
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene-based CO2 sensing and its cross-sensitivity with humidity
Anderson D. Smith *a,
Karim Elgammalbc,
Xuge Fand,
Max C. Lemme*e,
Anna Delinbcf,
Mikael Råsanderbg,
Lars Bergqvistbc,
Stephan Schröderd,
Andreas C. Fischerdh,
Frank Niklaus*d and
Mikael Östling*a
*a,
Karim Elgammalbc,
Xuge Fand,
Max C. Lemme*e,
Anna Delinbcf,
Mikael Råsanderbg,
Lars Bergqvistbc,
Stephan Schröderd,
Andreas C. Fischerdh,
Frank Niklaus*d and
Mikael Östling*a
aDepartment of Integrated Circuits and Devices, School of Information and Communication Technology, KTH Royal Institute of Technology, Electrum 229, SE-16440 Kista, Sweden. E-mail: andsmi@kth.se; ostling@kth.se
bDepartment of Materials and Nano Physics, School of Information and Communication Technology, KTH Royal Institute of Technology, Electrum 229, SE-16440 Kista, Sweden
cSwedish e-Science Research Center (SeRC), KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden
dDepartment of Micro- and Nanosystems, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden. E-mail: frank.niklaus@ee.kth.se
eRWTH Aachen, Otto-Blumenthal-Str., 52074 Aachen, Germany. E-mail: max.lemme@uni-siegen.de
fDepartment of Physics and Astronomy, Materials Theory Division, Uppsala University, Box 516, SE-75120 Uppsala, Sweden
gDepartment of Materials, Imperial College London, Exhibition Road, SW7 2AZ London, UK
hInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 21st April 2017
Abstract
We present graphene-based CO2 sensing and analyze its cross-sensitivity with humidity. In order to assess the selectivity of graphene-based gas sensing to various gases, measurements are performed in argon (Ar), nitrogen (N2), oxygen (O2), carbon dioxide (CO2), and air by selectively venting the desired gas from compressed gas bottles into an evacuated vacuum chamber. The sensors provide a direct electrical readout in response to changes in high concentrations, from these bottles, of CO2, O2, nitrogen and argon, as well as changes in humidity from venting atmospheric air. From the signal response to each gas species, the relative graphene sensitivity to each gas is extracted as a relationship between the percentage-change in graphene's resistance response to changes in vacuum chamber pressure. Although there is virtually no response from O2, N2 and Ar, there is a sizeable cross-sensitivity between CO2 and humidity occurring at high CO2 concentrations. However, under atmospheric concentrations of CO2, this cross-sensitivity effect is negligible – allowing for the use of graphene-based humidity sensing in atmospheric environments. Finally, charge density difference calculations, computed using density functional theory (DFT) are presented in order to illustrate the bonding of CO2 and water molecules on graphene and the alterations of the graphene electronic structure due to the interactions with the substrate and the molecules.
Introduction
The low cost and general scalability of solid state gas sensors has led to their increasing usage in a number of applications1,2 from breath sensors3 to smoke detectors.1 Previous and current gas sensor research has been primarily focused on sensors based on metal oxides,4 nanowires,5–8 nanotubes,5,6 graphene and graphene oxide.9–19 Its extraordinary electronic properties make graphene an attractive sensing material, which has demonstrated the ultimate level of sensitivity – the ability to detect a single molecule.20 Graphene's high electrical conductivity is formed by its dangling π-orbitals. These π-orbitals interact with the orbitals of neighboring atoms forming conduction bands9 and lead to high carrier mobility.21,22 However, these delocalized electrons are highly sensitive to changes in the local environment – allowing graphene to have a high sensitivity to a number of gases.23–27 When molecules adsorb onto the graphene surface, the bonding causes changes in the electronic structure of graphene. This correspondingly may change its effective carrier density or electron mobility, or both. There are numerous studies which have been performed on graphene's humidity sensing properties.13,17,18,28,29 Further, CO2 sensitivity has been both theorized and previously reported in graphene and carbon nanotubes.30–36 However, the possibility of cross-sensitivity of humidity with CO2 has not been addressed in graphene. This work analyzes the sensitivity and selectivity of both humidity and CO2 sensing for a graphene gas sensor and demonstrates that, while a cross-sensitivity between the two gases exists, the sensitivity of graphene to CO2 is negligible for typical CO2 concentration conditions in air.Experimental setup
The graphene device fabrication begins with a p-type doped silicon substrate chip which is diced to dimensions of 0.92 cm by 0.96 cm. SiO2 is then thermally grown to a thickness of 300 nm. 20 nm of titanium and 80 nm of gold are deposited onto the SiO2 using e-beam metal evaporation. This contact layer is then patterned using a lift-off process. By patterning contacts prior to the graphene transfer, the number of subsequent post-graphene transfer process steps is limited – thereby mitigating process-induced damage to the graphene layer. Next, graphene is transferred using a standard wet transfer technique.37–39 Commercially available chemical vapor deposited (CVD) graphene from Graphenea Inc., Spain is used. During the CVD growth process, monolayer graphene is uniformly grown on one side of a finely polished copper foil. Though the CVD process deposits carbon on both sides of the foil, the back side of the foil is lower quality and consists of primarily amorphous carbon rather than graphene. A layer of poly(bisphenol A) carbonate (PC) is applied to the side of the foil containing the high quality graphene. This acts as a protective layer for the graphene during its transfer from the copper onto the device substrate.37–41 Residues on the back side of the foil are subsequently removed using O2 plasma. The copper foil is then placed, graphene side up, into a solution of FeCl3 and left floating on the liquid surface. The FeCl3 etches the copper layer leaving only the polymer/graphene stack. Afterwards, the graphene is transferred from the FeCl3 solution into water followed by placing it in 8 vol% HCl in order to remove residues of FeCl3. Then, the graphene is transferred again to water as a final cleaning step. After cleaning, the graphene and supporting polymer layer are manually transferred onto a target chip and let dry on a hotplate for about 10 minutes at 45 °C. Once dried, the devices are placed into chloroform for about 12 hours in order to strip/dissolve the protective polymer layer.The transferred grapheme layer is patterned using standard photolithography and O2 plasma etching. Devices are then wire bonded and housed inside a dual-in-line-package (DIP). The packaged devices are electrically characterized inside a vacuum chamber. CO2 in the chamber is controlled by venting and pumping the desired gas into the evacuated chamber through the connection of a compressed CO2 gas bottle to the vacuum chamber. CO2 in the chamber is monitored by a commercial CO2 Engine K30 CO2 sensor from SenseAir AB, Sweden for concentrations of CO2 below 4000 ppm. In addition, the chamber pressure is monitored by a PDR 9000 (MKS Instruments, USA) digital vacuum transducer, the chamber humidity is monitored by an HIH-4000 (Honeywell, USA) humidity sensor and the chamber temperature is monitored by a LM35 temperature sensor (Texas Instruments, USA). In most experiments, since pure CO2 gas is introduced into the chamber, chamber pressure is used as an indicator for the chamber's CO2 molar volume. This allows for control of the CO2 molar volume ranging from approximately 730 dm3 mol−1 to 34 dm3 mol−1. Along with CO2, pure dry gases of O2, Ar, N2, as well as air are also pumped into the evacuated chamber in order to determine their effect on the device resistance. For the resistance measurements of the graphene, current is pulsed through the devices at 200 mV square wave pulses for durations of 500 μs in order to ensure that the effects of self-heating are minimal.3,42
Computational details
Ground-state density functional theory (DFT) calculations for graphene exposed to both humid and CO2-rich conditions have been performed in order to further understand the underlying sensing mechanism. The substrate configurations consist of a (111) plane cut in SiO2 cristobalite (with a Q03 defective Si surface atom). The first configuration is a monolayer of a 2 × 2 graphene supercell on top of the defected SiO2 substrate. The effect of the molecule on the graphene electronic structure is localized to the part of the graphene sheet close to the molecule and a 4 × 4 graphene supercell was seen to give virtually identical results to the 2 × 2 matrix with regard to bonding distances and charge density differences. The second configuration introduces a H2O molecule whose oxygen component is located mid-way between two carbon atoms within the graphene hexagon. Similarly, the third configuration is formed by adding a CO2 molecule in place of the H2O molecule (the carbon in the CO2 molecule has the same coordinates as the oxygen in the H2O molecule). Hamann, Schluter, Chiang and Vanderbilt (HSCV)43,44 norm-conserving pseudo-potentials were used for all the atoms together with the Quantum Espresso (QE)45 simulation package, with a plane-wave basis set and a kinetic energy cutoff of 130 Ry for the wave function. The Brillouin zone was sampled using a 16 × 16 × 1 Monkhorst–Pack46 k-point grid and a Methfessel–Paxton smearing of 0.0037 Ry. The Perdew, Burke and Ernzerhof (PBE) functional47 of the exchange–correlation part of the density functional was used with semi empirical Grimme's corrections48,49 for the van der Waals corrections. All calculations are spin polarized. The geometries for the substrates and graphene were converted from CIF formats to Quantum Espresso input files using the CIF2Cell code50 downloaded from ref. 51. The used CIF files (for graphene,52 cristobalite and quartz53) were downloaded from the materials project repositories,54,55 where they were fetched originally from the ICSD library56,57 then further relaxed as bulk structures via the materials project servers.Results and discussion
The sensing device is composed of a silicon substrate with a layer of SiO2 thermally grown on top (Fig. 1a). Contact pads are shown in gold and graphene is represented by a dark strip over the contacts. The active sensing region of the device consists of the region between the two innermost contacts. A close-up of the active region is represented by the area within the orange dash rectangle. The devices are packaged in order to ensure a reliable operation inside the vacuum chamber as shown in a color-enhanced scanning electron microscope (SEM) image (Fig. 1b). The graphene region is shaded blue, the contact pads are shaded yellow and wire bonds are shaded orange. After packaging, the devices are placed inside a vacuum chamber for characterization.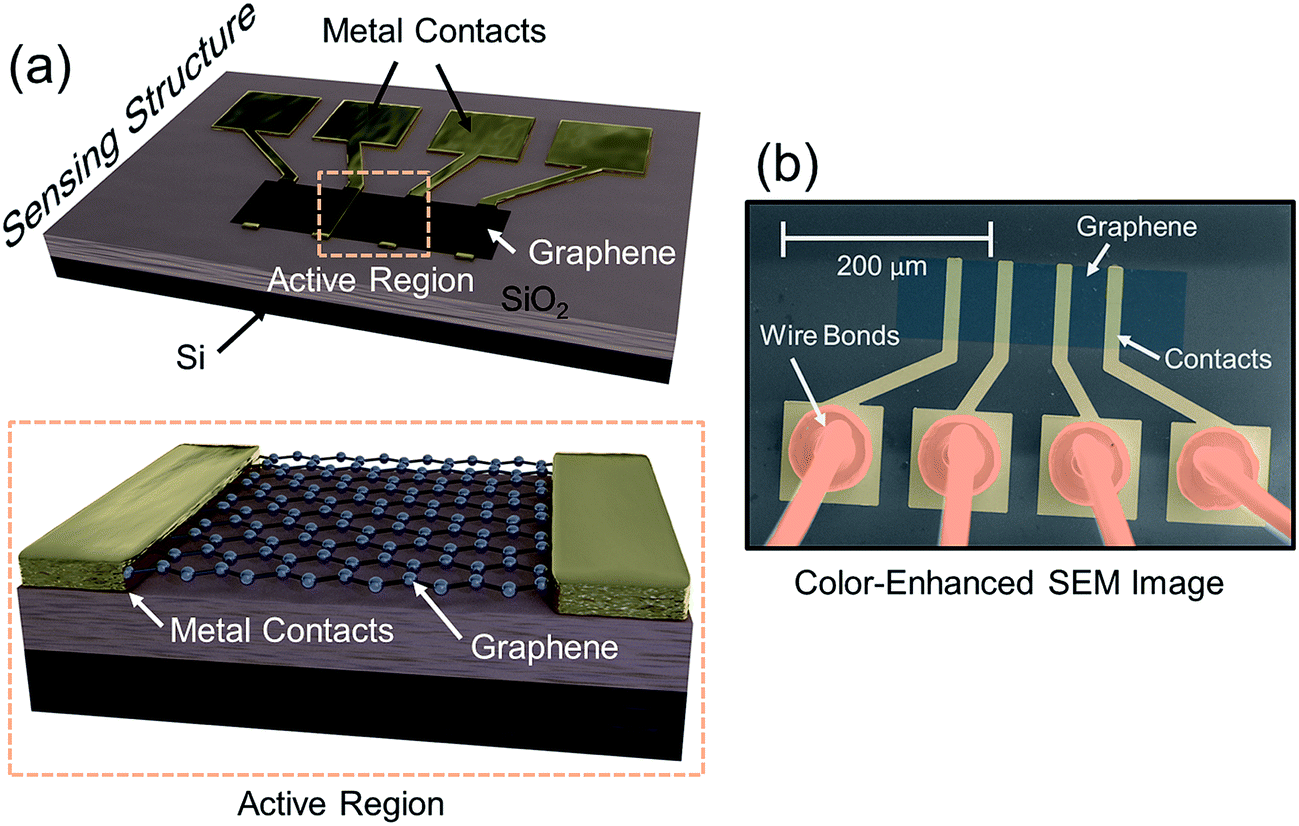 | ||
| Fig. 1 (a) Sensor setup with a layer of SiO2 thermally grown on top of Si. The contact pads are shown in gold and the graphene is represented by a dark strip over the contracts. The active sensing region of the device consists of the region between the two innermost contacts. A close-up of the active region is represented by the area within the orange dash rectangle. (b) Color-enhanced SEM image of the sensing device. The graphene region is shaded blue, the contact pads are shaded yellow and wire bonds are shaded orange. The device was wire bonded and housed inside a DIP package in order to electrically connect it inside the vacuum chamber. | ||
CO2 is vented into the chamber from a compressed CO2 bottle allowing the CO2 molecules to bind with the graphene surface as they diffuse into the chamber (Fig. 2a). By controlling the chamber pressure through a combination of a pump and an inlet valve for the gas, the CO2 molar volume in the chamber can likewise be controlled and the device response monitored. In the case of the device response shown in Fig. 2b, as the chamber pressure is modulated by the pump (changing the CO2 molar volume), there is a corresponding resistance response. During this pumping process, the chamber humidity is also closely monitored by the HIH-4000 commercial humidity sensor to ensure that the humidity is maintained at a near constant value. Chamber humidity is controlled by first evacuating the chamber to less than 100 mbar and then venting the chamber with argon. This is repeated 3 to 4 times until the chamber humidity is less than 1% relative humidity. This is done in order to mitigate humidity sensing effects during evaluation of graphene's CO2 sensing properties. The chamber is then evacuated to low pressure (less than 100 mbar) before the initial venting of CO2 gas. The CO2 is then kept in the chamber in order to allow the device response to reach a steady state value before pumping the CO2 from the chamber and monitoring the device response (initial response characteristics will be discussed in a following section). The graphene resistance response to changes in CO2 molar volume during pumping is linear with a coefficient of determination (R2) value of 0.9661. The R2 value estimates how linear the data is. A high R2 (close to 1) indicates highly linear behavior (Fig. 2b). In our case, the R2 value suggests that the data is highly linear. The CO2 resistance response of the graphene is very stable (Fig. 2c) when the chamber pressure is held constant. Further, the device has a fast response time (of less than 3 seconds) measured from the time it takes for the device to go from 10% to 90% of its final resistance change (Fig. 2d). As mentioned, the chamber pressure is used to estimate the chamber's CO2 molar volume using the ideal gas law (Fig. 2e). The molar volume is determined by first calculating the molar amount (nCO2) of CO2 from the ideal gas law (eqn (1)) where P is the pressure, V is the chamber volume, R is the gas constant and T is the temperature. The molar volume (ρmol) is then calculated by dividing the chamber volume by the molar amount (eqn (2)).
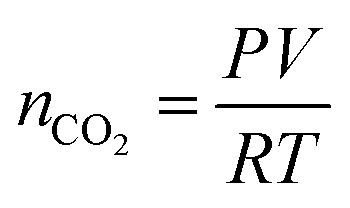 | (1) |
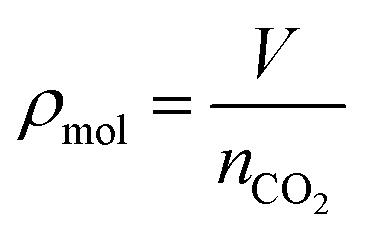 | (2) |
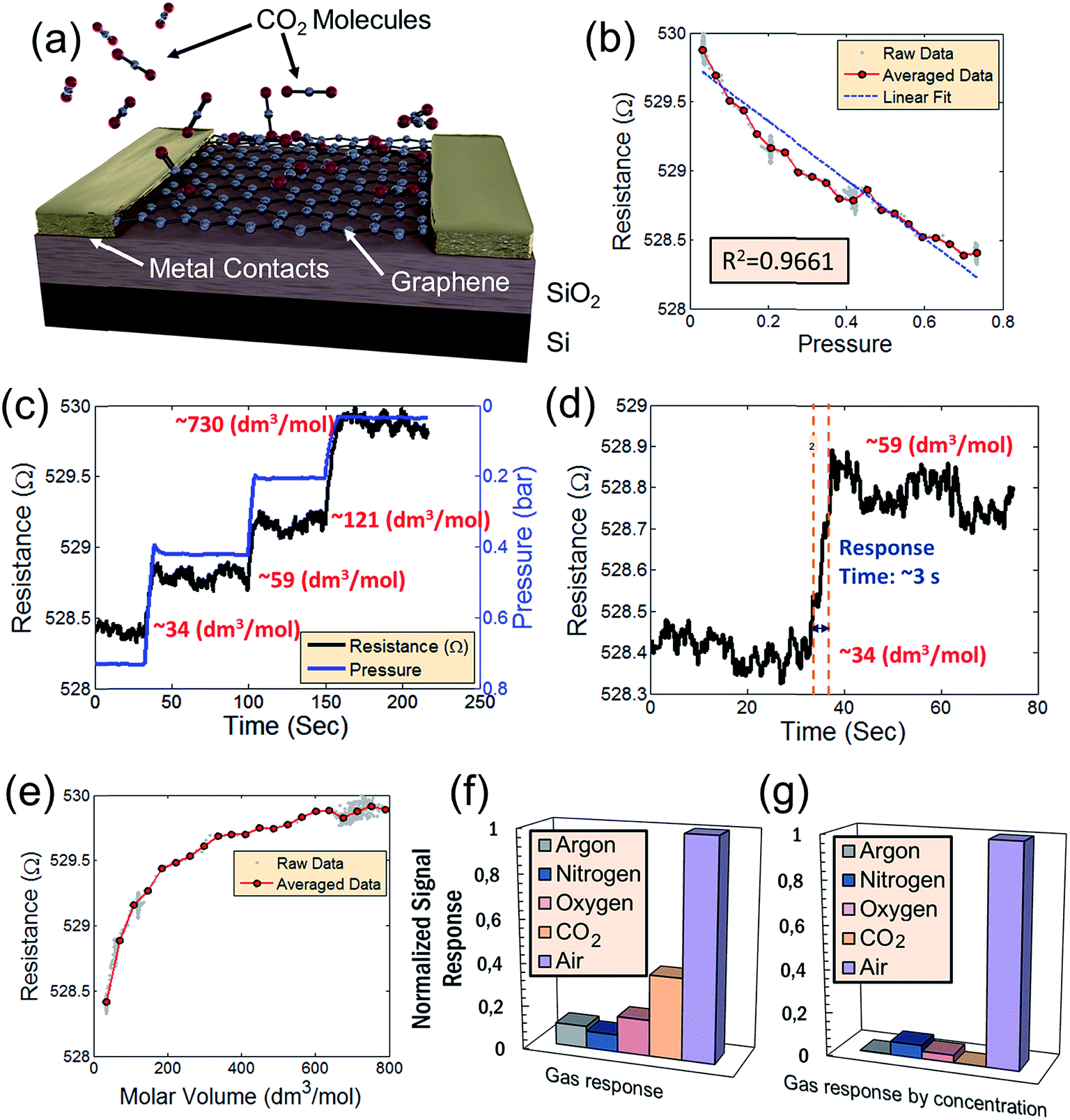 | ||
| Fig. 2 (a) Schematic of what occurs inside the vacuum chamber on the active graphene sensing area of the device. As CO2 is pumped into the evacuated chamber, the CO2 molecules bind with the dangling pi-orbitals of the graphene. (b) Resistance response of the device to changes in pressure. The resistance response is linear with an R2 value of 0.9661. (c) Resistance response to CO2 where the chamber pressure is varied by evacuating from a molar volume of 34 dm3 mol−1 of CO2 and subsequently held constant at different pressures (different CO2 molar volumes) – indicating reasonable device stability. (d) Response time of the CO2 sensor corresponding to a chance in molar volume from 34 dm3 mol−1 of CO2 to 59 dm3 mol−1 of CO2. (e) Relationship of the resistance versus the molar volume of CO2 in the chamber. (f) Device sensitivity of various constituent gases of air. (g) Normalized sensitivity of each gas with respect to its relative concentration in air. The combined response of all gases is insignificant in comparison to the response in air shown in previous reports.3 This suggests that the CO2 response, while present does not significantly impact the device performance of humidity sensors in air. | ||
As mentioned, by relating the pressure to device resistance, a comparison of the relative sensitivity of various gases (including air) to changes in pressure is performed (Fig. 2f). Graphene's resistance response to each individual gas, including CO2, is small relative to air. This suggests that the dominant effect is humidity sensing and not sensing of any constituent gases of air. Graphene's high selectivity to humidity is more apparent when considering the relative concentrations of each gas species in air (Fig. 2g). In both cases, the combined device resistance responses to all gases are insignificant in comparison to the resistance response in air – suggesting that the concentration of CO2 present in air has negligible impact on the device sensitivity to H2O. As a consequence, graphene-based humidity sensors may be operated in environmental conditions without concern for cross-sensitivity effects to atmospheric gases at typical concentrations.
The effect of CO2 and H2O molecules on graphene has been studied both experimentally3,13,17,18,29,30,32,35,36,58 and theoretically.31,34,59–64 Specifically, the influence of defects in the SiO2 substrate was analyzed by Wehling et al.63 in an H2O-rich environment. For both H2O and CO2, a change in resistance as the concentration of the gases in the chamber changes has been observed. This suggests a doping of the graphene layer by both CO2 and H2O, possibly facilitated by the substrate's atomic configuration. A control case with only graphene and the defective substrate is performed for comparison. In order to qualitatively demonstrate the effect of the two gas species, we performed DFT calculations for different cases of H2O and CO2 coverage with Q03 substrate defects65,66 present on the surface of the cristobalite SiO2 substrate for one case. An under-coordinated silicon at the surface of the α-quartz substrate is simulated for another case. The Q03 defect is chosen for the purpose of computational simplicity and being a well-established defect in SiO2 surfaces. In the calculations, the substrate defects give rise to an impurity band, similar to the results of Wehling et al.63 The electrostatic dipole moment of the H2O molecules may now shift this impurity band, leading to an effective doping and increased conductivity in the graphene layer, which complies with experimental observations.
Our calculated results are summarized in Fig. 3 for the case of cristobalite SiO2 substrate where the graphene/substrate distance is relaxed and found to be 3.40 Å above the substrate at the lowest total energy. Similarly, graphene/H2O and graphene/CO2 distances are found to have the lowest total energies at 3.21 Å and 3.20 Å respectively. Different orientations and configurations for the CO2 and H2O cases are examined and the lowest energy occurs when each adsorbate molecule lies parallel to the graphene sheet. The shown configurations (adsorbate atoms positioning relative to the graphene sheet and the substrate) give the lowest energies compared to other examined different cases. Fig. 3a shows graphene on top of the SiO2 substrates. The top Si atoms are displayed in green. Fig. 3b and c present the same graphene/SiO2 system in the presence of a H2O molecule and CO2 molecule respectively. The charge density difference (CDD) is defined according to eqn (3) where the (sc, s, g, ads) abbreviations stands for the supercell, substrate, graphene and adsorbates respectively. The charge density difference is displayed as a contour map with a 2D cut through the carbon dioxide molecule plane in each of the three unit cells, as indicated by the dashed lines in Fig. 3g–i.
| Δρ = ρsc − ρs − ρg − ρads | (3) |
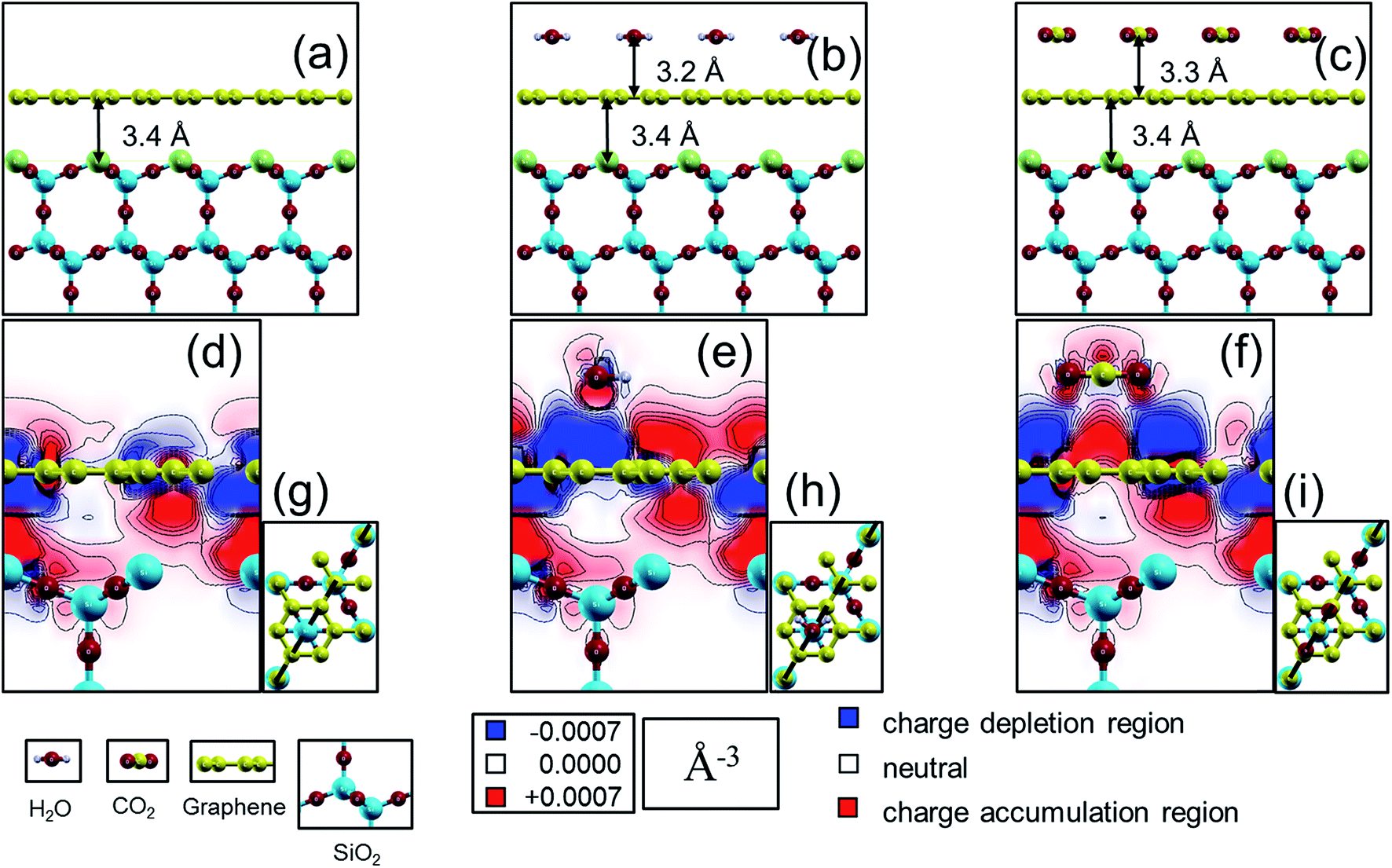 | ||
| Fig. 3 (a) Case for a monolayer of graphene 2 × 2 super cell (in yellow) on top of (111) cut in cristobalite SiO2 (oxygen in red, silicon in blue) forming a slab (with Q03 defective Si surface atom in green). Cases in (b) and (c) are the same in “a” with added molecules of water (hydrogen in grey) and carbon dioxide respectively. Charge density difference plots for the three systems in (a)–(c) are represented as 2D cuts in (d)–(f) according to the cutting planes demonstrated in (g)–(i) with a dashed black line passing through adjacent carbon atoms in the graphene hexagon as well as the oxygen in the water molecule and the center of the carbon dioxide molecule. Red/blue regions in (d)–(f) correspond to regions with electron charges changes greater/less than ±0.0007 (Å)−3, i.e. red regions are with higher electron carrier density and the opposite with the blue regions which have a higher hole carrier density. In both cases of water and carbon dioxide (e) and (f); charge accumulation/depletion is larger at the graphene sheet compared to the case in (d). Charge density difference plots were generated using xCrysden77 visualization program.78 | ||
Thus, with our electronic structure computations, it is possible to see how the charge density in and around the graphene sheet is altered in comparison to a pristine graphene sheet due to the presence of the substrate surface and the molecules. In Fig. 3d, we show how the charge density of graphene is affected by the silica substrate. Fig. 3e shows in addition the effect on the graphene charge density from an adsorbed water molecule, and finally Fig. 3f shows the corresponding effect from an adsorbed carbon dioxide molecule. Comparing Fig. 3d with Fig. 3e and f, we see that the physisorbed molecules clearly affect the charge density of the graphene sheet. For instance, in the cut shown, new charge depletion and accumulation regions around the graphene sheet develop. The changes in the graphene charge density result in corresponding changes of the Kohn–Sham eigenvalues, (i.e., the band structure in a single-particle picture of the electronic structure) and therefore band-structure related properties such as the band dispersion, the position of the Fermi level or the charge carrier effective mass may also change. Since it is well established that the transport properties of charge carriers in a material depend on the above mentioned band-structure related properties, it is reasonable to expect that the charge carrier transport properties will be affected by the observed changes in the charge density of the graphene sheet induced by the molecules. The effect on the graphene charge density from an adsorbed molecule is localized to the part of the graphene sheet in close proximity of the molecule. Therefore, a change in the concentration of adsorbed molecules (as long as saturation is not reached) will result in a change of how much of the graphene sheet that is affected by the molecules. It is also reasonable to expect that the transport properties of the graphene sheet will be more affected the higher the concentration of adsorbed molecules is (or, equivalently, less affected the lower the concentration of adsorbed molecules is) – as verified by experiments (Fig. 2).
Until now, both the data examined experimentally, as well as DFT simulations, focused on the graphene response due to direct CO2 adsorption onto the graphene surface. There is, however, a separate initial response to initial venting of the chamber with CO2 (as presented in Fig. 4a), which cannot be explained by surface adsorption and its implications for cross-sensitivity with the device response to humidity. As the pressure increases due to venting of the chamber with CO2 from a compressed CO2 bottle, (blue line) the graphene resistance shows a sharp initial decrease (black line). As previously discussed, the humidity is kept below 1% to isolate the device response to CO2. As mentioned, an increase in pressure is analogous to an increase in CO2 molar volume in the chamber. A white to orange gradient in Fig. 4a presents the increase in CO2 density over time in relation to an increase in pressure. After the chamber is filled with CO2 to 0.7 bar of pressure, it is maintained at a steady state pressure. Although the CO2 concentration remains at a stable value, the resistance slowly begins to recover from its initial response to roughly 67% of its original resistance value (Fig. 4b). This suggests a secondary effect, which is not due to direct adsorption of CO2 molecules. Evaluation of the device's CO2 sensitivity to the initial response is then performed to determine its possible cross-sensitivity effect with humidity. Normalization of device sensitivity is performed with respect to air by dividing the device sensitivity to the CO2 (Fig. 4c) by the device sensitivity to air (humidity). The initial response for pure CO2 is much higher than for air. However, CO2 typically is only about 0.039% of the relative concentration of air. Therefore, Fig. 4d shows the normalized sensitivity multiplied by the relative concentration in air. When the concentration of CO2 in air is taken into account, the effect of CO2 cross-sensitivity remains negligible compared to graphene's humidity response.
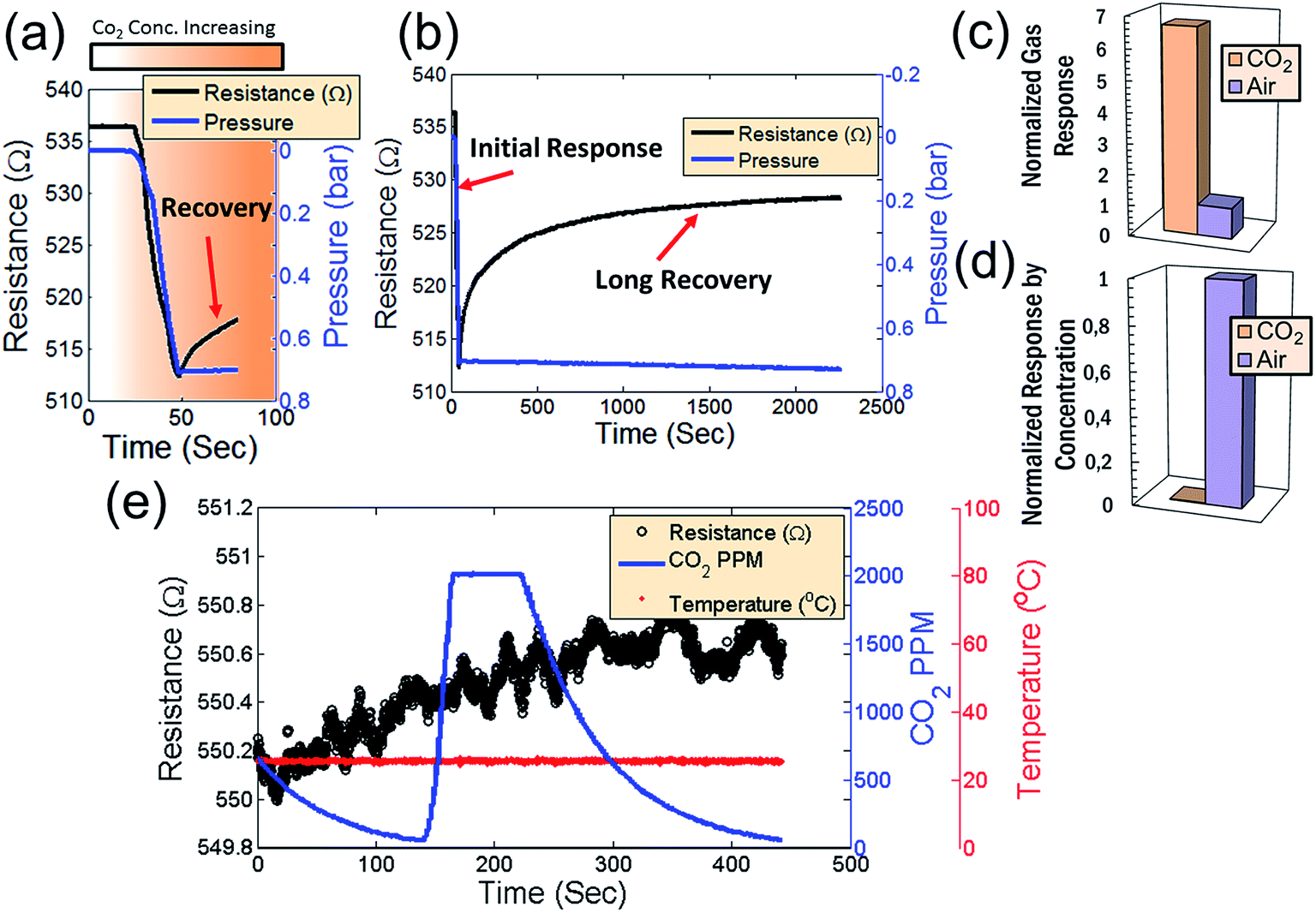 | ||
| Fig. 4 (a) Initial response of the graphene device to pure CO2. As the pressure increases (blue line) the resistance decreases (black line). An increase in pressure is also analogous to an increase in the amount of CO2 in the chamber. A white to orange gradient represents the increase in CO2 molar volume over time in relation to an increase in pressure. (b) After the chamber is filled with CO2, the graphene resistance slowly begins to recover to roughly 67% of its original deviation. (c) Normalized sensitivity of the initial gas response compared to the sensitivity of a humidity sensor. (d) Normalized sensitivity divided by the concentration. When the concentration of CO2 in the air is taken into account, the effect becomes negligible. (e) Signal from the commercial CO2 sensor in conjunction with the signal from the graphene sensor and temperature sensor in concentrations similar to air (roughly 400 ppm). Note that, although the commercial CO2 sensor reading varies from 0 to 2000 ppm as the chamber is evacuated and slightly vented with compressed CO2, there is no corresponding change in the graphene resistance response. | ||
In order to further address the possibility of cross-sensitivity of CO2 with humidity, a commercial Engine K30 CO2 sensor (with a detection range of 0 to 2000 ppm) was used for a secondary CO2 reading. Atmospheric CO2 concentration is approximately 400 ppm. If the CO2 value is maintained within this range, and there is no signal response from the sensor for changes in CO2 within this range, the effect of CO2 cross-sensitivity can be neglected under very conservative conditions (over 2000 ppm which over 5 times greater than atmospheric CO2 concentrations). Fig. 4e compares the measurement signal of the commercial CO2 sensor in conjunction with the signal of the graphene sensor. In addition, an LM35 temperature sensor was used to monitor the chamber temperature, demonstrating that the chamber temperature is maintained at room temperature during the measurements. For these measurements, humidity and CO2 concentrations are controlled in the same way as described for the measurements in Fig. 2. As the CO2 concentration varies from 0 to 2000 ppm (as the chamber is evacuated and slightly vented with high concentration CO2), there is no corresponding change in the graphene device resistance. This verifies that the response seen in Fig. 4a is suppressed at atmospheric concentrations of CO2 and confirms that the cross-sensitivity between CO2 and H2O is negligible for measuring humidity in air using these graphene devices.
In order to further illustrate this point, Table 1 compares device responsivities for a number of competing CO2 sensing technologies to our devices in the range from 0 to 2000 ppm.67–70 The comparison includes mechanically exfoliated (pristine) graphene,67 graphene oxide,68 carbon nanotubes (CNTs),69 printed graphene70 resistive sensors as well as several capacitive sensors.71–74 However, the majority of sensors in literature only investigate on CO2 concentrations ranging below 2000 ppm making comparison difficult at much larger concentrations. Our graphene sensor shows negligible responsivity to CO2 at these low ranges (the responsivity is below the noise threshold of our instrumentation) and two orders of magnitude below the responsivity of competing devices. These devices can therefore be operated as humidity sensors in atmospheric environments without concern for cross-sensitivity with CO2.
| Material | Mechanism | Response time (s) | Responsivity (% per ppm) | |
|---|---|---|---|---|
| a * calculated from reported results, — data not available. | ||||
| Smith et al. (this work) | CVD graphene | Res./Cap. | 3 | >0.0000091 |
| Yoon et al.67 | Pristine graphene | Resistive | 8 | 0.17 |
| Hafiz et al.68 | Graphene oxide | Resistive | 240 | 0.047* |
| Hafiz et al.68 | rGO | Resistive | 240 | 0.033* |
| Ong et al.69 | MWCNT | Resistive | 45 | — |
| Andò et al.70 | InkJet graphene | Resistive | — | 0.000235* |
| Mutschall et al.71 | AMO/PTMS | Capacitive | — | 0.000875* |
| Kim et al.74 | AMO/PTMS | Capacitive | — | 0.0004* |
As previously mentioned, the signal response of the graphene device to CO2 appears to be the result of two different phenomena (Fig. 2 and 4). In Fig. 2, the device behavior can be best explained by adsorption of CO2 onto the surface of the graphene (as supported by DFT analyses in Fig. 3). In order to further analyze a separate effect caused by the initial CO2 venting (behavior shown in Fig. 4), the graphene devices were passivated with a 25 nm layer of Al2O3 deposited over the entire device surface before packaging the devices.75 Fig. 5a displays a passivated device where the Al2O3 layer prevents CO2 molecules from being adsorbed directly onto the graphene surface. CO2 was introduced into the chamber in a series of venting and pumping cycles as shown in Fig. 5b1 in order to observe whether the passivated device shows the same behavior as the non-passivated. In Fig. 5, there remains an initial response when CO2 is introduced, despite the graphene device passivation. This confirms that the effect observed in Fig. 4 involves a different mechanism than direct surface adsorption. Just as in Fig. 4, after the initial response (red line), there is a recovery period. This recovery is not affected by subsequent removal of the CO2 during the pumping portion of the cycle (the chamber pressure is denoted by the blue line). In the non-passivated resistance response shown in Fig. 2b (which has already reached a recovery after the initial CO2 response), the mechanism is due to direct adsorption because the non-passivated device resistance varies linearly as CO2 is removed from the chamber – an effect which is not present during CO2 removal from the chamber in the passivated device. During repeated pumping and venting of the chamber with CO2, the same behavior is present as in Fig. 4a and b. The recovery period follows an exponential decay in each cycle (Fig. 5b1 as black dotted lines). Fig. 5b2 displays the same response and recovery with the exponential fits subtracted. This subtraction provides a more clear demonstration that the behavior of passivated devices is only affected by the initial introduction of CO2 molecules and not determined by the CO2 concentration in the chamber (as the direct adsorption case in Fig. 2 with non-passivated devices). The passivation layer thus provides effective isolation from CO2 adsorption onto the graphene surface.
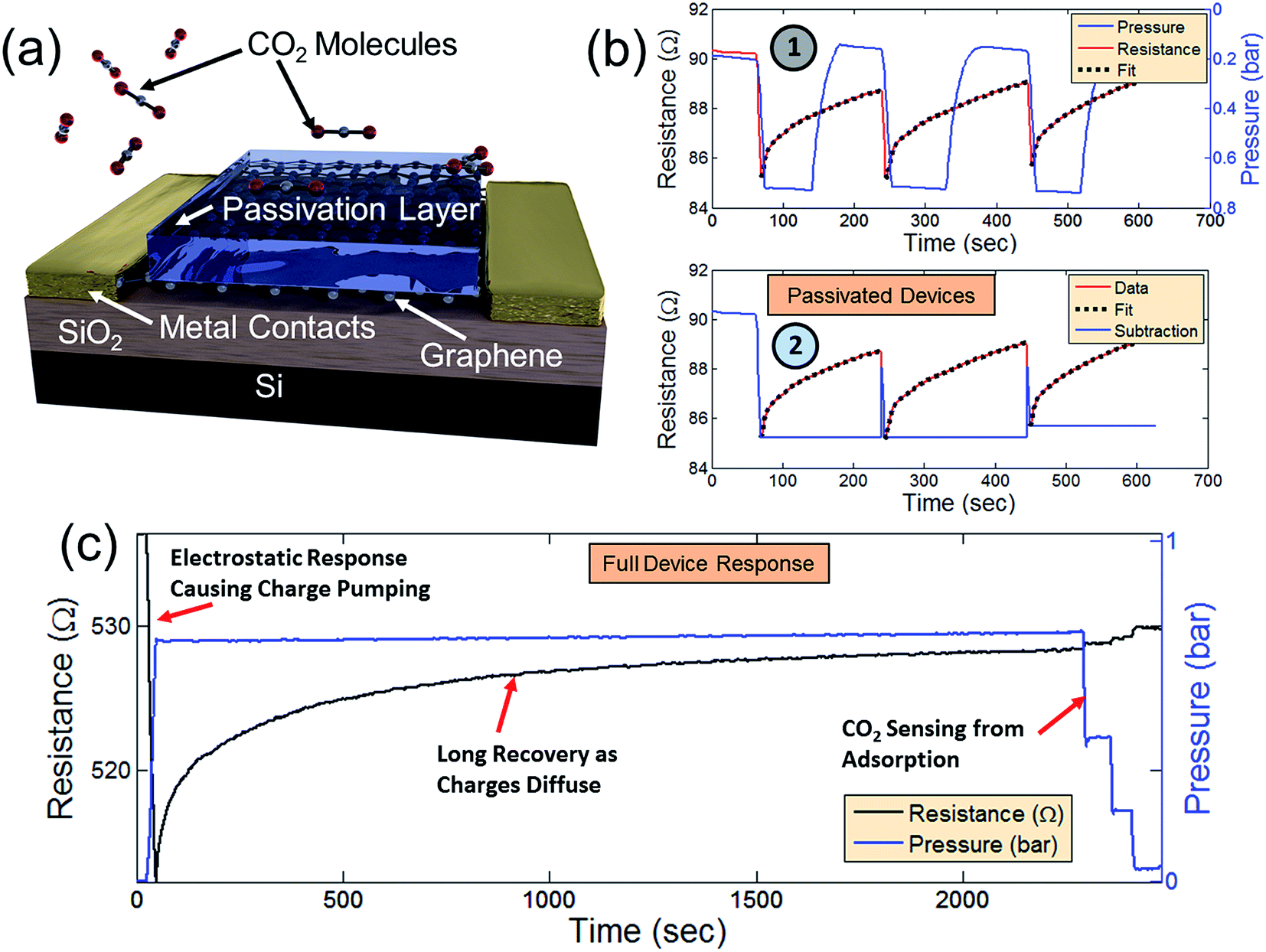 | ||
| Fig. 5 (a) Schematic of a passivated gas sensor effectively isolating the graphene channel region from the adsorption of CO2 molecules. (b) Signal response (red line) of the passivated device as CO2 is pumped and vented into and out of the previously evacuated chamber. An exponential fit to the recovery is performed (black dotted lines) and the fit subtracted from the signal response (2). (c) Combination of signal responses from Fig. 2 and 4 demonstrating both resistive and capacitive components to graphene's CO2 sensing properties. | ||
Although the precise mechanism of the initial graphene resistance response to CO2 is not presently understood, it may be the result of electrostatic charging of the CO2 as it is decompressed from the bottle and vented into the chamber. Electrostatic charging of compressed CO2 has been reported as a common feature of CO2 compression in other contexts.76 In addition, there are several reports of capacitive CO2 sensors which demonstrate a similar exponential decay behavior which we observe coupled with long recovery times.72–74 Although there is no consensus on the mechanism at work, these reports suggest that the primary effect is chemisorption with the substrate which changes the material dielectric constant. While this is an interesting possibility, it does not explain, in our case, the recovery of the devices while under a fixed CO2 concentration.
We therefore speculate that the observed effect is a capacitive coupling effect that causes an increase of charge carriers in the graphene that then diffuses as the CO2 molecules discharge through collisions with the grounded chamber walls. Such behavior would explain the exponential resistance decay in the graphene. Fig. 5c combines the data of Fig. 2 and 4. After an initial response and long recovery period, CO2 was pumped from the chamber in 4 steps (close-ups shown in Fig. 2f) in order to demonstrate the distinction between graphene's initial resistance response to CO2 from its response to direct CO2 adsorption. In devices without passivation layer, there is a definite signal response when CO2 is evacuated from the chamber. This effect is not present in passivated devices – signaling that the response in Fig. 2f is due to direct CO2 adsorption. Since the initial signal response and subsequent exponential decay is present in both passivated and non-passivated devices, this response is possibly due to capacitive coupling from electrostatically charged CO2. If the long recovery is due to a slow discharging of electrostatically charged CO2 molecules, the recovery may be drastically shortened by ensuring that the CO2 molecules are not electrostatically charged, thereby isolating the CO2 sensing effect due only to adsorption – an interesting topic for future investigation.
Conclusion
We demonstrate graphene-based CO2 sensing. In addition, we observe two distinct effects associated with the sensor – a resistive effect from direct adsorption onto the graphene surface and a possible capacitive effect. The direct adsorption effect may be understood by a change in the electronic structure. The capacitive effect is, at present, not understood. In addition, we primarily analyze the cross-sensitivity between CO2 and humidity – an important feature for implementating graphene-based humidity sensors under atmospheric conditions. The selectivity of the graphene gas sensor to humidity is determined by comparison of measurements performed in pure concentrations of O2, Ar, N2, CO2, and air. Although the response from O2, Ar, and N2 is minimal, there exists a cross sensitivity between CO2 and humidity. However, the cross-sensitivity of CO2 becomes negligible at typical CO2 concentrations present in air. This high selectivity to H2O in the presence of other gases is attractive for implementation of graphene-based humidity sensing in ambient air environments.Acknowledgements
We acknowledge financial support from Wavegraph, Erasmus Target II, the Göran Gustafsson Foundation, VR (Vetenskapsrådet), KVA (The Royal Swedish Academy of Sciences), KAW (the Knut and Alice Wallenberg Foundation), CTS (Carl Tryggers Stiftelse), STEM (Swedish Energy Agency), and SSF (Swedish Foundation for Strategic Research). The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC), Linköping University, the PDC Centre for High Performance Computing (PDC-HPC), KTH, and the High Performance Computing Center North (HPC2N), Umeå University.References
- P. T. Moseley, Solid State Gas Sensors, Meas. Sci. Technol., 1997, 8, 223–237 CrossRef CAS.
- S. Capone, A. Forleo, L. Francioso, R. Rella, P. Siciliano, J. Spadavecchia, D. S. Presicce and A. M. Taurino, Solid State Gas Sensors: State of the Art and Future Activities, J. Optoelectron. Adv. Mater., 2003, 5, 1335–1348 CAS.
- A. D. Smith, K. Elgammal, F. Niklaus, A. Delin, A. Fischer, S. Vaziri, F. Forsberg, M. Råsander, H. W. Hugosson and L. Bergqvist, et al., Resistive Graphene Humidity Sensors with Rapid and Direct Electrical Readout, Nanoscale, 2015, 19099–19109 RSC.
- G. Korotcenkov, Metal Oxides for Solid-State Gas Sensors: What Determines Our Choice?, Mater. Sci. Eng., B, 2007, 139, 1–23 CrossRef CAS.
- Q. Kuang, C. Lao, Z. L. Wang, Z. Xie and L. Zheng, High-Sensitivity Humidity Sensor Based on a Single SnO2 Nanowire, J. Am. Chem. Soc., 2007, 129, 6070–6071 CrossRef CAS PubMed.
- J. Kong, N. R. Franklin, C. Zhou, M. G. Chapline, S. Peng, K. Cho and H. Dai, Nanotube Molecular Wires as Chemical Sensors, Science, 2000, 287, 622–625 CrossRef CAS PubMed.
- X. T. Zhou, J. Q. Hu, C. P. Li, D. D. D. Ma, C. S. Lee and S. T. Lee, Silicon Nanowires as Chemical Sensors, Chem. Phys. Lett., 2003, 369, 220–224 CrossRef CAS.
- Y. Wang, X. Jiang and Y. Xia, A Solution-Phase, Precursor Route to Polycrystalline SnO2 Nanowires That Can Be Used for Gas Sensing under Ambient Conditions, J. Am. Chem. Soc., 2003, 125, 16176–16177 CrossRef CAS PubMed.
- S. Basu and P. Bhattacharyya, Recent Developments on Graphene and Graphene Oxide Based Solid State Gas Sensors, Sens. Actuators, B, 2012, 173, 1–21 CrossRef CAS.
- K. R. Ratinac, W. Yang, S. P. Ringer and F. Braet, Toward Ubiquitous Environmental Gas Sensors Capitalizing on the Promise of Graphene, Environ. Sci. Technol., 2010, 44, 1167–1176 CrossRef CAS PubMed.
- W. Yuan and G. Shi, Graphene-Based Gas Sensors, J. Mater. Chem. A, 2013, 1, 10078–10091 CAS.
- Y. Zhou, Y. Jiang, T. Xie, H. Tai and G. Xie, A Novel Sensing Mechanism for Resistive Gas Sensors Based on Layered Reduced Graphene Oxide Thin Films at Room Temperature, Sens. Actuators, B, 2014, 203, 135–142 CrossRef CAS.
- Y. Dan, Y. Lu, N. J. Kybert, Z. Luo and A. C. Johnson, Intrinsic Response of Graphene Vapor Sensors, Nano Lett., 2009, 9, 1472–1475 CrossRef CAS PubMed.
- S.-H. Hwang, D. Kang, R. S. Ruoff, H. S. Shin and Y.-B. Park, Poly(Vinyl Alcohol) Reinforced and Toughened with Poly (Dopamine)-Treated Graphene Oxide, and Its Use for Humidity Sensing, ACS Nano, 2014, 8, 6739–6747 CrossRef CAS PubMed.
- S. Borini, R. White, D. Wei, M. Astley, S. Haque, E. Spigone, N. Harris, J. Kivioja and T. Ryhänen, Ultrafast Graphene Oxide Humidity Sensors, ACS Nano, 2013, 7, 11166–11173 CrossRef CAS PubMed.
- H. Bi, K. Yin, X. Xie, J. Ji, S. Wan, L. Sun, M. Terrones and M. S. Dresselhaus, Ultrahigh Humidity Sensitivity of Graphene Oxide, Sci. Rep., 2013, 3, 2714 Search PubMed.
- Y. Yao, X. Chen, H. Guo, Z. Wu and X. Li, Humidity Sensing Behaviors of Graphene Oxide-Silicon Bi-Layer Flexible Structure, Sens. Actuators, B, 2012, 161, 1053–1058 CrossRef CAS.
- A. Ghosh, D. J. Late, L. S. Panchakarla, A. Govindaraj and C. N. R. Rao, NO2 and Humidity Sensing Characteristics of Few-Layer Graphenes, J. Exp. Nanosci., 2009, 4, 313–322 CrossRef CAS.
- M.-C. Chen, C.-L. Hsu and T.-J. Hsueh, Fabrication of Humidity Sensor Based on Bilayer Graphene, Electron Device Lett., 2014, 35, 590–592 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Detection of Individual Gas Molecules Adsorbed on Graphene, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Ultrahigh Electron Mobility in Suspended Graphene, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- S. V. Morozov, K. S. Novoselov, M. I. Katsnelson, F. Schedin, D. C. Elias, J. A. Jaszczak and A. K. Geim, Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer, Phys. Rev. Lett., 2008, 100, 016602 CrossRef CAS PubMed.
- E. Massera, V. La Ferrara, M. Miglietta, T. Polichetti, I. Nasti and G. Di Francia, Gas Sensors Based on Graphene, Chem. Today, 2011, 29, 39–41 CAS.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, Practical Chemical Sensors from Chemically Derived Graphene, ACS Nano, 2009, 3, 301–306 CrossRef CAS PubMed.
- M. Gautam and A. H. Jayatissa, Graphene Based Field Effect Transistor for the Detection of Ammonia, J. Appl. Phys., 2012, 112, 064304 CrossRef.
- Q. He, S. Wu, Z. Yin and H. Zhang, Graphene-Based Electronic Sensors, Chem. Sci., 2012, 3, 1764–1772 RSC.
- T. O. Wehling, K. S. Novoselov, S. V. Morozov, E. E. Vdovin, M. I. Katsnelson, A. K. Geim and A. I. Lichtenstein, Molecular Doping of Graphene, Nano Lett., 2008, 8, 173–177 CrossRef CAS PubMed.
- Y. Yang, K. Brenner and R. Murali, The Influence of Atmosphere on Electrical Transport in Graphene, Carbon, 2012, 50, 1727–1733 CrossRef CAS.
- J. Moser, A. Verdaguer, D. Jiménez, A. Barreiro and A. Bachtold, The Environment of Graphene Probed by Electrostatic Force Microscopy, Appl. Phys. Lett., 2008, 92, 123507 CrossRef.
- H. J. Yoon, J. H. Yang, Z. Zhou, S. S. Yang and M. M.-C. Cheng, Carbon Dioxide Gas Sensor Using a Graphene Sheet, Sens. Actuators, B, 2011, 157, 310–313 CrossRef CAS.
- K. K. Paulla and A. A. Farajian, Concentration Effects of Carbon Oxides on Sensing by Graphene Nanoribbons: Ab Initio Modeling, J. Phys. Chem. C, 2013, 117, 12815–12825 CAS.
- A. K. Mishra and S. Ramaprabhu, Carbon Dioxide Adsorption in Graphene Sheets, AIP Adv., 2011, 1, 032152 CrossRef.
- K. G. Ong and C. A. Grimes, A Carbon Nanotube-Based Sensor for CO2 Monitoring, Sensors, 2001, 1, 193–205 CrossRef CAS.
- K.-J. Lee and S.-J. Kim, Theoretical Investigation of CO2 Adsorption on Graphene, Bull. Korean Chem. Soc., 2013, 34, 3022–3026 CrossRef CAS.
- G. Lu, L. E. Ocola and J. Chen, Reduced Graphene Oxide for Room-Temperature Gas Sensors, Nanotechnology, 2009, 20, 445502 CrossRef PubMed.
- K. R. Nemade and S. A. Waghuley, Chemiresistive Gas Sensing by Few-Layered Graphene, J. Electron. Mater., 2013, 42, 2857–2866 CrossRef CAS.
- A. Reina, H. Son, L. Jiao, B. Fan, M. S. Dresselhaus, Z. Liu and J. Kong, Transferring and Identification of Single-and Few-Layer Graphene on Arbitrary Substrates, J. Phys. Chem. C, 2008, 112, 17741–17744 CAS.
- X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han, D. Chen, R. D. Piner, L. Colombo and R. S. Ruoff, Transfer of Large-Area Graphene Films for High-Performance Transparent Conductive Electrodes, Nano Lett., 2009, 9, 4359–4363 CrossRef CAS PubMed.
- Y.-C. Lin, C. Jin, J.-C. Lee, S.-F. Jen, K. Suenaga and P.-W. Chiu, Clean Transfer of Graphene for Isolation and Suspension, ACS Nano, 2011, 5, 2362–2368 CrossRef CAS PubMed.
- L. Jiao, B. Fan, X. Xian, Z. Wu, J. Zhang and Z. Liu, Creation of Nanostructures with Poly(Methyl Methacrylate)-Mediated Nanotransfer Printing, J. Am. Chem. Soc., 2008, 130, 12612–12613 CrossRef CAS PubMed.
- H. J. Park, J. Meyer, S. Roth and V. Skákalová, Growth and Properties of Few-Layer Graphene Prepared by Chemical Vapor Deposition, Carbon, 2010, 48, 1088–1094 CrossRef CAS.
- A. D. Smith, F. Niklaus, A. Paussa, S. Vaziri, A. C. Fischer, M. Sterner, F. Forsberg, A. Delin, D. Esseni and P. Palestri, et al., Electromechanical Piezoresistive Sensing in Suspended Graphene Membranes, Nano Lett., 2013, 13, 3237–3242 CrossRef CAS PubMed.
- D. Vanderbilt, Optimally Smooth Norm-Conserving Pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 8412 CrossRef CAS.
- The electronic structure laboratory website, http://www.http://fpmd.ucdavis.edu/potentials/.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al., QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, Role and Effective Treatment of Dispersive Forces in Materials: Polyethylene and Graphite Crystals as Test Cases, J. Comput. Chem., 2009, 30, 934–939 CrossRef CAS PubMed.
- T. Björkman, CIF2Cell: Generating Geometries for Electronic Structure Programs, Comput. Phys. Commun., 2011, 182, 1183–1186 CrossRef.
- CIF2Cell Generating cells for electronic structure calculations from CIF files, http://sourceforge.net/projects/cif2cell/.
- P. H. Gamlen and J. W. White, Structure and Dynamics of Microcrystalline Graphite, Graphon, by Neutron Scattering, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 446–455 RSC.
- T. Demuth, Y. Jeanvoine, J. Hafner and J. G. Angyan, Polymorphism in Silica Studied in the Local Density and Generalized-Gradient Approximations, J. Phys.: Condens. Matter, 1999, 11, 3833–3874 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, et al., Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation, APL Mater., 2013, 1, 011002 CrossRef.
- Materials Project, http://www.materialsproject.org.
- G. Bergerhoff, R. Hundt, R. Sievers and I. D. Brown, The Inorganic Crystal Structure Data Base, J. Chem. Inf. Comput. Sci., 1983, 23, 66–69 CrossRef CAS.
- F. Karlsruhe, The Inorganic Crystal Structure Database, http://icsd.fiz-karlsruhe.de Search PubMed.
- Y. Yang, K. Brenner and R. Murali, The Influence of Atmosphere on Electrical Transport in Graphene, Carbon, 2012, 50, 1727–1733 CrossRef CAS.
- Y. Yang and R. Murali, Binding Mechanisms of Molecular Oxygen and Moisture to Graphene, Appl. Phys. Lett., 2011, 98, 093116 CrossRef.
- R. R. Q. Freitas, R. Rivelino, F. B. Mota de and C. M. C. de Castilho, DFT Studies of the Interactions of a Graphene Layer with Small Water Aggregates, J. Phys. Chem. A, 2011, 115, 12348–12356 CrossRef CAS PubMed.
- O. Leenaerts, B. Partoens and F. M. Peeters, Water on Graphene: Hydrophobicity and Dipole Moment Using Density Functional Theory, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235440 CrossRef.
- T. O. Wehling, M. I. Katsnelson and A. I. Lichtenstein, Adsorbates on Graphene: Impurity States and Electron Scattering, Chem. Phys. Lett., 2009, 476, 125–134 CrossRef CAS.
- T. O. Wehling, A. I. Lichtenstein and M. I. Katsnelson, First-Principles Studies of Water Adsorption on Graphene: The Role of the Substrate, Appl. Phys. Lett., 2008, 93, 202110 CrossRef.
- J. Dai, J. Yuan and P. Giannozzi, Gas Adsorption on Graphene Doped with B, N, Al, and S: A Theoretical Study, Appl. Phys. Lett., 2009, 95, 232105 CrossRef.
- M. Wilson and T. R. Walsh, Hydrolysis of the Amorphous Silica Surface. I. Structure and Dynamics of the Dry Surface, J. Chem. Phys., 2000, 113, 9180–9190 CrossRef CAS.
- T. R. Walsh, M. Wilson and A. P. Sutton, Hydrolysis of the Amorphous Silica Surface. II. Calculation of Activation Barriers and Mechanisms, J. Chem. Phys., 2000, 113, 9191–9201 CrossRef CAS.
- H. J. Yoon, J. H. Yang, Z. Zhou, S. S. Yang and M. M.-C. Cheng, et al., Carbon Dioxide Gas Sensor Using a Graphene Sheet, Sens. Actuators, B, 2011, 157, 310–313 CrossRef CAS.
- S. M. Hafiz, R. Ritikos, T. J. Whitcher, N. M. Razib, D. C. S. Bien, N. Chanlek, H. Nakajima, T. Saisopa, P. Songsiriritthigul and N. M. Huang, et al., A Practical Carbon Dioxide Gas Sensor Using Room-Temperature Hydrogen Plasma Reduced Graphene Oxide, Sens. Actuators, B, 2014, 193, 692–700 CrossRef CAS.
- K. G. Ong, K. Zeng and C. A. Grimes, A Wireless, Passive Carbon Nanotube-Based Gas Sensor, IEEE Sens. J., 2002, 2, 82–88 CrossRef CAS.
- B. Andò, S. Baglio, G. Di Pasquale, A. Pollicino, S. D'Agata, C. Gugliuzzo, C. Lombardo and G. Re, An Inkjet Printed CO2 Gas Sensor, Procedia Eng., 2015, 120, 628–631 CrossRef.
- D. Mutschall and E. Obermeier, A Capacitive CO2 Sensor with on-Chip Heating, Sens. Actuators, B, 1995, 25, 412–414 CrossRef CAS.
- T. Ishihara and S. Matsubara, Capacitive Type Gas Sensors, J. Electroceram., 1998, 2, 215–228 CrossRef CAS.
- T. Ishihara, K. Kometani, Y. Mizuhara and Y. Takita, A New Type of CO2 Gas Sensor Based on Capacitance Changes, Sens. Actuators, B, 1991, 5, 97–102 CrossRef CAS.
- C. Kim, S. Pavlidis, M. Kim, O. Brand and H. Chen, Room Temperature CO2 Detection Using Interdigitated Capacitors with Heteropolysiloxane Sensing Films, in Sensors, 2016 IEEE, IEEE, 2016, pp. 1–3 Search PubMed.
- A. D. Smith, K. Elgammal, X. Fan, M. Lemme, A. Delin, F. Niklaus and M. Östling, Toward Effective Passivation of Graphene to Humidity Sensing Effects, in Solid-State Device Research Conference (ESSDERC), 2016 46th European, IEEE, 2016, pp. 299–302 Search PubMed.
- G. J. Butterworth and P. D. Dowling, Electrostatic Effects with Portable CO2 Fire Extinguishers, J. Electrost., 1981, 11, 43–55 CrossRef CAS.
- A. Kokalj, XCrySDen—a New Program for Displaying Crystalline Structures and Electron Densities, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS PubMed.
- X-window CRYstalline Structures and DENsities, http://www.xcrysden.org/ Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |