
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational investigation on the large energy gap between the triplet excited-states in acenes
Y. Y. Pan *ab,
J. Huanga,
Z. M. Wanga,
D. W. Yua,
B. Yang*b and
Y. G. Mac
*ab,
J. Huanga,
Z. M. Wanga,
D. W. Yua,
B. Yang*b and
Y. G. Mac
aSchool of Petrochemical Engineering, Shenyang University of Technology, 30 Guanghua Street, Liaoyang, 111003, P. R. China. E-mail: pyy39518768@163.com; Fax: +86-419-5319409; Tel: +86-419-5319409
bState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun, 130012, P. R. China. E-mail: yangbing@jlu.edu.cn; Fax: +86-431-85168502; Tel: +86-431-85193421
cState Key Laboratory of Luminescent Materials and Devices, Institute of Polymer Optoelectronic Materials and Devices, South China University of Technology, Guangzhou, 510640, P. R. China
First published on 18th May 2017
Abstract
The large energy gap between the two triplet excited-states in acenes has a huge impact on their optical and electronic properties. Accurate calculation and full use of this gap have always been a major challenge in the field of organic semiconductor materials. In the present study, we focus on the precise description of the large gap between the T1 and T2 states, and taking a series of acenes (benzene, naphthalene, anthracene, tetracene, and pentacene) as examples, investigate their excited state behavior to verify the energy gap structure. The results show that the symmetry of the transition molecular orbital and the excited state properties have a great influence on the transition energy, and may be the main cause of the large energy gap.
1. Introduction
In an electroluminescent device, electrons and holes driven by the electric field generate excitons. Generally, initially formed excitons tend to relax to the lowest singlet (S1) and triplet (T1) states before radiative decay in accordance with Kasha's rule, as a result of the much faster internal conversion (IC) rate than the intersystem crossing (ISC) rate.1 Of course, there are some rare exceptions that do not obey this rule. For instance, azulene emits from the S2 state instead of the S1 state as a result of the large energy gap between the S1 and S2 states which leads to the IC (S2 → S1) process becoming too slow to compete with the fluorescent radiation process of S2 → S0, as shown in Fig. 1(a).2,3 Similarly, a large energy gap between the triplet states can also lead to changes in the exciton radiation path. The TPA-NZP molecule displays the typical hot exciton mechanism. Its huge triplet state energy gap (ΔET1–T2) between the first (T1) and the second (T2) excited-state causes the IC (T2 → T1) rate to be so slow that it cannot compete with the RISC (T2 → S2) rate, thus ensuring the smooth channel of the high exciton and near 100% yield of singlet excitons harvested in the EL device, as shown in Fig. 1(b).4–10 In addition, P-type delayed fluorescence (triplet–triplet annihilation TTA) molecules annihilate the two triplets to form an excited singlet and a ground-state singlet to improve exciton utilization; however to completely ensure this mechanism, the energy of the T1 state is greater than the S1 but less than T2 (ES1 < 2ET1 < ET2), that is, there are energy gaps between the S1, T1 and T2, as