. This value of surface area was comparable to but slightly lower than that of the native CNTs in the original dispersion (250–300 m2 g−1) because of the aggregation of CNTs in the particles. However, when the original CNTs in the purchased dispersion were washed with DI water and dried on an evaporating dish, the surface area was measured to be 24 m2 g−1 and the average pore size was 38 nm. This result indicated that the CNTs were randomly and uncontrollably packed when the CNT dispersion was simply dried, but the particle formation process in this study maintained the relatively large surface area.
To further investigate if CNTs are closely packed inside of the particles, we prepared thin sections and directly observed them by TEM. As a result, almost the entire cross-section was densely packed with CNTs (Fig. 4(d)). Assuming the cylindrical shape of CNTs with a diameter of ∼6 nm, we estimated the volume content of CNTs by analyzing the total number and length of CNTs per unit area (with a thickness of 75 nm); the occupancy of the CNTs in the particles was determined to be ∼15%. There were small pores (diameter of 400–1000 nm) not only on the surface but also inside of the particles (Fig. 4(d)). We assumed that these pores were generated by the phase separation in the dissolving droplets; organic solvent incorporated into the droplets probably formed small droplets just before completing the droplet dehydration, which created the void space within the particles.
When the prepared particles are employed as supports for catalysts, the gas or liquid flux through the accumulated particles is a critical factor determining reaction efficiency. Therefore, we prepared accumulated particle sheets on a filter paper (Φ = 17 mm, 2 mg of CNT particles per cm2) and examined the flow rate generated through the sheet. As control samples, we prepared a sheet of native CNTs (so called Buckypaper) and that of graphite. DI water was flown through the sheet by aspiration (Fig. S2(b)†). The formed sheets of the CNT particles, native CNTs, and graphite are shown in ESI Fig. S2(c).† The fluxes of DI water through the sheets were calculated to be 0.77, 0.008, and 0.22 mL min−1 for the sheets made of CNT particles, native CNTs, and graphite, respectively. The value of 0.77 mL min−1 was comparable to that of the non-particle control (only the filter paper), which was 1.1 mL min−1, showing the significantly low fluid resistance through the CNT particle accumulation, especially compared with the native CNTs. This low resistance would be due to the inter-particle spaces. From these results, we consider that the obtained CNT particles are potentially useful as the support for catalysts working in a continuous gas or liquid phase reaction process, in which conventional CNT accumulation is not satisfying.
Finally, the electrical conductivity of the film of the accumulated CNT particles was measured and the value was compared to that of the accumulated native CNTs. As a result, the conductivities were 146 Ω−1 m−1 and 4340 Ω−1 m−1 for CNT particles and CNTs, respectively. This result indicates that the conductivity of accumulated CNT particles was ∼1/30 of the accumulated native CNTs, because of the lower density of CNTs in the accumulated bed of the CNT particles and the lower contact efficiency between the particles. This value was slightly lower than but comparable to previously reported values of those of MWCNT films (600–2000 Ω−1 m−1),40,41 showing the potential applicability of the particles to, for example, catalyst-loading electrodes for fuel cells. Future studies would include the characterization of the anisotropic physical strength of the obtained particles.
Shape control of MWCNT particles by additives
From the above-mentioned experiments, it was clarified that the dispersibility of CNTs in the shrinking droplets was a critical factor dominating the shape of the CNT particles. Therefore, we next investigated whether the particle shapes were also affected by chemicals added to the CNT suspension, using microfluidic devices and IPAc. First, we employed two types of polymers, Na-Alg and PEG. It was expected that Na-Alg reduces the dispersibility of CNTs, whereas PEG enhances the dispersibility as it is frequently used as a dispersion stabilizer for CNTs.42 When particles were prepared using a 0.1% (w/v) MWCNT suspension with 0.1% (w/v) Na-Alg, we obtained CNT particles (average diameter of 23 μm, CV of 10%) exhibiting a non-spherical, deflated beach ball-like morphology (Fig. 5(a)) as in the case of using MAc without employing additives (Fig. 1(f)), mainly because the dispersibility of CNTs was reduced by the presence of the alginate polymer. While in the case of using PEG as the additive (0.1% (w/v)), we obtained nearly spherical particles with an average diameter of 15 μm (CV of 5%, Fig. 5(b)). Considering that the initial droplet sizes were almost the same for these conditions, it was assumed that CNTs were more closely packed in the particles when PEG was added. | ||
| Fig. 5 SEM images of the CNT particles prepared using several types of additives; (a) 0.1% (w/v) Na-Alg, (b) 0.1% (w/v) PEG, (c) 0.3% (w/v) Triton X-100, and (d) 0.3% (w/v) SDS were added to the aqueous CNT dispersion, respectively. The initial CNT concentrations were (a, b) 0.1% (w/v) and (c, d) 0.5% (w/v), respectively. | ||
In addition, we investigated the effect of surfactant molecules on the particle shape. We added Triton X-100 and SDS at a concentration of 0.3% (w/v), which are frequently used as the stabilizers in MWCNT dispersions.43–46 The initial CNT concentration was 0.5% (w/v). As a result, we obtained nearly spherical particles (Fig. 5(c and d)) when these surfactants were used. The average diameters of the particles were 35 μm (CV of 11%) and 25 μm (CV of 4%) for particles prepared using Triton X-100 and SDS, respectively, indicating that the density of CNTs in the particles prepared using Triton-X 100 was lower than that of using SDS. Furthermore, the particle surface was not smooth when Triton X-100 was used. These results can be explained by the lower ability of Triton X-100 to disperse CNTs in an aqueous media than that of SDS.43,45 These results indicate that we can possibly control not only the shape but also the density and the surface topology of the particles by using different types of additives in the dispersed phase.
Preparation of SWCNT particles
Finally we attempted to prepare particles made of SWCNTs. In general, SWCNTs have a larger surface area than MWCNTs, and they are more suitable as materials for adsorbents and catalyst supports.47,48 However, there are only a few reports on the preparation of SWCNT particles, probably because SWCNTs easily form aggregates compared to MWCNTs.23,31 We prepared particles using an aqueous SWCNT dispersion (0.1% (w/v)) and EAc or IPAc as the continuous phase. Fig. 6 shows SEM images of the obtained SWCNT particles. We were able to successfully generate microparticles made of SWCNTs. When EAc was used, the average particle diameter was 10 μm (CV of 11%), and the particles exhibited a non-spherical, paper ball-like shape. On the other hand, differently shaped, concaved particles were obtained with an average diameter of 8 μm (CV of 15%) when IPAc was used. These particle shapes were similar with those shown in the above mentioned experiments on the production of MWCNT particles, and hence, the dispersibility of SWCNTs was also considered to be the primary factor dominating the morphology for SWCNT particles. It is worth noting that the homogenizing process of SWCNTs just before preparing these particles was highly important; we obtained particles like those shown in Fig. 6(a) even though we used IPAc as the continuous phase, when the homogenization process was not employed. | ||
| Fig. 6 SEM images of the SWCNT microparticles prepared using (a) EAc and (b) IPAc as the continuous phases, respectively. | ||
Conclusions
We have successfully demonstrated a facile process of producing CNT microparticles via rapid assembly of CNTs in droplets in a non-equilibrium state. From the experimental results using different types of solvents and additives, we revealed that the dispersibility of CNTs in the droplets is the most critical factor dominating the morphology of the particles. Because the droplet dissolution rapidly progresses as we used polar organic solvents as the continuous phase, one-step production of CNT particles is possible without any needs for additional chemical crosslinking or removal of solid core particles. Two processes were demonstrated to produce particles, droplet microfluidics and membrane emulsification; the former is advantageous in terms of high controllability in size and the latter is suitable for a larger-scale particle production. The obtained particles were stable both in aqueous and non-aqueous medium, and had good physicochemical characteristics including relatively high specific surface area, low fluid resistance through the accumulated particle bed, and tunability in shape. In recent years, microparticles made of CNTs have drawn attention as new materials for catalyst supports,18 adsorbents for chemicals,33 and electrodes,49 and hence, the presented particles would also be applicable as functional materials for these purposes.Acknowledgements
This study was supported in part by KAKENHI (23106007 and 16H04571) from MEXT, Japan. We thank Mr Masayuki Tamba and Ms Eri Asano for technical assistance.References
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535 CrossRef CAS PubMed.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337 CrossRef CAS.
- Q. Liao, J. Sun and L. Gao, Colloids Surf., A, 2009, 345, 95 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797 RSC.
- K. Pyrzynska and M. Bystrzejewski, Colloids Surf., A, 2010, 362, 102 CrossRef CAS.
- V. K. Gupta, R. Kumar, A. Nayak, T. A. Saleh and M. A. Barakat, Adv. Colloid Interface Sci., 2013, 193, 24 CrossRef PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787 CrossRef CAS PubMed.
- J. Wang, Electroanalysis, 2005, 17, 7 CrossRef CAS.
- Y. Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. H. Lin, Electroanalysis, 2010, 22, 1027 CrossRef CAS.
- C. Li, E. T. Thostenson and T. W. Chou, Compos. Sci. Technol., 2008, 68, 1227 CrossRef CAS.
- P. Brochu and Q. B. Pei, Macromol. Rapid Commun., 2010, 31, 10 CrossRef CAS PubMed.
- H. S. Liu, C. J. Song, L. Zhang, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95 CrossRef CAS.
- M. Kumar and Y. Ando, J. Nanosci. Nanotechnol., 2010, 10, 3739 CrossRef CAS PubMed.
- L. S. Ying, M. A. B. M. Salleh, H. B. M. Yusoff, S. B. A. Rashid and J. B. Abd Razak, J. Ind. Eng. Chem., 2011, 17, 367 CrossRef CAS.
- Q. Zhang, J. Q. Huang, W. Z. Qian, Y. Y. Zhang and F. Wei, Small, 2013, 9, 1237 CrossRef CAS PubMed.
- X. C. Chen, Y. Q. Hou, H. Wang, Y. Cao and J. H. He, J. Phys. Chem. C, 2008, 112, 8172 CAS.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. P. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef PubMed.
- X. C. Chen, J. H. He, C. X. Yan and H. M. Tang, J. Phys. Chem. B, 2006, 110, 21684 CrossRef CAS PubMed.
- K. Nakagawa, H. Oda, A. Yamashita, M. Okamoto, Y. Sato, H. Gamo, M. Nishitani-Gamo, K. Ogawa and T. Ando, J. Mater. Sci., 2009, 44, 221 CrossRef CAS.
- S. Ko, Y. Takahashi, A. Sakoda, Y. Sakai and K. Komori, Langmuir, 2012, 28, 8760 CrossRef CAS PubMed.
- M. Sano, A. Kamino, J. Okamura and S. Shinkai, Nano Lett., 2002, 2, 531 CrossRef CAS.
- M. A. Correa-Duarte, A. Kosiorek, W. Kandulski, M. Giersig and L. M. Liz-Marzan, Chem. Mater., 2005, 17, 3268 CrossRef CAS.
- H. J. Jin, H. J. Choi, S. H. Yoon, S. J. Myung and S. E. Shim, Chem. Mater., 2005, 17, 4034 CrossRef CAS.
- B. Kim, Y. H. Lee, S. J. Han, J. H. Ryu and K. D. Suh, Colloids Surf., A, 2007, 298, 245 CrossRef CAS.
- A. M. Yashchenok, D. N. Bratashov, D. A. Gorin, M. V. Lomova, A. M. Pavlov, A. V. Sapelkin, B. S. Shim, G. B. Khomutov, N. A. Kotov, G. B. Sukhorukov, H. Mohwald and A. G. Skirtach, Adv. Funct. Mater., 2010, 20, 3136 CrossRef CAS.
- J. F. Feller, J. Lu, K. Zhang, B. Kumar, M. Castro, N. Gatt and H. J. Choi, J. Mater. Chem., 2011, 21, 4142 RSC.
- M. I. H. Panhuis and V. N. Paunov, Chem. Commun., 2005, 1726 RSC.
- H. X. Yi, H. H. Song and X. H. Chen, Langmuir, 2007, 23, 3199 CrossRef CAS PubMed.
- H. Xu, Z. Q. Tan, H. Abe and M. Naito, J. Ceram. Soc. Jpn., 2011, 119, 180 CrossRef CAS.
- D. Gueon and J. H. Moon, ACS Appl. Mater. Interfaces, 2015, 7, 20083 CAS.
- X. J. Cao, L. L. Zang, Z. P. Bu, L. G. Sun, D. C. Guo and C. Wang, J. Mater. Chem. A, 2016, 4, 10479 CAS.
- D. J. Han, J. H. Jung, J. S. Choi, Y. T. Kim and T. S. Seo, Lab Chip, 2013, 13, 4006 RSC.
- S. Sugaya, M. Yamada, A. Hori and M. Seki, Biomicrofluidics, 2013, 7, 054120 CrossRef PubMed.
- T. Ono, M. Yamada, Y. Suzuki, T. Taniguchi and M. Seki, RSC Adv., 2014, 4, 13557 RSC.
- M. Yamada, A. Hori, S. Sugaya, Y. Yajima, R. Utoh, M. Yamato and M. Seki, Lab Chip, 2015, 15, 3941 RSC.
- M. Mizuno, T. Toyota, M. Konishi, Y. Kageyama, M. Yamada and M. Seki, Langmuir, 2015, 31, 2334 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS PubMed.
- M. D. Rein, O. Breuer and H. D. Wagner, Compos. Sci. Technol., 2011, 71, 373 CrossRef CAS.
- L. Hussein, G. Urban and M. Krüger, Phys. Chem. Chem. Phys., 2011, 13, 5831 RSC.
- P. Zhang and D. B. Henthorn, AICHE J., 2010, 56, 1610 CrossRef CAS.
- J. Liu, A. G. Rinzler, H. J. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, Y. S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253 CrossRef CAS PubMed.
- L. Vaisman, H. D. Wagner and G. Marom, Adv. Colloid Interface Sci., 2006, 128, 37 CrossRef PubMed.
- M. A. Chappell, A. J. George, K. M. Dontsova, B. E. Porter, C. L. Price, P. H. Zhou, E. Morikawa, A. J. Kennedy and J. A. Steevens, Environ. Pollut., 2009, 157, 1081 CrossRef CAS PubMed.
- W. H. Duan, Q. Wang and F. Collins, Chem. Sci., 2011, 2, 1407 RSC.
- H. J. Dai, Acc. Chem. Res., 2002, 35, 1035 CrossRef CAS PubMed.
- S. Niyogi, M. A. Hamon, H. Hu, B. Zhao, P. Bhowmik, R. Sen, M. E. Itkis and R. C. Haddon, Acc. Chem. Res., 2002, 35, 1105 CrossRef CAS PubMed.
- J. H. Choi, C. L. Lee, K. S. Park, S. M. Jo, D. S. Lim and I. D. Kim, RSC Adv., 2014, 4, 16062 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental results and setups. See DOI: 10.1039/c7ra01846k |
| This journal is © The Royal Society of Chemistry 2017 |