
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The positive effect of water on photo-induced step transfer-addition & radical-termination (START) polymerization†
Tianchi Xu,
Lifen Zhang*,
Zhenping Cheng * and
Xiulin Zhu
* and
Xiulin Zhu
Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: chengzhenping@suda.edu.cn; zhanglifen@suda.edu.cn; Fax: +86-512-65882787
First published on 24th March 2017
Abstract
Step-growth radical copolymerization between α,ω-diiodoperfluoroalkanes (A) and α,ω-unconjugated dienes (B) proceeds efficiently through a photo-induced Step Transfer-Addition & Radical-Termination (START) strategy in aqueous/organic biphasic system. The addition of water in our polymerization strategy enhanced the overall polymerization efficiency and inhibited the function loss (C–I) significantly, which has been illustrated through UV-vis tests. Therefore, most of the functional groups (C–I) in the polymer chain end have been preserved in the final polymer product (AB)n based on 19F NMR analysis. After polymerization, we could erase the iodine atoms in the main chain of (AB)n, which generates semifluorinated polyolefins with enhanced thermal stability.
1. Introduction
Since the foundation of macromolecular engineering, olefins have always been an important kind of polymerizable monomer, and the corresponding polyolefin materials have been applied worldwide.1 The appearance of ionic polymerization realized the living polymerization of not only olefin monomers (e.g., ethylene, propylene), but also active conjugated ones (e.g., methyl methacrylate (MMA) and styrene (St)).2 Although the ionic polymerization strategy is a powerful polymerization tool, it is highly sensitive to impurities existing in the polymerization system. Contrasted to ionic ones, the polymerization conditions in a radical process were rather mild, which greatly eased the practical operation difficulties. Recently, reversible deactivation radical polymerizations (RDRPs) have progressed greatly, such as initiator-transfer agent-terminator (iniferter) polymerization,3 nitroxide-mediated radical polymerization (NMP),4 atom transfer radical polymerization (ATRP),5 reversible addition–fragmentation chain-transfer (RAFT),6 and iodine transfer polymerization (ITP).7 However, most RDRPs were dealt with active conjugated monomers, which were ineffective for unconjugated ones, such as 1,7-octadiene. Considering both the advantages of radical polymerization process, and the huge application values of polyolefin materials,8 it is of high demand to realize the controlled radical polymerization of unconjugated olefin monomers.As is well known to all, functional polymers with well-defined microstructure were widely investigated in high-tech areas,9 such as self-assembly,10 single-chain folding,11 and drug delivery.12 Fluorine-containing polymers were also one kind of such functional polymers.13 The incorporation of fluorine could endow polymers with superior material performances, such as high thermal stability, chemical inertness, and so on.14 That's also the reason why so many scientists devoted themselves to the fluorine chemistry in spite of investigation difficulties.15 Semifluorinated hydrocarbons (F(CF2)m(CH2)nH) were the simplest self-assembly molecules, widely used as surfactants.16 Recently, fluorocarbon/hydrocarbon diblock materials, (CnF2n+1CH2)(Cm−2H2m−3)CH–CH(CnF2n+1CH2)(Cm−2H2m−3) (abbreviated as di(FnHm)), have been synthesized, the arrangement of which was in the manner of supramolecular organization.17 Traced back to the literature, through the utilization of α,ω-diiodoperfluoroalkanes and α,ω-unconjugated dienes, fluorocarbon–hydrocarbon microblocks have been polymerized through free radical mechanism.18 After the polymerization, iodine atoms in the polymer chain could be erased in high efficiency for the generation of fluorocarbon–hydrocarbon microblocks, which could be deemed as the primitive sequence-controlled polymers.19 The merits of the well-ordered semifluorinated polyethylenes mentioned above deserved our further exploration.
Recently, we have successfully utilized photo-mediated atom transfer radical addition (ATRA) reaction for the construction of perfluorocarbon-containing alternating copolymers (AB)n under irradiation of blue LED at room temperature (25 °C).20 At the same time, the newly-developed polymerization method could also be deemed as a new sequence-controlled radical polymerization strategy for α,ω-unconjugated dienes. In most photo-mediated RDRPs involved with iodine-containing agent, loss of functional groups (C–I) in the polymer chain end was common with the generation of I2, and inhibited the further proceeding of the polymerization.21 Similar circumstance has also occurred in our polymerization system due to the presence of methanol (MeOH). So we tried to suppress the functional loss (C–I) to ensure the smooth proceeding of our polymerization strategy in this work.
Water has long been a popular solvent for free radical polymerization, especially of fluorinated monomers.22 The polar solvent stabilized the transition state of perfluoroalkyl radicals in the reaction process, considering their high electrophilicity.23 Dolbier' group first reported the positive effect of water on the radical addition reaction between alkenes (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and fluorinated primary alkyl radicals (RF˙).24 So we speculated water also had some similar positive effect on our polymerization strategy. Herein, we carried out the copolymerization between α,ω-diiodoperfluoroalkanes A and α,ω-unconjugated dienes B in aqueous/organic biphasic system for the preparation of (AB)n. Based on the analysis of UV-vis tests, the whole polymerization proceeded smoothly with no generation of I2. Thus, the presence of water could provide a strong polar environment and promote the proceeding of our polymerization strategy, consistent with the literature report. What's more, functional group (C–I) in the polymer chain end has been highly preserved through 19F NMR analysis, which was a great progress compared with our previous report. After the polymerization, we erased the iodine atoms in the main chain of (AB)n, generating semifluorinated polyethylenes with enhanced thermal stability.
C) and fluorinated primary alkyl radicals (RF˙).24 So we speculated water also had some similar positive effect on our polymerization strategy. Herein, we carried out the copolymerization between α,ω-diiodoperfluoroalkanes A and α,ω-unconjugated dienes B in aqueous/organic biphasic system for the preparation of (AB)n. Based on the analysis of UV-vis tests, the whole polymerization proceeded smoothly with no generation of I2. Thus, the presence of water could provide a strong polar environment and promote the proceeding of our polymerization strategy, consistent with the literature report. What's more, functional group (C–I) in the polymer chain end has been highly preserved through 19F NMR analysis, which was a great progress compared with our previous report. After the polymerization, we erased the iodine atoms in the main chain of (AB)n, generating semifluorinated polyethylenes with enhanced thermal stability.
2. Experimental section
2.1. Materials
Diallyl adipate (TCl, 98%) and diallyl 1,4-cyclohexanedicarboxylate (cis- and trans-mixture) (TCl, 98%), 1,6-diiodoperfluorohexane (TCl, 98%), 1,4-diiodoperflourobutane (TCl, 98%), 1,8-diiodoperfluorooctane (Sigma-Aldrich, 98%) and Ru(bpy)3Cl2 (Adamas-beta, +99%) were used as received and stored in light-resistant container at 0 °C. 1,7-Octadiene (Alfa Aesar, >97%) was passed through a neutral alumina column for the removal of inhibitor and stored in light-resistant container at 0 °C. L(+)-Ascorbic acid sodium salt (AsAc-Na) (Wako, +99%) and tetrabutylammonium iodide (TBAI) (J&K, 99%) were used as received and stored in desiccator for a dry environment. 1,4-Bis(bromomethyl) benzene (Apollo Scientific, 98%), and allyl bromide (J&K, 98%) were used as received. Deionized water was purchased from Hangzhou Wahaha Co. Ltd. and used as received. All the regular solvents (Analytical reagent) were used directly without any further treatment.2.2. Methods
Both the number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) of all the polymers were determined by TOSOH HLC-8320 gel permeation chromatograph (GPC) equipped with a refractive-index detector (Waters 2414), using TSK gel guardcolumn Super AWM-H columns (4.6 mm I.D. × 15 cm × 2) with measurable molecular weights ranging from 1 × 103 to 10 × 105 g mol−1. THF was used as the eluent at a flow rate of 0.35 mL min−1 at 40 °C. All the GPC samples were injected using a TOSOH plus autosampler and calibrated with PMMA standards purchased from TOSOH. 1H NMR and 13C NMR spectra of 1,4-bis(allyloxy)benzene (B1) were recorded on a Bruker 300 MHz nuclear magnetic resonance (NMR) instrument using CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard at room temperature (25 °C). All the 1H NMR, 19F NMR and 13C NMR data of the semifluorinated polyolefins were recorded on a Brucker 600 MHz Advance III instrument using CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard for 1H and 13C chemical shifts at room temperature (25 °C). The sampling for the in situ NMR spectra was as follows: After the predetermined period of time, the ampoule was opened, then 100 μL mixture was sucked to the NMR tube and dissolved with 0.5 mL of CDCl3 for the NMR test instantly. Thermogravimetric analysis (TGA) was carried out on a TG/DTA 6300 Instruments with a heating rate of 10 °C min−1 from the room temperature to 650 °C under nitrogen atmosphere. Ultraviolet-visible (UV-vis) absorption spectra of the samples were recorded on a Shimadzu UV-2600 spectrophotometer at room temperature.2.3. Reaction appratus
The photoredox polymerizations were carried out at room temperature under visible light irradiation provided by blue LED Flex strip (λmax = 458 nm, 0.85 mW cm−2) which was fixed around the reaction vessel. The polymerization temperature (25 °C) was maintained by control of fanning.2.4. Synthesis and characterization of α,ω-unconjugated diene 1,4-bis(allyloxy)benzene (B1)
In an inert atmosphere, magnesium chips (2.4 g, 0.1 mol) was added to the solution of Et2O (75 mL) with continuing stirring, then a solution of 3-bromopropene (12.2 g, 0.1 mol) in Et2O (75 mL). The whole mixture was refluxed for 2 hours at room temperature with careful exclusion of light. Then 1,4-bis(bromomethyl) benzene (12.2 g, 0.1 mol) was dissolved in THF (50 mL), which was slowly dropped to the mixture. After the completion of dropping, the mixture was refluxed for another 4 hours, then hydrolyzed by water (15 mL) and aq. NH4Cl (25 mL). The organic layer was separated, dried by sodium sulfate. Then it was vacuum filtered to remove the sodium sulfate and the solvent was removed by rotary evaporation. The crude product was purified by means of column chromatography (silica gel; EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:20) to obtain a colorless liquid product (3.5 g, 0.02 mol, 64.0%). 1H NMR (CDCl3, 300 MHz), δ ppm: 7.09 (s, 4H, –C6H4–), 5.86 (m, 2H, –CH2–CHCH2), 5.01 (m, 4H, –CH2–CHCH2), 2.67 (t, 4H, –C6H4–CH2–CH2–), 2.36 (m, 4H, –C6H4–CH2–CH2–). 13C NMR (CDCl3, 300 MHz), δ ppm: 139.4 (s, –C6H4–), 138.3 (s, CH2CH–), 129.3 (s, –C6H4–), 114.9 (s, CH2CH–), 35.6 (s, C6H4–CH2–CH2), 35.0 (s, C6H4–CH2–CH2).
:hexane = 1:20) to obtain a colorless liquid product (3.5 g, 0.02 mol, 64.0%). 1H NMR (CDCl3, 300 MHz), δ ppm: 7.09 (s, 4H, –C6H4–), 5.86 (m, 2H, –CH2–CHCH2), 5.01 (m, 4H, –CH2–CHCH2), 2.67 (t, 4H, –C6H4–CH2–CH2–), 2.36 (m, 4H, –C6H4–CH2–CH2–). 13C NMR (CDCl3, 300 MHz), δ ppm: 139.4 (s, –C6H4–), 138.3 (s, CH2CH–), 129.3 (s, –C6H4–), 114.9 (s, CH2CH–), 35.6 (s, C6H4–CH2–CH2), 35.0 (s, C6H4–CH2–CH2).
2.5. General procedure for the polymerization
A typical polymerization procedure with the molar ratio of [A]0:[B]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5 was described as follows. Mixture of α,ω-diiodoperfluoroalkane (A) (0.5 mmol), α,ω-unconjugated diene (B), Ru(bpy)3Cl2 and AsAc-Na were added to a dried ampoule with a stir bar. Then 7.0 mL of solution mixture of 1,4-dioxane, MeOH and water with the feed ratio of 3:1:3 (v/v/v) was added for the thorough dissolution of the mixture. The reaction mixture was degassed by at least four freeze–pump–thaw cycles to totally eliminate the dissolved oxygen, and then the ampoule was flame-sealed and placed in the presence of blue LED light with stirring. The whole system was fanned for the maintenance of temperature 25 °C. After the predetermined period of time, the ampoule was opened and then the mixture was precipitated into a large amount of methanol (250 mL) to remove the unreacted monomer and Ru(bpy)3Cl2. The polymers obtained by filtration were dried under vacuum at 35 °C overnight until a constant weight. The polymer yield was determined gravimetrically.
2.6. General post-polymerization modification process for the perfluorocarbon-containing alternating copolymers (A2B3)n
General procedure for the reduction of the iodine atom along the polymer chain (Fig. S3(a) in ESI†): a dried 10 mL ampoule was equipped with a magnetic stir bar and was charged with 0.02 mmol of macromolecule P1 (the structure shown in Fig. S3 in ESI†), 0.2 mmol of AIBN and 6.0 mmol of Bu3SnH. Then 5.0 mL of toluene was added. The reaction mixture was degassed by at least four freeze–pump–thaw cycles to totally eliminate the dissolved oxygen, and then the ampoule was flame-sealed and placed in oil bath with the temperature at 70 °C. After the predetermined period of time, the ampoule was opened and then the mixture was precipitated into a large amount of methanol (250 mL) to remove the unreacted components and solvent. The polymers obtained by filtration were dried under vacuum at 35 °C overnight until a constant weight. The polymer yield was determined gravimetrically. And the polymer generated P2 (the structure shown in Fig. S3 in ESI†) was examined thoroughly by NMR (1H and 19F) and GPC tests.3. Results and discussion
The initial construction of photo-induced Step Transfer-Addition & Radical-Termination (START) strategy was carried out in the mixed solvent system (1,4-dioxane/MeOH). MeOH was essential for the thorough dissolution of the catalysis system and ensured the polymerization proceeding. However, with a deep insight into the polymerization mechanism, we came to realise MeOH caused chain transfer reaction in the polymerization system.25 As a result, part of the functional group (C–I) has been lost with the generation of I2. Considering the positive effect of water on the radical addition reaction between alkenes (CC) and fluoroalkyl radicals (RF˙), we carried out the polymerization in aqueous/organic biphasic system in this work. According to Table 1, the addition of 3.0 mL water (entry 7, Table 1) could generate (AB)n with both relative high polymer yield and molecular weight (Mn,GPC). Further increase of water volume (entry 8, Table 1) would only reduce the total polymerization efficiency. The high tolerance of the whole polymerization process to water also well illustrated the radical character of the polymerization mechanism.
| Entry | Water added (mL) | Time (h) | Conv. (%) | Mn,GPC (g mol−1) | Mw/Mn |
|---|---|---|---|---|---|
| a Polymerization conditions: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5, n(B3) = 0.5 mmol, V(1,4-dioxane):V(MeOH) = 3:1, V(organic solvent) = 4.0 mL, time = 24 h, irradiation under blue LED at room temperature. |
|||||
| 1 | 0 | 24 | 70.9 | 6200 | 1.65 |
| 2 | 0.05 | 24 | 67.9 | 6200 | 1.71 |
| 3 | 0.1 | 24 | 70.5 | 6200 | 1.71 |
| 4 | 0.5 | 12 | 57.3 | 9500 | 2.10 |
| 5 | 1.0 | 12 | 62.5 | 14900 |
2.11 |
| 6 | 2.0 | 12 | 72.6 | 10500 |
1.85 |
| 7 | 3.0 | 12 | 81.5 | 9200 | 1.85 |
| 8 | 4.0 | 12 | 74.3 | 7600 | 1.76 |
Thorough comparison of the polymerization behaviour in different conditions (pure organic solvent system and aqueous/organic biphasic one) through UV-vis tests have been presented in Fig. 1. Based on the comparison of curves A, B, and C in Fig. 1(a), the absorption peak around 365.0 nm was attributed to I2, which was generated during the polymerization process in pure organic solvent system. When the polymerization was carried out in aqueous/organic biphasic system, the characteristic signal for I2 didn't appear throughout the polymerization process (Fig. 1(b)). Combined with the literature report, the aqueous/organic biphasic system created a strong polar environment to stabilize the transition state of perfluoroalkyl radicals (RF˙) in the polymerization system, which preferred to react with alkene (CC) rather than chain transfer from MeOH. As a result, the loss of functional group (C–I) in the polymerization system has been greatly reduced with no generation of I2 in the presence of water.
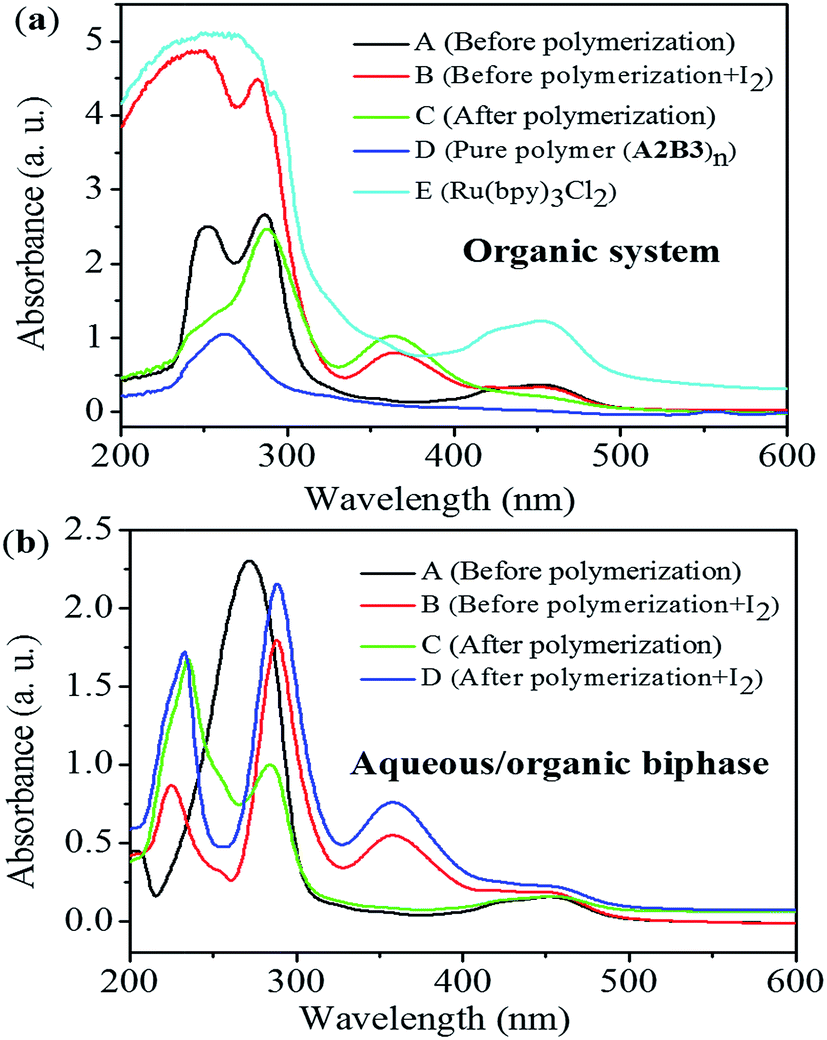 | ||
| Fig. 1 UV-vis absorption spectra of polymerization systems carried out in different conditions. (a) Polymerization conditions: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5, n(B3) = 0.5 mmol, V(1,4-dioxane):V(MeOH) = 3:1, V(total solvent) = 4.0 mL, irradiation under blue LED at room temperature. Polymer sample (A2B3)n (Mn,GPC = 7400 g mol−1; Mw/Mn = 1.71) (CA, CB = 1.2 mmol L−1, CC = 0.2 mmol L−1, CD, CE = 0.04 mmol L−1); (b) polymerization conditions: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5, n(B3) = 0.5 mmol, V(1,4-dioxane):V(MeOH):V(water) = 3:1:3, V(total solvent) = 7.0 mL, irradiation under blue LED at room temperature (CA, CB = 1.2 mmol L−1, CC, CD = 0.2 mmol L−1). | ||
After the successful construction of our polymerization system in aqueous/organic biphasic system, we wondered whether we could further enhance the polymerization efficiency through addition of phase transfer catalyst (PTC). It is well known that 15-crown-5 was a high efficient PTC for the complexation with Na+.26 However the addition of 15-crown-5 (entry 1, Table S1 in ESI†) did not enhance the reducing catalysis function of AsAc-Na. Although tetrabutyl ammonium bromide (TBABr) was another common PTC in chemical reaction, the addition of TBABr didn't have positive effect on the whole polymerization process, either (entry 2, Table S1 in ESI†). Some other attempts (entries 3 and 4, Table S1 in ESI†) to convert the polymerization process into emulsion polymerization also failed. These failed trials meant we could only utilize the positive polar effect of water on our polymerization system and realize the polymerization in aqueous/organic biphasic system, rather than as emulsion polymerization.
As the polymerization was carried out in the presence of visible light using Ru(bpy)3Cl2 as the photocatalyst with the aid of AsAc-Na, it was very necessary to evaluate the roles of the three components (light, Ru(bpy)3Cl2, and AsAc-Na) played in the polymerization system. No polymers were generated in the absence of light and Ru(bpy)3Cl2 (entries 8 and 9, Table 2), which elaborated the intrinsic features of photo-redox catalysis mechanism. In addition, in the absence of AsAc-Na (entry 10, Table 2), no polymers were generated, which perfectly manifested the photo-reducing function of AsAc-Na for Ru(bpy)3Cl2. The amount of metal catalyst could be reduced to 0.5 mol% compared to the amount of monomer (entries 1 to 3, Table 2), on the premise of both high polymer yield and molecular weight. Based on the comparison of entries 1, 4, 5 in Table 2, 0.5 equiv. of AsAc-Na was essential for the continuous reducing catalysis of the photocatalyst Ru(bpy)3Cl2. To further prove the intrinsic character of the polymerization process, we also carried out the polymerization with unequal monomer feed ratio. No matter the feed ratio of A2 and B3 was fixed as 1:1.2 or 0.8:1 (entries 6 and 7, Table 2), polymers with only low yield could be generated. That is to say, once all the C–I bonds in A2 were initiated in the polymerization process, the presence of unequal fed B3 could terminate the “living” character of the whole polymerization process instantly and only oligomers would be generated. When only one kind of monomer was added, no polymers were generated (entries 13 and 14, Table 2), which precluded the possibility of radical coupling between A and chain growth between B. Based on the discussion above, we could conclude the polymerization process was catalyzed by the photocatalyst Ru(bpy)3Cl2 with the aid of AsAc-Na and controlled by visible light, consistent with our previous report.
| Entry | Feed ratio | Time (h) | Conv. (%) | Mn,GPC (g mol−1) | Mw/Mn |
|---|---|---|---|---|---|
| a Polymerization conditions: the feed ratio was fixed as: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0, V(1,4-dioxane):V(MeOH):V(water) = 3:1:3, V(total solvent) = 7.0 mL, n(B3) = 0.5 mmol, irradiation under blue LED at room temperature. n(A2) = 0.5 mmol.b Polymerization conditions: the feed ratio was fixed as: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0, V(1,4-dioxane):V(MeOH):V(water) = 3:1:3, V(total solvent) = 7.0 mL, n(B3) = 0.5 mmol, irradiation under blue LED at room temperature. In dark (no irradiation).c Not determined. |
|||||
| 1 | 1:1:0.02:0.5 |
12 | 73.1 | 8900 | 1.72 |
| 2 | 1:1:0.01:0.5 |
12 | 65.8 | 8400 | 1.76 |
| 3 | 1:1:0.005:0.5 |
12 | 73.7 | 8400 | 1.77 |
| 4 | 1:1:0.02:0.3 |
12 | 74.1 | 6700 | 1.59 |
| 5 | 1:1:0.02:0.1 |
12 | 51.1 | 5100 | 1.46 |
| 6a | 1:1.2:0.02:0.5 |
12 | 29.3 | 4500 | 1.28 |
| 7 | 0.8:1:0.02:0.5 |
12 | 44.0 | 6000 | 1.36 |
| 8b | 1:1:0.2:0.5 |
36 | 0 | —c | —c |
| 9 | 1:1:0:0.5 |
36 | 0 | —c | —c |
| 10 | 1:1:0.2:0 |
36 | 0 | —c | —c |
| 11 | 1:1:0.02:0.5 |
6 | 71.2 | 6800 | 1.51 |
| 12 | 1:1:0.02:0.5 |
8 | 73.1 | 8800 | 1.72 |
| 13 | 1:0:0.02:0.5 |
36 | 0 | —c | —c |
| 14 | 0:1:0.02:0.5 |
36 | 0 | —c | —c |
In order to further estimate the stability of the newly-generated C–I bonds in our polymerization system, we assumed the new C–I bond was active,27 and utilized (A2B3)n (Mn,GPC = 6200 g mol−1, Mw/Mn = 1.72, Mn,NMR = 5900 g mol−1) as the macromolecular initiator. The macromolecular initiator (A2B3)n was chain ended by –CF2H, detailed structure analysis of which were provided in Fig. S2 in ESI.† The polymerization for MMA was carried out under irradiation of blue LED at room temperature through the utilization of catalysis system Ru(bpy)3Cl2/AsAc-Na. No polymers were generated (entries 1 to 4, Table 3), even when we prolonged the polymerization process to 72 h. On the contrary, when we utilized IC6F12I (A2) as initiator for the polymerization of MMA in the same conditions, PMMA with high molecular weight (Mn,GPC = 32300 g mol−1, Mw/Mn = 1.30) was successfully generated in 24 h (entry 5, Table 3). The results well illustrated the significant activity difference between the original C–I bonds in A2 and the newly-generated ones along the polymer chain. The inertness of the newly-generated C–I bonds in our catalysis system was the prerequisite for the step growth polymerization strategy with no branching or chain growth.
| Entry | Feed ratio | Time (h) | Conv. (%) | Mn,GPC (g mol−1) | Mw/Mn |
|---|---|---|---|---|---|
| a Polymerization conditions: feed ratio was fixed as: [MMA]0:[initiator]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0, irradiation under blue LED at room temperature. VMMA = 1.0 mL. All the initiator applied was (A2B3)n (Mn,GPC = 6200 g mol−1, Mw/Mn = 1.72, Mn,NMR = 5900 g mol−1) and V(1,4-dioxane):V(MeOH) = 3:1, V(total solvent) = 2.0 mL, except for entry 5, where the initiator applied was IC6F12I (A2), bulk polymerization. The structure analysis for macromolecular initiator (A2B3)n were provided in Fig. S2 in ESI.b Not determined. |
|||||
| 1 | 500:1:0.02:0 |
12 | 0 | —b | —b |
| 2 | 500:1:0.02:0 |
72 | 0 | —b | —b |
| 3 | 500:1:0.02:5 |
12 | 0 | —b | —b |
| 4 | 500:1:0.02:5 |
72 | 0 | —b | —b |
| 5 | 500:1:0.01:1 |
24 | 74.0 | 32300 |
1.30 |
With the inertness of newly-generated C–I bonds guaranteed, polymerization kinetics experiments have also been carried out to further investigate the polymerization behaviors. As could be seen from Fig. 2(a), the whole polymerization corresponded with step-growth radical process.28 By the efficient catalysis of the photo-redox agent Ru(bpy)3Cl2, most of the C–I bonds in monomer A2 were opened up and the reaction was initiated, thus oligomers with relatively high yield were generated quickly (∼10 min). Then the molecular weights of the oligomers were doubled or tripled through further addition between functional groups (Fig. 2(b) and (c)). The resultant polymers were alternating copolymers (A2B3)n with no branching, which has been discussed vide infra.
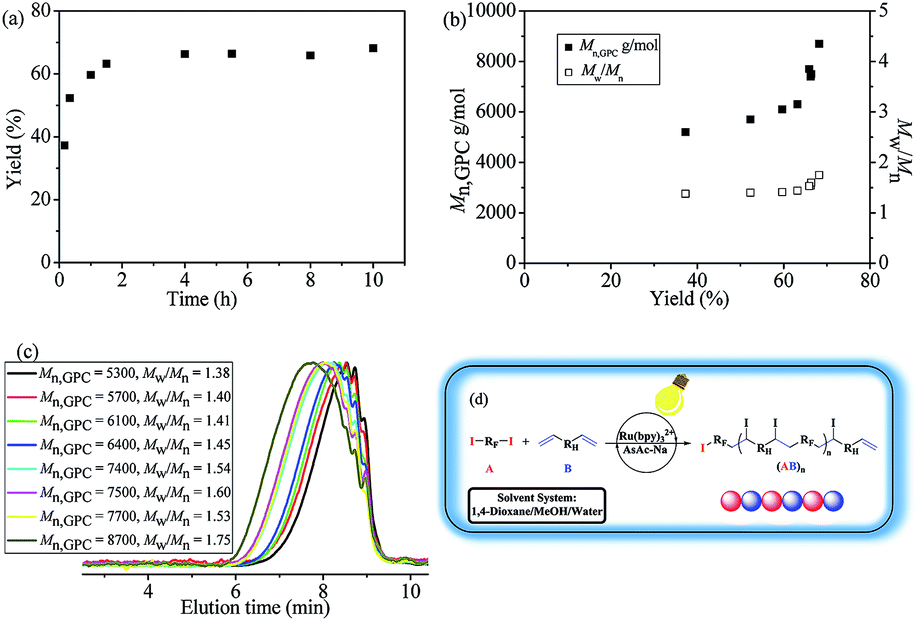 | ||
| Fig. 2 Polymerization kinetics analysis for the polymerization of A2 and B3 catalyzed by Ru(bpy)3Cl2/AsAc-Na in aqueous/organic biphase under irradiation of blue LED at room temperature. (a) Time-monomer conversion curve; (b) number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus monomer conversion; (c) corresponding gel permeation chromatogram (GPC) curves; (d) proposed polymerization pathway. Polymerization conditions: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5, n(B) = 0.5 mmol, V(1,4-dioxane):V(MeOH):V(water) = 3:1:3, V(total solvent) = 7.0 mL, irradiation under blue LED light at room temperature. | ||
Due to the specificity of the resultant polymer structure, only based on the combined analysis of both 1H NMR (Fig. 3(a)) and 19F NMR (Fig. 3(b)) spectra could exact structure identification be available. The integration ratio between the peak area of CH2CH– bond (a + b) and CH2CH– (c) was exactly 2:1, which perfectly elucidated the existence of CC bond in the polymer chain end and could be deemed as the characteristic peak for the calculation of the polymerization degree. When we set the integration of signal c as 1.0 for the estimation of the molecular weight, the corresponding integration of signal (a + b) was 2.0. The triplet of triplets centered at 6.1 was the characteristic peak assigned to HCF2CF2 (j) caused by chain transfer from MeOH, the generation of which was unavoidable due to the presence of MeOH in the polymerization system. The integration of signal –CH2CH(I)CH2 (h) was 16.4. And once a pair of monomer A2 and B3 was polyadded, a new C–I bond would be generated, which corresponded to the “n − 1” law. So the total incorporation number of monomer was 17.4, generating an alternating copolymer (A2B3)n. The polymerization degree n of the resultant (A2B3)n was 8.7 for the time being as the molecular weight calculation based on 1H NMR was only the average one. If we calculate the degree of polymerization based on the integration of the signal i + i′ according to eqn (1), the same results could be obtained, which also manifested that all the newly generated C–I bond was stable enough along the polymer chain in the present catalysis condition and the repeat unit was in the middle of (A2B3)n.
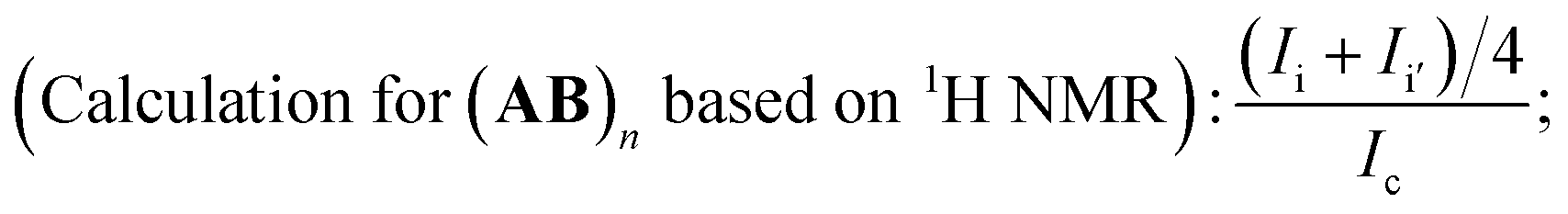 | (1) |
 | (2) |
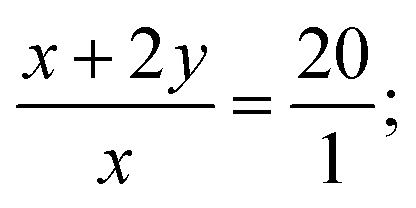 | (3) |
| x + y = 1. | (4) |
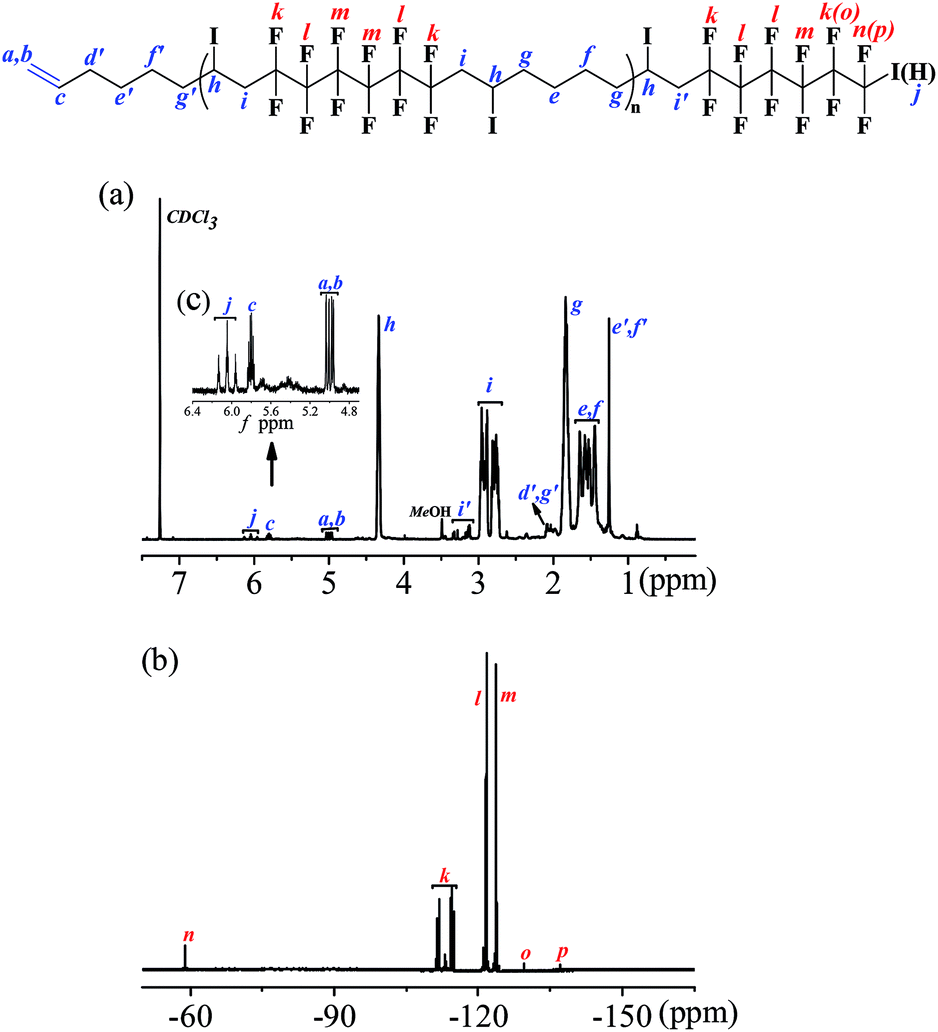 | ||
| Fig. 3 1H and 19F NMR spectra of the resultant alternating copolymer (A2B3)n in CDCl3. (a) 1H NMR spectrum of the resultant alternating copolymer (A2B3)n. (b) 19F NMR spectrum of the resultant alternating copolymer (A2B3)n. (c) Magnification of 1H NMR spectrum from 4.70 ppm to 6.40 ppm (inserted in (b)). Sample: Mn,GPC = 6100 g mol−1, Mw/Mn = 1.41, yield% = 59.7; the alternating copolymer (A2B3)n was obtained through the polymerization process below: [A2]0:[B3]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5, n(B3) = 0.5 mmol, V(1,4-dioxane):V(MeOH):V(water) = 3:1:3, V(total solvent) = 7.0 mL, time = 1 h, irradiation under blue LED at room temperature. | ||
As for the perfluorocarbon part of the polymers, analysis of the 19F NMR was necessary. As mentioned above, the chemical shift at −60.8 ppm was attributed to ICF2CF2– (n) in the polymer chain end, part of which has underwent chain transfer reaction and the corresponding chemical shift HCF2CF2– (p) has been shifted to the high field (δ = −137.5 ppm). So there existed two different perfluorocarbon chain end, ICF2CF2–, and HCF2CF2–. If we set the integration of HCF2CF2– (p) as 2.0, the corresponding chemical shifts for HCF2CF2– (o), and ICF2CF2– (n) were 2.0 and 18.3, respectively. According to eqn (2), the exact incorporation number of A2 was 8.1, consulted from the 19F NMR spectrum of A2 (Fig. S4 in ESI†). As mentioned above, when only one kind of monomer (A or B) was added into the polymerization system (entries 13 and 14, Table 2), no polymers could be generated. So the final polymers were consisted of (A2B3)n1, B3(A2B3)n2 and (A2B3)n3A2 (n1 was not necessarily equal to n2 or n3). There existed three possibilities for the functional groups in the polymer chain end, CH2CH–, HCF2CF2–, and ICF2CF2–, and the ratio was 1:2:18. So the corresponding ratio between the two different chain end, A and B, was 20:1, no matter A was in the form of HCF2CF2–, or ICF2CF2–. Considering the low content of B in the polymer chain end, we assumed there only existed (A2B3)n1 and (A2B3)n3A2 to simplify the calculation process. According to eqn (3) and (4), we could know the molar percent of (A2B3)n1, and (A2B3)n3 A2 were 9.5% and 90.5%, respectively. The low molar percent of (A2B3)n1 in the final polymer matched well with our assumption above. And the final polymer was constructed in strict alternating manner of A and B. What's more, most part of the functional group (C–I) in the polymer chain end has been preserved after the polymerization based on the analysis of 19F NMR spectrum, which was a great progress compared with our previous investigation. So the alternating copolymer (AB)n generated here was potential macromolecular initiator for the construction of more complex functional polymers, which are under investigation in our laboratory.
In most cases, the polymerization was terminated with the final polymer yield up to 80.0%, we wondered what remained in the precipitant (MeOH). So we studied one polymer sample thoroughly through NMR (1H and 19F) tests (Fig. S2 in ESI†), including in situ NMR for the calculation of function consumption (C–I polymerization conditions: feed ratio was fixed as: [MMA]0:[initiator]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0, irradiation under blue LED at room temperature. VMMA = 1.0 mL, and CC (B)), polymer structure analysis, and residue analysis. In order to ensure the homogeneity of sampling for in situ NMR experiment, we carried out the polymerization in organic solvent system (1,4-dioxane/MeOH). Based on the analysis of in situ NMR spectra ((1H (Fig. S2(a) in ESI†), and 19F (Fig. S2(b) in ESI†)), we could know all the functional groups in the polymerization system have been consumed up when the polymerization was terminated. After the completion of the polymerization, we also analyzed the purified polymer sample (A2B3)n. As there existed no signals for CH2CH– in 1H NMR (Fig. S2(c) in ESI†), the polymer was chain-ended by HCF2− in both ends. We could firstly calculate the exact incorporation number of A2 based on 19F NMR. The chemical shifts i, and j in 19F NMR (Fig. S2(d) in ESI†) were attributed to HCF2– in the polymer chain end and could be deemed as the characteristic signals. If we set the integration of chemical shift of j as 1.0, the corresponding integration for chemical shift g was 23.9. Based on eqn (5), the exact number of A2 incorporated into the polymer chain was 10.0, which meant the polymer sample was in the form of (A2B3)n A2 (n = 9). As for the analysis of 1H NMR spectrum (Fig. S2(c) in ESI†), the chemical shift a (HCF2–) could be deemed as the characteristic signal for the calculation of the polymerization degree, and we set the integration of a as 2.0. Then the integration for signal –CH2CH(I)CH2– (c) was 17.3. Based on eqn (6), the total incorporation number of monomer was 18.3, the analysis result of which corresponded with 19F NMR one. The chemical shifts k and l were structure faults caused by the elimination of HF, which is a common phenomenon in fluorine-containing polymers.29 As for the residues in the precipitant (MeOH), we firstly assumed the residues were consisting of oligomers, and calculated the polymerization degree. Based on eqn (5), the exact number of A2 incorporated in the polymer was 0.5 (Fig. S2(f) in ESI†). In the analysis of 1H NMR spectrum (Fig. S2(e) in ESI†), the polymerization degree n of (A2B3)n was 1.0 based on eqn (6). The analysis results above meant the average polymerization degree for the residues was less than 1, so there only existed dimers A2B3, A2, and B3 in residues, instead of polymers. So no oligomers remained in the precipitant, and all the polymers have been collected successfully.
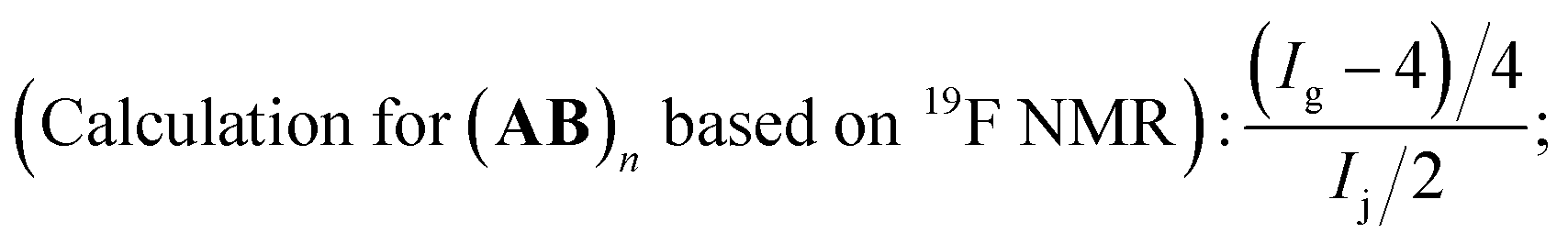 | (5) |
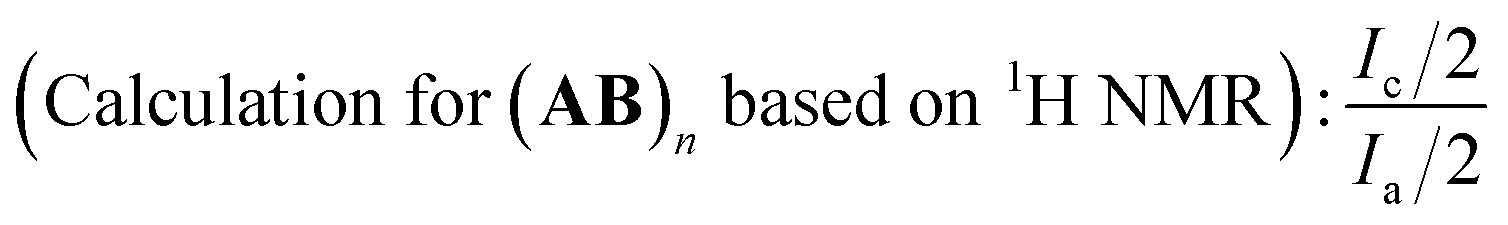 | (6) |
More experiments have also been carried out to inspect the monomer scope applicable in the novel polymerization process (Table 4). Satisfied results have been achieved for all the applied α,ω-diiodoperfluoroalkanes A and α,ω-unconjugated dienes B. As long as we ensured the inertness of CC bonds in α,ω-unconjugated dienes B, various functional perfluorocarbon-containing alternating copolymers (AB)n could be efficiently prepared through the utilization of different pairs of A and B.
|
||||||
|---|---|---|---|---|---|---|
| Entry | A | B | Time (h) | Conv. (%) | Mn,GPC (g mol−1) | Mw/Mn |
| a Polymerization conditions: [A]0:[B]0:[Ru(bpy)3Cl2]0:[AsAc-Na]0 = 1:1:0.02:0.5, n(B) = 0.5 mmol, V(1,4-dioxane):V(MeOH):V(water) = 3:1:3, V(total solvent) = 7.0 mL, irradiation under blue LED at room temperature. |
||||||
| 1 | A1 | B3 | 24 | 47.8 | 8200 | 1.38 |
| 2 | A2 | B1 | 12 | 59.9 | 4100 | 1.48 |
| 3 | A2 | B2 | 24 | 48.1 | 9000 | 2.17 |
| 4 | A2 | B3 | 12 | 81.5 | 9200 | 1.85 |
| 5 | A2 | B4 | 12 | 54.4 | 17200 |
1.99 |
| 6 | A3 | B2 | 12 | 90.9 | 8400 | 1.47 |
| 7 | A3 | B3 | 12 | 79.4 | 4800 | 1.48 |
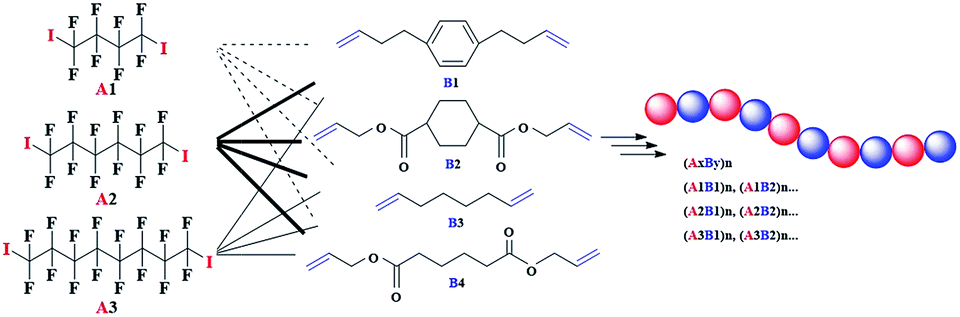
In addition, some post-polymerization experiments have been carried out to obtain pure semifluorinated polyolefins and the detailed modification procedures are presented in the experimental part above. Based on the comparison of NMR analysis (Fig. S3 in ESI†), the signal attributed to –CH2CH(I)CH2– has disappeared thoroughly after the reduction of iodine atoms in the polymer chains. Therefore, the final polymer P2 (Mn,GPC = 2700 g mol−1, Mn,NMR = 4200 g mol−1) was composed of alkane chain and perfluorocarbon chain in strict alternating manner. Considering the successful generation of different functional perfluorocarbon-containing alternating copolymers (AB)n shown in Table 3, we could also obtain various kinds of functional semifluorinated polyolefins in high efficiency, not restricted to simple semifluorinated polyolefins. And the corresponding different functional semifluorinated polyolefins could be applied in different occasions.
TGA experiments were carried out to investigate the thermal stability of resultant polymers (Fig. 4). The decomposition process for raw perfluorocarbon-containing alternating copolymer (A2B3)n (P3, Mn,GPC = 6100 g mol−1, Mw/Mn = 1.41) could be divided into two stages, similar with literature report.30 The first stage in decomposition process for (A2B3)n corresponded to the loss of HI, which began to degrade at 223.9 °C. And the second stage stood for the autocatalytic chain excision and decomposition of the polymers. The common halogen-containing polyolefins almost decomposed completely when the temperature reached 400.0 °C. On the contrary, the final residual char for (A2B3)n was 20.7%, when the temperature reached 482.1 °C. The improved thermal stability was due to the presence of perfluorocarbon segment in (A2B3)n. It was worthy to be mentioned the thermal stability for the pure semifluorinated polyolefin sample P4 (Mn,GPC = 5200 g mol−1, Mw/Mn = 1.47) has been further improved, which did not decompose until the temperature reached 350.0 °C, comparable with flame-retardant polymer materials.31 And the final residual char was about 27.6% when the temperature reached 485.5 °C. TGA tests well illustrated the thermal stability properties of both raw perfluorocarbon-containing alternating copolymer (A2B3)n, and the semifluorinated polyolefin sample P4.
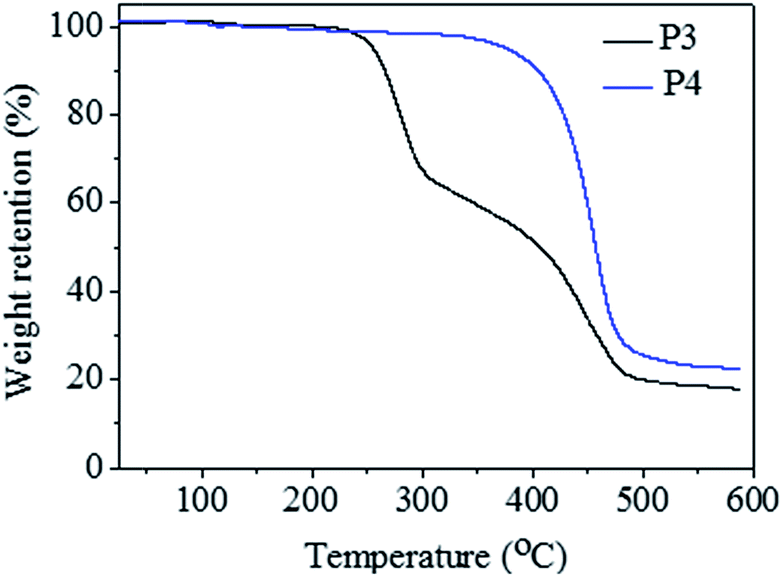 | ||
| Fig. 4 TGA for the alternating copolymer (A2B3)n before and after the reduction modification process. Polymer sample P3 (Mn,GPC = 6100 g mol−1, Mw/Mn = 1.41) was the raw perfluorocarbon-containing alternating copolymer (A2B3)n without any modification; polymer sample P4 (Mn,GPC = 5200 g mol−1, Mw/Mn = 1.47) was the semifluorinated polyolefins after the reduction modification (detailed modification process was shown in Fig. S3 in ESI†). | ||
Based on the combined analyses of polymerization kinetics and NMR (1H and 19F) tests, the whole polymerization mechanism has been revealed, which is presented in Scheme 1. Firstly, in the presence of visible light, the photo-redox catalyst Ru(bpy)3Cl2 was irradiated to the excited state *Ru(bpy)3Cl2. In the presence of AsAc-Na, the excited photo-catalyst *Ru(bpy)3Cl2 was reductively quenched to [Ru(bpy)3]+, which was highly reductive to open up the C–I bond of α,ω-diiodoperfluoroalkane (A). Then the initial carbon radicals (–CF2˙) were generated, to which α,ω-unconjugated diene B was added at both carbon radicals. Since the α,ω-unconjugated diene B was also symmetrical, the probability of the opening up for the two CC bonds was same. The single-addition step was completed by the addition of iodine atom to the newly-generated carbon radical. The mixed solvent system (1,4-dioxane/MeOH/water) provided a strong polar environment and promoted the radical addition between perfluoroalkyl radicals (RF˙) and alkenes (CC) and greatly inhibited the chain transfer reaction of –RF˙ from MeOH. As a result, the copolymerization between α,ω-diiodoperfluoroalkanes A and α,ω-unconjugated dienes B progressed smoothly and efficiently for the generation of perfluorocarbon-containing alternating copolymers (AB)n with both high polymer yield and molecular weight in aqueous/organic biphase.
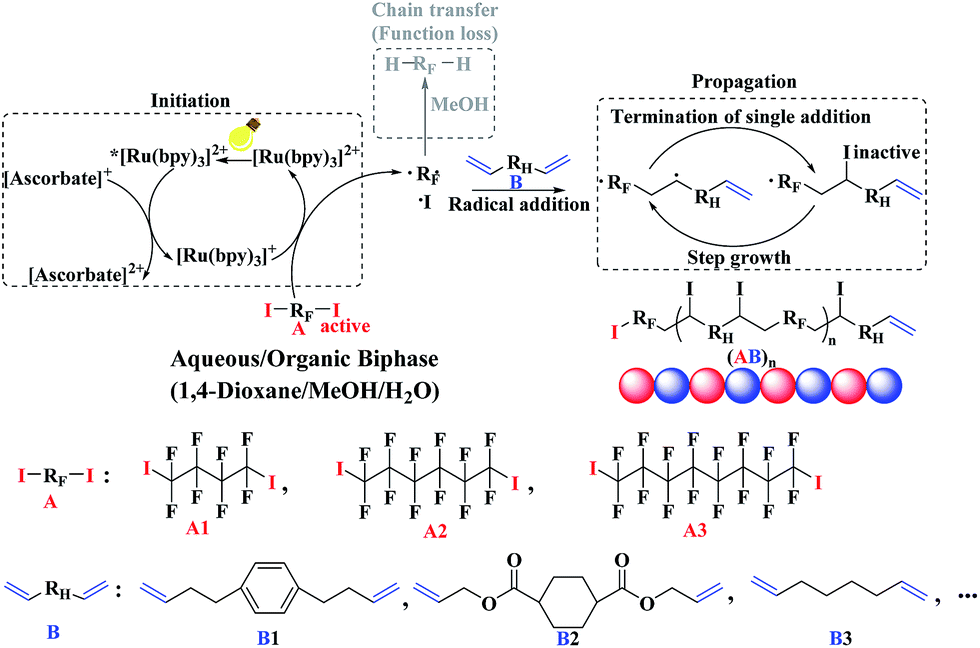 | ||
| Scheme 1 Proposed polymerization mechanism of photo-induced step tranfer-addition & radical-termination (START) strategy in aqueous/organic biphase. | ||
4. Conclusions
In summary, in aqueous/organic biphasic system, the step-growth radical polymerization between α,ω-diiodoperfluoroalkanes A and α,ω-unconjugated dienes B proceeded efficiently, generating perfluorocarbon-containing alternating copolymers (AB)n with both high polymer yield and molecular weight. The mixed solvent system (1,4-dioxane/MeOH/water) provided a strong polar environment, which promoted the radical reaction between perfluoroalkyl radicals (RF˙) and alkene (CC). The polymerization proceeded efficiently and smoothly with no generation of I2, based on the analysis of UV-vis tests. Most part of the functional group (C–I) in the polymer chain end has been preserved through 19F NMR analysis, which was a great progress compared with our previous investigation. In addition, some post-polymerization modification about the polymers have been carried out for the generation of semifluorinated polymers with enhanced thermal stability. Further investigations are underway for the property exploration of the resultant polymers and their practical applications.
Acknowledgements
The financial support from the National Natural Science Foundation of China (No. 21674071), the Project of Science and Technology Development Planning of Suzhou (No. ZXG201413), and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) is gratefully acknowledged.Notes and references
- R. Mülhaupt, Macromol. Chem. Phys., 2003, 204, 289–327 CrossRef.
- (a) H. Hsieh and R. P. Quirk, Anionic polymerization: principles and practical applications, CRC Press, 1996 Search PubMed; (b) K. Matyjaszewski, Cationic polymerizations: mechanisms, synthesis & applications, CRC Press, 1996 Search PubMed.
- (a) T. Otsu and M. Yoshida, Makromol. Chem., Rapid Commun., 1982, 3, 127–132 CrossRef CAS; (b) T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS; (c) X. Gao, X. Hu, P. Guan, C. Du, S. Ding, X. Zhang, B. Li, X. Wei and R. Song, RSC Adv., 2016, 6, 110019–110031 RSC.
- (a) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS; (b) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed; (c) V. Sciannamea, R. Jérôme and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS PubMed; (d) J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS; (e) X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- (a) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS; (b) J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS; (c) K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed; (d) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS PubMed; (e) L. J. Bai, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2012, 3, 2685–2697 RSC; (f) W. W. He, H. J. Jiang, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2013, 4, 2919–2938 RSC; (g) J. L. Pan, Z. Li, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Chin. J. Polym. Sci., 2014, 32, 1010–1018 CrossRef CAS; (h) L. J. Bai, W. X. Wang, M. H. Wang, J. M. Sun and H. Chen, Chin. J. Polym. Sci., 2015, 33, 1260–1270 CrossRef CAS; (i) B. J. Zhang, X. W. Jiang, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2015, 6, 6616–6622 RSC; (j) M. Q. Ding, X. W. Jiang, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Macromol. Rapid Commun., 2015, 36, 1702–1721 CrossRef CAS PubMed; (k) X. W. Jiang, Y. J. Luo, Z. Li, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2015, 6, 6394–6401 RSC; (l) J. Y. Peng, M. Q. Ding, Z. P. Cheng, L. F. Zhang and X. L. Zhu, RSC Adv., 2015, 5, 104733–104739 RSC; (m) X. D. Liu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2016, 7, 689–700 RSC.
- (a) J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS; (b) C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed; (c) D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC; (d) Z. Li, W. J. Chen, Z. B. Zhang, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2015, 6, 1937–1943 RSC; (e) J. Qin, L. F. Zhang, H. J. Jiang, J. Zhu, Z. B. Zhang, W. Zhang, N. C. Zhou, Z. P. Cheng and X. L. Zhu, Chem.–Eur. J., 2012, 18, 6015–6021 CrossRef CAS PubMed; (f) S. G. Niu, L. F. Zhang, J. Zhu, W. Zhang, Z. P. Cheng and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2013, 53, 1197–1204 CrossRef; (g) Z. Li, W. J. Chen, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2015, 6, 5030–5035 RSC; (h) L. F. Zhang, Z. P. Cheng, N. C. Zhou, S. P. Shi, X. R. Su and X. L. Zhu, Polym. Bull., 2009, 62, 11–22 CrossRef CAS; (i) C. Tian, T. C. Xu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, RSC Adv., 2016, 6, 34659–34665 RSC; (j) Q. L. Li, L. Li, H. S. Wang, R. Wang, W. Wang, Y. J. Jiang, Q. Tian and J. P. Liu, Chin. J. Polym. Sci., 2017, 35, 66–77 CrossRef CAS.
- (a) M. Guerre, B. Campagne, O. Gimello, K. Parra, B. Ameduri and V. Ladmiral, Macromolecules, 2015, 48, 7810–7822 CrossRef CAS; (b) T. Segura, M. Menes-Arzate, F. León, A. Ortega, G. Burillo and R. D. Peralta, Polymer, 2016, 102, 183–191 CrossRef CAS; (c) M. Guerre, S. M. W. Rahaman, B. Ameéduri, R. Poli and V. Ladmiral, Macromolecules, 2016, 49, 5386–5396 CrossRef CAS; (d) X. D. Liu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2016, 7, 3576–3588 RSC; (e) X. D. Liu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Chem. Commun., 2016, 52, 10850–10853 RSC.
- (a) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS PubMed; (b) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H. W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 419, 384–387 CrossRef CAS PubMed.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- T. Terashima, T. Mes, T. F. A. Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- D. Rochambeau, M. Barłóg, T. G. W. Edwardson, J. J. Fakhoury, R. S. Stein, H. S. Bazzi and H. F. Sleiman, Polym. Chem., 2016, 7, 4998–5003 RSC.
- T. Soulestin, V. Ladmiral, T. Lannuzel, F. D. D. Santos and B. Ameduri, Macromolecules, 2015, 48, 7861–7871 CrossRef CAS.
- N. O. Brace, J. Fluorine Chem., 1999, 93, 1–25 CrossRef CAS.
- A. Vitale, R. Bongiovanni and B. Améduri, Chem. Rev., 2015, 115, 8835–8866 CrossRef CAS PubMed.
- M. P. Turberg and J. E. Brady, J. Am. Chem. Soc., 1988, 110, 7797–7801 CrossRef CAS.
- C. G. Lux, B. Donnio, B. Heinrich and M. P. Kraff, Langmuir, 2013, 29, 5325–5336 CrossRef PubMed.
- L. M. Wilson and A. C. Griffin, Macromolecules, 1993, 26, 6312–6314 CrossRef CAS.
- T. Davidson, A. C. Griffin, L. M. Wilson and A. H. Windle, Macromolecules, 1995, 28, 354–357 CrossRef CAS.
- T. C. Xu, H. N. Yin, X. H. Li, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Macromol. Rapid Commun., 2016 DOI:10.1002/marc.201600587.
- (a) A. Goto, T. Suzuki, H. Ohfuji, M. Tanishima, T. Fukuda, Y. Tsujii and H. Kaji, Macromolecules, 2011, 44, 8709–8715 CrossRef CAS; (b) X. D. Liu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2016, 3576–3588 RSC.
- O. Hassager, Comprehensive polymer science, Pergamon Press, 1992 Search PubMed.
- L. Zhang, W. R. Dolbier Jr, B. Sheeller and K. U. Ingold, J. Am. Chem. Soc., 2002, 124, 6362–6366 CrossRef CAS PubMed.
- (a) L. Zhang, J. Cradlebaugh, G. Litwinienko, B. E. Smart, K. U. Ingold and W. R. Dolbier Jr, Org. Biomol. Chem., 2004, 2, 689–694 RSC; (b) J. Wu, Z. Liu, Q. Chen and C. Liu, J. Fluorine Chem., 2016, 184, 45–49 CrossRef CAS.
- M. Duc, B. Ameduri, B. Boutevin, M. Kharroubi and J. M. Sage, Macromol. Chem. Phys., 1998, 199, 1271–1289 CrossRef CAS.
- (a) S. Ida, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2010, 132, 14748–14750 CrossRef CAS PubMed; (b) D. M. Rosenbaum and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 13924–13925 CrossRef CAS PubMed.
- (a) A. Ohtsuki, A. Goto and H. Kaji, Macromolecules, 2013, 46, 96–102 CrossRef CAS; (b) A. Ohtsuki, L. Lei, M. Tanishima, A. Goto and H. Kaji, J. Am. Chem. Soc., 2015, 137, 5610–5617 CrossRef CAS PubMed.
- K. Satoh, M. Mizutani and M. Kamigaito, Chem. Commun., 2007, 1260–1262 RSC.
- R. Saint-Loup and B. Améduri, J. Fluorine Chem., 2002, 116, 27–34 CrossRef CAS.
- (a) E. Boz, K. B. Wagener, A. Ghosal, R. Fu and R. G. Alamo, Macromolecules, 2006, 39, 4437–4447 CrossRef CAS; (b) M. Guerre, S. M. W. Rahaman, B. Ameduri, R. Poli and V. Ladmiral, Macromolecules, 2016, 49, 5386–5396 CrossRef CAS.
- T. C. Xu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2015, 6, 2283–2289 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01925d |
| This journal is © The Royal Society of Chemistry 2017 |