shown in Fig. 1(c).11 The third example is singlet fission (SF) in which two triplet excitations are produced from an excited singlet. Suitable candidates for SF should have S1 state energy more than twice the T1 energy and for the efficient formation of triplets by singlet fission, it is important to ensure that neither 2ET1 − ES1 nor 2ET1 − ET2 are distinctly positive, as shown in Fig. 1(d).12–14 The above three examples show that the energy gap between the triplet states can slow down the IC rate which is comparable with the ISC rate. This will give rise to a change in the resultant ratio of excitons generated between the singlet and triplet state in the EL device, which further causes a change in fluorescent efficiency.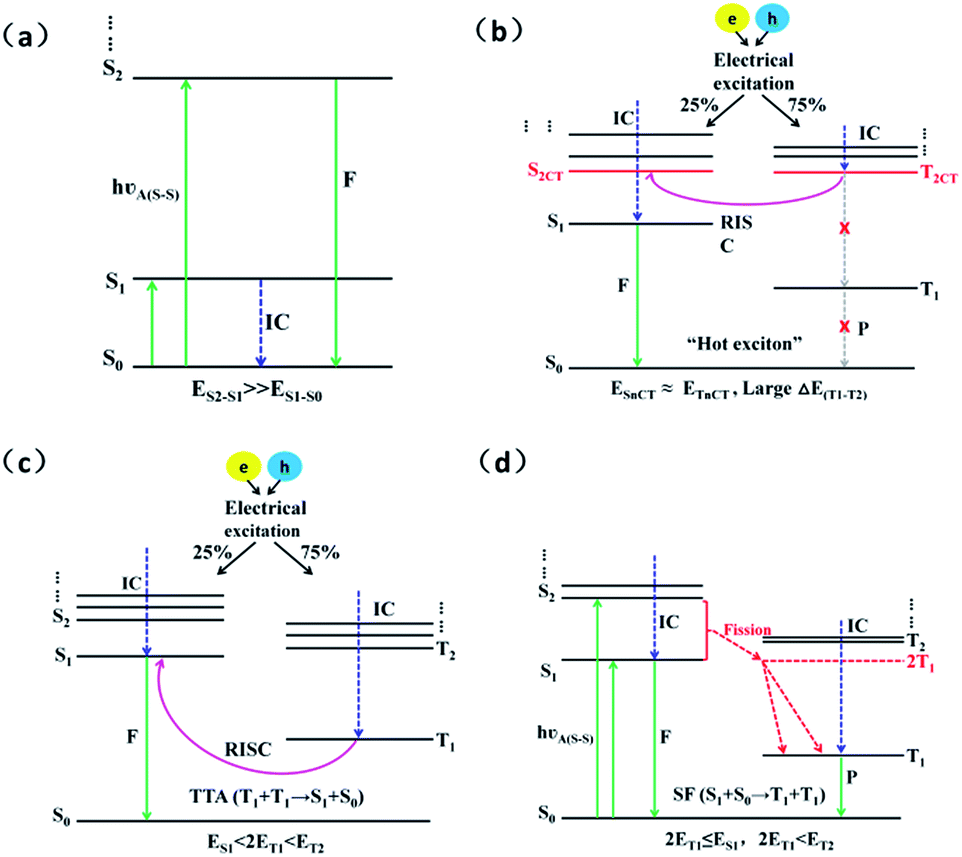 | ||
| Fig. 1 Simple schematic of the exciton decay and the electroluminescence process of (a) azulene molecules; (b) hot exciton mechanism; (c) TTA mechanism; and (d) SF mechanism. Where, S: singlet state; T: triplet state; F: fluorescence; P: phosphorescence; KIC: internal conversion rate; KRISC: reverse intersystem crossing rate; CT: charge-transfer state; and ΔEST: singlet–triplet energy splitting. | ||
Among the many electroluminescent materials, due to their simple structure, easy synthesis and high luminous efficiency, acenes have received extensive attention as potential candidate materials for TTA, hot exciton and singlet fission; thus the study of acenes triplet excited-state has also gradually increased.15–21 For instance, Lewis and Kasha,22 McClure,23 Hunziker,24,25 and Meyer et al.26 have probed experimentally the triplet states of naphthalene and the lowest triplet state in longer stable oligomers has also been determined experimentally.27–31 However, the energy of the higher triplet states and the energy gap between them have not been given any attention both experimentally and theoretically.
In the present study, we first aim to ascertain the accuracy of the method applied to describe the energy level of the triplet state, and on this basis, we further focus on the large transition energy gap between the T1 and T2 state taking a series of acenes (benzene, naphthalene, anthracene, tetracene, and pentacene) (as shown in Scheme 1) as examples to investigate the transition properties of their triplet excited states to explore the huge resulting ΔET1–T2. The study of the excited state behaviour of these molecules to verify their high level triplet excited state energy gap structure is very helpful for a rational molecular design to adjust and control the gap between the excited states with the aim of developing new ideas on the adjustable energy gap of high performance organic photoelectric functional materials.
 | ||
| Scheme 1 Representation of the acenes investigated herein. | ||
2. Methodology and computational details
As is known, the ground-state electronic structures for most chromophores can be conveniently obtained using various quantum chemistry computational methods. However, it is difficult to calculate the electronic structures of the electronic excited state for molecular systems, especially the triplet state. Nowadays, the equation of motion coupled cluster with single and double excitation model32 (EOM-CCSD) methods and time-dependent density functional theory33–38 (TD-DFT) are very popular computational methods to determine excited-state electronic structures. TD-DFT is the most widely-used method to describe the excited-state properties of medium to large molecular systems. The EOM-CCSD method takes into account both single and double substitutions and can give relatively accurate results, but the calculation cost is very high; whereas TD-DFT is a more cost-effective method to calculate the excited-state. In the present study, in order to explore the energy gap between the triplet excited states of acenes, the excited state energy needs to be calculated accurately first. To assess the performances of the TD-DFT approaches, we tried to cover a broad and diverse selection of functionals. We use the typical acenes molecule anthracene, tetracene and pentacene (Scheme 1) as examples to investigate the most suitable functionals. The performance of 2 local functionals (SVWN39 and PBE40), 7 hybrid functionals (BLYP (0%HF),41 B3LYP (20%HF),42 PBE0 (25%HF),43 BMK (42%HF),44 BH and HLYP (50%HF),45 M06-2X (56%HF),46 and M06HF (100%HF),47) and 2 long-range-corrected functionals CAM-B3LYP48 and ωB97X49 in combination with 6-31+G(d,p) basis sets50 is explored. The EOM-CCSD method is also included for comparison purposes.In this part, we focus on describing the transition energy level of the triplet states (T1 and T2) accurately. As shown in Fig. 2, we used different methods and functionals as abscissa, and the difference between the experimental value as the ordinate for mapping histograms.51 On the whole, each functional of the DFT method shows a tendency to gradually underestimate the transition energy as the conjugate length increases. When the conjugate length is small (anthracene), the difference between the calculated values of T1 and T2 and the experimental values is not as large as the whole, where the maximum value is 0.83 eV and the minimum value is only 0.01 eV, among which the BLYP, B3LYP and ωB97X functionals are controlled within the range of 0.1 eV. When the conjugate length increases to tetracene, the difference increases and the maximum value reaches −1.96 eV, where the BLYP and ωB97X functionals remain within the range of 0.1. When the conjugate length continues to increase to pentacene, the difference is further increased, where the maximum value is −2.08 eV, as calculated by the BH and HLYP functional. As can be seen in Fig. 2(e) and (f), the difference values are systematically negative, which indicates that the TD-DFT functional tends to underestimate the transition energies in the relatively long conjugate systems. Only the ωB97X functional continues to maintain a difference of less than 0.1. This situation may be due to the conjugate length growth, where the distance of the electron transition increases, and similar to the charge transfer process, the traditional local functionals and most hybrid functionals underestimate the transition energy with an increase in conjugate length. However, the ωB97X functional optimizes the range separation parameter ω through the self-consistent restriction system, which aims at physically motivated correction for the long-range Coulomb interaction. This functional can be established to enable the accurate and quantitative description of the intramolecular charge-transfer (CT) state. At the same time, the EOM-CCSD method also exhibits the trend that with conjugate length growth the transition energy gradually goes from being overvalued to underestimated. Considering the computational accuracy and cost, the TD-DFT/ωB97X method was finally chosen to describe the excited-state properties of acenes. Earlier benchmarks have shown that different functionals have almost no effect on the geometries of the ground states and the minimum structures are similar in accuracy to the structures obtained from experiment.52 Herein, all ground state structures were optimized using the B3LYP functional combined with the 6-31+G(d,p) basis set. The optimized structures were confirmed to be local energy minima by calculating vibrational frequencies. All DFT/TD-DFT and EOM-CCSD calculations were performed with Gaussian09.D.01 Revision.53
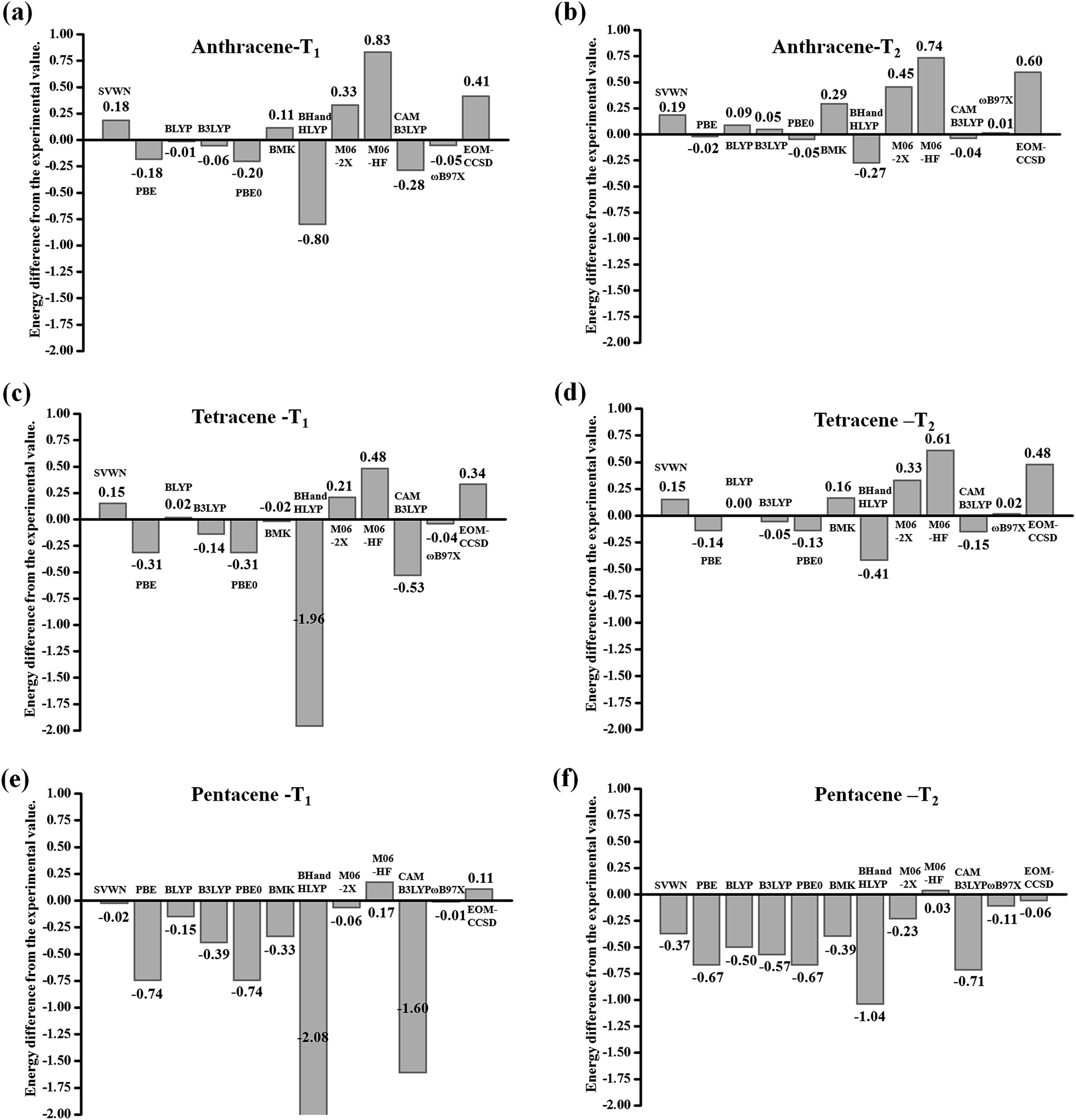 | ||
| Fig. 2 Histograms of the difference from the experimental value for the triplet vertical excitation energies (eV) calculated using TD-DFT and the EOM-CCSD method with respect to the acene: (a) T1 for anthracene; (b) T2 for anthracene; (c) T1 for tetracene; (d) T2 for tetracene; (e) T1 for pentacene and (f) T2 for pentacene. | ||
3. Results and discussion
3.1 Transition energy gap between T1 and T2 (ΔET1–T2)
We take the conjugate length (number of benzene ring) as the abscissa and triplet excitation energy as the ordinate in drawing presented in Fig. 3. Overall, with the growth of the conjugated length and the excitation energies of T1 and T2 show a downward trend, which can be explained by the fact that the excitation energy of the triplet states is associated with the conjugate length, and the longer the length the lower the transition energy. However, there is a special phenomenon that from benzene to anthracene T1 decreases by about 1.0 eV for each benzene added and it decreases by only about 0.2 eV from tetracene to pentacene. At the same time, the excitation energy of T2 reduces by a similar value of about 0.8 eV from benzene to anthracene, until from tetracene to pentacene the gap between T2 is reduced to 0.2 eV. Thus, the trend of the gap between T1 and T2 (ΔET1–T2) first increases (from 1.10 eV to 1.44 eV) then decreases (from 1.44 eV to 1.33 eV) and finally stabilizes (from 1.33 eV to 1.37 eV).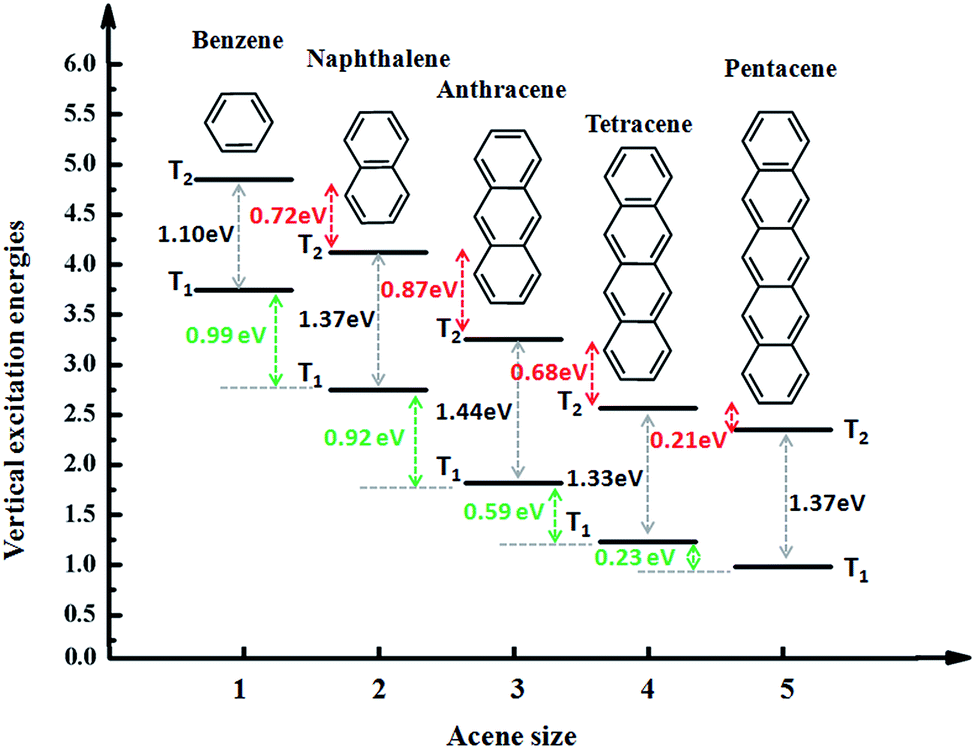 | ||
| Fig. 3 Energy diagrams of the acenes (benzene, naphthalene, anthracene (An), tetracene, and pentacene). | ||
Anthracene has the largest ΔET1–T2 and benzene has the smallest ΔET1–T2. Is the large ΔET1–T2 related to its structural characteristics? To facilitate a comparison, we chose a series of common anthracene derivatives, for which their triplet excitation energy was calculated using the TD-DFT/ωB97X method, as presented in Fig. 4. The results show that due to the introduction of heteroatoms, the energy changes a lot, where ΔET1–T2 both increases and decreases, which indicates that the cause of the large ΔET1–T2 is the structure and other reasons. In the next section we will discuss the cause of the large more ΔET1–T2 by analyzing the nature of the transitional or excited states.
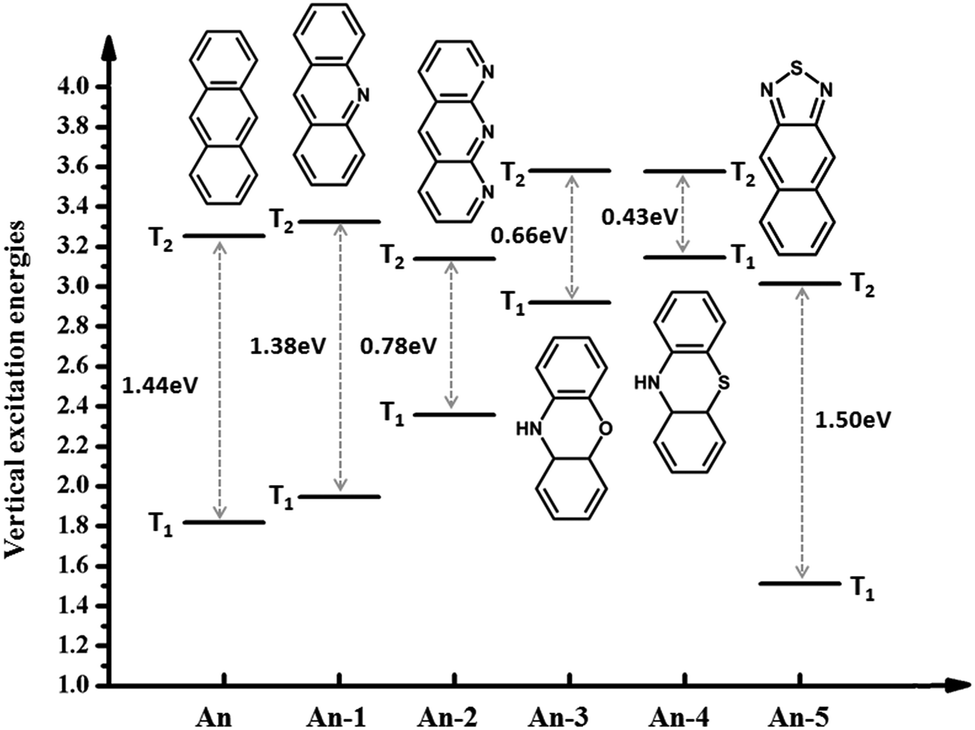 | ||
| Fig. 4 Energy diagrams of anthracene derivatives of the first two triplet excited-states from calculation. | ||
3.2 The molecular orbitals (MOs)
We analyzed the transition configurations of the T1 and T2 excited-state of the acenes. As shown in Table 1, the T1 transition is mainly contributed from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), and the coefficient increases with the conjugate length. It is implied that the longer the conjugate length, the more intense the transition from the HOMO to LUMO. On the contrary, the transition of the T2 state presents other modes, since it is a transition from the inner MOs of the HOMO to the LUMO. For example the transition of the T2 state in anthracene is HOMO−2 → LUMO, tetracene is HOMO−2 → LUMO and pentacene is HOMO−1 → LUMO and molecules with such transitions are accompanied by a large ΔET1–T2 in energy levels. Upon further analysis of the orbital energy, it was noted that the energy differences between HOMO−1 and HOMO in anthracene, tetracene and pentacene are very large, which are 1.28 eV, 1.62 eV and 1.44 eV, respectively. Therefore, the electron transition energies from these inner occupied orbitals to the LUMO are much larger than from the HOMO to LUMO. We also analyzed the symmetry of the molecular orbital, as listed in Table 1. As is known, the electronic transition is in accordance with the rules of parity, which requires that the molecules must change their symmetry after the transition. In other words, the transition is allowed if the symmetry of the orbital is changed after the electron transition, for example u → g and g → u, whereas the transition is prohibited for u → u and g → g. As shown in Table 1, from naphthalene to pentacene, the T2 all show the transition between the same parity MO, which means that these transitions are parity-forbidden. This feature may lead to the emergence of a large energy difference between T1 and T2.| MOs character and coefficient | Orbital symmetry | Transition probability (YES or NO) | ΔET1–T2 (eV) | |
|---|---|---|---|---|
| Benzene | T1 HOMO → LUMO 0.48 | e1g → e2u | YES | 1.10 |
| T2 HOMO → LUMO + 1 0.47 | e1g → e2u | YES | ||
| T3 HOMO−1 → LUMO + 1 0.47 | e1g → e2u | YES | ||
| Naphthalene | T1 HOMO → LUMO 0.66 | au → b1g | YES | 1.37 |
| T2 HOMO−1 → LUMO 0.50 | b2g → b1g | NO | ||
| T3 HOMO−2 → LUMO 0.49 | b3u → b1g | YES | ||
| Anthracene | T1 HOMO → LUMO 0.69 | b2g → b3u | YES | 1.44 |
| T2 HOMO−2 → LUMO 0.50 | au → b3u | NO | ||
| T3 HOMO−1 → LUMO 0.61 | b1g → b3u | YES | ||
| Tetracene | T1 HOMO → LUMO 0.71 | au → b1g | YES | 1.33 |
| T2 HOMO−2 → LUMO 0.50 | b2g → b1g | NO | ||
| T3 HOMO−1 → LUMO 0.67 | b3u → b1g | YES | ||
| Pentacene | T1 HOMO → LUMO 0.77 | b2g → b3u | YES | 1.37 |
| T2 HOMO−1 → LUMO 0.51 | au → b3u | NO | ||
| T3 HOMO−3 → LUMO 0.43 | b2g → b3u | YES |
3.3 Excited state properties
Excited-state configurations from the stationary points of the ground-state energy surfaces were then obtained at the TD-DFT/ωB97X/6-31+G(d,p) level. We analyzed the natural transition orbital (NTOs)54 of the T1 and T2 excited-states of acenes. From Fig. 5, the nature of the two excited states, T1 and T2, deserves some other comments. In the transition process, the “particles” are very similar but the “holes” are different for T1 and T2. From benzene to pentacene the T1 state is largely represented by singly excited configurations, and among them, the HOMO → LUMO has the largest weight. The π → π* density is mostly centered between the C atoms so that the T1 transition may be classified as a covalent state. On the contrary, the contributions to the T2 state from all singly excited configurations are smaller and more balanced, which correspond to the promotion of an electron from HOMO−1 or HOMO−2 to LUMO, where the location of the electron density distribution changes in different repeat units, such as the charge-transfer state. On the other hand, the NTOs of the T1 and T2 transition have extremely similar electron cloud distributions with the frontier molecular orbitals (FMOs). The NTOs of the T1 state indicate the character of the local excited state, and the T2 state shows some characteristics of the charge-transfer state. The two excited states show different characteristics of the excited state and the orbital symmetry, thus there is a big difference in energy.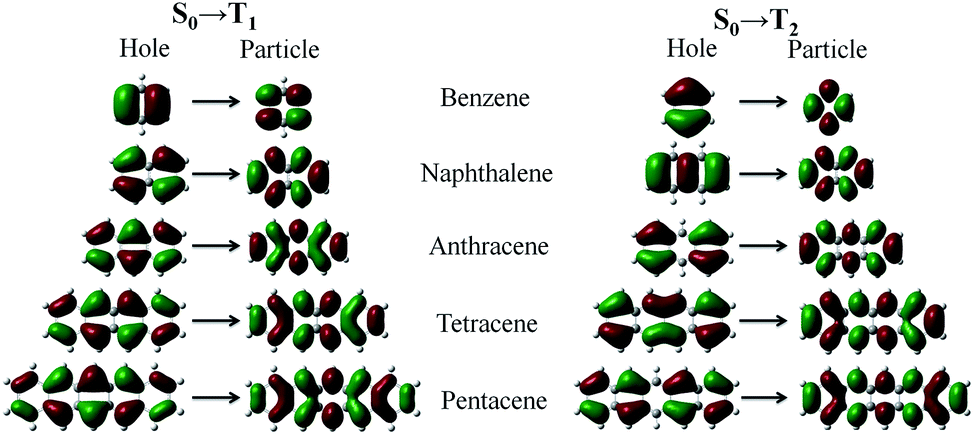 | ||
| Fig. 5 Natural transition orbital (NTOs) of T1 and T2 for the acenes (benzene, naphthalene, anthracene, tetracene, and pentacene). | ||
Since the transition of the triplet state is forbidden, we could not calculate the transition matrix, which cannot be intuitive to determine whether the transition properties will affect its energy difference. Hence, for further verification, we selected a few molecules that have large singlet energy differences, ΔES1–S2, and quantified the composition of these excited states.
We calculated the wave functions of the electron–hole pairs from the transition density matrix and plotted them in a two-dimensional (2D) color-filled map, which is related to the probability of finding the electron and hole in the atomic orbitals localized on each non-hydrogen atom, using the Multiwfn software 3.3.9 Revision.55 From this map we can understand which atoms are mainly affected by the electron transition and which atom pairs are strongly coherent when the electron transits. If the effect is stronger during the transition the value is brighter in the map. The diagonal part of the map represents the LE component localized on the main backbone, whereas the off-diagonal region denotes the CT component. The map in Fig. 6 shows that the values of the S1 and the S2 transition have a great difference, for example, the values of the S1 state of azulene (ΔES1–S2 = 1.48 eV) are only concentrated in the lower left quarter of the map in the diagonal, but the values of the S2 state transition are distributed throughout the graph. Both the diagonal and the off-diagonal are included, which indicates that the transition of the S1 state are LE and localized on one of the group of the azulene molecule. On the contrary, the S2 state exhibit both CT and LE properties. The same trend is displayed for the other molecules in Fig. 6. This means that the transition properties of the S1 state and the S2 state are different in mechanism. It is also seen from the distribution of electron clouds of NTOs that the S1 and S2 states of pentacene and An-5 are the same as that of the T1 and T2 states, which indicates that the transitions of the T1 and T2 states also have different transition properties. This may also be one of the reasons that lead to the energy difference.
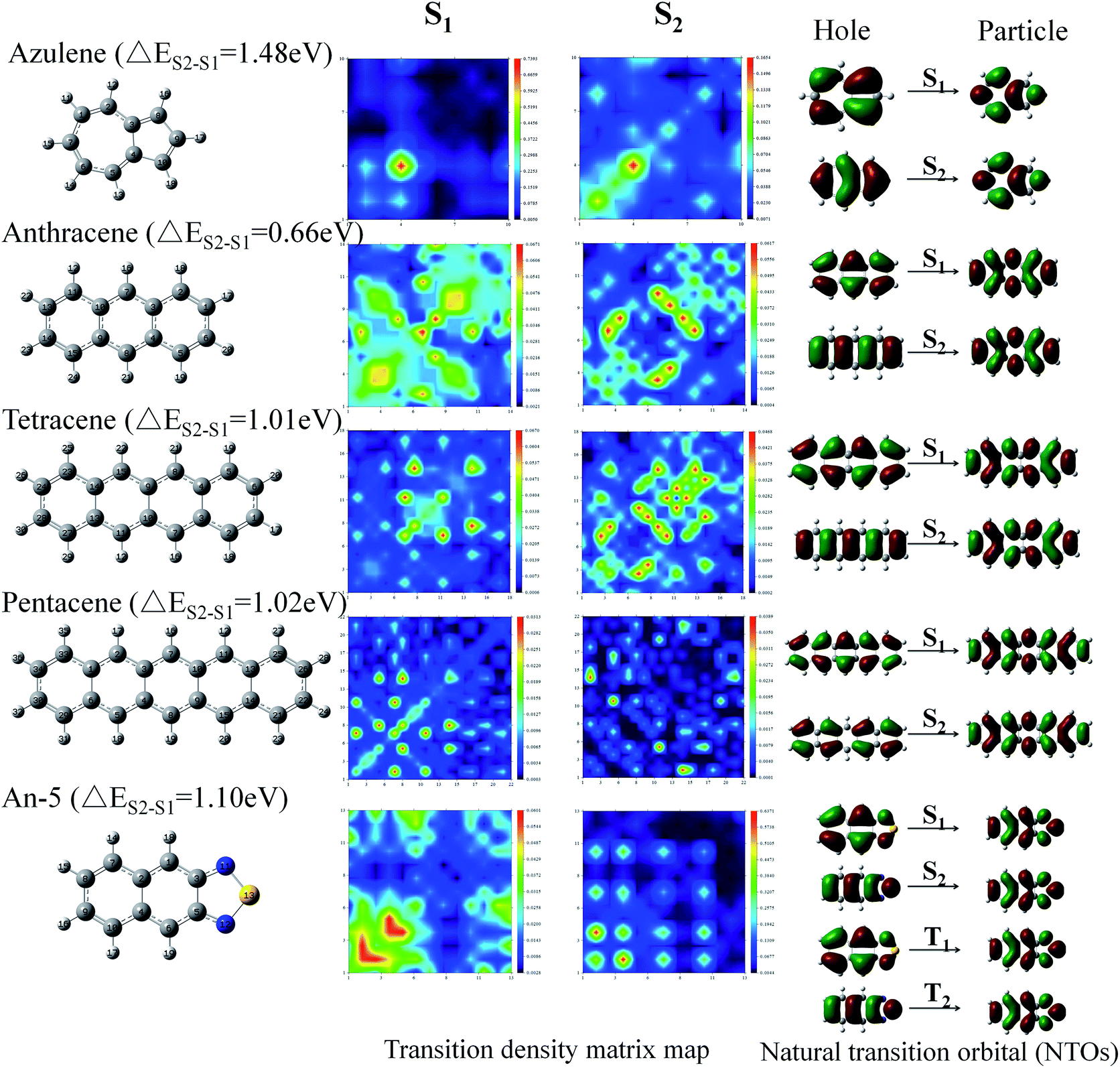 | ||
| Fig. 6 Transition density matrix map and the natural transition orbital (NTO) of S1 and S2 for some molecules (azulene, anthracene, tetracene, pentacene and An-5), that have a large ΔES1–S2. | ||
4. Conclusions
The description of the triplet excited state is highly challenging for standard density functionals. Using the typical acenes molecules anthracene, tetracene and pentacene, we benchmarked the EOM-CCSD method and 2 local functionals (SVWN and PBE), 7 hybrid functionals (BLYP, B3LYP, PBE0, BMK, BHHLYP, M06-2X and M06HF), and two long-range-corrected functional CAM-B3LYP and ωB97X of the DFT method in combination with the 6-31+G(d,p) basis sets in the framework of the simulation of the triplet excited state properties. The overall results suggest that the ωB97X functional is the closest to the experimental values. On the basis of calculating the transition energy of the excited state accurately, we find that the ΔET1–T2 of the acenes molecules has a certain trend and anthracene has the largest. We further studied the triplet transition molecular orbital and excited state properties of the acene molecules. The results show that the symmetry of the transition molecular orbital and the excited state properties are very different in the molecule with a relatively large energy gap. This finding will provide an approach for the design of adjustable energy molecules and provide a reference for the application of energy levels of triplet excited states in the future.Acknowledgements
We are grateful for financial support from National Science Foundation of China (grant number 51603127, 91233113, 51273078, 51473063, 51203091) and National Basic Research Program of China (973 Program grant number 2013CB834705, 2013CB834801, 2015CB655003).References
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14 RSC.
- M. Damm, F. Deckert, H. Hippler and J. Troe, J. Phys. Chem., 1991, 95, 2005 CrossRef CAS.
- P. Foggi, F. V. R. Neuwahl, L. Moroni and P. R. Salvi, J. Phys. Chem. A, 2003, 107, 1689 CrossRef CAS.
- W. J. Li, D. D. Liu, F. Z. Shen, D. G. Ma, Z. M. Wang, Y. Xu, B. Yang, Y. G. Ma and T. Feng, Adv. Funct. Mater., 2012, 22, 2797 CrossRef CAS.
- W. J. Li, Y. Y. Pan, R. Xiao, Q. M. Peng, S. T. Zhang, D. G. Ma, F. Li, F. Z. Shen, Y. H. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2014, 24, 1609 CrossRef CAS.
- S. Tang, W. J. Li, F. Z. Shen, D. D. Liu, B. Yang and Y. G. Ma, J. Mater. Chem., 2012, 22, 4401 RSC.
- S. T. Zhang, W. J. Li, L. Yao, Y. Y. Pan, B. Yang, Y. G. Ma, F. Shen and R. Xiao, Chem. Commun., 2013, 49, 11302 RSC.
- L. Yao, S. T. Zhang, R. Wang, W. J. Li, F. Z. Shen, B. Yang and Y. G. Ma, Angew. Chem., 2014, 126, 2151 CrossRef.
- Y. Y. Pan, W. J. Li, S. T. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. G. Ma, Adv. Opt. Mater., 2014, 2, 510 CrossRef CAS.
- W. J. Li, Y. Y. Pan, L. Yao, H. C. Liu, S. T. Zhang, C. Wang, F. Z. Shen, B. Yang, Y. G. Ma and P. Lu, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- Y. C. Luo and H. Aziz, Adv. Funct. Mater., 2010, 20, 1285 CrossRef CAS.
- C. E. Swenberg and N. E. Geacintov, Org. Mol. Photophys., 1973, 18, 489 CAS.
- M. Pope and C. E. Swenberg, Electronic Processes in Organic Crystals and Polymers, Oxford University Press, Oxford, U.K., 2nd edn, 1999, p. 134 Search PubMed.
- M. Smith and J. Michl, Chem. Rev., 2010, 110, 6891 CrossRef CAS PubMed.
- T. Zeng, R. Hoffmann and N. Ananth, J. Am. Chem. Soc., 2014, 136, 5755 CrossRef CAS PubMed.
- S. F. Nelson, Y. Y. Lin, D. J. Gundlach and T. N. Jackson, Appl. Phys. Lett., 1998, 72, 1854 CrossRef CAS.
- A. Skotheim and J. E. Reynolds, Conjugated Polymers: Theory, Synthesis, Properties, and Characterization, CRC Press, 2006 Search PubMed.
- K. Müllen and G. Wegner, Electronic Materials: The Oligomer Approach, Wiley-VCH, 1998 Search PubMed.
- J. H. Burroughes, D. D. C. Bradeley, A. R. Brown, R. N. Marks, K. Machay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- M. Berggren, O. Inganas, J. Rasmusson, G. Gustafsson, M. R. Andersson, O. Wennerstrom and T. Hjertberg, Nature, 1994, 372, 444 CrossRef CAS.
- T. Dobbertin, M. Kroeger, D. Heithecker, D. Schneider, D. Metzdorf, H. Neuner, E. Becker, H.-H. Johannes and W. Kowalsky, Appl. Phys. Lett., 2003, 82, 284 CrossRef CAS.
- G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1994, 66, 2100 CrossRef.
- D. S. McClure, J. Chem. Phys., 1949, 17, 905 CrossRef CAS.
- H. Hunziker, Chem. Phys. Lett., 1969, 3, 504 CrossRef CAS.
- H. E. Hunziker, J. Chem. Phys., 1972, 56, 400 CrossRef CAS.
- Y. H. Meyer, R. Astier and J. M. Leclercq, J. Chem. Phys., 1972, 56, 801 CrossRef CAS.
- S. P. McGlynn, T. Azumi and M. Kasha, J. Chem. Phys., 1964, 40, 507 CrossRef CAS.
- T. G. Pavlopoulos, J. Chem. Phys., 1972, 56, 227 CrossRef CAS.
- J. Burgos, M. Pope, C. E. Swenberg and R. R. Alfano, Phys. Status Solidi B, 1977, 83, 249 CrossRef CAS.
- C. Hellner, L. Lindqvist and P. C. Roberge, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 1928 RSC.
- H. Angliker, E. Rommel and J. Wirz, Chem. Phys. Lett., 1982, 87, 208 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer III, J. Chem. Phys., 1989, 90, 3700 CrossRef CAS.
- Y. Y. Pan, J. Huang, S. T. Zhang, D. W. Yu, B. Yang and Y. G. Ma, RSC Adv., 2016, 6, 108404 RSC.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
- C. Van Caillie and R. D. Amos, Chem. Phys. Lett., 1999, 308, 249 CrossRef CAS.
- C. Van Caillie and R. D. Amos, Chem. Phys. Lett., 2000, 317, 159 CrossRef CAS.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- A. D. Boese and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126 CrossRef CAS PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef CAS.
- H. Chakraborty and A. Shuklab, J. Chem. Phys., 2014, 141, 164301 CrossRef PubMed.
- Y. Y. Pan, J. Huang, Z. M. Wang, D. W. Yu, B. Yang and Y. G. Ma, RSC Adv., 2017, 7, 19576 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 477 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |