
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniform core–shell Cu6Sn5@C nanospheres with controllable synthesis and excellent lithium storage performances†
Liwei Su a,
Jianghao Fua,
Pinjie Zhangb,
Lianbang Wang*a,
Yuanhao Wang*c and
Manman Rend
a,
Jianghao Fua,
Pinjie Zhangb,
Lianbang Wang*a,
Yuanhao Wang*c and
Manman Rend
aState Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: wanglb99@zjut.edu.cn
bJuhua Group Technology Center, Quzhou 324004, China
cXinjiang Technical Institute of Physics & Chemistry, Chinese Academy of Sciences, Urumqi, 830011, China. E-mail: yuanhaowang@yahoo.com
dInstitute of Materials Science and Engineering, Qilu University of Technology, Jinan 250353, China
First published on 30th May 2017
Abstract
Metallic tin (Sn) is one of the most promising alternatives to graphite anodes for lithium ion batteries due to its higher theoretical capacity, higher packing density and safer thermodynamic potential, while the huge volume transformation during repeated cycling leads to rapid pulverization and consequently poor capacity retention. This work provides an easy-to-control method to prepare uniform core–shell Cu6Sn5@C nanospheres in which Cu@Sn cores (40–50 nm in diameter) are well encapsulated by PANI-derived carbon layers with a thickness of ∼5 nm. The obtained Cu6Sn5@C exhibits an excellent cycling ability and good rate capabilities. Both the reversible capacity (518 mA h g−1) after 100 cycles and the initial coulombic efficiency (89.2%) are the highest values in Cu6Sn5-based materials. The impressive cycling performance is believed to result from the carbon coating that not only prevents particle agglomeration during the synthesis but also accommodates the vast structural transformation of the Cu6Sn5 nanocores during the electrochemical (de)lithiation process, so ensuring good ionic and electronic transport to the core. The effect of synthesis conditions on the composition are also investigated systematically.
1. Introduction
Low volumetric capacity and safety issues limit the performances of graphite-based anodes in high energy/power applications, such as electric vehicles (EVs) and hybrid electric vehicles (HEVs).1–4 Metallic tin (Sn) is one of the most promising alternatives to graphite due to its high theoretical capacity (990 mA h g−1, 7200 mA h cm−3) (cf. graphite: 372 mA h g−1, 800 mA h cm−3), high packing density, and safe thermodynamic potential.5–8 However, the large specific volume change that occurs during charging and discharging leads to rapid pulverization of the Sn electrode and consequently a decrease of electrical conductivity, resulting in poor capacity retention upon extended cycling.9In order to overcome this problem, an effective approach is to alloy Sn with inactive metals such as Fe,10,11 Co,12–17 Cu,18–21 Ni,22–25 and Zn26,27 that do not react as readily with lithium and thus provide a buffer matrix that can absorb the extensive volume expansion and contraction of Sn. In this respect, many efforts have focused on Sn–Cu alloys, which react with lithium at a few hundred milli-volts above the potential of metallic lithium and lithiated graphite electrodes LixC6 (x ≤ 1).28–30 One of key issues in the synthesis of Cu–Sn alloys is to ensure intimate contact between Sn and Cu and at the same time inhibit the agglomeration of products due to the melting of Sn (and alloying with Cu) during heat treatment.
Ideally, the alloy should be synthesized in a nanostructured form to enhance electronic conduction and shorten the Li-ion migration paths.31 Various methods including high energy ball milling,32 direct melting,33 electro-deposition,34,35 and carbothermal reduction36 have been used to prepare such Cu–Sn materials. However, it is difficult to prepare small/isolated particles with controlled size and morphology using these synthetic methods. Further, these relatively complicated synthesis routes are often not convenient for scaled-up production. Thus the efficiency of such approaches has been relatively limited. In recent years, electro-less deposition has been used to prepare Sn thin films on Cu substrates, with annealing leading to the formation of Cu–Sn thin film anodes.37,38 The advantage of this method is that an intimate contact between Sn and Cu can be achieved, regardless of the shape of the Cu substrate.
It is believed that carbon surface modification presents significant advantages to enhance the cycleability of active materials, as it can prevent the aggregation of active particles, accommodate the strain of volume change, and enhance the surface electronic conductivity of such materials.39,40 For example, Wang et al. found that polyamine (PANI) coating FePO4 as opposed to the final LiFePO4 product could effectively restrict the particle growth during the reaction of FePO4 and CH3COOLi to form LiFePO4. PANI decomposed into carbon over the course of the reaction.41 The resultant LiFePO4 nanoparticles are coated with conductive carbon nanolayers and exhibits close-to-theoretical capacity and much improved rate capabilities.42 This indicates that an intermediate coating can effectively control the size of final particles and in the process form a protective conducting shell around the particle. Similarly, pomegranate-like Sn@C,43 Si@C,44,45 Sb@C,46 and Fe3O4@C47 hierarchical nanocomposites are also reported recently and exhibited extraordinary performance for Li/Na storage.
Motivated by the above findings, herein we propose a new strategy to prepare uniform core–shell Cu6Sn5@C nanospheres. Our strategy involves the electro-less preparation of Cu–Sn powder and the formation of double-shelled Cu@Sn@PANI intermediate compounds by fabricating Sn and PANI double layers around Cu nanoparticles, followed by an annealing step to generate core–shell Cu6Sn5@C nanospheres. The role of PANI is thus not only to act as the carbon source, but also to suppress the particle aggregation by isolating the Cu@Sn@PANI intermediate particles, ensuring that there is no growth beyond the nanoscale at each step. Hence, we were able to control fully the synthesis of high quality core–shell Cu6Sn5@C nanospheres at the nanoscale level. The Cu6Sn5@C nanospheres were electrochemically tested as anodes for Li-ion batteries to verify its applicability. As shown below, the nanospheres demonstrated excellent Li+ storage properties over extended (de)lithiation cycling and exhibited high rate capabilities.
2. Experimental
2.1 Materials synthesis
All the reagents were of analytical grade and used without further purification. The Cu–Sn precursor powder was synthesized by electro-less deposition. The synthesis procedure of Cu–Sn@C nanospheres can be schematically illustrated in Scheme 1 and include three main steps: (i) the electro-less deposition of Sn on Cu nanoparticles, (ii) the polymerization of AN molecules on Cu@Sn surface to obtain Cu@Sn@PANI precursors, and (iii) the formation of Cu–Sn alloy cores and the carbonization of PANI shells via heating at 300 °C in an inert atmosphere.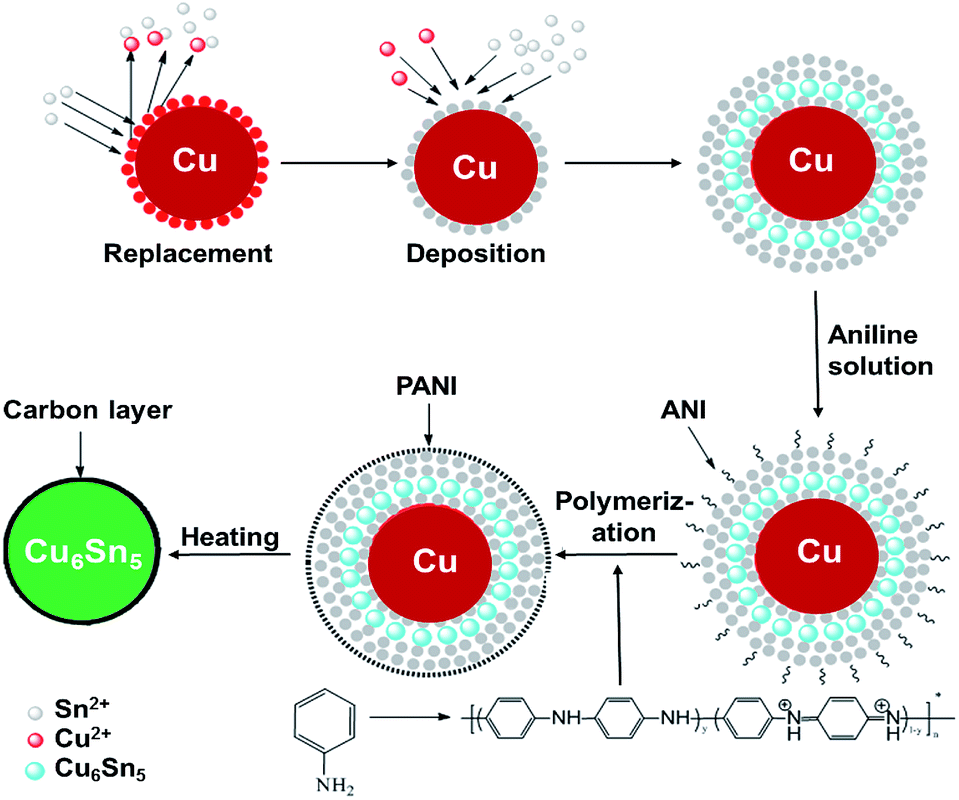 | ||
| Scheme 1 Schematic of the preparation of the core–shell Cu6Sn5@C nanospheres. | ||
In a typical synthesis, the plating solution was prepared using reagent grade chemicals containing stannous sulfate for as a source of Sn2+ ions (SnSO4 0.32 g L−1), citric acid for buffering the solution, sulphuric acid for pH adjustment to pH = 1.0, which was monitored by pH meter (Shanghai Jingke, PHS-3C). Thiourea (100 g L−1) was added as a complexing agent to form soluble [Cu(NH2CSNH2)4]2+ ions. First, Cu powder (20 nm, purity 99.99%, Suzhou Canfuo Nanotechnology Company) was cleaned using dilute hydrochloric acid (1%) and acetone before the coating process. Then, the pretreated Cu power was slowly added to the plating solution under vigorous stirring. Plating was performed for 10 min at 20 °C. After plating, 0.5 mL of aniline (AN) solution (2 mol L−1) was added drop-wise to the Sn/Cu suspension. The solution immediately became grey/green in colour when slowly adding the a few drops of H2O2 (2 wt%), indicating the formation of PANI. The residual Cu2+ in the solution was analyzed by ICP-MS (PerkinElmer Elan DRC-e spectrometer). The powder product was dried under vacuum (10−5 Pa) at room temperature and heated under nitrogen (to avoid the oxidation of the Cu and Sn metals) to 300 °C at a heating rate of 5 °C min−1 for 10 h to form the final Cu6Sn5@C product. Additional Cu–Sn@C composites with different Cu to Sn ratios were prepared for comparison. Expect for the (NH2)2SC and SnSO4 concentrations, the synthesis procedures used for these additional samples were the same as those previously described.
2.2 Materials characterization
The structures and morphology of the samples were characterized by X-ray diffraction (XRD) (PANalytical X'Pert Pro with Cu Kα radiation, λ = 1.5418 Å, using a scan rate of 0.03° min−1), scanning electronic microscope (SEM, Hitachi S-4700 operated at 15 kV) and transmission electronic microscopy (TEM, Tecnai G2 F30 S-Twin operated at 300 kV). The carbon contents of Cu6Sn5@C materials were measured using a ThermoFisher Flash EA 1112 analyzer. Fourier transform infrared spectroscopy (FTIR) analysis of Cu@Sn@PANI nanospheres was performed using a Nicolet 6700 spectrometer. Thermogravimetric analysis (TGA) of Cu@Sn@PANI and Cu6Sn5@C nanospheres was performed with a Perkin Elmer thermobalance at a heating rate of 5 °C min−1 in a 50 mL min−1 flow of argon and in air, respectively. Raman spectra were measured in a back scattering configuration using a micro-Raman spectrometer HR 800 (Jobin Yvon Horiba).2.3 Electrochemical measurements
For electrochemical characterization, electrodes were prepared by mixing 80 wt% of active materials, 10 wt% of acetylene black (Changzhou dafu), and 10 wt% of polyvinylidene fluoride (PVdF) (Shanghai Sanaifu) in a 1-methyl-2-pyrrolidinone (NMP) solution (Shenzhen Huachang). The as-prepared slurry was coated onto a Cu foil and dried at 80 °C for 10 h under vacuum (10−5 Pa). The average loading mass of the electrodes was 1.0–1.1 mg cm−2. Electrochemical cells used Li metal sheet as the counter electrode. The electrolyte was 1 mol dm−3 LiPF6 in ethylene carbonate (EC), dimethyl carbonate (DMC), ethylmethyl carbonate (EMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, v/v/v) (from Zhangjiagang GuotaiRonghua). Cycling tests were performed at 298 K with various charge–discharge current densities between 0.01–1.2 V and with a LAND CT2001A battery tester. Except noted, the presented capacities are calculated on the basis of the total weight of composites. Cyclic voltammetry (CV) was performed with a Solartron 1287A potentiostat/galvanostat and 1260A impedance/gain-phase analyzer at a scan rate of 0.1 mV s−1. Electrochemical impedance spectra (EIS) were performed on a CHI 660B electrochemical workstation and recorded over a frequency range from 10−2 to 105 Hz, with voltage amplitude of 5 mV at room temperature.
:1:1, v/v/v) (from Zhangjiagang GuotaiRonghua). Cycling tests were performed at 298 K with various charge–discharge current densities between 0.01–1.2 V and with a LAND CT2001A battery tester. Except noted, the presented capacities are calculated on the basis of the total weight of composites. Cyclic voltammetry (CV) was performed with a Solartron 1287A potentiostat/galvanostat and 1260A impedance/gain-phase analyzer at a scan rate of 0.1 mV s−1. Electrochemical impedance spectra (EIS) were performed on a CHI 660B electrochemical workstation and recorded over a frequency range from 10−2 to 105 Hz, with voltage amplitude of 5 mV at room temperature.
3. Results and discussion
3.1 Synthesis mechanism, compositions, and performances of Cu–Sn@C nanocomposites
The formation mechanism of Sn shell on Cu nanoparticles by the electro-less deposition can be described as follows: (NH2)2SC forms stable complexes with Cu2+, e.g. [Cu(NH2CSNH2)4]2+/Cu and so by its addition one would expect the electrochemical potential of [Cu(NH2CSNH2)4]2+/Cu to move to a more negative value (e.g. −0.157 V when the concentration of (NH2)2SC is 1 mol L−1).48 Thus some Cu on the surface was replaced by Sn2+ with the addition of (NH2)2SC (Cu2+/Cu and Sn2+/Sn is 0.337 V and −0.136 V, respectively). The electro-less deposition process was initiated. The solvated Cu2+ would return to the Cu particle by combining the Sn2+ (to form Cu6Sn5), and the residual Cu2+ concentration was found to be as low as 0.0001 mol L−1 after synthesis indicating that the previously replaced Cu2+ could be deposited to the Cu particles. Afterwards, Sn started to be deposited to form Sn layer around Cu particles.The concentrations of (NH2)2SC and SnSO4 were modified in an effort to exert a greater control over the final compositions of the Cu–Sn@C materials. Table 1 summarizes the effect of the preparation conditions on the phase properties for the obtained products (according to XRD analysis as shown in Fig. S1†). Diffraction data show that the concentrations of (NH2)2SC and Sn2+ greatly affect the final phase composition due to different Cu:Sn ratios resulting from the electro-less reduction. For instance, when the concentration of (NH2)2SC is lower than 100 g L−1 and the concentration of Sn2+ fixes at 0.1 mol L−1, metallic copper and other Cu-rich phases (e.g. Cu3Sn) are found in the final product (samples a–d). The occurrence of these phases can be ascribed to the relatively small quantity of Sn plated in the Cu@Sn@PANI precursors. By increasing the concentration of Sn2+ ions, the Cu and Cu3Sn phases are gradually removed and the reflections from Cu6Sn5 become dominant (samples d–f). The Cu:Sn mole ratio of sample f is 5.9:5.1 confirmed by EDX. Inevitably, a high concentration of Sn2+ ions resulted in a large amount of Sn in the product (e.g. sample g).
| Samples | C(NH2)2SCa | CSnSO4b | Cu | Sn | Cu3Sn | Cu6Sn5 | Mole ratio of Cu:Sn by EDX |
|---|---|---|---|---|---|---|---|
| a Concentrations of (NH2)2SC, g L−1.b Concentrations of SnSO4, g L−1. | |||||||
| Sample a | 40 | 0.15 | ● | ● | 6.3:0.7 |
||
| Sample b | 80 | 0.15 | ● | ● | ● | 3.3:1.1 |
|
| Sample c | 100 | 0.15 | ● | ● | ● | ● | 2.1:0.9 |
| Sample d | 120 | 0.15 | ● | ● | ● | ● | 2.9:1.1 |
| Sample e | 100 | 0.25 | ● | ● | 3.1:2.2 |
||
| Sample f | 100 | 0.32 | ● | 5.9:5.1 |
|||
| Sample g | 100 | 0.50 | ● | ● | 6.1:7.4 |
||
Table 2 summarizes the electrochemical performances of Cu–Sn@C nanospheres (samples a–g). Cu-Rich samples (e.g. a–e) exhibit an extended reversible capacity of <370 mA h g−1 after 100 cycles. For Sn-rich samples (e.g. sample g), however, the capacity fading over 100 cycles is significant although the initial capacity is high, presumably due to presence of unprotected Sn. Given that the sample f is with the best electrochemical properties, we therefore devoted our efforts to this sample to study its electrochemical properties in detail.
| Samples | Initial DCa | 100th DC | Cycling retention | CCb at 1C | CC at 2C | CC at 5C |
|---|---|---|---|---|---|---|
| a Discharge capacity.b Charge capacity. | ||||||
| Sample a | 113 | 105 | 92.9% | 87 | 73 | 69 |
| Sample b | 138 | 118 | 85.5% | 104 | 81 | 75 |
| Sample c | 220 | 179 | 81.4% | 137 | 94 | 82 |
| Sample d | 361 | 273 | 75.6% | 217 | 138 | 91 |
| Sample e | 510 | 369 | 72.4% | 251 | 143 | 91 |
| Sample f | 686 | 490 | 71.4% | 334 | 275 | 206 |
| Sample g | 828 | 405 | 48.9% | 175 | 118 | 101 |
3.2 Materials characterization of core–shell Cu6Sn5@C
The electro-less deposition of Sn2+ ions and polymerization of AN were conducted in sequence on Cu nanoparticles to fabricate the Cu@Sn@PANI precursors. The phase composition of the starting and intermediate compounds for sample f was determined by XRD and the results are shown in Fig. 1. All the reflections of the Cu nanoparticles could be readily indexed to cubic Cu (JCPDS no. 01-1241). Reflections in the XRD pattern of Cu@Sn@PANI could also be attributed to Cu, with additional reflections that could be assigned to Sn (JCPDS no. 04-0673), implying that a Cu–Sn composite was formed. Note that, due to the high surface energy of the Cu nanoparticles, some of the deposited Sn at Cu/Sn interface would also readily diffuse into the Cu lattice to form Cu6Sn5, which appears in the Cu@Sn@PANI nanospheres. XRD patterns of the Cu6Sn5@C nanospheres obtained after heat treatment were indexed to monoclinic Cu6Sn5 (C2/c, JCPDS no. 45-1488) with an average crystalline size of ∼40 nm, (using the Scherrer equation: D = Kλ/βcosθ). The absence of a diffraction reflection at 2θ = 23° indicates that carbon resulting from the PANI decomposition may exist in an amorphous form.
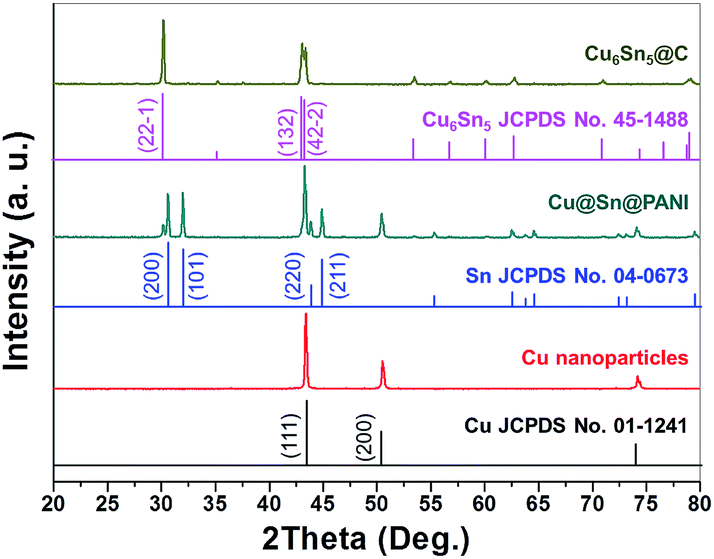 | ||
| Fig. 1 XRD patterns of Cu nanoparticles, Cu@Sn@PANI precursors, and core–shell Cu6Sn5@C nanospheres. | ||
The formation of PANI in the Cu@Sn@PANI precursors can be observed via FTIR analysis as shown in Fig. 2a. The absorption bands at 1548 and 1497 cm−1 are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching deformation of the quinoid and benzoid rings of PANI, respectively. The bands at 1300 cm−1 belongs to the C–N stretching of the intermediate aromatic amine, while the bonds at 1136 and 814 cm−1 correspond to the C–H deformations. After heat treatment, the characteristic peaks of PANI disappear as would be expected as PANI decomposes into carbon. TGA (Fig. 2b) in flow argon of Cu@Sn@PANI shows that the removal of adsorbed water and the carbonization process mainly occur before 300 °C and lead to a mass loss of 10.5 wt%. This observation can explain why we adopt 300 °C as the annealing temperature, as higher temperatures trend to result in agglomeration of alloy nanoparticles.
C stretching deformation of the quinoid and benzoid rings of PANI, respectively. The bands at 1300 cm−1 belongs to the C–N stretching of the intermediate aromatic amine, while the bonds at 1136 and 814 cm−1 correspond to the C–H deformations. After heat treatment, the characteristic peaks of PANI disappear as would be expected as PANI decomposes into carbon. TGA (Fig. 2b) in flow argon of Cu@Sn@PANI shows that the removal of adsorbed water and the carbonization process mainly occur before 300 °C and lead to a mass loss of 10.5 wt%. This observation can explain why we adopt 300 °C as the annealing temperature, as higher temperatures trend to result in agglomeration of alloy nanoparticles.
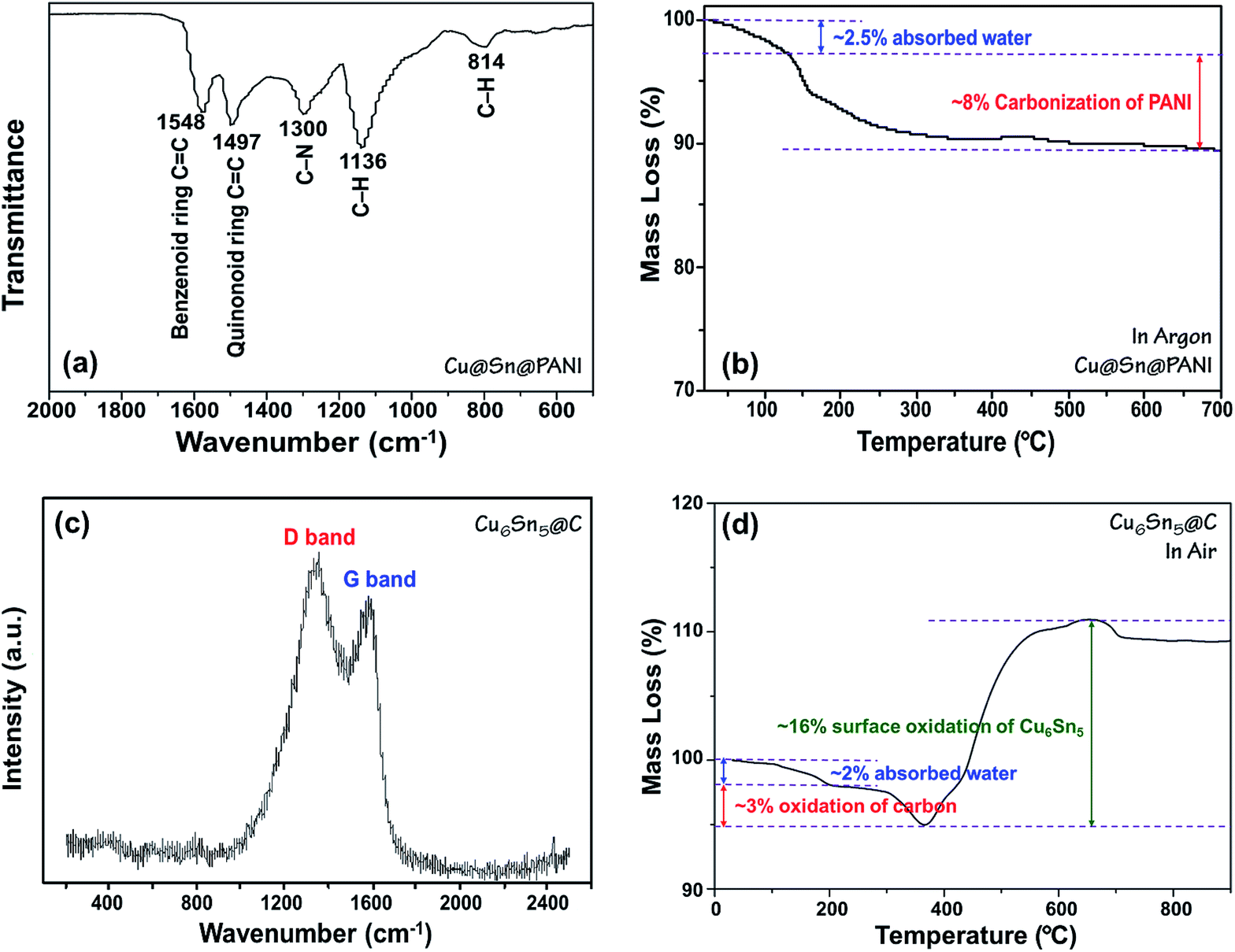 | ||
| Fig. 2 (a) FTIR spectrum and (b) TGA curve in Ar of Cu@Sn@PANI precursors; (c) Raman spectrum and (d) TGA curve in air of Cu6Sn5@C nanospheres. | ||
After heat treatment, the PANI converted into amorphous carbon confirmed by Raman spectroscopy, as shown in Fig. 2c, which is recognized as predictive for sp2 bonded carbons and physical properties of carbon materials. The strong bands at 1340 and 1590 cm−1 are respectively attributed to the D-band and G-band of carbon.49 The ID/IG ratio is calculated to be 1.59, implying that the carbon obtained from the carbonization of PANI is almost amorphous. Fig. 2d shows TGA curve for Cu6Sn5@C nanospheres in air. The mass loss of ∼2 wt% before 200 °C can be mainly ascribed to the releasing of absorbed water, while the loss of ∼3 wt% at 200–380 °C belongs to the preliminary oxidation of carbon coating. However, the complete removal of carbon generally happens in the temperature range of 400–500 °C, where a significant mass increasing of over 10 wt% appears due to the surface oxidation of Cu6Sn5. Therefore, it is inaccurate to estimate the detail carbon content. Elemental analysis was conducted instead and showed the carbon content was 9.8 wt%.
TEM images of the Cu@Sn@PANI and Cu6Sn5@C nanospheres are shown in Fig. 3. The average size of the Cu6Sn5@C nanoparticles are in the range of 40–50 nm, in good agreement with the values calculated from XRD patterns and SEM image (Fig. S2†). The sizes of the Cu6Sn5@C nanoparticles are almost identical to those of the Cu@Sn@PANI intermediate (Fig. 3a and b). This observation indicates that the PANI layer can effectively prevent agglomeration during the melting and alloying of Sn with Cu to make Cu–Sn intermetallic phases while maintaining the nanoscale morphology of the composite. The transformation from Cu@Sn@PANI to Cu6Sn5@C is thus essentially pseudomorphic. A TEM image of a selected Cu6Sn5@C particle clearly reveals that a coarse carbon layer forms around the core particle (Fig. 3c), where the carbon layer originates from the decomposition of the previously deposited PANI shell. It also demonstrates that the thickness of the carbon layer is ca. 5 nm. Selected area electron diffraction (SAED) patterns further confirms the crystalline nature of the core material and could be indexed to the monoclinic Cu6Sn5. At high resolution, TEM image (Fig. 3d) shows that the distance between neighboring fringes is 2.1 Å, very close to the (132) d-spacing in the monoclinic structure of Cu6Sn5. Therefore, based on all the results discussed above, it can be deduced that core–shell Cu6Sn5@C nanospheres were successfully prepared from Cu@Sn@PANI precursors.
 | ||
| Fig. 3 TEM images of (a) Cu@Sn@PANI precursors and (b) resultant Cu6Sn5@C nanospheres. (c) HRTEM image and (d) SAED patterns of Cu6Sn5@C. Scale bars are 50, 20, 5, and 2 nm separately. | ||
3.3 Electrochemical performances of core–shell Cu6Sn5@C nanospheres
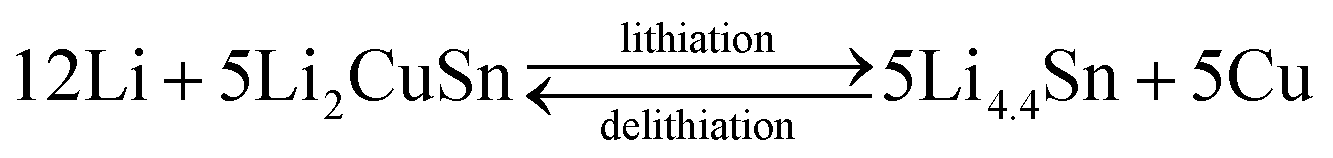
The core–shell Cu6Sn5@C nanospheres were electrochemically characterized by CV, charge–discharge profiles, and cycling performances at different rates in CR-2032 coin cells (shown in Fig. 4). CV curves for the Cu6Sn5@C sample are shown in Fig. 4a. Over the course of the lithiation process the CV profiles show two apparent reduction peaks at ca. 0.35 and 0.55 V, corresponding to the formation of Li2CuSn and Li4.4Sn as shown in eqn (1) and (2), respectively. In contrast to previous studies, irreversible peaks at potentials above 1.0 V due to catalytic decomposition of the electrolyte on the exposed active Sn surface were not observed in our data.50 The absence of these features at higher potential could be ascribed to the protection of the Cu6Sn5 core by the carbon shell, which inhibits such side reactions.
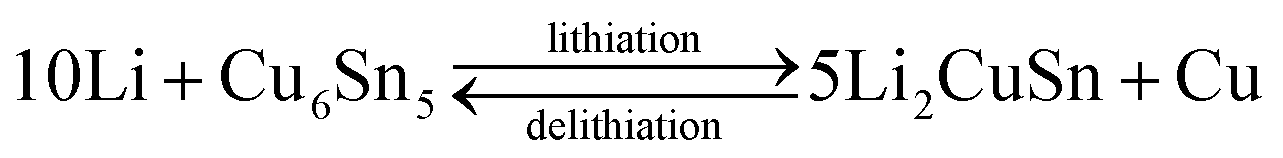 | (1) |
 | (2) |
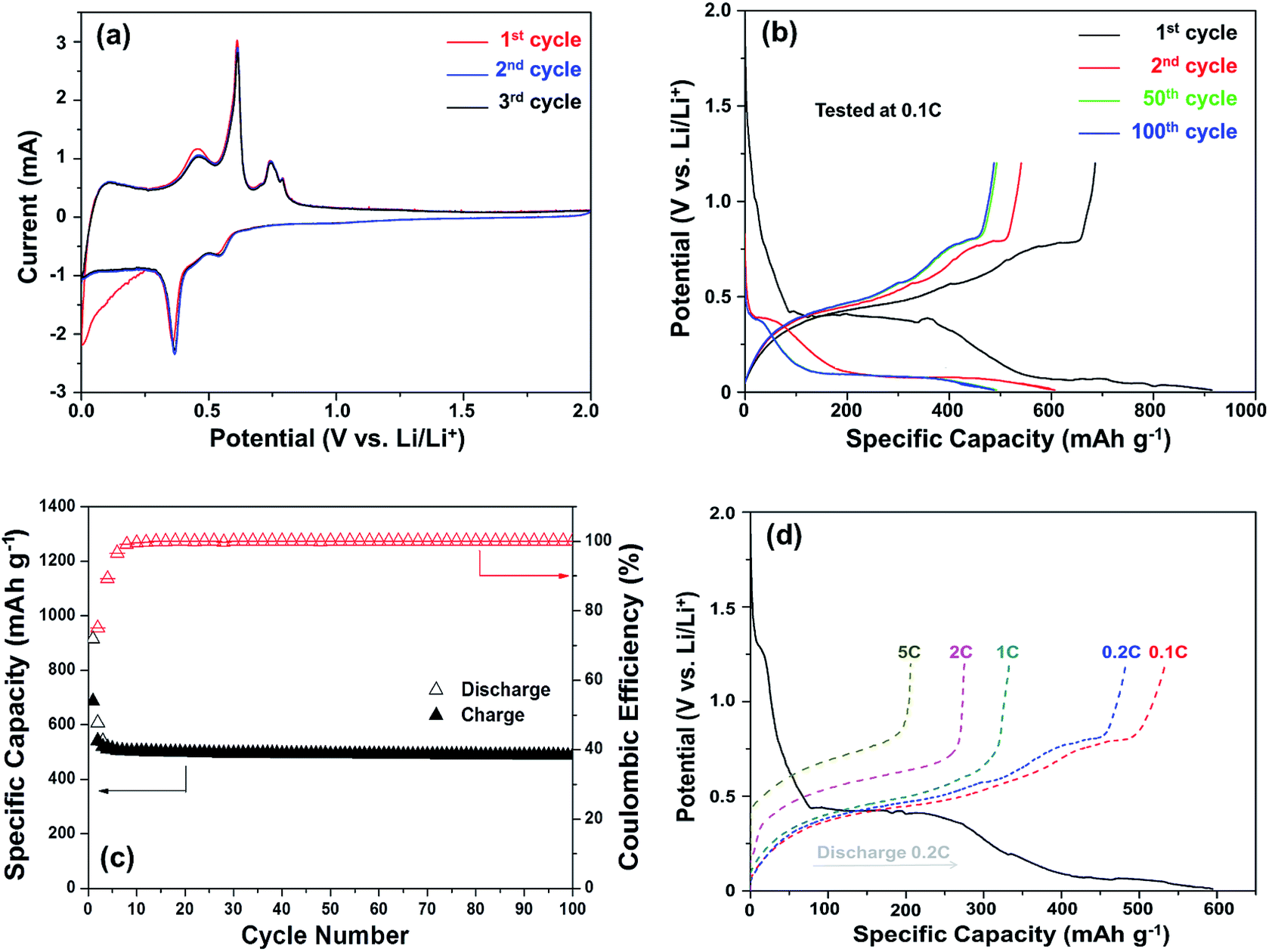 | ||
| Fig. 4 Electrochemical performances of Li/Cu6Sn5@C cells: (a) CV curves at a scan rate of 0.1 mV s−1, (b) charge/discharge profiles at 0.1C (1C = 605 mA g−1), (c) the cycleability and coulombic efficiency at 0.1C, and (d) high-rate performances. | ||
Typical charge–discharge voltage curves of the Cu6Sn5@C nanospheres cycled between 0.01 and 1.20 V were given in Fig. 4b. Several different potential regions can be identified in the discharge profiles between 0.4 and 0.1 V. In the first cycle, the discharging and charging capacity were 914.2 and 815 mA h g−1, respectively, resulting in a coulombic efficiency of 89.2%. The irreversible capacity observed in the first cycle can be attributed to the formation of a solid electrolyte interface (SEI) film. The level of SEI formation is proportional to the surface area and the quality of coating, and a homogeneous carbon coating would effectively suppress the formation of coarse SEI layer. The initial coulombic efficiency (ICE) of the Cu6Sn5@C nanospheres is 89.2% higher than the uncoated nanosized Cu–Sn anode materials (usually less than 75%, see details in Table 3). In the following cycles, Cu6Sn5@C demonstrates a coulombic efficiency of more than 99%. The charge–discharge profiles are in accord with the CV results and the close overlap of the curves offer further evidence for the observed high coulombic efficiency.
| Materials | Carbon (%) | RC (mA h g−1) | ICEc (%) | Current (mA g−1) | Cycles | Ref. |
|---|---|---|---|---|---|---|
| a Not given in references.b Calculated according to the given data.c ICE: initial coulombic efficiency. | ||||||
| Porous Cu6Sn5 | 0 | 404 | 73 | 100 | 100 | 38 |
| Porous Cu6Sn5 | 0 | 420 | 83b | 0.5 mA cm−2 | 20 | 21 |
| Microporous Cu6Sn5–Sn | 0 | 400 | 58 | 666 | 54 | 51 |
| Cu6Sn5/carbon fiber | —a | 250 | 68 | 131 | 30 | 37 |
| Cu6Sn5@C on carbon | 53.7 | 366 | 56.5 | 1000 | 200 | 18 |
| Cu6Sn5/C | 8.86 | 400 | — | 140b | 100 | 52 |
| Cu6Sn5/GNS | 52.6b | 411 | 70 | 500 | 1600 | 20 |
| Sn rich Cu6Sn5 | 0 | 440 | 98 | 200 | 50 | 53 |
| Cu6Sn5@SnO2–C | —a | 619 | 65 | 200 | 500 | 54 |
| Cu6Sn5 nanoparticles | 0 | 490 | 71.4 | 60 | 100 | This work |
| Core–shell Cu6Sn5@C | 9.8 | 518 | 89.2 | 60 | 100 | This work |
The cycling performance of the Cu6Sn5@C nanospheres is shown in Fig. 4c. It shows a ability of maintaining a reversible capacity of 518 mA h g−1 without significant fading after an initial decrease in the first few cycles. If excluding the contribution of 9.8 wt% carbon, the exact capacity of Cu6Sn5 cores is ca. 570 mA h g−1, that is very close to its theoretical value of 605 mA h g−1 calculated according to eqn (1) and (2). Rate capability is an important factor in evaluating the potential application of anode materials. This motivated us to investigate the rate performance of the Cu6Sn5@C nanospheres further. Fig. 4d displays the rate capability of the Cu6Sn5@C nanospheres at rates of 0.1–5C. The Cu6Sn5@C nanospheres maintain a steady capacity output at high current densities, e.g. 488 mA h g−1 at 0.2C, 331 mA h g−1 at 1C, 275 mA h g−1 at 2C, and 206 mA h g−1 at 5C. Fig. S3† demonstrates that the Cu6Sn5@C nanospheres still maintain a structural integrity and a reversible crystalline phase of Cu6Sn5 after 30 cycles at 0.2C.
Table 3 compares the lithium storage performance of the core–shell Cu6Sn5@C nanospheres with representative Cu6Sn5-based materials reported previously. Totally speaking, the core–shell Cu6Sn5@C nanospheres in this work exhibit comparable or even better performance especially at a relatively low rate. To the best of our knowledge, both the reversible capacity of 518 mA h g−1 and the initial coulombic efficiency of 89.2% are the highest value in pure Cu6Sn5 and Cu6Sn5/C materials. Note that, with the help of high-capacity materials such as Sn53 and SnO2,54 the counterpart performance can be further enhanced. The impressive performance can be ascribed to the reduced Li+ ion diffusion pathways in the nanoscale material and the enhanced conductivity imparted by the carbon shell.
4. Conclusions
In conclusion, a controllable electro-less reduction strategy was reported to prepare uniform core–shell Cu6Sn5@C nanospheres from Cu@Sn@PANI precursors. The composition and lithium storage performance of the Cu–Sn@C nanospheres were heavily influenced by the synthesis parameters. Isolated, approximately isotropic Cu6Sn5/C nanospheres (40–50 nm in diameter) exhibited a demonstrably superior electrochemical performance to those composites prepared with alternative Sn:Cu ratios and offers promise as a candidate high power anode material.
Acknowledgements
This work was supported by the NSFC (21403195), the Key Research and Development Program (2016C01SA500898, 2015C01001, and 2017C01023) of Science and Technology Department of Zhejiang Province, the Shenzhen Peacock Plan (KQTD2015071616442225), and the Chinese Government “Thousand Talent” Program (Y62HB31601).References
- A. Vlad, N. Singh, C. Galande and P. M. Ajayan, Chem.–Eur. J., 2016, 22, 1–7 CrossRef.
- S. Pan, J. Ren, X. Fang and H. Peng, Adv. Energy Mater., 2016, 6, 1501867 CrossRef.
- X. L. Sun, C. L. Yan, Y. Chen, W. P. Si, J. W. Deng, S. Oswald, L. F. Liu and O. G. Schmidt, Green Chem., 2016, 18, 2078–2088 RSC.
- X. Sun, W. Si, X. Liu, J. Deng, L. Xi, L. Liu, C. Yan and O. G. Schmidt, Nano Energy, 2014, 9, 168–175 CrossRef CAS.
- J. E. Cloud, Y. Wang, T. S. Yoder, L. W. Taylor and Y. Yang, Angew. Chem., Int. Ed. Engl., 2014, 53, 14527–14532 CrossRef CAS PubMed.
- Y. Zhong, M. Yang, X. Zhou, J. Wei and Z. Zhou, Part. Part. Syst. Charact., 2015, 32, 104–111 CrossRef CAS.
- H. Kim, G. Jeong, Y. U. Kim, J. H. Kim, C. M. Park and H. J. Sohn, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC.
- M. Cao, M. Zhang, L. Xing, Q. Wang and X.-Y. Xue, J. Alloys Compd., 2017, 694, 30–39 CrossRef CAS.
- L. Su, Y. Jing and Z. Zhou, Nanoscale, 2011, 3, 3967–3983 RSC.
- U. G. Nwokeke, R. Alcántara, J. L. Tirado, R. Stoyanova, M. Yoncheva and E. Zhecheva, Chem. Mater., 2010, 22, 2268–2275 CrossRef CAS.
- U. G. Nwokeke, R. Alcántara, J. L. Tirado, R. Stoyanova and E. Zhecheva, J. Power Sources, 2011, 196, 6768–6771 CrossRef CAS.
- L. Su, Y. Xu, J. Xie, L. Wang and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 35172–35179 CAS.
- N. Mahmood, C. Z. Zhang, F. Liu, J. H. Zhu and Y. L. Hou, ACS Nano, 2013, 7, 10307–10318 CrossRef CAS PubMed.
- B. Liu, A. Abouimrane, Y. Ren, M. Balasubramanian, D. P. Wang, Z. G. Z. Fang and K. Amine, Chem. Mater., 2012, 24, 4653–4661 CrossRef CAS.
- P. Chen, L. Guo and Y. Wang, J. Power Sources, 2013, 222, 526–532 CrossRef CAS.
- G. Ferrara, C. Arbizzani, L. Damen, M. Guidotti, M. Lazzari, F. G. Vergottini, R. Inguanta, S. Piazza, C. Sunseri and M. Mastragostino, J. Power Sources, 2012, 211, 103–107 CrossRef CAS.
- F. Nacimiento, R. Alcántara and J. L. Tirado, J. Power Sources, 2011, 196, 2893–2898 CrossRef CAS.
- Z. Wang, S. Luo, F. Chen, D. Wang, Y. Liu, X. Qi, C. Shi and N. Zhao, RSC Adv., 2016, 6, 54718–54726 RSC.
- W. J. Cui, F. Li, H. J. Liu, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2009, 19, 7202–7207 RSC.
- J. Chen, L. Yang, S. Fang, Z. Zhang and S.-i. Hirano, Electrochim. Acta, 2013, 105, 629–634 CrossRef CAS.
- H. C. Shin and M. L. Liu, Adv. Funct. Mater., 2005, 15, 582–586 CrossRef CAS.
- H. Zhang, T. Shi, D. J. Wetzel, R. G. Nuzzo and P. V. Braun, Adv. Mater., 2016, 28, 742–747 CrossRef CAS PubMed.
- H.-R. Jung, E.-J. Kim, Y. J. Park and H.-C. Shin, J. Power Sources, 2011, 196, 5122–5127 CrossRef CAS.
- M. Tian, W. Wang, Y. Wei and R. Yang, J. Power Sources, 2012, 211, 46–51 CrossRef CAS.
- C. Leonhardt, A. Seifert, S. Csihony, H. Sommer and M. Mehring, RSC Adv., 2016, 6, 3091–3098 RSC.
- L. Wang, S. Kitamura, T. Sonoda, K. Obata, S. Tanase and T. Sakai, J. Electrochem. Soc., 2003, 150, A1346–A1351 CrossRef CAS.
- L. Wang, S. Kitamura, K. Obata, S. Tanase and T. Sakai, J. Power Sources, 2005, 141, 286–292 CrossRef CAS.
- M. M. Thackeray, J. T. Vaughey, A. J. Kahaian, K. D. Kepler and R. Benedek, Electrochem. Commun., 1999, 1, 111–115 CrossRef CAS.
- D. Larcher, L. Beaulieu, D. MacNeil and J. Dahn, J. Electrochem. Soc., 2000, 147, 1658–1662 CrossRef CAS.
- J. T. Vaughey, K. D. Kepler, R. Benedek and M. M. Thackeray, Electrochem. Commun., 1999, 1, 517–521 CrossRef CAS.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336–2340 CrossRef CAS.
- G. X. Wang, L. Sun, D. H. Bradhurst, S. X. Dou and H. K. Liu, J. Alloys Compd., 2000, 299, L12–L15 CrossRef CAS.
- K. D. Kepler, J. T. Vaughey and M. M. Thackeray, J. Power Sources, 1999, 81, 383–387 CrossRef.
- X.-Y. Fan, F.-S. Ke, G.-Z. Wei, L. Huang and S.-G. Sun, Electrochem. Solid-State Lett., 2008, 11, A195–A197 CrossRef CAS.
- F. C. Walsh and C. T. J. Low, Surf. Coat. Technol., 2016, 304, 246–262 CrossRef CAS.
- K. Wang, X. M. He, L. Wang, J. G. Ren, C. Y. Jiang and C. R. Wan, J. Electrochem. Soc., 2006, 153, A1859–A1862 CrossRef CAS.
- H. S. Hwang, T. Yoon, J. Jang, J. J. Kim, J. H. Ryu and S. M. Oh, J. Alloys Compd., 2017, 692, 583–588 CrossRef CAS.
- L. Xue, Z. Fu, Y. Yao, T. Huang and A. Yu, Electrochim. Acta, 2010, 55, 7310–7314 CrossRef CAS.
- J. Xie, L. Tong, L. Su, Y. Xu, L. Wang and Y. Wang, J. Power Sources, 2017, 342, 529–536 CrossRef CAS.
- M. Zhang, M. Cao, Y. Fu, L. Xing, Q. Wang and X. Xue, Mater. Lett., 2016, 185, 282–285 CrossRef CAS.
- P. Zhang, L. Wang, J. Xie, L. Su and C. a. Ma, J. Mater. Chem. A, 2014, 2, 3776–3782 CAS.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed. Engl., 2008, 47, 7461–7465 CrossRef CAS PubMed.
- N. Zhang, Q. Zhao, X. Han, J. Yang and J. Chen, Nanoscale, 2014, 6, 2827–2832 RSC.
- N. Liu, Z. D. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. T. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- L. Zhang, R. Rajagopalan, H. Guo, X. Hu, S. Dou and H. Liu, Adv. Funct. Mater., 2016, 26, 440–446 CrossRef CAS.
- S. Qiu, X. Wu, L. Xiao, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 1337–1343 CAS.
- L. Su, Y. Zhong and Z. Zhou, J. Mater. Chem. A, 2013, 1, 15158–15166 CAS.
- T. N. Khoperia, T. J. Tabatadze and T. I. Zedgenidze, Electrochim. Acta, 1997, 42, 3049–3055 CrossRef CAS.
- L. W. Su, Z. Zhou and M. M. Ren, Chem. Commun., 2010, 46, 2590–2592 RSC.
- Y. S. Jung, K. T. Lee, J. H. Ryu, D. Im and S. M. Oh, J. Electrochem. Soc., 2005, 152, A1452–A1457 CrossRef CAS.
- L. Trahey, J. T. Vaughey, H. H. Kung and M. M. Thackeray, J. Electrochem. Soc., 2009, 156, A385–A389 CrossRef CAS.
- J. S. Thorne, J. R. Dahn, M. N. Obrovac and R. A. Dunlap, J. Electrochem. Soc., 2011, 158, A1328–A1334 CrossRef CAS.
- H. Algul, M. Uysal, M. Tokur, S. Ozcan, T. Cetinkaya, H. Akbulut and A. Alp, Int. J. Hydrogen Energy, 2016, 41, 9819–9827 CrossRef CAS.
- R. Hu, G. H. Waller, Y. Wang, Y. Chen, C. Yang, W. Zhou, M. Zhu and M. Liu, Nano Energy, 2015, 18, 232–244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, components, and cycle performance of Cu–Sn@C composites synthesized under different conditions; SEM image of Cu6Sn5@C nanospheres. See DOI: 10.1039/c7ra02214j |
| This journal is © The Royal Society of Chemistry 2017 |