
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of Mn-containing oxide catalysts for low temperature selective catalytic reduction of NOx with NH3: reaction mechanism and catalyst deactivation
Shengen Zhang *,
Bolin Zhang,
Bo Liu and
Shuailing Sun
*,
Bolin Zhang,
Bo Liu and
Shuailing Sun
Institute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, PR China. E-mail: zhangshengen@mater.ustb.edu.cn
First published on 16th May 2017
Abstract
Atmospheric pollutants of nitrogen oxides (NOx) can be reduced by selective catalytic reduction (SCR). SCR of NOx with ammonia (NH3) at low temperatures has attracted much interest for high nitric oxide (NO) conversion, and this method is dominated by catalysts. Manganese (Mn)-containing oxide catalysts exhibit high activity and selectivity for the unique redox property of manganese oxides (MnOx). The reaction mechanisms and deactivation processes are summarized in this review. SCR of NOx with NH3 follows both the Langmuir–Hinshelwood and the Eley–Rideal mechanisms, which also contribute to the nitrous oxide formation. Fast SCR has a higher reaction rate than standard SCR. Mn-containing catalysts could also be deactivated by sulfur oxides and water vapor. The deactivation process of sulfur dioxide can be classified into two categories: deposition of (NH4)2SO4 and sulfation of active sites. The deactivation caused by water vapor can be attributed to the competitive adsorption. The adsorption of water on catalysts' surface blocked the active sites, which are provided for the adsorption of NH3 and NO. Alkali, alkaline earth and heavy metal ions existing in fine fly ash can also damage the catalysts' acid sites. A notable improvement on performance was obtained when Mn-containing catalysts were doped with a transition metal, for these enhanced its adsorption capacity and oxidation ability. Furthermore, this review gives a comprehensive discussion of the synergistic mechanism between bi-metal or multi-metal oxides. Major conclusions and several possible directions for further research are presented finally.
1. Introduction
Nitrogen oxides (NOx) are a series of active gases, and include nitrogen dioxide (NO2), nitrogen oxide (NO) and nitrous oxide (N2O), and so on. Human activities cause a huge emission rate of NOx, which is double that of the biotic and abiotic nitrogen fixation rates. Released NOx can cause a series of environmental issues, such as photochemical smog, acid rain, and ozone depletion, and it can affect global tropospheric chemistry.1–4 Great efforts have been devoted to abating the emission of NOx.The technologies used to control NOx emission can be categorized as combustion controls and post-combustion controls.5,6 Combustion controls, which aim to control the production of NOx, include low NOx burners,7 air graded burning and staged fuel combustion.8 Post-combustion controls aim to decrease the NOx produced by reducing active N to fixed nitrogen gas (N2). The technologies for reducing NOx from flue gas can be divided into: direct decomposition,9,10 selective catalytic reduction (SCR),11,12 selective non-catalytic reduction (SNCR),13,14 hybrid SNCR/SCR15 and NOx storage-reduction catalysis.16 With the advantages of high efficiency and low cost, NOx emitted from stationary sources (e.g., thermal plants or industrial boilers) has been predominantly controlled by SCR of NO with ammonia (NH3-SCR) in the presence of excess oxygen (O2) for decades.17
The catalyst to be used is a decisive factor in the process of decreasing NOx (deNOx). The common catalysts include noble metal catalysts,18 metal-exchanged zeolite catalysts,19 metal oxide catalysts,20,21 heteropoly acid catalysts,22 and so on. Metal oxide catalysts are widely applied in NH3-SCR. Nowadays, the most widely used catalysts are vanadium(V)-based catalysts and tungsten trioxide (WO3) and/or molybdenum trioxide (MoO3) doped vanadium(V) oxide (V2O5)/titanium dioxide (TiO2) catalysts. These are usually installed at the upstream of flue gas because they require a higher working temperature of 300–400 °C.23 However, some tough problems have not been solved, such as the effect of excessive dust pollution to the catalysts upstream of the flue, the deactivation by sulfur dioxide (SO2) and alkali metal ions, the poor thermal stability at high temperatures and the toxicity of vanadium from the disabled catalysts.24 One of the efficient ways to overcome these obstacles is transferring the SCR reactor from upstream to downstream of the flue gas, where there is relatively less dust and sulfur oxides in the flue gas but a lower temperature below 300 °C.25
A series of metal oxide catalysts have been investigated to adapting low temperature, such as cerium (Ce), cobalt (Co), copper (Cu), iron (Fe), manganese (Mn), molybdenum (Mo), nickel (Ni) and V.26–30 Of these, manganese oxides (MnOx) catalysts show a notable NO conversion and N2 selectivity for its multi oxidation state, high valence state and characteristic crystallinity. Peña et al.26 advocated that MnOx/TiO2 had the highest activity among Co, chromium (Cr), Cu, Fe, Mn, Ni and V oxides supported on TiO2 at low temperatures. Manganese dioxide (MnO2) and manganese(III) oxide (Mn2O3) show the highest activity and N2 selectivity, respectively, among several MnOx.31 The activity and poison tolerance can be improved by doping with other transition metals. Ceria (CeO2) provides sufficient oxygen in the reaction of redox NOx, and improves the activity of MnOx catalysts.32–34 Mn–Fe spinel shows an excellent SCR performance at low temperature.35 Other Mn containing catalysts, such as MnOx–CoOx/TiO2,28,36 MnOx–CrOx/TiO2,37,38 MnOx–CuOx,39,40 lanthanum manganite (LaMnO3),41 have been investigated by many researchers. Mn containing catalysts have been recognized as the potential alternative for industrial applications.
To date, advances in low temperature NH3-SCR of NOx have been reviewed.5,6,42 A review by Li et al.43 summarized the use of metal oxides and zeolite catalysts and focused on the catalysts' components, preparation process and catalytic performance, however, the reaction mechanisms were not clarified clearly. A recent review in 2016 by Liu et al.44 summarized the use of MnOx-based catalysts and concentrated on the technological processes and improvement methods, however, little effort was made to summarize the reaction mechanisms and catalyst deactivation processes.
In this review, the advances in the use of Mn containing oxide catalysts are summarized. The focal point of this review is to address the reaction mechanisms and deactivation processes of Mn containing oxide catalysts. The N2 selectivity and side reactions are discussed together. This review gives a comprehensive discussion of the synergistic effects between bi-metal or multi-metal oxides. The deactivation process using sulfur oxides, water vapor, alkali metal and heavy metal ions and the regeneration methods are summarized. Finally, the major conclusions and several possible directions of research are presented.
2. Reaction mechanisms
To meet the newest and stringent emission standards, (NOx concentration ≤ 50 mg m−3),45 academic researchers and engineers are more interested in use of low temperature SCR, which is one of the efficient ways to install a processor downstream of the flue. A number of metal oxide catalysts have been investigated so far. Transition metal oxides play an important role in low temperature SCR catalysts, such as V2O5, MnOx, CeO2 and copper oxide (CuO). Of these, MnOx shows an excellent performance because of its different crystallinity, special surface area and multi oxidation. It is vital to elucidate the reaction mechanisms for future research. In this section, the reaction mechanisms of NH3-SCR over Mn-containing oxide catalysts are summarized.2.1 Standard SCR
The NH3-SCR of NO aims to reduce active N to fixed N2, which is harmless to the atmosphere. In the presence of excess O2, the main overall reaction is eqn (1).46 A great number of studies have proposed that eqn (1) shows the reaction stoichiometry in typical SCR conditions.47–50 In the absence of O2, reaction in eqn (1) would convert into the reaction in eqn (2):51| 4NH3 + 4NO + O2 → 4N2 + 6H2O(g), ΔG0298 = −1651 kJ mol−1 | (1) |
| 4NH3 + 6NO → 5N2 + 6H2O(g), ΔG0298 = −1821 kJ mol−1 | (2) |
Because the content of NO is more than 90% among NOx, eqn (1) is proposed as the standard SCR reaction and dominates the reaction stoichiometry. It is reported widely that the NH3-SCR of the NO reaction when comparing the stoichiometric conditions follows both the Langmuir–Hinshelwood (L–H) mechanism and the Eley–Rideal (E–R) mechanism.52,53 Through the L–H mechanism, both NH3 and NO are adsorbed on the surface of catalysts. However, via the E–R mechanism, adsorbed NH3 reacts with gaseous NO. It is suggested that the gaseous NH3 could be adsorbed on both Lewis acid sites and Brønsted acid sites, however, the gaseous NO is mainly adsorbed by a physical adsorption process.54 The adsorption of NH3 has been recognized as the first step of the SCR reaction because it is easier for NH3 to be adsorbed on acid sites rather than NO, O2 and the reaction products.55
The SCR process over MnOx catalysts via the L–H mechanism can be approximately described as follows:23,53,56
| NH3(g) → NH3(ad) | (3) |
| NO(g) → NO(ad) | (4) |
Mnn+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + NO(ad) → Mn(n−1)+–O–NO O + NO(ad) → Mn(n−1)+–O–NO
| (5) |
| NH3(ad) + Mn(n−1)+–O–NO → Mn(n−1)+–O–NO–NH3 → Mn(n−1)+–OH + N2 + H2O | (6) |
|
Mn(n−1)+–OH + 1/4O2 → Mnn+O + 1/2H2O
| (7) |
Eqn (3) and (4) are the adsorption of gaseous NH3 and NO. NH3 is usually adsorbed on the Lewis acid sites and Brønsted acid sites to form adsorbed NH3 species of coordinated NH3 and ionic NH4+, respectively.57 Nevertheless, the coordinated NH3 on the Lewis acid sites possesses a higher thermal stability than the ionic NH4+ on Brønsted acid sites. Manganese cations can provide a great number of Lewis acid sites.49,58
Fang et al.59,60 investigated the adsorption of NH3 on the Mn2O3 (222), manganese(II,II) oxide (Mn3O4) (211) and MnO2 (110) surfaces using density functional theory. It is claimed that, with more negative adsorption energy values and the shorter Mn–N bonds, Mn2O3 (222) and Mn3O4 (211) surfaces were more active for NH3 adsorption than the MnO2 (110) surface, which contributed to a higher performance (Table 1). Kapteijn et al.31 proposed that the highest NO conversion is exhibited by MnO2, followed by Mn5O8, Mn2O3 and Mn3O4.
The adsorbed NO is oxidized by the high valency state Mnn+ cations, (e.g., Mn4+) on the catalysts' surface to form adsorbed monodentate nitrite (Mn(n−1)+–O–NO) and the very metal cations are reduced as Mn(n−1)+ [eqn (5)]. Furthermore, Mn(n−1)+–O–NO reacts with adsorbed NH3 species to form Mn(n−1)+–O–NO–NH3, which decomposes subsequently to N2 and water (H2O) [eqn (6)]. Then, the reduced Mn(n−1)+ ions are regenerated by gaseous O2 [eqn (7)].
The SCR process over MnOx catalysts via the E–R mechanism can be described approximately as follows:35,48,61
| NH3(g) → NH3(ad) | (8) |
|
NH3(ad) + Mnn+O → NH2(ad) + Mn(n−1)+–OH
| (9) |
| NH2(ad) + NO(g) → NH2NO → N2 + H2O | (10) |
|
Mn(n−1)+–OH + 1/4O2 → Mnn+O + 1/2H2O
| (11) |
The adsorption of NH3 on the Lewis acid sites is recognized as the first step of NO reduction via the E–R mechanism. Coordinated NH3 could be deprived of a hydrogen and be activated by the labile oxygen or the lattice oxygen of metal oxides to form an amine (NH2) species [eqn (9)]. Labile oxygen can be released via the change of the valence states of Mn. Activated NH2 species on the catalysts' surface reacted with gaseous NO to form the most important intermediate of NH2NO, which subsequently decomposes to N2 and H2O [eqn (10)]. Then, the reduced Mn(n−1)+ cations could be oxidized by O2.
Furthermore, the formation of NH4NO2 is a typical SCR mechanism for Mn-containing catalysts. Qi and Tang,56 and Eigenmann et al.62 proposed an amide–nitrosamine type mechanism, which is actually similar to the E–R mechanism. An extra species of NH4NO2 was presented in this mechanism. NH4NO2 could be decomposed to NH2NO and H2O, and is then decomposed to N2 and H2O [eqn (12)–(14)]:
| OH(ad) + NO2(ad) → O(ad) + HNO2(ad) | (12) |
| NH3(ad) + HNO2(ad) → NH4NO2(ad) → NH2NO(ad) + H2O | (13) |
| NH2NO(ad) → N2 + H2O | (14) |
In accordance with the transient eqn (3)–(11), Mn3+–O–NO–NH3 and NH2NO are the most important intermediate in the reaction of the L–H mechanism and E–R mechanism, respectively. There is a quite similarity between these two different mechanisms. A comproportionation, (i.e., N3+ and N3−, N2+and N2−) occurs on both the L–H and E–R mechanism (eqn (6) and (10)).53,63
2.2 Fast SCR
A fast SCR reaction of NH3 with NO + NO2 over Mn-containing oxide catalysts has been reported. It is suggested that the fast SCR has a higher reaction rate than standard SCR.64 Fast SCR was firstly investigated by Koebel et al., and Madia et al.65–67 The general reaction can be described as follows:68,69| 4NH3 + 2NO2 + O2 → 3N2 + 6H2O(g), ΔG0298 = −1412 kJ mol−1 | (15) |
| 4NH3 + 2NO + 2NO2 → 4N2 + 6H2O, ΔG0298 = −1581 kJ mol−1 | (16) |
In the presence of O2, NO can be oxidized by active oxygen to form NO2 [eqn (17)].70 Judged by the Gibbs free energy, the reaction shown in eqn (15) does not occur easily and consequently limits the rate of eqn (15) or (16). Mn-containing metal oxide catalysts could catalyze this reaction in some extent:71,72
| 2NO + O2 → 2NO2, ΔG0298 = −70 kJ mol−1 | (17) |
NO2 is the difference between fast SCR standard SCR. NO2 acts as a more efficient oxidizing agent than O2 in the redox process of the SCR reaction. NO2 can form surface nitrites and nitrates via dimerization:73
| 2NO2 → N2O4 | (18) |
| N2O4 + H2O → HNO2 + HNO3 | (19) |
NH4NO3 is formed by the reaction between NH3 and HNO3. NH4NO3 or its related surface species is the key intermediate in the fast SCR process. The reaction processes can be described as follows:
| 2NH3 + 2NO2 → N2 + NH4NO3 + H2O | (20) |
| NH4NO3 + NO → N2 + NO2 + 2H2O | (21) |
Many researchers considered that NH4NO3 would be solid below 170 °C. NH4NO3 could be reduced by NO at a higher temperatures [eqn (21)].64,74 It is pointed out that NH3 can restrain fast SCR by inhibiting the formation of NO2 at 150–170 °C.75 Actually, eqn (21) can be described as two intermediate reactions:
| NH4NO3 ↔ NH3 + HNO3 | (22) |
| 2HNO3 + NO → 3NO2 + H2O | (23) |
There is a chemical equilibrium in the fast SCR process [eqn (22)]. The formation of HNO3 will be restrained while the NH3 concentration is raised, and that inhibits the formation of NO2 [eqn (23)]. Among the fast SCR processes, the vital process is the redox reaction between NO and HNO3, which dominates the rate of fast SCR.
The performance of low temperature SCR has been extensively investigated. Excellent NO conversion and N2 selectivity has been observed using simulated flue gas in the laboratory. Qi and Yang76 obtained more than 99% of NO conversion on the MnOx(0.3)–CeO2 catalyst sintered at 120 °C. Long et al.77 investigated the Fe–Mn-based catalysts. These catalysts showed nearly 100% NO conversion at 100–180 °C. Recently, France et al.78 studied the CeO2 modified FeMnOx catalysts, and more than 95% NO conversion was obtained at 90–135 °C without the influence of SO2 and H2O. Zhu et al.79 studied the holmium (Ho) modified Fe–Mn/TiO2 catalysts, which revealed good performance for NO conversion and high SO2 tolerance. However, more attempts need to be made to understand the fundamental mechanism of low temperature SCR, such as surface chemistry, crystal structure, kinetics and scientific reaction mechanism. These have a great influence on the performance of catalysts and knowledge of them would be beneficial in designing a new catalyst.
2.3 Side reactions
As the reductant, NH3 is a vital resource in the SCR reaction. NH3 is consumed mainly via N2 and N2O formation and the oxidation of the catalyst to NOx.52 The wastage of NH3 is a huge additional cost of the deNOx process. To decrease the wastage of NH3, an appropriate NH3/NO ratio is necessary. Authors agree that a NH3/NO ratio near to 1 is good. Furthermore, undesired reactions can occur during the SCR process. Eqn (24) and (25) show the undesired ammonia loss:| 4NH3 + 4O2 → 2N2O + 6H2O(g), ΔG0298 = −1102 kJ mol−1 | (24) |
| 4NH3 + 3O2 → 2N2 + 6H2O(g), ΔG0298 = −1310 kJ mol−1 | (25) |
These are the thermodynamically favored reactions but they occur rarely in practice.80 In addition, there is another undesired reaction during the NH3-SCR process:
| 4NH3 + 5O2 → 4NO + 6H2O, ΔG0298 = −964 kJ mol−1 | (26) |
Wang et al.81 claim that eqn (26) may replace eqn (1) as the dominant reaction over MnOx/TiO2 catalysts when the temperature was raised higher than 175 °C. This was proved by the determination of the components of outlet flue gas. This oxidization of NH3 gives a decline in NO conversion and extra consumption of NH3.
When the concentration of NH3 is appropriate, the formation of N2O is the primary waste of NH3 and this decreases the N2 selectivity [eqn (27)].35
| 4NH3 + 4NO + 3O2 → 4N2O + 6H2O(g), ΔG0298 = −1240 kJ mol−1 | (27) |
Adsorbed NH3 is oxidized on the catalyst surface to form an amine species (NH2), which subsequently reacts with NO to form N2 and H2O. However, when a further hydrogen atom is abstracted from NH2 to form an NH species, a N2O species will be formed by the reaction of the NO and NH species.82 Both the L–H mechanism and the E–R mechanism pathways contribute to N2O formation.
As previously mentioned, in the L–H mechanism, physically adsorbed NO can be oxidized by Mnn+ to Mn(n−1)+–O–NO, which can be further oxidized to monodentate nitrate (Mn(n−1)+–O–NO2) [eqn (28)]. The Mn(n−1)+–O–NO2 can react with adsorbed NH3 to form Mn(n−1)+–O–NO2–NH3. Subsequently, Mn(n−1)+–O–NO2–NH3 will be decomposed to N2O [eqn (29)]:17,83
| Mn(n−1)+–O–NO + (1/2)O2 → Mn(n−1)+–O–NO2 | (28) |
| Mn(n−1)+–O–NO2 + NH3(ad) → Mn(n−1)+–O–NO2–NH3 → Mn(n−1)+–OH + N2O + H2O | (29) |
As previously mentioned in Section 2.2, the reaction of NH4NO3 with NO is a vital step in the fast SCR process. Zhu et al.74 speculated that NH4NO3 could be decomposed to N2O and H2O via the L–H mechanism [eqn (30)]. Referring to eqn (28) and (29), the formation of N2O could be attributed to the better capacity for NH3 activation and adsorbed active nitrate species.
| NH4NO3 → N2O + 2H2O | (30) |
As mentioned previously, in the E–R mechanism, NH2 species can react with gaseous NO to form N2 and H2O. While the NH2 species is further oxidized on the metal cation to NH species, N2O will be formed by the reaction of the NH species and gaseous NO [eqn (31) and (32)].55,63,84 It is obvious that the formation of NH2NO is a crucial step of NO reduction, which is directly related to the NO conversion and N2 selectivity.58
|
NH2 + Mnn+O → NH + Mn(n−1)+–OH
| (31) |
| NH + NO(g) → N2O + H+ | (32) |
Whether the adsorbed NO is oxidized to monodentate nitrate or the NH2 species is dehydrated to NH, the N2 selectivity will be restrained and N2O is formed.85 This is an important difference from the standard SCR. The formation of N2 and N2O during the SCR process is illustrated in Fig. 1.
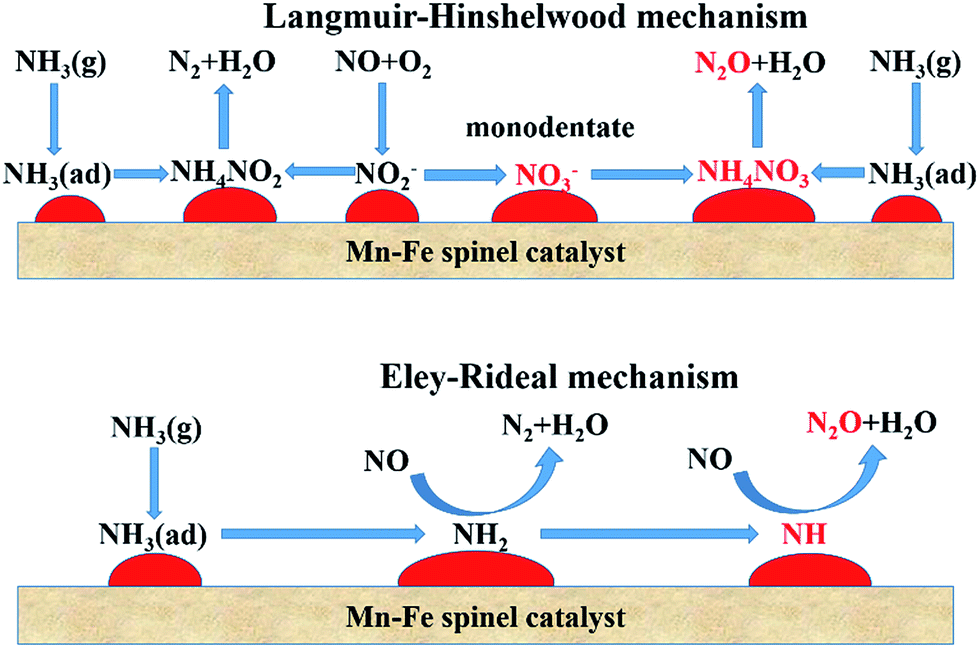 | ||
| Fig. 1 The scheme of the SCR reaction through L–H and E–R mechanisms over Mn–Fe spinel catalyst. (Reprinted with permission from ref. 35. Copyright 2014 American Chemical Society.) | ||
Hinted at by the previous equations, it is obvious that the two N atoms in N2O originate from NO and NH3, respectively. Suárez et al.86 pointed out that N2O did not primarily originate from the NH3 oxidation reaction. The feasible main reaction path is that between the coordinated NO3− (generated from NO/NO2 in the presence of O2) and the adsorbed NHx. Tang et al.63 demonstrated that the N2O selectivity of the SCR reaction over β-MnO2 was higher than that over α-Mn2O3 at 150 °C. The N2O is generated directly from the reaction of NO with NH3 via the E–R mechanism. Use of calcium (Ca) modification improves the performance of N2 selectivity for Mn-containing catalysts.87
It is suggested that N2O formation mainly resulted via the E–R mechanism.53 Yang et al.35 studied the mechanism of N2O formation over Mn–Fe spinel catalysts. N2O formation via the E–R mechanism was much more than via the L–H mechanism over the Mn–Fe spinel catalysts. In addition, N2O selectivity was not promoted by increasing the NO concentration, but it was increased with the increase in NH3 concentration. N2O selectivity is also related to the gas hourly space velocity (GHSV). It was also found that N2O in the SCR reaction over Mn–Ce catalysts was generated via the E–R mechanism, not the L–H mechanism.88 The choice of E–R or L–H mechanisms ways will vary with the changes of temperature. It is reported that the L–H mechanism plays the main role below 150 °C, and the E–R mechanism way dominates the SCR reaction at higher temperatures.55,89
2.4 Synergistic effect
A pure metal oxide may not be suitable for practical applications because of its defects. However, the property of one metal oxide can be improved by introducing foreign metal cations into its lattice. There will also be an interaction between different metal oxides. For example, reports in the literature indicate that Mn–Ce mixed oxide catalysts demonstrated the best performance among a multitude of metal oxide catalysts. Ceria can enhance the adsorption of NO and O2, which benefits the oxidation of NO to NO2 and improve sulfur resistance. Qi and Tang76 found that the oxidation of NO to NO2 was increased significantly after addition of ceria to MnOx and that it speeded up the overall process. Actually, pure CeO2 cannot be applied in industry because of its small specific surface area and low thermal stability.90 Meanwhile, as is reported,91 modification with titanium (Ti) or tin (Sn) can improve the SCR property of cerium oxides. Qi et al. and Imamura et al.27,92 found using X-ray diffraction patterns that there was no manganese oxide phase in the calcined Mn–Ce catalyst prepared by a co-precipitation method. This indicated that strong interactions exist between manganese and cerium oxides, because Mn2O3 and MnO2 can be detected in pure manganese oxide calcined at the same temperature.The redox property of catalysts is the key factor of the NH3-SCR processes.29 Electronic transfer, showing as oxidation and reduction, plays quite an important role in catalytic reactions. The redox couples exist over the metal oxide catalysts, such as Mn4+/Mn3+, Ce4+/Ce3+ and Fe3+/Fe2+, which provide the redox cycles with excess oxygen. The activity of bi-metal and multi-metal oxide catalysts could be promoted by dual redox cycles. The general formula can be described as follows:
| Mn+ + Nm+ ↔ M(n−1)+ + N(m+1)+ | (33) |
There is a typical SCR reaction process via the E–R mechanism on Mn–Ce/TiO2 and Mn–Ce/aluminium oxide (Al2O3) catalysts.27,93 Manganese oxides and ceria oxides also interact. They can form a solid solution because of the similarity of their structure.94 Ceria has a superior oxygen storage performance. Thus, the process of oxidizing Mn3+ to Mn4+ is enhanced by using ceria.95
Liu et al.96 investigated a Mn–Ce–Ti mixed oxide catalyst prepared using a hydrothermal method, and found that there were dual redox cycles, such as Mn4+ + Ce3+ ↔ Mn3+ + Ce4+ and Mn4+ + Ti3+ ↔ Mn3+ + Ti4+. These dual redox cycles can promote each other and facilitate the electron transfer between Mn, Ce and Ti active sites by decreasing the migration energy. The proposed schemes are as follows (Fig. 2).
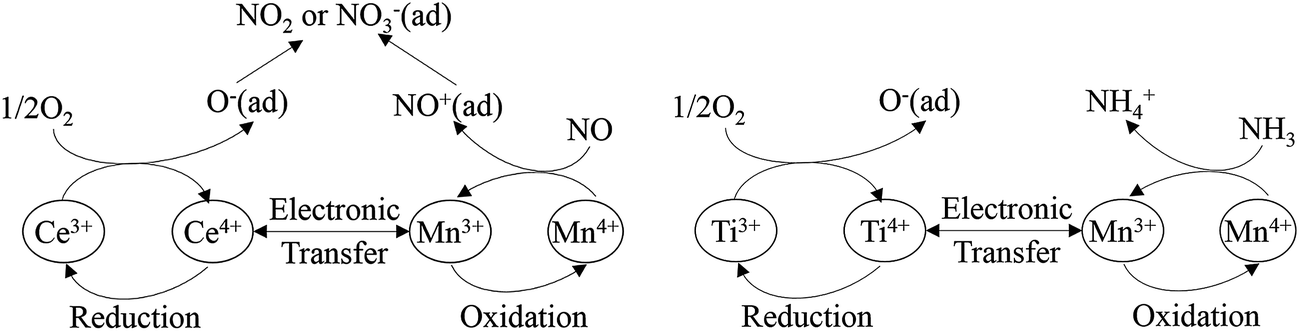 | ||
| Fig. 2 The scheme of dual redox cycle during SCR process. (Reprinted with permission from ref. 96. Copyright 2014 American Chemical Society.) | ||
The scheme shows that Mn cation sites may be the main active site for the adsorption of N. Furthermore, the addition of Ce, Fe, Cu, Ni and so on, may show a synergistic effect, which facilitates the generation of Mn4+ from Mn3+. Kwon et al.97 studied the MnOx/CeO2–TiO2 catalyst system. When Ce was added to Mn/Ti, an oxygen bridge of Mn–O–Ce was formed and, thus enhanced the binding between Mn and O2. This oxygen bridge provided a channel for the electron transfer between manganese and cerium cations, and particularly accelerated the oxidation of Mn3+ to Mn4+ by Ce4+.23
| Mn2O3 + 2CeO2 → 2MnO2 + Ce2O3 | (34) |
Among the Mn–Fe mixed oxide catalysts, electronic transfer occurs between the different oxidation states of Fe3+, Fe2+, Mn4+ and Mn3+.98 The performance of the Mn/TiO2 catalyst was improved by the addition of Fe.99 The process can be described approximately as follows:
| Fe3+ + Mn3+ ↔ Fe2+ + Mn4+ | (35) |
| NO + Mn4+ → NO+(ad) + Mn3+ | (36) |
| 1/2O2 + Fe2+ → Fe3+ + O−(ad) | (37) |
| NO+(ad) + O−(ad) → NO2 | (38) |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe2+ + Mn4+ → Fe3+ + Mn3+ Fe2+ + Mn4+ → Fe3+ + Mn3+
| (39) |
Liu et al.100,101 investigated a series of WO3-doped Mn–zirconium (Zr) mixed oxide catalysts. Using catalyst performance measurements, the SCR performance and poisoning tolerance of the Mn–Zr catalyst doped with WO3 was higher than that for the Mn–Zr catalyst alone. There were redox couples of Mn4+/Mn3+ and W6+/W5+, (i.e., W5+ + Mn4+ ↔ W6+ + Mn3+). The redox property and the electron transfer was improved using these dual redox couples (Fig. 3). Thus, the electron transfer between Mn and W active sites was promoted and this contributes to the activation of NH3 and an improvement of the NO conversion (Fig. 4).
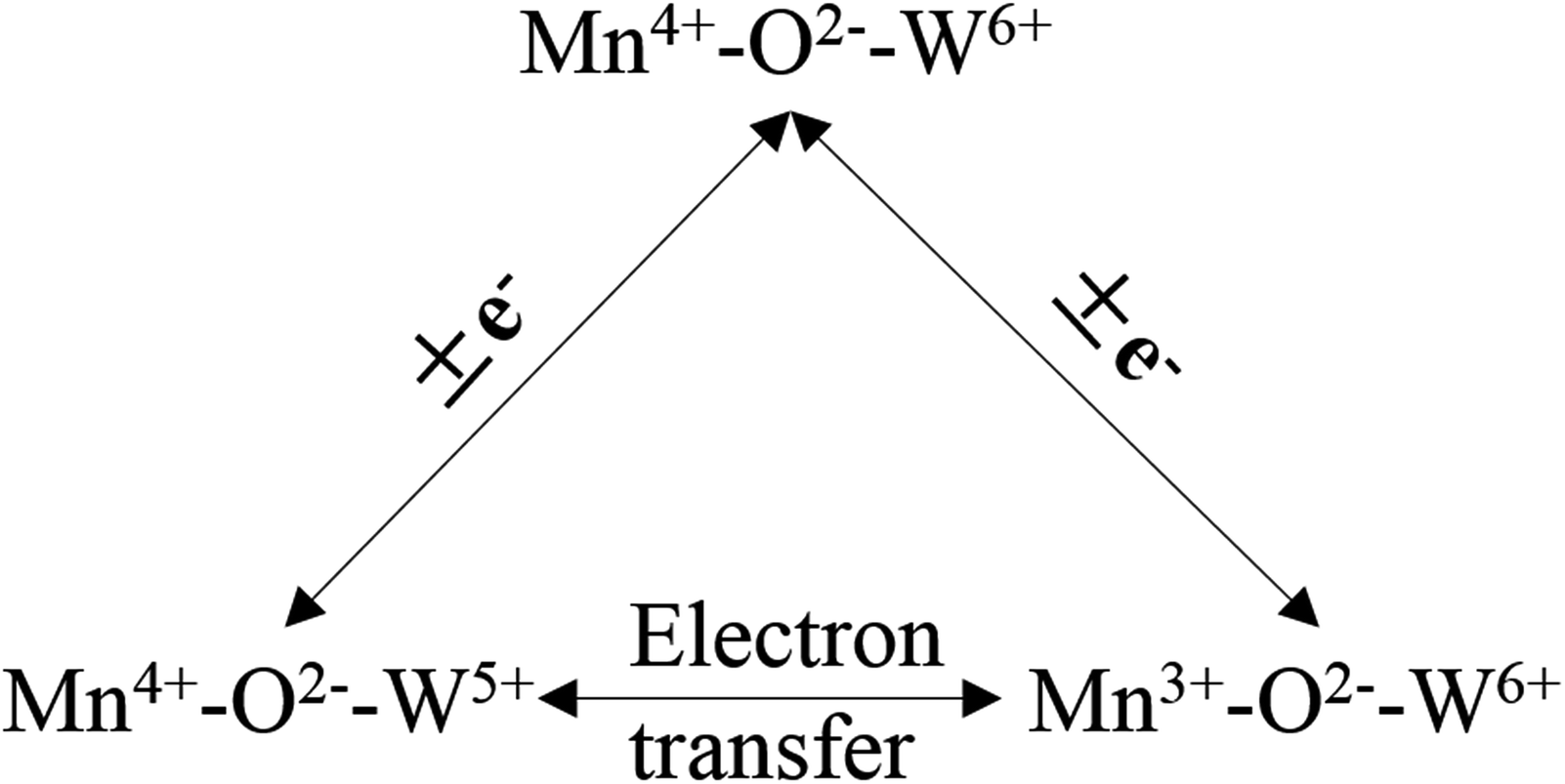 | ||
| Fig. 3 The electron transfer of redox couples of Mn4+/Mn3+ and W6+/W5+. | ||
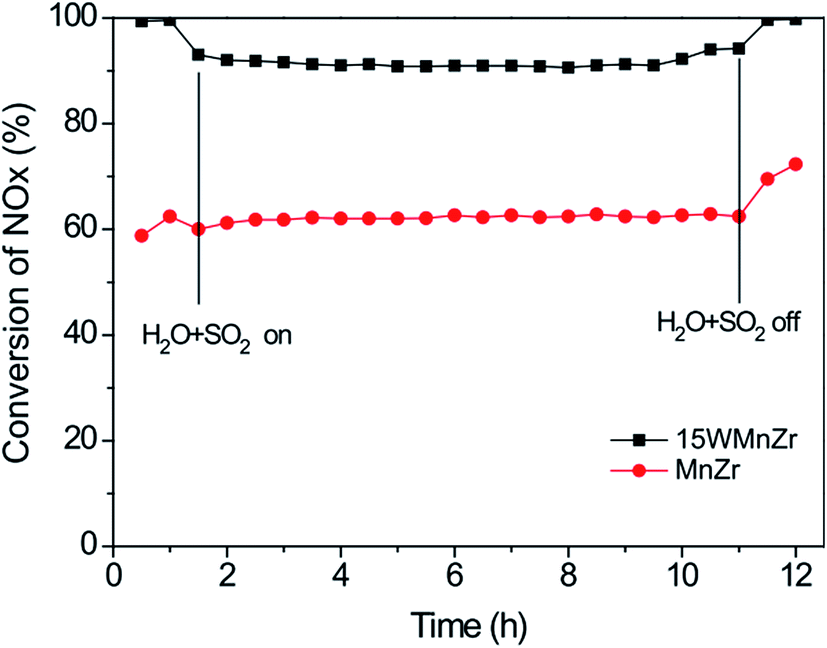 | ||
Fig. 4 NOx conversion over MnZr and WMnZr catalysts at 300 °C. Reaction conditions: [NO] = [NH3] = 500 ppm, [O2] = 5%, [H2O] = 5%, [SO2] = 50 ppm, GHSV = 128![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. (Reprinted from ref. 100. Copyright 2015, with permission from Elsevier.) 000 h−1. (Reprinted from ref. 100. Copyright 2015, with permission from Elsevier.) | ||
Metal oxides could catalyze the reduction of NO with NH3 via the transfer of electrons.102,103 As is known, catalysts play a role in accelerating the reaction rate. Referring to Fig. 2, it can be seen that metal cations provide the adsorption sites and function as the transfer station of electrons in the SCR process. Manganese mainly acts as the adsorption center for nitrogen. Mn4+ receives an electron from NO or NH3 and will be reduced into Mn3+. Then the reduced Mn3+ would be restored to Mn4+ by an extra oxygen and then the next redox cycle starts. However, a faster pathway is via the transfer of an electron between metal oxides, such as Ce, Fe, W and so on. Therefore, to design a catalyst, it is necessary to introduce an element for the role of the adsorption and oxidation of nitrogen. Simultaneously, another element is required for superior oxygen storage to quickly restore the reduced element. The coordination of these two types of elements will improve the performance of SCR.
It is essential to characterize the catalysts' structure in order to design an excellent catalyst. The current technology for treating the exhaust gas is supported vanadium-based catalysts on TiO2 modified by W or Mo addition. Depending on the coverage, different polymeric vanadium oxides (VOx) could segregate at the surface and these exhibited different turnover frequency (TOF) and selectivity.104,105 This could be interesting if the same trend existed for MnOx species, however, there has been little research proposed on use of different polymeric MnOx corresponding to their different performances. Ettireddy et al.106 studied TiO2 supported manganese oxide catalysts. Different TOFs were obtained on the Mn/TiO2 loaded with different amounts of manganese (Table 2). It was proposed that the polymeric or microcrystalline form of MnOx was envisaged at higher loadings. As a general trend, the TOF and selectivity decreased with the polymeric form increasing at higher loadings. However, further study should be done to confirm which kind of polymeric manganese was formed and its TOF and selectivity should also be determined.
In this section, the reaction mechanisms have been summarized. It was supposed originally that NO2 could be the reactant of SCR process. However, it is widely agreed that the main reactant for the SCR process is NO, while NO2 is reduced by the fast SCR process.107 The synergistic effect among the different metal cations is essential to improve the catalysts' performance, such as NO conversion, selectivity and poisons' tolerance. According to various reports, the low resistance to different poisons is the greatest obstacle for the application of low temperature SCR catalysts.
3. Catalyst deactivation
Because of the demands of high temperature operation, conventional SCR catalysts suffer a huge amount of damage from the sulfur oxides, water vapor, heavy metal ions and alkali and alkaline earth metal ions in the upstream of the flue gas.108 Installing the reactor downstream of the desulfurizer and precipitator is an excellent way to avoid deactivation. Many metal oxide catalysts have been reported as being low temperature SCR catalysts,99,109,110 however, commercial low temperature SCR catalysts have narrow fields of application because they are not immune to the residual SO2 and H2O contained in real flue gas. The poor tolerance of SO2 and H2O has been a major obstacle for practical applications.111 Therefore, it is significant to illuminate the poisoning mechanisms of SO2, H2O and so on.3.1 SO2 and H2O
Sulfur oxides are mainly generated from the combustion of fossil fuels and the sintering of ore. Residual SO2 after desulfurization can still damage the metal oxide catalysts. The deposition of ammonium sulfates, such as ammonium bisulfate (NH4HSO4) and ammonium sulfate [(NH4)2SO4], is the primary cause for the deactivation of metal oxide catalysts at low temperature.112 The decomposition temperature of ammonium sulfite [(NH4)2SO3] and (NH4)2SO4 salts is higher than the operation temperature of the catalysts. Most researchers regard the poisoning of SO2 as a major problem. The deactivation of SO2 can be classified into two categories: deposition of (NH4)2SO4 and sulfation of active sites. The undesired metal sulfates and (NH4)2SO4 would occupy active sites on the surface and gradually deactivate the catalyst. The deactivation caused by water vapor can contribute to the competitive adsorption. The adsorption of H2O on the catalysts' surface blocks the active sites, which are provided for the adsorption of NH3 and NO.| SO2 + 1/2O2 → SO3 | (40) |
| NO2 + SO2 → NO + SO3 | (41) |
Gaseous NH3 was assisted by the Brønsted acid sites to form NH4+, which could react with SO2 or SO3 to form (NH4)2SO3 or (NH4)2SO4, respectively. In addition, NH4HSO4 species were also generated in the flue gas. The formation of NH4HSO4, (NH4)2SO3 and (NH4)2SO4 can be described as follows:114
| SO3 + H2O → H2SO4 | (42) |
| H2SO4 + NH3 → NH4HSO4 | (43) |
| 2NH3 + SO2 + H2O → (NH4)2SO3 | (44) |
| 2NH3 + SO3 + H2O → (NH4)2SO4 | (45) |
Actually, (NH4)2SO3 and (NH4)2SO4 can be decomposed at a relatively higher temperature. However, low temperature SCR of NOx is usually requested at a low operation temperature, which is lower than the decomposition temperature of the (NH4)2SO3 and (NH4)2SO4. Therefore, removing the undesired side-products of (NH4)2SO4 salts is a big challenge to researchers.
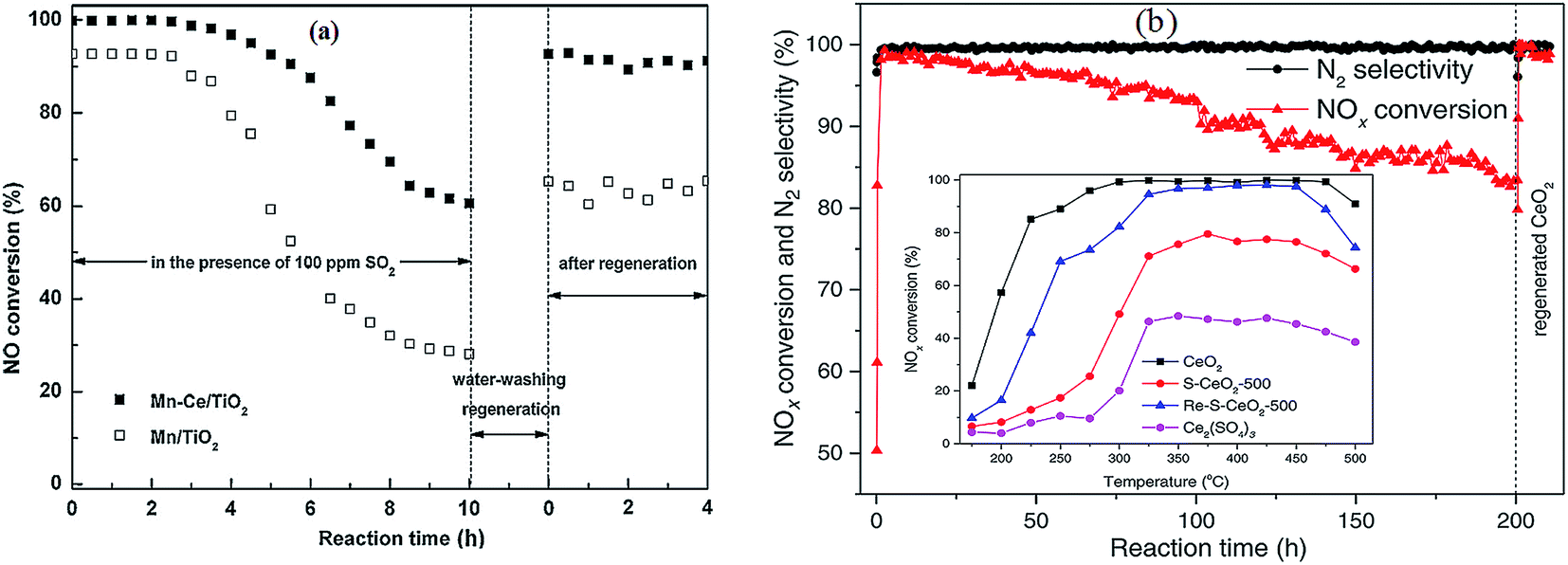
Almost all of reported MnOx catalysts were affected by the introduction of SO2 in the feed gas.115 Zhang et al.116 introduced 100 ppm SO2 in the feed gas, which induced an apparent decrease of NO conversion over the Mn–Ce metal oxide catalysts supported on carbon nanotubes. Lu et al.117 fed 200 ppm SO2 to the flue gas, and then the NOx conversion of Mn–Ce/TiO2 catalyst decreased from an initial value of 99% to about 78%. Jiang et al.118 investigated the effect of SO2 on MnOx(0.4)/TiO2 catalysts prepared by three methods, sol–gel, impregnation and co-precipitation. The NO conversions had an apparent decrease for these catalysts (Fig. 5a).
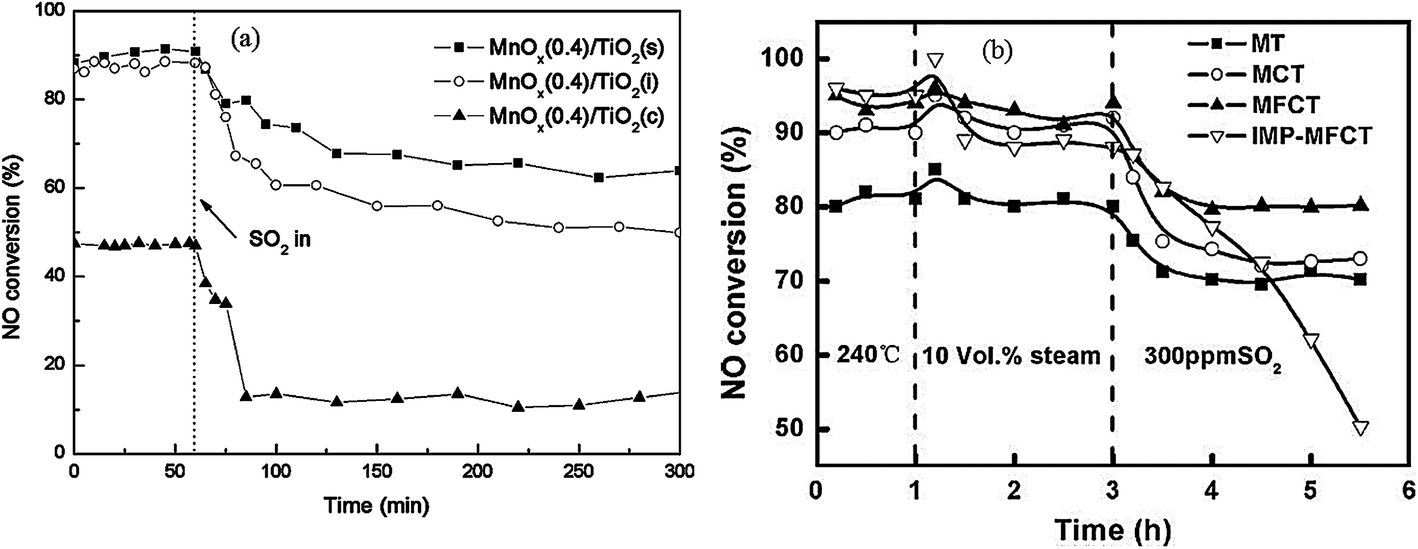 | ||
| Fig. 5 The effect of SO2 on NO conversion. (a) Reaction conditions: [NO] = [NH3] = 1000 ppm, [O2] = 3%, [SO2] = 200 ppm, balance N2, temperature: 150 °C, GHSV = 30000 h−1. (Reprinted from ref. 118. Copyright 2008, with permission from Elsevier.) (b) Reaction conditions: [NO] = 600 ppm, [NH3] = 480 ppm, [O2] = 2%, [SO2] = 300 ppm, [H2O] = 10 vol%, balance N2, temperature: 240 °C, GHSV = 24000 h−1. (Reprinted from ref. 111. Copyright 2009, with permission from Elsevier.) | ||
Yu et al.111 prepared MnO2–Fe2O3–CeO2–TiO2 catalysts. The performance of this catalyst was decreased by introducing SO2. The NH4+ species and the SO42− species were determined from Fourier-transform infrared spectra. The NH4+ species were chemisorbed on to the Brønsted acid sites.119 This means that the poisoning of SO2 can be via the formation and deposition of (NH4)2SO4, which blocks the active channels of the catalyst. The NO conversion was decreased to 50% from 90% (Fig. 5b).
Xu et al.120 also found that NH4HSO3 and NH4HSO4 formed via the reaction of SO2 and NH3 could be deposited on catalysts' surface and blocked the active sites [eqn (34)–(39)]. Furthermore, more Brønsted acid sites will be generated while the sulfates are formed by SO2 adsorption on surface. The Lewis acid site could be transformed to the Brønsted acid site by adsorption of a water molecule.121 This means that a wet atmosphere would promote the formation of the Brønsted acid sites, which facilitates the sorption of NH4+.122 In terms of diffuse reflectance infrared Fourier transform (DRIFT) spectra, Jiang et al.123 proved that the formation of NH4+ was promoted after introducing SO2. However, even though Brønsted acid sites were formed by the sulfatization, NO conversion was decreased because SO2 occupied the NO adsorption sites.
Therefore, to obtain high NO conversion, it is necessary to prevent the formation of (NH4)2SO4. Actually, it is nearly impossible to eliminate the residual SO2 completely. Efficient ways to do it may be preventing the oxidation of SO2 and decreasing the decomposition temperature of (NH4)2SO4 and NH4HSO4 on the catalysts' surface.
Jin et al.25 studied the Mn–Ce/TiO2 and Mn/TiO2 catalysts. In terms of the thermogravimetry/differential scanning calorimetry (TG/DSC) results, the decomposition temperatures of (NH4)2SO4 and NH4HSO4 on the Mn/TiO2 catalyst was determined to be 213 °C and 361 °C, respectively. However, in the case of the Mn–Ce/TiO2 catalyst, the decomposition temperature of NH4HSO4 was approximately 286 °C, which was much lower than 361 °C. This indicated that the thermal stability of NH4HSO4 on the catalyst was greatly reduced after introducing cerium. This inference was also proved by the DRIFT results. Therefore, ceria improved the performance of Mn/TiO2 catalyst.
There is a universal agreement that residual SO2 damages the metal oxide catalysts and decreases the NO conversion. (NH4)2SO3 and (NH4)2SO4 were formed on catalysts' surface by the reaction of SO2. Researchers found that the NO conversion would increase for a while when SO2 was introduced and then finally decrease. The adsorption of SO2 improved the amount of Lewis acid sites, and thus the capacity of NH3 was improved. However, the sulfation damages the manganese cations, which are the active sites of NO.
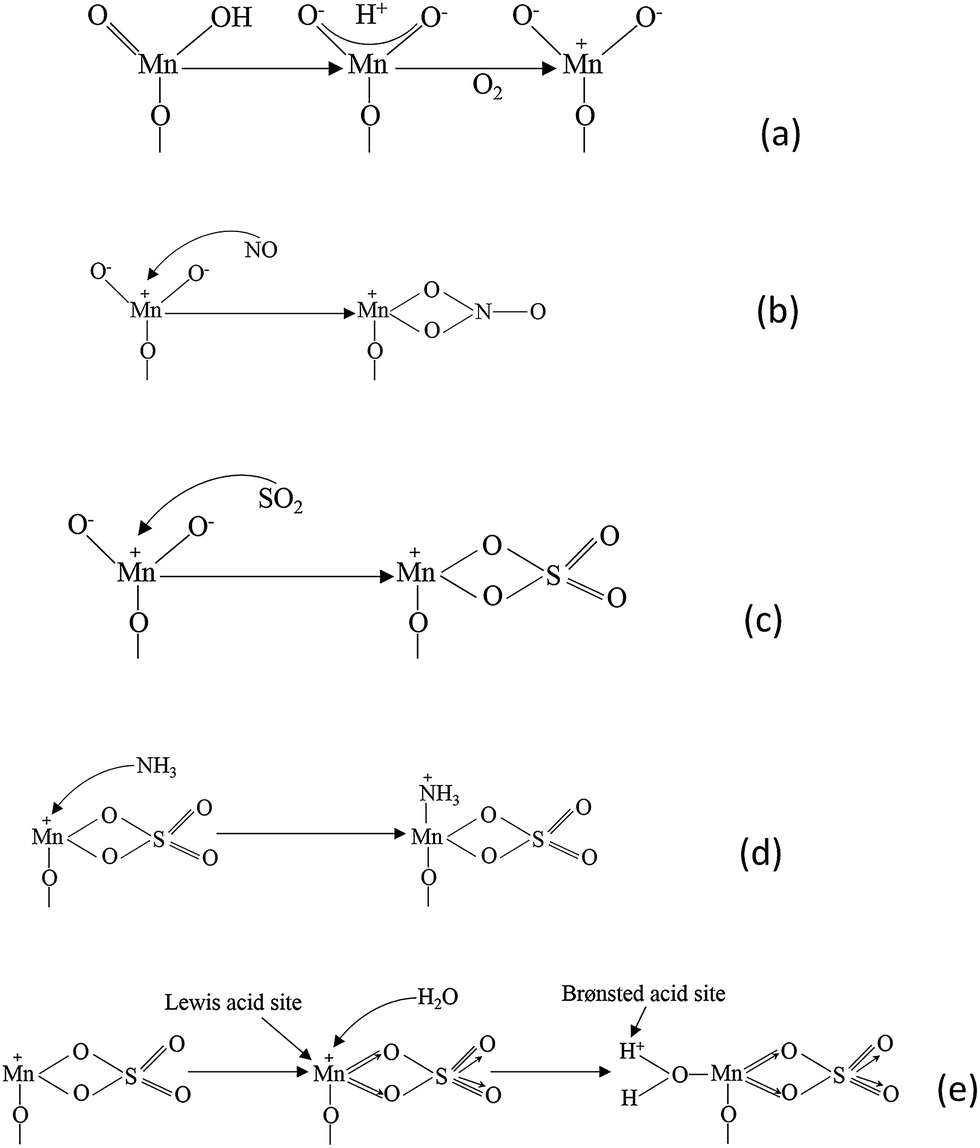 | ||
| Fig. 6 The proposed mechanism of SO2 deactivation effect on the SCR reaction. (Reprinted with permission from ref. 123. Copyright (2010) American Chemical Society.) | ||
Yu et al.111 investigated the formation of metal sulfation on fresh Mn–Fe–Ce–Ti catalyst impregnated (NH4)2SO4. In terms of the TG curve, SO3 was released from (NH4)2SO4 decomposition and then combined with Mn species to form manganese sulfate (MnSO4). They claimed that the MnSO4 could not be formed directly by the reaction of oxidized SO2 and Mn species.57 Kijlstra et al.125 proved that the transformation of MnO to MnSO4 on MnOx/Al2O3 catalyst significantly deactivated the catalyst's activity.
Efforts have been made to facilitate the SO2 tolerance of metal oxide catalysts. Ceria may trap SO2 for NOx storage catalysts to limit the sulfation of the dominating active phase and inhibit the formation of (NH4)2SO4 and NH4HSO4.117,126 After pre-treatment with SO2, Ce doped Mn/TiO2 catalysts had more Lewis acid sites than Mn/TiO2 catalysts. This result implied that the addition of ceria could prevent the Lewis acid sites from the sulfation of SO2.
Liu et al.127 compared the performance of Mn–Ce mixed oxide catalysts prepared using the surfactant template method and the conventional co-precipitation method. Referring to the catalytic activity measurement, the Mn5–Ce5 catalyst prepared using the surfactant template method showed the highest NOx conversion whether SO2 and H2O were introduced or not. The catalysts prepared using the surfactant template method possessed a higher surface area and smaller active sites, which contributed to a higher NOx reduction.
In terms of in situ DRIFT analysis, Jin et al.25 found that the Lewis acid sites could be preserved effectively with the doping of Ce while the SO2 was added. SO2 was oxidized to SO3 or sulfation species on MnOx, however, SO3 and sulfation species move into ceria to form bulk like sulfate species. Therefore, ceria trapped SO2 and protected the dominant active manganese cations (Fig. 7). Furthermore, in terms of the DRIFT and TG-DSC results, it was indicated that the thermal stability of sulfation species over the Mn–Ce catalyst was lower than that over the MnOx catalyst. Referring to the study of Kylhammar et al.,128 it is assumed that the bulk sulfation species in ceria reveals a high mobility, which facilitates their desorption.
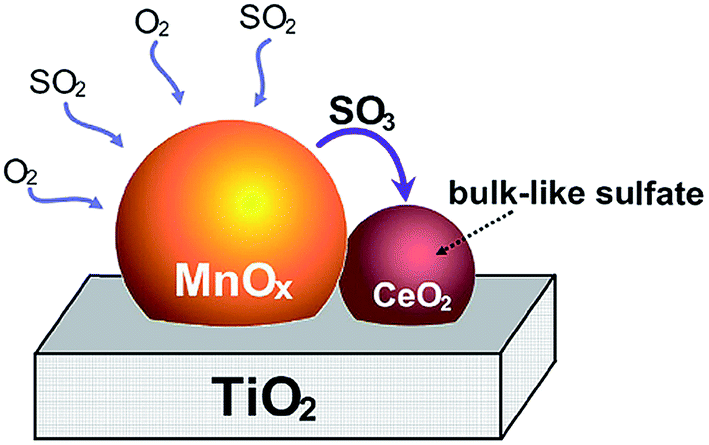 | ||
| Fig. 7 The formation schematic of bulk like sulfate on Mn–Ce/Ti catalysts. (Reprinted from ref. 25. Copyright 2013, with permission from Elsevier.) | ||
Wang et al.,113 Xu et al.,120 and Shi et al.129 proved that the active manganese cation was reserved for ceria, which finally sulfated it. Ce4+ distributed on the catalysts' surface transformed into Ce3+ after sulfation. The reaction can be described as follows:
| 2CeO2 + 3SO2 + O2 → Ce2(SO4)3 | (46) |
Furthermore, it is reported that Zr could optimize the redox property and strengthen SO2 tolerance.89 Chang et al.130,131 reported that Sn modification could further improve the tolerance of the Mn–Ce catalyst to SO2 and H2O. They compared the NO conversion of Sn(0.1)–Mn(0.4)–Ce(0.5)–O and Mn(0.4)–Ce(0.6)–O mixed oxide catalysts. It was obvious that the NO conversion of the Mn(0.4)–Ce(0.6)–O catalyst was decreased more significantly than that of the Sn(0.1)–Mn(0.4)–Ce(0.5)–O catalyst when 200 ppm of SO2 and 3% O2 was fed in to the system at 220 °C.
Shi et al.132 compared the resistance of the Mn/TiO2 catalyst and the hierarchically macro-mesoporous Mn/TiO2 (HM-Mn/TiO2) catalyst prepared by the sol–gel method. After feeding 30 ppm SO2 to the system, the NO conversion of the Mn/TiO2 catalyst decreased sharply from 57% to 15%, however, the NO conversion of the HM-Mn/TiO2 catalyst kept a higher value of more than 84%. The result indicated that maybe the SO2 resistance could be improved by using a hierarchically macro-mesoporous structure.
As previously, because NH3 could be adsorbed on both the Lewis acid sites and the Brønsted acid sites, there is little influence on the adsorption of NH3. However, the adsorption ability of SO2 was higher than that of NO. Residual SO2 would be adsorbed on Mn cations, which are the active sites for the adsorption of NO. The damage caused by sulfation would be permanent and irreversible. Doping with ceria should be a good choice to divert this damage from Mn. More research should be done to investigate the reaction mechanism between SO2 and Mn cations. The correlations should be established between the extent of sulfation and the degree of dispersion of MnOx species at the surface.
The main reason for the decrease of activity can be attributed to the competitive adsorption of H2O. Many researchers reported that the adsorption of H2O on the catalysts' surface blocked the active sites, which are provided for the adsorption of NH3 and NO.109,133 Chen et al.134 studied a MnOx–niobium oxides (NbOx)–CeO2 catalyst prepared by a sol–gel method and found that the adsorption of H2O inhibited the adsorption of NOx. Xiong et al.108 compared the SCR performance of Mn–Fe spinel catalysts in the presence and absence of H2O. They proposed that the effect of H2O can be attributed to the competitive adsorption, the decrease of oxidation ability and the inhibition of interface reactions.135,136 The temperature programmed desorption (TPD) profiles of NH3 and NOx were obtained, and the NOx and NH3 adsorption capacity of Mn–Fe spinel in the absence of H2O and in the presence of 5% H2O are shown in Table 3.
| Condition | NH3 (μmol−1 g−1) | NOx (μmol−1 g−1) |
|---|---|---|
| In the absence of H2O | 122 | 82 |
| In the presence of 5% H2O | 105 | 46 |
Fig. 8 shows that the NOx conversion apparently decreased when 5% H2O was fed in to the flue gas, especially at the lower temperature, e.g., below 160 °C. The adsorption of H2O vapor on the catalyst's active sites deprived the sites of NH3 adsorption, which apparently decreased the NO conversion. There is a summary of Mn-containing catalysts' performance in the presence and in the absence of SO2 and H2O (Table 4).
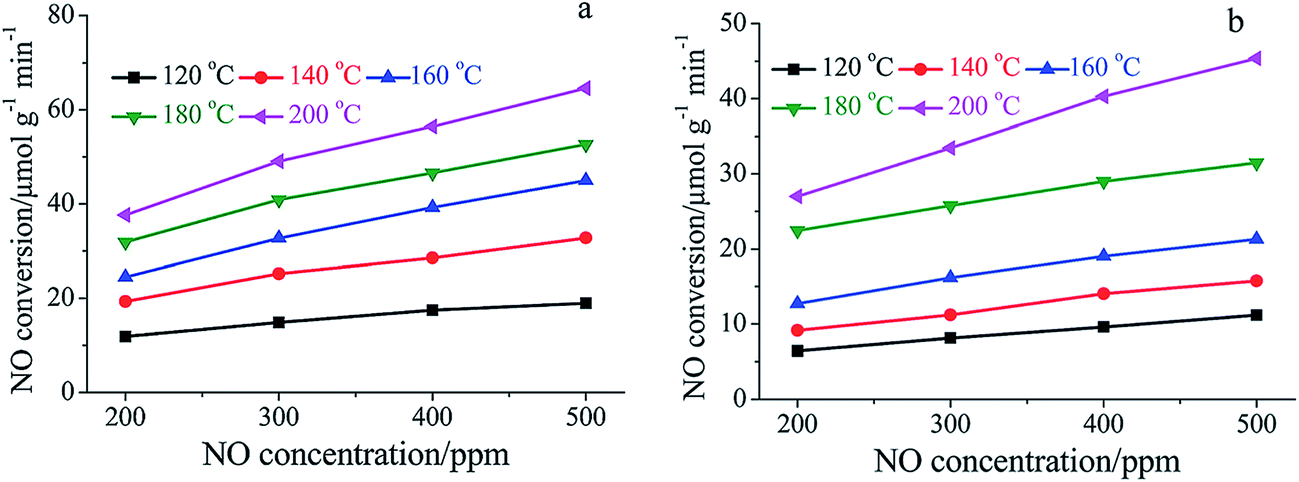 | ||
| Fig. 8 Dependence of NO conversion rate on gaseous NO concentration over Mn–Fe spinel: (a) in the absence of H2O; (b) in the presence of 5% H2O. (Reproduced from ref. 108 with permission from the Royal Society of Chemistry.) | ||
| Catalysts | Preparation processa | Reaction conditionsb | NOx conversion 1c | Poison conditiond | NOx conversion 2e | Ref. |
|---|---|---|---|---|---|---|
| a Preparation process means of preparation method, calcination temperature and time.b Reaction gas mixture and GHSV.c NO conversion at a specified temperature.d The concentration of SO2 and H2O introduced on the basis of reaction gas.e NO conversion at a certain temperature after introducing SO2 and/or H2O for a specified time. | ||||||
| Mn0.4–Ce0.5–Sn0.1–O | Co-precipitation/500 °C/6 h | 0.1% NO, 0.1% NH3, 2% O2/35000 h−1 |
100% (110–230 °C) | 0.01% SO2, 9% H2O | 62% (110 °C) | 130 |
| Mn0.2–Ce0.1–Ti0.7–O | Hydrothermal/500 °C/6 h | 0.05% NO, 0.05% NH3, 5% O2/64000 h−1 |
>92% (150–250 °C) | 0.005% SO2, 5% H2O | ∼90% (200 °C/10 h) | 96 |
| Mn0.3–Ce0.7–O | Citric acid/650 °C/6 h | 0.1% NO, 0.1% NH3, 2% O2/42000 h−1 |
>95% (100–150 °C) | 0.01% SO2, 2.5% H2O | ∼95% (120 °C/4 h) | 76 |
| Mn5–Ce5–O | Surfactant template/500 °C/4 h | 0.05% NO, 0.05% NH3, 5% O2/64000 h−1 |
>95% (100–200 °C) | 0.005% SO2, 5% H2O | >90% (150–200 °C/-) | 127 |
| Mn0.28–Ce0.05–Ti 0.67–O | Co-precipitation/400 °C/2 h | 0.06% NO, 0.06% NH3, 3% O2/40000 h−1 |
>92% (120–180 °C) | 0.07% SO2, 3% H2O | 35% (120 °C/13 h) | 137 |
| Mn0.4–Ce0.07–Ti1–O | Sol–gel/500 °C/6 h | 0.08% NO, 0.08% NH3, 3% O2/40000 h−1 |
∼100% (100–180 °C) | 0.01% SO2, 3% H2O | ∼60% (100 °C/10 h) | 138 |
| Sn0.1–Mn0.4–Ce0.5–O | Co-precipitation/500 °C/6 h | 0.1% NO, 0.1% NH3, 2% O2/35000 cm3 g−1 h−1 |
∼100% (110–230 °C) | 0.01% SO2, 12% H2O | ∼70% (110 °C/9 h) | 131 |
| Mn1–Ce0.3/TiO2–graphene | Impregnation/500 °C/6 h | 0.05% NO, 0.05% NH3, 7% O2/67000 h−1 |
>90% (140–180 °C) | 0.02% SO2, 10% H2O | ∼75% (180 °C/3 h) | 117 |
| Mn0.6/Ce0.5–Zr0.5–O | Impregnation/500 °C/6 h | 0.06% NO, 0.06% NH3, 3% O2/30000 h−1 |
>90% (140–180 °C) | 0.01% SO2, 3% H2O | ∼90% (180 °C/3 h) | 139 |
| Mn0.4–Ce0.07–Ti1–O | Sol–gel/500 °C/6 h | 0.1% NO, 0.1% NH3, 3% O2/40000 h−1 |
∼100% (120–220 °C) | 0.01% SO2, 3% H2O | ∼82% (150 °C/7 h) | 140 |
| Mn0.4–Ce0.5–W0.1–O | Sol–gel/600 °C/3 h | 0.05% NO, 0.05% NH3, 5% O2/40000 h−1 |
>80% (140–300 °C) | 0.006% SO2, 5% H2O | ∼55% (150 °C/3 h) | 141 |
| Mn0.3–Ce0.7–O | Citric acid/650 °C/6 h | 0.1% NO, 0.1% NH3, 2% O2/42000 h−1 |
∼100% (120–150 °C) | 0.01% SO2, 6% H2O | ∼92% (120 °C/4 h) | 23 |
| Mn0.2–Ce0.1–Ti0.7–O | Hydrothermal/500 °C/6 h | 0.05% NO, 0.05% NH3, 5% O2/64000 h−1 |
>95% (150–350 °C) | 0.005% SO2, 5% H2O | ∼90% (200 °C/10 h) | 96 |
| Mn–Ce–W–Ti–O | Impregnation/400 °C/4 h | 0.02% NO, 0.02% NH3, 8% O2/30000 h−1 |
∼100% (160–200 °C) | 0.01% SO2, 8% H2O | ∼85% (180 °C/10 h) | 97 |
| Mn0.4–Ce0.07–Ti1–O | Co-precipitation/400 °C/2 h | 0.06% NO, 0.06% NH3, 3% O2/40000 h−1 |
>92% (120–180 °C) | 0.07% SO2, 3% H2O | 61% (120 °C/2.5 h) | 142 |
| Mn0.2–Fe0.15–Ce0.3–Ti1–O | Sol–gel/500 °C/6 h | 0.06% NO, 0.06% NH3, 3% O2/50000 h−1 |
>95% (160–260 °C) | 0.01% SO2, 3% H2O | ∼85% (180 °C/6 h) | 143 |
| Mn0.6–Ce0.5–Zr0.5–O | Impregnation/500 °C/6 h | 0.06% NO, 0.066% NH3, 6% O2/45000 h−1 |
>95% (140–220 °C) | 0.01% SO2, 3% H2O | ∼90% (180 °C/7 h) | 144 |
| Mn0.4–Fe0.1–Ce0.5–O | Co-precipitation/500 °C/6 h | 0.1% NO, 0.1% NH3, 2% O2/84000 h−1 |
>82% (150–180 °C) | 0.01% SO2, 2.5% H2O | >90% (150 °C/4 h) | 27 |
| Mn0.4–Ce0.1–Ti1–O | Sol–gel/500 °C/6 h | 0.08% NO, 0.08% NH3, 3% O2/40000 h−1 |
∼100% (150 °C) | 0.01% SO2, 3% H2O | ∼60% (150 °C/10 h) | 25 |
| Mn0.6–Fe0.4–O | Citric acid/500 °C/3 h | 0.1% NO, 0.1% NH3, 3% O2/30000 h−1 |
>95% (90–220 °C) | 0.01% SO2, 5% H2O | ∼88% (120 °C/6 h) | 98 |
| 10% Mn/Fe–Ti spinel | Impregnation/500 °C/3 h | 0.05% NO, 0.05% NH3, 2% O2/24000 cm3 g−1 h−1 |
>95% (150–250 °C) | 0.006% SO2, 8% H2O | ∼80% (200 °C/13 h) | 53 |
| Mn0.4–Fe0.1/Ti1–Zr0.5–O | Sol–gel/500 °C/6 h | 0.1% NO, 0.1% NH3, 4% O2/30000 h−1 |
>95% (80–180 °C) | 0.01% SO2, 8% H2O | ∼70% (150 °C/5 h) | 89 |
| Mn0.4–Fe0.1/Ti0.5–O | Sol–gel/500 °C/6 h | 0.1% NO, 0.1% NH3, 3% O2/30000 h−1 |
∼100% (150 °C) | 0.02% SO2 | ∼65% (150 °C/6 h) | 123 |
| Mn0.6–Ti1–O | Sol–gel/500 °C/6 h | 0.1% NO, 0.1% NH3, 3% O2/30000 h−1 |
∼100% (110 °C) | 0.003% SO2 | ∼85% (120 °C/9 h) | 132 |
| Mn0.4–Ti1–O | Sol–gel/500 °C/6 h | 0.1% NO, 0.1% NH3, 3% O2/30000 h−1 |
>90% (180–250 °C) | 0.02% SO2, 3% H2O | ∼70% (150 °C/6 h) | 118 |
| Mn–Fe–Ce–Ti–O | Sol–gel/500 °C/6 h | 0.06% NO, 0.048% NH3, 2% O2/24000 h−1 |
>80% (200–300 °C) | 0.03% SO2, 10% H2O | ∼80% (240 °C/5 h) | 111 |
| 7% Mn/Ti1–graphene | Impregnation/450° C/6 h | 0.05% NO, 0.05% NH3, 7% O2/67000 h−1 |
>80% (120–180 °C) | 0.02% SO2, 10% H2O | ∼72% (180 °C/3 h) | 115 |
| Mn0.5–Zr0.5–O | Citric acid/450 °C/3 h | 0.1% NO, 0.1% NH3, 3% O2/30000 h−1 |
∼100% (100–200 °C) | 0.01% SO2, 5% H2O | ∼40% (150 °C/13 h) | 124 |
| Mn2.5–Cu0.1–Ti1–O | Co-precipitation/350 °C/6 h | 0.05% NO, 0.05% NH3, 5% O2/30000 h−1 |
∼100% (60–280 °C) | 0.01% SO2, 11% H2O | ∼60% (125 °C/10 h) | 39 |
| Mn4/Co0.6–Ce2.7–Zr2.7 | Impregnation/500 °C/6 h | 0.06% NO, 0.06% NH3, 6% O2/45000 h−1 |
>97% (120–220 °C) | 0.01% SO2, 3% H2O | ∼93% (180 °C/7 h) | 109 |
| Mn0.6–Cr0.4–O | Citric acid/650 °C/3 h | 0.1% NO, 0.1% NH3, 3% O2/30000 h−1 |
∼100% (120–220 °C) | 0.01% SO2 | ∼82% (120 °C/5 h) | 37 |
| Mn0.3–Ce0.7–O | Citric acid/650 °C/6 h | 0.05% NO, 0.05% NH3, 2% O2/30000 cm3 g−1 h−1 |
>95% (120–160 °C) | 5% H2O | ∼70% (120–140 °C) | 61 |
| Mn0.3–Ce0.7–O | Citric acid/650 °C/6 h | 0.05% NO, 0.05% NH3, 2% O2/120000 cm3 g−1 h−1 |
>90% (140–200 °C) | 5% H2O | >70% (160–200 °C) | 88 |
| Mn–Ce/W–Zr–O | Impregnation/550 °C/3 h | 0.1% NO, 0.1% NH3, 5% O2/10000 h−1 |
>90% (150–250 °C) | 0.01% SO2, 10% H2O | <80% (140–240 °C) | 145 |
| Mn–Ce/activated carbon honeycomb | Impregnation/400 °C/3 h | 0.05% NO, 0.05% NH3, 5% O2/1910 h−1 | ∼84% (160 °C) | 0.03% SO2 | ∼44% (160 °C/7 h) | 113 |
| Mn0.23–Nb0.23–Ce0.54–O | Co-precipitation/650 °C/5 h | 0.1% NO, 0.1% NH3, 10% O2/52000 h−1 |
— | 5% H2O | >80% (200–300 °C) | 146 |
| Mn0.23–Nb0.23–Ce0.54–O | Co-precipitation/650 °C/5 h | 0.1% NO, 0.1% NH3, 10% O2/52000 h−1 |
>78% (200–300 °C) | 0.005% SO2, 5% H2O | ∼20% (250 °C/0.5 h) | 147 |
| Mn2.5–La2.5–Ce1–Ni1 | 500 °C/6 h | 0.06% NO, 0.06% NH3, 6% O2/20000 h−1 |
∼98% (150–350 °C) | 0.03% SO2 | ∼85% (200 °C/4 h) | 148 |
 | ||
| Fig. 9 SCR activities of Mn/Ti and Mn–Ce/Ti in the presence of SO2. (a) Reaction conditions: [NO] = [NH3] = 800 ppm, [O2] = 3%, [SO2] = 100 ppm, [H2O] = 3 vol%, balance N2, temperature: 150 °C, GHSV = 40000 h−1. (Reprinted from ref. 25. Copyright 2013, with permission from Elsevier.) (b) Regeneration of sulfur poisoned CeO2 catalyst using a thermal treatment. Reaction conditions: [NO] = [NH3] = 500 ppm, [O2] = 5%, [SO2] = 25 ppm, balance N2, temperature: 350 °C, GHSV = 175000 h−1. (Reprinted from ref. 129. Copyright 2016, with permission from Elsevier.) | ||
Huang et al.152 investigated a series of Fe–Mn oxide catalysts supported on mesoporous silica (MPS), which showed good activity. When H2O and SO2 was fed in to the system at 190 °C, the NO conversion over Mn–Fe/MPS was finally decreased to 85.3% from 99.2%. This was attributed to the formation of the NH4HSO4 and (NH4)2SO4 in the presence of both H2O and SO2. However, the deactivated catalyst could be regenerated using a heating treatment, because the deactivation was because of the catalyst pore plugging and surface area loss by the deposition of (NH4)2SO4. When the temperature is above 140 °C, H2O has no negative effect on its activity.
Guan et al.153 investigated the resistance to deactivation by H2O and SO2 of Ti0.9Ce0.05V0.05O2−x catalysts, which showed a high NO conversion and N2 selectivity. After feeding 400 ppm SO2 for 26 h at 150 °C, the surface of catalyst was deposited with significant agglomeration and bulk NH4NO3 and (NH4)2SO4 with a size of 30–50 μm. Then, the NH4NO3 and (NH4)2SO4 was decomposed when the catalyst was calcined at 200 °C and 400 °C, because the decomposition temperatures were 170 °C and 300 °C, respectively. The surfaces were scanned using scanning electron microscopy (SEM), and the transformation of the surface is shown in Fig. 10.
 | ||
| Fig. 10 SEM images (a) feeding with 400 ppm SO2 at 150 °C for 26 h, (b) calcined at 200 °C and 400 °C for 2 h. (Reprinted from ref. 153. Copyright 2011, with permission from Elsevier.) | ||
3.2 Alkali and alkaline earth metal ions
Fine fly ash still exists in the downstream of the flue gas after desulfurizing and dedusting. Amounts of alkali and alkaline earth metals were released from the raw materials or coal, such as in the cement production process. Alkali salts are important components in fine fly ash, which not only plugs the pores of catalysts, but also decreases SCR activity by reacting with the active phase.154–156 In addition, because of the water solubility or ion exchange, alkali metal has a high liquidity to neutralize the acid sites.157 For the traditional V2O5-based SCR catalysts, alkali metal deactivated these by affecting the acid sites on the surface.154,158 Alkali metals could lower MnOx reducibility, decrease specific surface areas and damage the acid sites of low temperature catalysts.Zhou et al.159 reported that sodium sulfate, used to simulate the combined effects of alkali metal and SO2 in the flue gas, had strong effects on the activity of the Mn–Ce/TiO2 catalyst, such as simultaneous pore occlusion and sulfation effect. Guo et al.160 investigated the deactivation effect of sodium (Na) and potassium (K) on a Mn/TiO2 catalyst. The catalyst was prepared using a sol–gel method and Na and K were doped via an impregnation method. The Mn/TiO2 catalysts exhibited a high activity of 90% NO conversion. However, when Na or K was doped, the conversion was decreased from 95% to 78% and 27%, respectively. In this study, the effect of K was apparently more serious than that of Na.161 Furthermore, Chen et al.155 found that on the catalysts' surface chemisorbed oxygen was reduced by alkali and alkaline earth ions together with a decrease of SCR activity. The downward trend was K > Na > Ca > Mg.
Shen et al.162,163 studied the effects of K, Na and Ca on a Mn–Ce/Zr catalyst. From the NH3-TPD measurements, the adsorption of NH3 was decreased when the catalyst was doped with alkali metal ions. This may indicate that the alkali metal on the surface of the catalysts may destroy the surface acidic sites, and decrease the redox property and chemisorbed oxygen. Furthermore, they also found that K was more harmful to the catalyst compared to Na or Ca. However, Kustov et al.164 found that V2O5 supported on sulfated zirconium dioxide showed a good resistance towards alkali ions. Chen et al.70 reported that the K resistance of the Mn/TiO2 catalyst could be improved by doping it with Co, which increased the adsorption of NH3 and NOx species.
3.3 Heavy metal ions
Heavy metal ions, regarded as hazardous pollutants, can deactivate the SCR catalysts. Heavy metal ions in the flue gas are mainly generated from coal used as fuel.165 It has been proved that heavy metals could lead to the deactivation of vanadium-based SCR catalysts.166 Kong et al.167 found that the Brønsted acid sites of a V–W/TiO2 catalyst were impacted when mercury chloride was introduced. Actually, there is little heavy metal ions found in the downstream of the precipitator because the heavy metal ions usually exist in the fly ash. Moreover, water vapor exists in the flue gas all along. For the water solubility of heavy metal ions, it is necessary to take the effect of heavy metals into consideration.Lead (Pb) and zinc (Zn) are typical heavy metals found in the flue gas of coal fired power plants. Guo et al.156,168, and Li et al.169 compared the poisoning effect of Pb and Zn on a Mn/TiO2 catalyst. The Pb or Zn was loaded on to the Mn/TiO2 catalyst using impregnation. As a result, both Pb and Zn were found to have a negative effect on the Mn/TiO2 catalyst (Fig. 11a). From the characterization experiments, the redox ability of Zn–Mn/TiO2 and Pb–Mn/TiO2 was found to be decreased because of the drop of Mn4+ and chemisorbed oxygen. Zhou et al.170 investigated the deactivation effects of lead(II) oxide (PbO) on the Mn–Ce/TiO2 catalyst. It was proposed that the surface area, the concentration of Mn4+, Ce3+ and chemisorbed oxygen was decreased after introducing PbO. Consequently, the performance of the Mn–Ce/TiO2 catalyst was greatly decreased because of the poisoning of PbO (Fig. 11b).
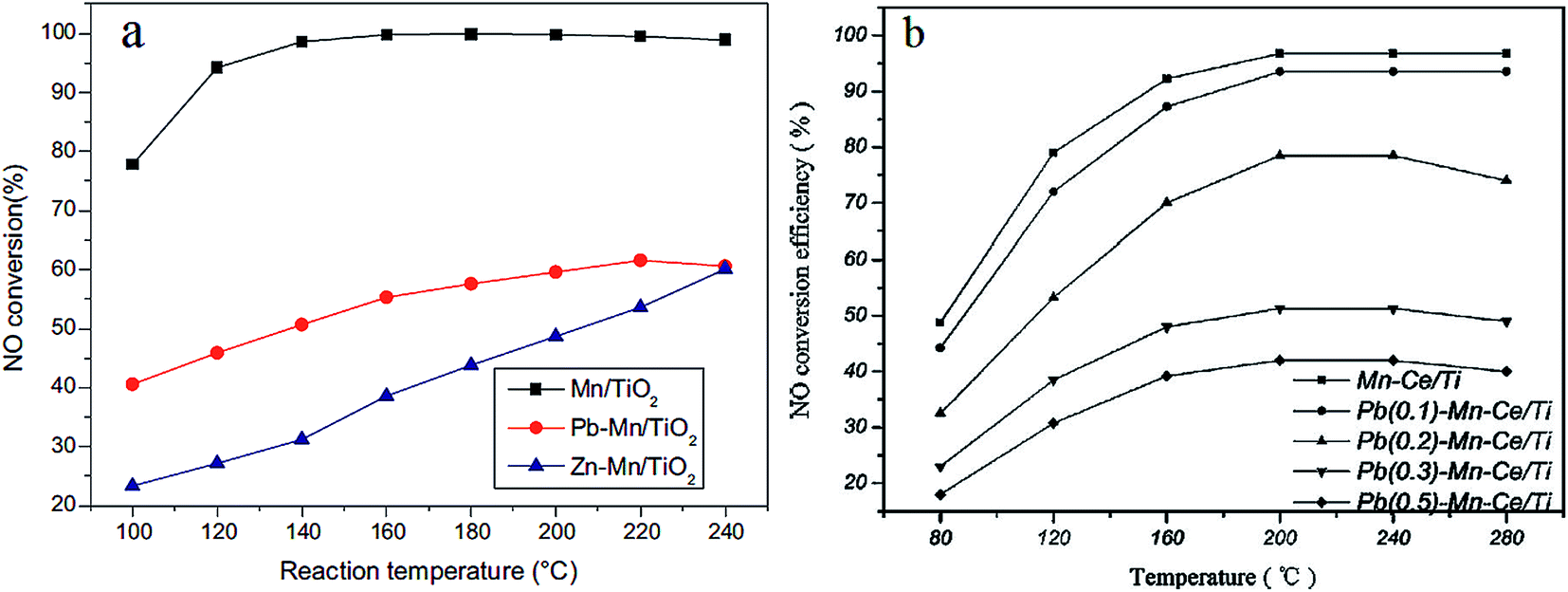 | ||
| Fig. 11 NO conversion over pure and poisoned catalysts. (a) Reaction conditions: [NO] = [NH3] = 600 ppm, [O2] = 5%, balance N2, GHSV = 108000 h−1. (Reprinted from ref. 156. Copyright 2015, with permission from Elsevier.) (b) Reaction conditions: [NO] = [NH3] = 800 ppm, [O2] = 5%, balance N2, GHSV = 200000 h−1. (Reprinted from ref. 170. Copyright 2016, with permission from Elsevier.) | ||
Mercury (Hg0) is a toxic trace element in the atmosphere and has a high concentration in coals used in China, such as anthracite, bituminous coal and lignite.171 Researchers have attempted to remove the NO and Hg0 simultaneously. However, Hg0 is harmful to the catalysts of SCR of NO because it will compete with NH3 for adsorption on the active sites.172 Xu et al.41 investigated the influence of Hg0 on the NO conversion over a LaMnO3 catalyst. The NO conversion had a slight decrease in the presence of Hg0 (Fig. 12).
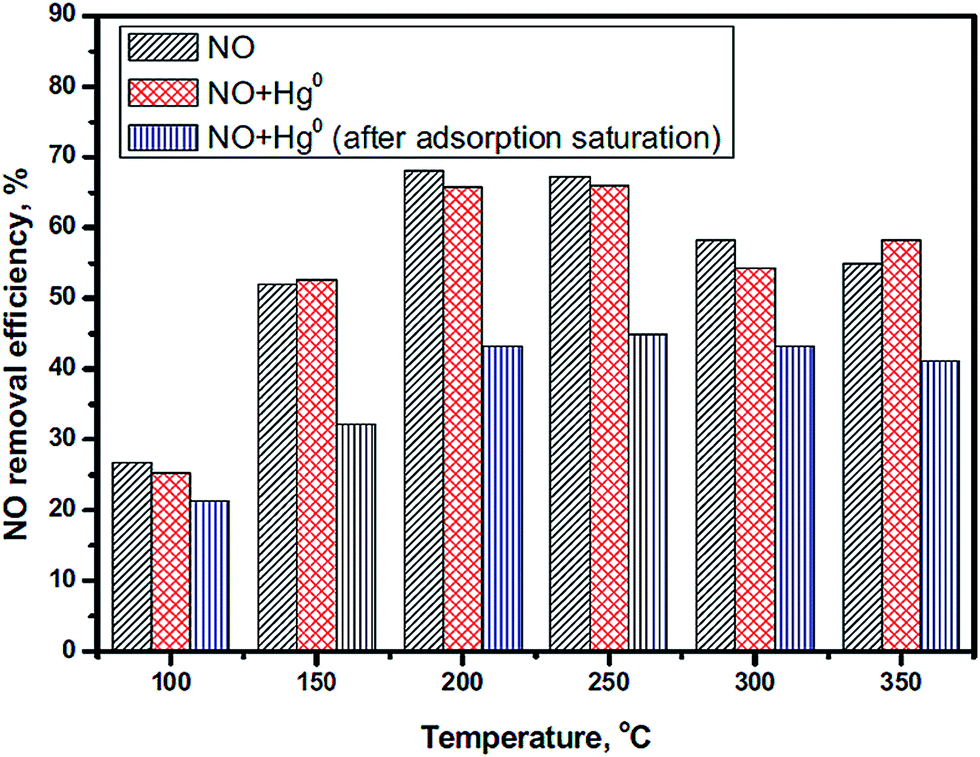 | ||
| Fig. 12 The effect of Hg0 on NO conversion. Reaction conditions: [NO] = [NH3] = 500 ppm, [Hg0] = 500 μg m−3, [O2] = 4%, balance N2, GHSV = 478000 h−1. (Reprinted from ref. 41. Copyright 2016, with permission from Elsevier.) | ||
4. Conclusions and perspectives
NH3-SCR of NOx in the presence of O2 is one of the important strategies in controlling NOx emissions. Low temperature SCR has been investigated for several decades. Mn-containing metal oxide catalysts generally gave the preferable performance. SCR of NOx with NH3 follows both the L–H and the E–R mechanisms. There is quite a similarity between these two different mechanisms. A comproportionation occurs in both the L–H and E–R mechanisms. Fast SCR has a higher reaction rate than standard SCR and it depends on the formation of NO2. N2O formation can mainly be explained using the E–R mechanism. A synergistic mechanism is vital for designing a remarkable metal oxide catalyst. Multi-metal cations will promote the performance mutually. Manganese cations mainly serve as the adsorption center for nitrogen. Thus, it is necessary to introduce an element for the adsorption of oxygen and to provide a redox cycle.A big challenge in the industrial use of Mn-containing oxide catalysts is their durability. They are vulnerable to the effects of both SO2 and H2O. Sulfur oxides and water vapor cause the deactivation of Mn-containing catalysts. Alkali metals could lower manganese oxide reducibility, decrease specific surface areas and damage the acid sites of low temperature catalysts. The poisoning process of SO2 can be classified into two categories: deposition of (NH4)2SO4 and sulfation of the active phase. For the low temperature downstream of the flue gas, the deposition of (NH4)2SO4 or NH4HSO4 occurs more easily and NH3 is evidently adsorbed by H2O in comparison with the operation upstream. Many efforts have been made to improve the durability. Nonetheless, few techniques have been useful in practical industrial applications.
On the basis of the previous analysis, some conclusions can be drawn as follows:
(1) Most research is related to the performance of the catalysts, such as NO conversion, N2 selectivity and poisons' tolerance, as well as the mechanism of this process. An excellent NO conversion of catalysts has been obtained, however, the N2 selectivity is not satisfactory.
(2) Less effort has been made on determining the relationship of metal oxide crystal structure and its performance, which is required for the design of catalysts. More attention should be given to the relationship between the catalysts' structure and its reaction mechanism, which guides us exactly to design a low temperature SCR catalyst for different flue gases.
(3) Mn-containing metal oxide catalysts show a notable SCR performance at low temperature. However, the single manganese oxide catalysts have a poor tolerance of SO2 and H2O, which has been improved by modifying other elements in bench scale experiments. Researchers have been engaged in improving Mn-containing catalysts by modifying them with different metal oxides. Ce can enhance the adsorption of NO and O2 which benefits the oxidization of NO to NO2 and improves sulfur resistance, and inhibits the formation of (NH4)2SO4 and NH4HSO4. Ce has good selectivity for improving the catalysts' performance. More research efforts should be made on the activity and poisoning tolerance.
(4) Most catalysts were powder rather than monolith catalysts, such as honeycomb or slab. A laboratory study is a small scale test that will react differently to industrial tests. Specific surface area is important to the activity and closely related to the particles' size, shape and aggregation. The preparation method is also important to the catalysts' performance. Researchers should give more attention to pilot scale tests or industrial tests.
(5) The low temperature SCR catalysts have been investigated for several decades. Lots of elements have been studied in the catalysts. To avoid repetitive work and waste of resources, a low temperature SCR catalysts' materials database should be built.
(6) Heaps of disabled SCR catalysts should be regenerated and reused. The regeneration and recycling of SCR catalysts is another big task for researchers. This problem should be taken into consideration while researchers are designing new SCR catalysts.
Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (Grants U1360202, 51472030, 51672024 and 51502014) and the 111 Project (No. B17003). The authors would like to thank the editor for editing the manuscript and the anonymous reviewers for their detailed and helpful comments.References
- H. Amini, S.-M. Taghavi-Shahri, S. B. Henderson, V. Hosseini, H. Hassankhany, M. Naderi, S. Ahadi, C. Schindler, N. Künzli and M. Yunesian, Sci. Rep., 2016, 6, 32970 CrossRef CAS PubMed.
- A. Richter, J. P. Burrows, H. Nusz, C. Granier and U. Niemeier, Nature, 2005, 437, 129–132 CrossRef CAS PubMed.
- J. N. Galloway, F. J. Dentener, D. G. Capone, E. W. Boyer, R. W. Howarth, S. P. Seitzinger, G. P. Asner, C. C. Cleveland, P. A. Green, E. A. Holland, D. M. Karl, A. F. Michaels, J. H. Porter, A. R. Townsend and C. J. Vöosmarty, Biogeochemistry, 2004, 70, 153–226 CrossRef CAS.
- J. Zhu and A. Thomas, Appl. Catal., B, 2009, 92, 225–233 CrossRef CAS.
- M. Fu, C. Li, P. Lu, L. Qu, M. Zhang, Y. Zhou, M. Yu and Y. Fang, Catal. Sci. Technol., 2014, 4, 14–25 CAS.
- C. Tang, H. Zhang and L. Dong, Catal. Sci. Technol., 2016, 6, 1248–1264 CAS.
- B. Liu, Y. H. Wang and H. Xu, J. Energy Eng., 2016, 142, 10 Search PubMed.
- B. Liu, B. Bao, Y. Wang and H. Xu, J. Energy Inst., 2016, 90, 441–451 CrossRef.
- T. Ishihara, J. Catal., 2003, 220, 104–114 CrossRef CAS.
- D. G. Streets and S. T. Waldhoff, Atmos. Environ., 2000, 34, 363–374 CrossRef CAS.
- P. Forzatti, Appl. Catal., A, 2001, 222, 221–236 CrossRef CAS.
- M. Koebel, G. Madia and M. Elsener, Catal. Today, 2002, 73, 239–247 CrossRef CAS.
- M. T. Javed, N. Irfan and B. M. Gibbs, J. Environ. Manage., 2007, 83, 251–289 CrossRef CAS PubMed.
- S. W. Bae, S. A. Roh and S. D. Kim, Chemosphere, 2006, 65, 170–175 CrossRef CAS PubMed.
- J. O. L. Wendt, W. P. Linak, P. W. Groff and R. K. Srivastava, AIChE J., 2001, 47, 2603–2617 CrossRef CAS.
- S. Roy and A. Baiker, Chem. Rev., 2009, 109, 4054–4091 CrossRef CAS PubMed.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- K.-I. Shimizu, J. Shibata, H. Yoshida, A. Satsuma and T. Hattori, Appl. Catal., B, 2001, 30, 151–162 CrossRef CAS.
- S. Brandenberger, O. Kroecher, A. Tissler and R. Althoff, Catal. Rev.: Sci. Eng., 2008, 50, 492–531 CAS.
- L. Casagrande, L. Lietti, I. Nova, P. Forzatti and A. Baiker, Appl. Catal., B, 1999, 22, 63–67 CrossRef CAS.
- K. Bourikas, C. Fountzoula and C. Kordulis, Appl. Catal., B, 2004, 52, 145–153 CrossRef CAS.
- S. Hodjati, K. Vaezzadeh, C. Petit, V. Pitchon and A. Kiennemann, Top. Catal., 2001, 16, 151–155 CrossRef.
- G. Qi and R. T. Yang, Chem. Commun., 2003, 848–849, 10.1039/b212725c.
- R. Qu, X. Gao, K. Cen and J. Li, Appl. Catal., B, 2013, 142–143, 290–297 CrossRef CAS.
- R. Jin, Y. Liu, Y. Wang, W. Cen, Z. Wu, H. Wang and X. Weng, Appl. Catal., B, 2014, 148–149, 582–588 CrossRef CAS.
- D. A. Peña, B. S. Uphade and P. G. Smirniotis, J. Catal., 2004, 221, 421–431 CrossRef.
- G. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS.
- B. Thirupathi and P. G. Smirniotis, Appl. Catal., B, 2011, 110, 195–206 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Appl. Catal., B, 2012, 115, 100–106 CrossRef.
- N. Apostolescu, B. Geiger, K. Hizbullah, M. T. Jan, S. Kureti, D. Reichert, F. Schott and W. Weisweiler, Appl. Catal., B, 2006, 62, 104–114 CrossRef CAS.
- F. Kapteijn, L. Singoredjo, A. Andreini and J. A. Moulijn, Appl. Catal., B, 1994, 3, 173–189 CrossRef CAS.
- F. ç. Larachi, J. Pierre, A. Adnot and A. Bernis, Appl. Surf. Sci., 2002, 195, 236–250 CrossRef CAS.
- B. Shen, F. Wang and T. Liu, Powder Technol., 2014, 253, 152–157 CrossRef CAS.
- F. Liu, H. He, C. Zhang, Z. Feng, L. Zheng, Y. Xie and T. Hu, Appl. Catal., B, 2010, 96, 408–420 CrossRef CAS.
- S. Yang, S. Xiong, Y. Liao, X. Xiao, F. Qi, Y. Peng, Y. Fu, W. Shan and J. Li, Environ. Sci. Technol., 2014, 48, 10354–10362 CrossRef CAS PubMed.
- H. Hu, S. Cai, H. Li, L. Huang, L. Shi and D. Zhang, ACS Catal., 2015, 5, 6069–6077 CrossRef CAS.
- Z. Chen, Q. Yang, H. Li, X. Li, L. Wang and S. Chi Tsang, J. Catal., 2010, 276, 56–65 CrossRef CAS.
- M. A. Zamudio, N. Russo and D. Fino, Ind. Eng. Chem. Res., 2011, 50, 6668–6672 CrossRef CAS.
- M. Kang, E. D. Park, J. M. Kim and J. E. Yie, Catal. Today, 2006, 111, 236–241 CrossRef CAS.
- D. Fang, J. Xie, D. Mei, Y. Zhang, F. He, X. Liu and Y. Li, RSC Adv., 2014, 4, 25540 RSC.
- H. Xu, Z. Qu, C. Zong, F. Quan, J. Mei and N. Yan, Appl. Catal., B, 2016, 186, 30–40 CrossRef CAS.
- Z. Liu and S. Ihl Woo, Catal. Rev., 2006, 48, 43–89 CAS.
- J. Li, H. Chang, L. Ma, J. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS.
- C. Liu, J.-W. Shi, C. Gao and C. Niu, Appl. Catal., A, 2016, 522, 54–69 CrossRef CAS.
- http://www.sdpc.gov.cn/gzdt/<?ccdc_no 201409?><?ccdc_no 201409?>201409<?ccdc END?><?ccdc END?>/t20140919_626240.html, accessed Dec. 7th, 2016.
- P. R. Ettireddy, N. Ettireddy, T. Boningari, R. Pardemann and P. G. Smirniotis, J. Catal., 2012, 292, 53–63 CrossRef CAS.
- G. Busca, M. A. Larrubia, L. Arrighi and G. Ramis, Catal. Today, 2005, 107–108, 139–148 CrossRef CAS.
- G. Zhou, B. Zhong, W. Wang, X. Guan, B. Huang, D. Ye and H. Wu, Catal. Today, 2011, 175, 157–163 CrossRef CAS.
- L. Qiu, J. Meng, D. Pang, C. Zhang and F. Ouyang, Catal. Lett., 2015, 145, 1500–1509 CrossRef CAS.
- D. Ye, Practical Inorganic Thermodynamic Data Manual, Metallurgical Industry Press, Beijing, 1st edn, 1981 Search PubMed.
- I. Nam, J. Catal., 1989, 119, 269 CrossRef CAS.
- S. Yang, F. Qi, Y. Liao, S. Xiong, Y. Lan, Y. Fu, W. Shan and J. Li, Ind. Eng. Chem. Res., 2014, 53, 5810–5819 CrossRef CAS.
- S. Yang, F. Qi, S. Xiong, H. Dang, Y. Liao, P. K. Wong and J. Li, Appl. Catal., B, 2016, 181, 570–580 CrossRef CAS.
- T. Chen, B. Guan, H. Lin and L. Zhu, Chin. J. Catal., 2014, 35, 294–301 CrossRef CAS.
- S. Yang, C. Wang, J. Li, N. Yan, L. Ma and H. Chang, Appl. Catal., B, 2011, 110, 71–80 CrossRef CAS.
- G. Qi and R. T. Yang, J. Phys. Chem. B, 2004, 108, 15738–15747 CrossRef CAS.
- F. Liu and H. He, Catal. Today, 2010, 153, 70–76 CrossRef CAS.
- Z. Wu, B. Jiang, Y. Liu, H. Wang and R. Jin, Environ. Sci. Technol., 2007, 41, 5812–5817 CrossRef CAS PubMed.
- D. Fang, F. He, D. Li and J. Xie, Appl. Surf. Sci., 2013, 285, 215–219 CrossRef CAS.
- D. Fang, J. Xie, H. Hu, H. Yang, F. He and Z. Fu, Chem. Eng. J., 2015, 271, 23–30 CrossRef CAS.
- S. Xiong, Y. Liao, X. Xiao, H. Dang and S. Yang, J. Phys. Chem. C, 2015, 119, 4180–4187 CAS.
- F. Eigenmann, M. Maciejewski and A. Baiker, Appl. Catal., B, 2006, 62, 311–318 CrossRef CAS.
- X. Tang, J. Li, L. Sun and J. Hao, Appl. Catal., B, 2010, 99, 156–162 CrossRef CAS.
- I. Nova, C. Ciardelli, E. Tronconi, D. Chatterjee and B. Bandl-Konrad, Catal. Today, 2006, 114, 3–12 CrossRef CAS.
- M. Koebel, G. Madia, F. Raimondi and A. Wokaun, J. Catal., 2002, 209, 159–165 CrossRef CAS.
- M. Koebel, M. Elsener and G. Madia, Ind. Eng. Chem. Res., 2001, 40, 52–59 CrossRef CAS.
- G. Madia, M. Koebel, M. Elsener and A. Wokaun, Ind. Eng. Chem. Res., 2002, 41, 3512–3517 CrossRef CAS.
- M. Stanciulescu, P. Bulsink, G. Caravaggio, L. Nossova and R. Burich, Appl. Surf. Sci., 2014, 300, 201–207 CrossRef CAS.
- W. Zhao, C. Li, P. Lu, Q. Wen, Y. Zhao, X. Zhang, C. Fan and S. Tao, Environ. Technol., 2013, 34, 81–90 CrossRef CAS PubMed.
- Q.-L. Chen, R.-T. Guo, Q.-S. Wang, W.-G. Pan, N.-Z. Yang, C.-Z. Lu and S.-X. Wang, J. Taiwan Inst. Chem. Eng., 2016, 64, 116–123 CrossRef CAS.
- X. Zeng, X. Huo, T. Zhu, X. Hong and Y. Sun, Molecules, 2016, 21, 1491 CrossRef PubMed.
- Y. Liang, Y. Huang, H. Zhang, L. Lan, M. Zhao, M. Gong, Y. Chen and J. Wang, Environ. Sci. Pollut. Res. Int., 2017, 24, 9314–9324 CrossRef CAS PubMed.
- A. Grossale, I. Nova and E. Tronconi, J. Catal., 2009, 265, 141–147 CrossRef CAS.
- L. Zhu, Z. Zhong, H. Yang and C. Wang, Water, Air, Soil Pollut., 2016, 227, 476 CrossRef.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, J. Catal., 2008, 256, 312–322 CrossRef CAS.
- G. Qi and R. T. Yang, J. Catal., 2003, 217, 434–441 CrossRef CAS.
- R. Q. Long, R. T. Yang and R. Chang, Chem. Commun., 2002, 452–453, 10.1039/b111382h.
- L. J. France, Q. Yang, W. Li, Z. Chen, J. Guang, D. Guo, L. Wang and X. Li, Appl. Catal., B, 2017, 206, 203–215 CrossRef CAS.
- Y. Zhu, Y. Zhang, R. Xiao, T. Huang and K. Shen, Catal. Commun., 2017, 88, 64–67 CrossRef CAS.
- S. M. Mousavi, A. Niaei, M. J. Illán Gómez, D. Salari, P. Nakhostin Panahi and V. Abaladejo-Fuentes, Mater. Chem. Phys., 2014, 143, 921–928 CrossRef CAS.
- T. Wang, K. Sun, Z. Lu and Y. Zhang, React. Kinet., Mech. Catal., 2010, 101, 153–161 CrossRef CAS.
- Y. Liu, T. Gu, X. Weng, Y. Wang, Z. Wu and H. Wang, J. Phys. Chem. C, 2012, 116, 16582–16592 CAS.
- S. Xiong, Y. Liao, H. Dang, F. Qi and S. Yang, RSC Adv., 2015, 5, 27785–27793 RSC.
- L. Xu, X.-S. Li, M. Crocker, Z.-S. Zhang, A.-M. Zhu and C. Shi, J. Mol. Catal. A: Chem., 2013, 378, 82–90 CrossRef CAS.
- B. Jiang, Z. Li and S.-C. Lee, Chem. Eng. J., 2013, 225, 52–58 CrossRef CAS.
- S. Suarez, J. Martin, M. Yates, P. Avila and J. Blanco, J. Catal., 2005, 229, 227–236 CrossRef CAS.
- M. A. Zamudio, N. Russo and D. Fino, Ind. Eng. Chem. Res., 2011, 50, 6668–6672 CrossRef CAS.
- S. Yang, Y. Liao, S. Xiong, F. Qi, H. Dang, X. Xiao and J. Li, J. Phys. Chem. C, 2014, 118, 21500–21508 CAS.
- B. Jiang, B. Deng, Z. Zhang, Z. Wu, X. Tang, S. Yao and H. Lu, J. Phys. Chem. C, 2014, 118, 14866–14875 CAS.
- T.-Y. Li, S.-J. Chiang, B.-J. Liaw and Y.-Z. Chen, Appl. Catal., B, 2011, 103, 143–148 CrossRef CAS.
- R. Farra, M. García-Melchor, M. Eichelbaum, M. Hashagen, W. Frandsen, J. Allan, F. Girgsdies, L. Szentmiklósi, N. López and D. Teschner, ACS Catal., 2013, 3, 2256–2268 CrossRef CAS.
- S. Imamura, A. Doi and S. Ishida, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 75–80 CrossRef CAS.
- D. A. Peña, B. S. Uphade, E. P. Reddy and P. G. Smirniotis, J. Phys. Chem. B, 2004, 108, 9927–9936 CrossRef.
- H. Chang, J. Li, J. Yuan, L. Chen, Y. Dai, H. Arandiyan, J. Xu and J. Hao, Catal. Today, 2013, 201, 139–144 CrossRef CAS.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Chemosphere, 2010, 78, 1160–1166 CrossRef CAS PubMed.
- Z. Liu, J. Zhu, J. Li, L. Ma and S. I. Woo, ACS Appl. Mater. Interfaces, 2014, 6, 14500–14508 CAS.
- D. W. Kwon, K. B. Nam and S. C. Hong, Appl. Catal., A, 2015, 497, 160–166 CrossRef CAS.
- Z. Chen, F. Wang, H. Li, Q. Yang, L. Wang and X. Li, Ind. Eng. Chem. Res., 2012, 51, 202–212 CrossRef CAS.
- Y. J. Kim, H. J. Kwon, I.-S. Nam, J. W. Choung, J. K. Kil, H.-J. Kim, M.-S. Cha and G. K. Yeo, Catal. Today, 2010, 151, 244–250 CrossRef CAS.
- Z. Liu, Y. Liu, Y. Li, H. Su and L. Ma, Chem. Eng. J., 2016, 283, 1044–1050 CrossRef CAS.
- Z. Liu, Y. Li, T. Zhu, H. Su and J. Zhu, Ind. Eng. Chem. Res., 2014, 53, 12964–12970 CrossRef CAS.
- P. Lu, C. Li, G. Zeng, L. He, D. Peng, H. Cui, S. Li and Y. Zhai, Appl. Catal., B, 2010, 96, 157–161 CrossRef CAS.
- Y. Shen, S. Zhu, T. Qiu and S. Shen, Catal. Commun., 2009, 11, 20–23 CrossRef CAS.
- G. Ramis, J. Catal., 1990, 124, 574–576 CrossRef CAS.
- G. Busca, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- P. R. Ettireddy, N. Ettireddy, S. Mamedov, P. Boolchand and P. G. Smirniotis, Appl. Catal., B, 2007, 76, 123–134 CrossRef CAS.
- M. Inomata, J. Catal., 1980, 62, 140–148 CrossRef CAS.
- S. Xiong, Y. Liao, X. Xiao, H. Dang and S. Yang, Catal. Sci. Technol., 2015, 5, 2132–2140 CAS.
- X. Zhang, B. Shen, K. Wang and J. Chen, J. Ind. Eng. Chem., 2013, 19, 1272–1279 CrossRef CAS.
- X. Wang, X. Li, Q. Zhao, W. Sun, M. Tade and S. Liu, Chem. Eng. J., 2016, 288, 216–222 CrossRef CAS.
- J. Yu, F. Guo, Y. Wang, J. Zhu, Y. Liu, F. Su, S. Gao and G. Xu, Appl. Catal., B, 2010, 95, 160–168 CrossRef CAS.
- G. Xie, Z. Liu, Z. Zhu, Q. Liu, J. Ge and Z. Huang, J. Catal., 2004, 224, 36–41 CrossRef CAS.
- Y. Wang, X. Li, L. Zhan, C. Li, W. Qiao and L. Ling, Ind. Eng. Chem. Res., 2015, 54, 2274–2278 CrossRef CAS.
- Y.-J. Shi, H. Shu, Y.-H. Zhang, H.-M. Fan, Y.-P. Zhang and L.-J. Yang, Fuel Process. Technol., 2016, 150, 141–147 CrossRef CAS.
- X. Lu, C. Song, C.-C. Chang, Y. Teng, Z. Tong and X. Tang, Ind. Eng. Chem. Res., 2014, 53, 11601–11610 CrossRef CAS.
- D. Zhang, L. Zhang, L. Shi, C. Fang, H. Li, R. Gao, L. Huang and J. Zhang, Nanoscale, 2013, 5, 1127–1136 RSC.
- X. Lu, C. Song, S. Jia, Z. Tong, X. Tang and Y. Teng, Chem. Eng. J., 2015, 260, 776–784 CrossRef CAS.
- B. Jiang, Y. Liu and Z. Wu, J. Hazard. Mater., 2009, 162, 1249–1254 CrossRef CAS PubMed.
- Z. Zhu, Z. Liu, S. Liu and H. Niu, Appl. Catal., B, 2001, 30, 267–276 CrossRef CAS.
- W. Q. Xu, H. He and Y. B. Yu, J. Phys. Chem. C, 2009, 113, 4426–4432 CAS.
- R. Q. Long and R. T. Yang, J. Catal., 1999, 186, 254–268 CrossRef CAS.
- H. H. Phil, M. P. Reddy, P. A. Kumar, L. K. Ju and J. S. Hyo, Appl. Catal., B, 2008, 78, 301–308 CrossRef CAS.
- B. Q. Jiang, Z. B. Wu, Y. Liu, S. C. Lee and W. K. Ho, J. Phys. Chem. C, 2010, 114, 4961–4965 CAS.
- J. Zuo, Z. Chen, F. Wang, Y. Yu, L. Wang and X. Li, Ind. Eng. Chem. Res., 2014, 53, 2647–2655 CrossRef CAS.
- W. Sjoerd Kijlstra, M. Biervliet, E. K. Poels and A. Bliek, Appl. Catal., B, 1998, 16, 327–337 CrossRef CAS.
- Z. Wu, R. Jin, H. Wang and Y. Liu, Catal. Commun., 2009, 10, 935–939 CrossRef CAS.
- Z. Liu, Y. Yi, S. Zhang, T. Zhu, J. Zhu and J. Wang, Catal. Today, 2013, 216, 76–81 CrossRef CAS.
- L. Kylhammar, P.-A. Carlsson, H. H. Ingelsten, H. Grönbeck and M. Skoglundh, Appl. Catal., B, 2008, 84, 268–276 CrossRef CAS.
- Y. Shi, S. Tan, X. Wang, M. Li, S. Li and W. Li, Catal. Commun., 2016, 86, 67–71 CrossRef CAS.
- H. Chang, X. Chen, J. Li, L. Ma, C. Wang, C. Liu, J. W. Schwank and J. Hao, Environ. Sci. Technol., 2013, 47, 5294–5301 CrossRef CAS PubMed.
- H. Chang, J. Li, X. Chen, L. Ma, S. Yang, J. W. Schwank and J. Hao, Catal. Commun., 2012, 27, 54–57 CrossRef CAS.
- Y. Shi, S. Chen, H. Sun, Y. Shu and X. Quan, Catal. Commun., 2013, 42, 10–13 CrossRef CAS.
- M. D. Amiridis, I. E. Wachs, G. Deo, J.-M. Jehng and D. S. Kim, J. Catal., 1996, 161, 247–253 CrossRef CAS.
- L. Chen, Z. Si, X. Wu, D. Weng and Z. Wu, J. Environ. Sci., 2015, 31, 240–247 CrossRef PubMed.
- Z. Lei, B. Han, K. Yang and B. Chen, Chem. Eng. J., 2013, 215–216, 651–657 CrossRef CAS.
- S. Pan, H. Luo, L. Li, Z. Wei and B. Huang, J. Mol. Catal. A: Chem., 2013, 377, 154–161 CrossRef CAS.
- Z. Sheng, Y. Hu, J. Xue, X. Wang and W. Liao, J. Rare Earths, 2012, 30, 676–682 CrossRef CAS.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Catal. Today, 2010, 153, 84–89 CrossRef CAS.
- B. Shen, Y. Wang, F. Wang and T. Liu, Chem. Eng. J., 2014, 236, 171–180 CrossRef CAS.
- Z. Wu, R. Jin, Y. Liu and H. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS.
- Z. Ma, X. Wu, Y. Feng, Z. Si and D. Weng, Catal. Commun., 2015, 69, 188–192 CrossRef CAS.
- Z. Sheng, Y. Hu, J. Xue, X. Wang and W. Liao, Environ. Technol., 2012, 33, 2421–2428 CrossRef CAS PubMed.
- B. Shen, T. Liu, N. Zhao, X. Yang and L. Deng, J. Environ. Sci., 2010, 22, 1447–1454 CrossRef CAS.
- B. Shen, X. Zhang, H. Ma, Y. Yao and T. Liu, J. Environ. Sci., 2013, 25, 791–800 CrossRef CAS.
- H. Xu, Q. Zhang, C. Qiu, T. Lin, M. Gong and Y. Chen, Chem. Eng. Sci., 2012, 76, 120–128 CrossRef CAS.
- M. Casapu, O. Kröcher, M. Mehring, M. Nachtegaal, C. Borca, M. Harfouche and D. Grolimund, J. Phys. Chem. C, 2010, 114, 9791–9801 CAS.
- M. Casapu, O. Kröcher and M. Elsener, Appl. Catal., B, 2009, 88, 413–419 CrossRef CAS.
- B. Yang, D.-H. Zheng, Y.-S. Shen, Y.-S. Qiu, B. Li, Y.-W. Zeng, S.-B. Shen and S.-M. Zhu, J. Ind. Eng. Chem., 2015, 24, 148–152 CrossRef CAS.
- B. Yang, Y. Shen, S. Shen and S. Zhu, J. Rare Earths, 2013, 31, 130–136 CrossRef CAS.
- Y. Yu, C. He, J. Chen, L. Yin, T. Qiu and X. Meng, Catal. Commun., 2013, 39, 78–81 CrossRef CAS.
- M. Pourkhalil, A. Z. Moghaddam, A. Rashidi, J. Towfighi and Y. Mortazavi, Appl. Surf. Sci., 2013, 279, 250–259 CrossRef CAS.
- J. Huang, Z. Tong, Y. Huang and J. Zhang, Appl. Catal., B, 2008, 78, 309–314 CrossRef CAS.
- B. Guan, H. Lin, L. Zhu and Z. Huang, J. Phys. Chem. C, 2011, 115, 12850–12863 CAS.
- R. Khodayari and C. U. I. Odenbrand, Appl. Catal., B, 2001, 30, 87–99 CrossRef CAS.
- L. Chen, J. Li and M. Ge, Chem. Eng. J., 2011, 170, 531–537 CrossRef CAS.
- R.-T. Guo, Q.-S. Wang, W.-G. Pan, Q.-L. Chen, H.-L. Ding, X.-F. Yin, N.-Z. Yang, C.-Z. Lu, S.-X. Wang and Y.-C. Yuan, J. Mol. Catal. A: Chem., 2015, 407, 1–7 CrossRef CAS.
- L. Zhang, S. Cui, H. Guo, X. Ma and X. Luo, J. Mol. Catal. A: Chem., 2014, 390, 14–21 CrossRef CAS.
- F. Moradi, J. Brandin, M. Sohrabi, M. Faghihi and M. Sanati, Appl. Catal., B, 2003, 46, 65–76 CrossRef CAS.
- A. Zhou, D. Yu, L. Yang and Z. Sheng, Appl. Surf. Sci., 2016, 378, 167–173 CrossRef CAS.
- R.-T. Guo, Q.-S. Wang, W.-G. Pan, W.-L. Zhen, Q.-L. Chen, H.-L. Ding, N.-Z. Yang and C.-Z. Lu, Appl. Surf. Sci., 2014, 317, 111–116 CrossRef CAS.
- X. S. Du, X. Gao, R. Y. Qu, P. D. Ji, Z. Y. Luo and K. F. Cen, Chemcatchem, 2012, 4, 2075–2081 CrossRef CAS.
- S. Boxiong, Y. Yan, C. Jianhong and Z. Xiaopeng, Microporous Mesoporous Mater., 2013, 180, 262–269 CrossRef.
- B. Shen, L. Deng and J. Chen, Front. Environ. Sci. Eng., 2013, 7, 512–517 CrossRef CAS.
- A. L. Kustov, M. Y. Kustova, R. Fehrmann and P. Simonsen, Appl. Catal., B, 2005, 58, 97–104 CrossRef CAS.
- R. Stolle, H. Koeser and H. Gutberlet, Appl. Catal., B, 2014, 144, 486–497 CrossRef CAS.
- Y. Jiang, X. Gao, Y. Zhang, W. Wu, H. Song, Z. Luo and K. Cen, J. Hazard. Mater., 2014, 274, 270–278 CrossRef CAS PubMed.
- M. Kong, Q. Liu, L. Jiang, F. Guo, S. Ren, L. Yao and J. Yang, Catal. Commun., 2016, 85, 34–38 CrossRef CAS.
- R.-T. Guo, C.-Z. Lu, W.-G. Pan, W.-L. Zhen, Q.-S. Wang, Q.-L. Chen, H.-L. Ding and N.-Z. Yang, Catal. Commun., 2015, 59, 136–139 CrossRef CAS.
- W. Li, R.-T. Guo, S.-X. Wang, W.-G. Pan, Q.-L. Chen, M.-Y. Li, P. Sun and S.-M. Liu, Fuel Process. Technol., 2016, 154, 235–242 CrossRef CAS.
- L. Zhou, C. Li, L. Zhao, G. Zeng, L. Gao, Y. Wang and M. E. Yu, Appl. Surf. Sci., 2016, 389, 532–539 CrossRef CAS.
- C. Zhu, H. Tian, K. Cheng, K. Liu, K. Wang, S. Hua, J. Gao and J. Zhou, J. Cleaner Prod., 2016, 114, 343–351 CrossRef CAS.
- P. Wang, S. Su, J. Xiang, H. You, F. Cao, L. Sun, S. Hu and Y. Zhang, Chemosphere, 2014, 101, 49–54 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |