
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liver uptake of cefditoren is mediated by OATP1B1 and OATP2B1 in humans and Oatp1a1, Oatp1a4, and Oatp1b2 in rats
Hepeng Wanga,
Pengyuan Sun*ab,
Changyuan Wang ab,
Qiang Mengab,
Zhihao Liuab,
Xiaokui Huoab,
Huijun Sunab,
Xiaodong Maab,
Jinyong Pengab and
Kexin Liu*ab
ab,
Qiang Mengab,
Zhihao Liuab,
Xiaokui Huoab,
Huijun Sunab,
Xiaodong Maab,
Jinyong Pengab and
Kexin Liu*ab
aDepartment of Clinical Pharmacology, College of Pharmacy, Dalian Medical University, 9 West Section, Lvshun South Road, Lvshunkou District, Dalian 116044, China. E-mail: pysun@dlmedu.edu.cn; Fax: +86-411-8611-0406; Fax: +86-411-8611-0407; Tel: +86-411-8611-0406 Tel: +86-411-8611-0407
bProvincial Key Laboratory for Pharmacokinetics and Transport, Dalian Medical University, Liaoning, China. E-mail: kexinliu@dlmedu.edu.cn
First published on 9th June 2017
Abstract
Cefditoren, a β-lactam antibiotic, is widely used in respiratory tract and skin infections in the clinic. This study was aimed at investigating the mechanism underlying hepatic uptake of cefditoren in rats and humans. Cmax of cefditoren plasma exposure was increased (0.80 ± 0.06 to 1.02 ± 0.16 μg mL−1) when co-administrated with rifampicin. Extraction ratio of cefditoren was decreased (34.70% ± 4.61% to 18.30% ± 2.89%) by combining with rifampicin in perfused rat livers in situ. Uptake of cefditoren by rat liver slices was temperature-dependent and significantly inhibited by Oatp modulators, such as ibuprofen, digoxin, cyclosporin A and glycyrrhizic acid, but not by tetraethylammonium or p-aminohippurate. Uptake of cefditoren in hOATP1B1- and hOATP2B1-human embryonic kidney (HEK) 293 cells indicated a saturable process with a Km of 189.7 ± 60.4 μmol L−1 and 122.7 ± 37.37 μmol L−1, respectively. Sartans inhibited the transport of cefditoren in hOATP1B1- and hOATP2B1-HEK293 cells. Moreover, cefditoren could increase the gene and protein expression levels of Oatp1a1 in rat liver, while Oatp1a4 and Oatp1b2 were unchanged. These results indicated that OATP1B1 and OATP2B1 are involved in hepatic uptake of cefditoren in humans, and multiple Oatps (Oatp1a1, Oatp1a4 and Oatp1b2) might participate in this process in rats. In addition, cefditoren could prompt the up-regulation of Oatp1a1 expression. In the clinic, additional attention should be paid to the alternative exposure of cefditoren when co-administrated with sartans or other drugs that are substrates or inhibitors of OATP1B1 and/or OATP2B1.
1. Introduction
Cefditoren (Fig. 1), developed by Meiji Seika Kaisha Ltd, Japan, is a third-generation, broad spectrum, orally administered cephalosporin. It has documented bactericidal activity against many Gram-positive and -negative pathogens, and stability against clinically important β-lactamases.1–4 In the clinic, cefditoren has been used for the treatment of streptococcal pharyngitis, acute maxillary sinusitis, acute otitis media, sinusitis, group A beta-hemolytic streptococcal pharyngotonsillitis, acute exacerbation of chronic bronchitis, and uncomplicated skin and skin structure infections in adult and adolescent patients.1,5 In order to increase the bioavailability, cefditoren is often converted into cefditoren pivoxil (a pivoxil ester). After oral administration, cefditoren pivoxil is hydrolysed by intestinal esterases to form cefditoren (an active metabolite).6 Clinically, the mean maximum plasma concentration (Cmax) was 3.8–4.6 mg L−1 after 2.4–3.1 hours, via a single 400 mg oral dose of cefditoren administered with food in humans. The mean area under the plasma concentration–time curve (AUC) values were 11.4–17.4 mg h L−1. Binding of cefditoren to plasma proteins averages 88%, and the mean volume of distribution (Vd) of cefditoren at steady state was 9.3 L.6,7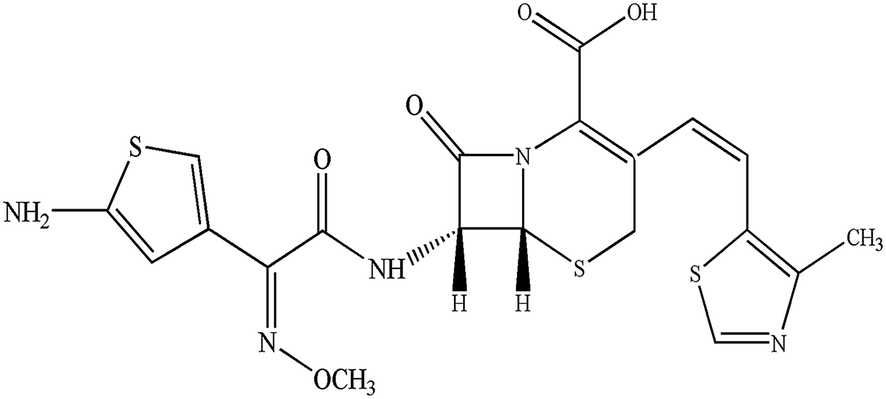 | ||
| Fig. 1 Chemical structure of cefditoren. | ||
The disposition of drugs in the liver is affected by several processes, such as hepatic sinusoidal uptake into the hepatocytes, intracellular transport and excretion into the bile, which may relate to carrier-mediated mechanisms that are saturable and rate limiting. Uptake transporters regulate the transport of drugs from the basolateral membrane into the hepatocytes.8–10 Various transporters, such as organic anion-transporting polypeptide 1B1 (OATP1B1) (OATP-C, OATP2, LST-1 and SLC21A6), OATP2B1 (OATPB and SLC21A9), OATP1B3 (OATP8, LST-2, and SLC21A8), organic anion transporter 2 (OAT2, SLC22A7), organic cation transporter 1 (OCT1, SLC22A1), and Na+-taurocholate cotransporting polypeptide, are expressed on the sinusoidal membrane of hepatocytes. These proteins participate in the transport of various compounds, including clinical drugs from blood into hepatocytes.11–20 Remarkably, OATP1B1, OATP2B1 and OATP1B3, selectively expressed on the basolateral side of the membrane of human hepatocytes with broad substrate specificities,12,16,21,22 are thought to develop a vital impact on the elimination of various xenobiotics such as sartans,23–25 statins,17,26,27 fexofenadine,28 endogenous substances such as bile salt29 and neutral steroids30,31 from the bloodstream into hepatocytes,9,32,33 in a Na+-independent manner. Furthermore, the substrate specificity of each transporter shares a broad spectrum, suggesting that a substrate can be recognized by one transporter or multiple transporters.10,34,35
Previous studies indicated that cefditoren was a substrate of PEPT1,4 and both Mrp2 and Bcrp were involved in the biliary excretion of cefditoren.3 However, whether the uptake transporters participate in the hepatic transport of cefditoren in rats and humans has not been sufficiently investigated yet. Therefore, the purpose of this study is to clarify the involvement of transporters in the hepatic uptake process of cefditoren. The investigation of transporters participating in the hepatic transport of cefditoren is significant for the prediction of potential cefditoren-related drug–drug interactions (DDIs). Also it may contribute to understanding the mechanisms of hepatic uptake of β-lactam antibiotics. In the present study, we, for the first time, used in vivo experiments in rat, perfused rat liver in situ, rat liver slices, human hOATP1B1-, hOATP2B1- and hOATP1B3-transfected cells, RT-PCR relative quantification and western blotting to investigate the underlying mechanism of OATP-mediated hepatic uptake of cefditoren in rats and humans, and to clarify the regulation of rat Oatps by cefditoren.
2. Materials and methods
2.1 Chemicals
Cefditoren was obtained from Meiji Seika Kaisha Ltd (Tokyo, Japan). Cyclosporin A and rifampicin were purchased from the National Institute for the Control of Pharmaceutical and Biologic Products (Beijing, China). Tetraethyl ammonium (TEA), digoxin, and glycyrrhizic acid were purchased from Hubei Saibo Chemical Co. Ltd (Wuhan, China) and Nanjing ZeLang Medical Technology Co. Ltd (Nanjing, China), respectively. p-Aminohippurate (PAH), (RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid (ibuprofen), estradiolum-17β-glucuro acid (E17β), estrone-3-sulfate (E3S) and hydrochlorothiazide [internal standard (IS)] were purchased from Sigma-Aldrich (St Louis, MO). Valsartan and irbesartan were obtained from Lunan Pharmaceutical Group Company Ltd (Linyi, China). Telmisartan was provided by Zhejiang Kinglyuan Pharmaceutical Company Ltd (Shangyu, China). Eprosartan was purchased from Dalian Meilun Medical Science and Technology Company Ltd (Dalian, China). All other chemicals were of analytical grade and were commercially available.2.2 Animals
Male Wistar rats (220–250 g, 6–8 weeks of age) were purchased from the Experimental Animal Center of Dalian Medical University (Dalian, China; permit number SCXK 2013-0003). The rats were allowed free access to water and food, but were in abrosia 12 hours (with water continuously) before the experiments. All experiments were approved by the Animal Care and Use Committee of Dalian Medical University, and the experimental procedures were strictly carried out in accordance with Legislation Regarding the Use and Care of Laboratory Animals of China. No experiments involved human subjects.2.3 Cell culture
The stable transfected OATP1B3-HEK293 and vector-HEK293 cells were a grateful gift from Yuichi Sugiyama (Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan). OATP1B1- and OATP2B1-HEK293 cells were a generous gift from researcher Xiulin Yi (State Key Laboratory of Drug Delivery Technology and Pharmacokinetics, Tianjin Institute of Pharmaceutical Research, Tianjin, China). The cells were cultured in high-glucose Dulbecco's Vogt modified Eagle's medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 units/100 mg mL−1, Invitrogen) at 37 °C with 95% relative humidity and 5% (v/v) CO2 atmosphere. Cell culture reagents were purchased from Gibco (Grand Island, NY).2.4 In vivo experiment in rats
Rats were orally administered with cefditoren or combined with rifampicin (50 mg kg−1) at a dose of 18 mg per kg body weight once a day (n = 3). Before the experiment, the rats were fasted overnight (about 12 h). Cefditoren (18 mg kg−1) was dissolved in normal saline. Then blood samples (0.2 mL) were collected via orbital blood at 1, 5, 10, 30, 60, 90, 120, 180, 240, 360, 480, 600 and 720 min in heparin tubes for cefditoren determination as described in a previous study.36 And the common bile duct was cannulated with polyethylene tubing (PE-10). Bile samples were collected at 2, 4, 6, 8, 10, 12 h after oral dose. The samples were frozen immediately at −20 °C until the determination of cefditoren.2.5 In situ perfused rat liver experiment
Livers of rats were prepared by standard techniques according to a previous study.37 After anesthesia, the bile duct was cannulated, and the liver (n = 3) was perfused in situ in a single pass manner via the portal vein with oxygenated Krebs–Henseleit buffer (pH 7.4), including 4.7 mmol L−1 KCl, 118 mmol L−1 NaCl, 1.2 mmol L−1 KH2PO4, 25 mmol L−1 NaHCO3, 2.5 mmol L−1 CaCl2, 5.0 mmol L−1 glucose, 1.2 mmol L−1 MgSO4.3,20,37 Following balancing with Krebs–Henseleit buffer for approximately 10 min, the liver was permitted to perfuse with oxygenated Krebs–Henseleit buffer including 20% (v/v) washed bovine erythrocytes at a flow rate of 10 mL min−1.38 Then, the superior vena cava was cannulated, and the effluent perfusate was collected. The initial bile flow (>2 μL min−1) was confirmed as the reference of normal liver viability. After reaching a balance, the liver was perfused for 45 minutes after addition of cefditoren 1 μmol L−1, cefditoren 1 μmol L−1 + rifampicin 10 μmol L−1 (final concentration, n = 3). The effluent perfusate (500 μL) was collected at 1, 3, 5, 10, 15, 20, 25, 45 min and the liver was rapidly removed at the end. All the samples were stored at −20 °C until analysis.2.6 In vitro uptake in rat liver slices
Rat liver slices were prepared as previously described.37 In short, the liver was rapidly excised and promptly released into ice-cold buffer, saturated with 95% O2/5% CO2 (carbogen; pH 7.4). Then, livers were sliced into thin pieces with a ZQP-86 tissue slicer (10–14 mg wet weight; 200–300 mm thickness; Zhixin Co. Ltd, Shanghai, China), and gassed with carbogen in 6-well culture plates with continuous shaking. The liver slices were shifted to 24-well culture plates including fresh carbogen-saturated cefditoren for further incubation (37 °C and 4 °C) after incubation for 3 minutes at 37 °C. Moreover, the liver uptake of cefditoren (1 μmol L−1) and digoxin (5 μmol L−1) was determined at 0, 1, 3, 5, 10, 15, and 30 minutes. After that, the liver slices were rinsed with ice-cold Hanks' balanced salt solution (pH 7.5) three times and dried on filter paper. The uptake of cefditoren at different time points was basically linear; according to this, the overall rate of uptake time was selected as 15 min. Also, this time was applied to investigate the effects of inhibitors, containing ibuprofen (100 μmol L−1), TEA (1 mmol L−1), PAH (1 mmol L−1), glycyrrhizic acid (100 μmol L−1) and digoxin (10 μmol L−1), on the uptake of cefditoren. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was used to determine the accumulation of cefditoren in homogenized liver slices.2.7 Uptake experiments by transporter expression systems
The transport investigation was accomplished as in a previous study.17 Cells were cultured for 48 h (almost confluent) before the uptake experiment, in 24-well culture plates. Cells had been rinsed twice and incubated with Krebs–Henseleit buffer at 37 °C for 15 minutes, and then the uptake experiment was started by adding Krebs–Henseleit buffer including drugs. The uptake experiment was ended at prescribed times by ice-cold Krebs–Henseleit buffer, following the elimination of the incubation buffer. Thereafter, cells were rinsed twice with ice-cold Krebs–Henseleit buffer (1 mL), lysed with 0.1% (v/v) Triton X-100 (0.3 mL), and placed at 37 °C for 1 h, and then shifted into a polyethylene tube for quantization by LC-MS/MS. The cell protein was determined by the bicinchoninic acid procedure (Solarbio, Beijing, China) employing bovine serum albumin as the standard. The uptake of cefditoren (5 μmol L−1) was measured in hOATP1B1-, hOATP2B1-, hOATP1B3- and vector-HEK293 cells at designated times (0.5, 1, 2, 3, 5 and 10 min). In the light of the results, the time point of 1 minute was chosen as representative of the uptake rate and was applied to investigate the concentration-dependent uptake and the influences of transporter modulators on the uptake of cefditoren.2.8 Quantitative real-time PCR
The RNA of total tissues was extracted using TRIzol® Reagent (Invitrogen, Shanghai, China) on the basis of the manufacturer's protocol in all groups. Moreover, the RNA was examined by qRT-PCR as previously. The sequences of the specific primers for rats are shown in Table 1. The cDNA was amplified by using SYBR® Premix Ex Taq™ kit (Takara, Dalian, China) and ABI PRISM® 7500 system (Applied Biosystems, Foster City, CA). The requirements for PCR were: 1 cycle of denaturation at 95 °C for 30 s, and 40 cycles of denaturation at 95 °C for 5 s, and combined annealing/extension (60 °C, 20 s). Gene expressions were calculated by the comparative Ct method as described previously and the values were normalized to endogenous reference β-actin.39,40| Gene | GenBank no. | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|---|
| Oatp1a1 | NM_017111 | AGACCTGTGGTGATTGGTATTGGA | AGTTGCCTGTAGGTGAGATCGTTG |
| Oatp1a4 | U88036 | TGCGGAGATGAAGCTTACC | TCCTCCGTCACTTTCGACCTT |
| Oatp1b2 | AJ 271682 | AGACGTTCCCATCACAACCAC | GCCTCTGCAGCTTTCCTTGA |
| β-Actin | NM_031144 | GGAGATTACTGCCCTGGCTCCTA | GACTCATCGTACTCCTGCTTGCTG |
2.9 Western blotting analysis
Western blotting analysis was conducted as in a previous study.37 The protein extracted from tissues was completely homogenized by using radioimmunoprecipitation assay (RIPA) lysis buffer. The concentration of protein was determined by the procedure of bicinchoninic acid (Solarbio, Beijing, China). Proteins (50 μg) were resuspended in electrophoresis sample buffer and separated by sodium dodecylsulfate-polyacrylamide (SDS-PAGE) (Bio-Rad, Hercules, CA), and blotted onto PVDF membranes (Millipore, Bedford, MA). Then 5% non-fat milk was used to block the PVDF membranes at 37 °C for 2 h (milk dissolved in Tris-buffered saline with 0.1% Tween-20, TBST). Next the PVDF membranes were incubated overnight at 4 °C with primary antibody for Oatp1a1, Oatp1a4, Oatp1b2 and β-actin (Santa Cruz Biotechnology, Santa Cruz, USA), rinsed with TBST three times on the second day and incubated for 2 h at 37 °C with secondary antibody. Finally the PVDF membranes were exposed to enhanced chemiluminescence-plus reagents (ECL) and were visualized with Chemi-Doc XRS Molecular Imager.40,412.10 LC-MS/MS analysis
An Agilent LC system (Agilent HP1200, Agilent Technology Inc., Palo Alto, CA, USA) and an API 3200 triple-quadrupole mass spectrometer (Applied Biosystems, Concord, ON, Canada) were used for LC-MS/MS analysis. Isocratic chromatographic separation column: Hypersil BDS-C18 column. Mobile phase: 0–2 min, 85% (v/v) water with 0.1% (v/v) formic acid, 15% (v/v) acetonitrile; 2–5.5 min, 10% (v/v) water with 0.1% (v/v) formic acid, 90% (v/v) acetonitrile; 5.5–10 min, 85% (v/v) water with 0.1% (v/v) formic acid, 15% (v/v) acetonitrile for cefditoren (flow rate 0.4 mL min−1); 70% (v/v) methyl alcohol, 30% (v/v) water with 0.1% formic acid for digoxin (flow rate: 0.5 mL min−1). The ionization was realized with a TurboIonSpray interface in negative ion mode. Multiple reaction monitoring (MRM) was used to control the equipment, data acquisition and analysis. The MRM fragmentation transitions were m/z 505.0 → 239.0 for cefditoren, m/z 779.0 → 649.0 for digoxin, and m/z 295.6 → 204.8 for hydrochlorothiazide (IS).2.11 Data analysis
The main pharmacokinetic parameters were calculated using 3P97 program which is edited by the Chinese Mathematical Pharmacological Society (Table 2).| Parameters | Drugs | |
|---|---|---|
| Cefditoren | Cefditoren + rifampicin | |
| a Pharmacokinetic parameters of cefditoren by oral co-administration with rifampicin in rats. A significant difference from the control group in the pharmacokinetic parameters of cefditoren (*P < 0.05, **P < 0.01; mean ± SD, n = 5). | ||
| Cmax (μg mL−1) | 0.80 ± 0.06 | 1.02 ± 0.16** |
| AUC0→∞ (μg h mL−1) | 5.29 ± 0.95 | 6.38 ± 0.62** |
| Tmax (h) | 1.50 ± 0.50 | 1.67 ± 0.30 |
| t1/2 (h) | 3.13 ± 0.93 | 3.46 ± 0.99* |
The hepatic extraction ratio (Eh) of cefditoren in perfused rat liver was calculated as:
| Eh = 1 − Fh | (1) |
| Fh = Cout/Cin | (2) |
Specific uptake was obtained by subtracting the uptake into vector-transfected cells from the uptake into OATP-transfected cells.
The specific uptake was calculated using the following equation:
| Uptakespecific = uptaketransporter expressed cells − uptakemock cells | (3) |
Kinetic parameters were obtained using nonlinear, least-squares regression analysis via the Michaelis–Menten equation:
| v = (VmaxS)/(Km + S) | (4) |
Each experimental point represents the mean ± SD of 3–6 measurements. All statistical analyses were conducted with the SPSS11.5 package (IBM SPSS Inc., Chicago, IL). Test results are expressed as the mean ± SD, and differences with P ≤ 0.05 are considered to be statically significant.
3. Results
3.1 Pharmacokinetics of cefditoren in rat in vivo
Initially, the pharmacokinetics experiment was performed to investigate the hepatic uptake process of cefditoren. Cefditoren was orally administered alone (control group) and co-administered with rifampicin (an inhibitor of Oatps)42 to investigate the pharmacokinetic changes in plasma concentration. The plasma exposure of cefditoren in co-administered group was increased significantly compared with that of control group (0.80 ± 0.06 to 1.02 ± 0.16 μg mL−1, P = 0.009) (Fig. 2A). Remarkably, Cmax was altered when cefditoren was orally co-administered with rifampicin. Moreover, other pharmacokinetic parameters such as AUC and t1/2 were also changed significantly (Table 2). Nevertheless, the cumulative biliary excretion of cefditoren was unchanged compared with that of control group (Fig. 2B).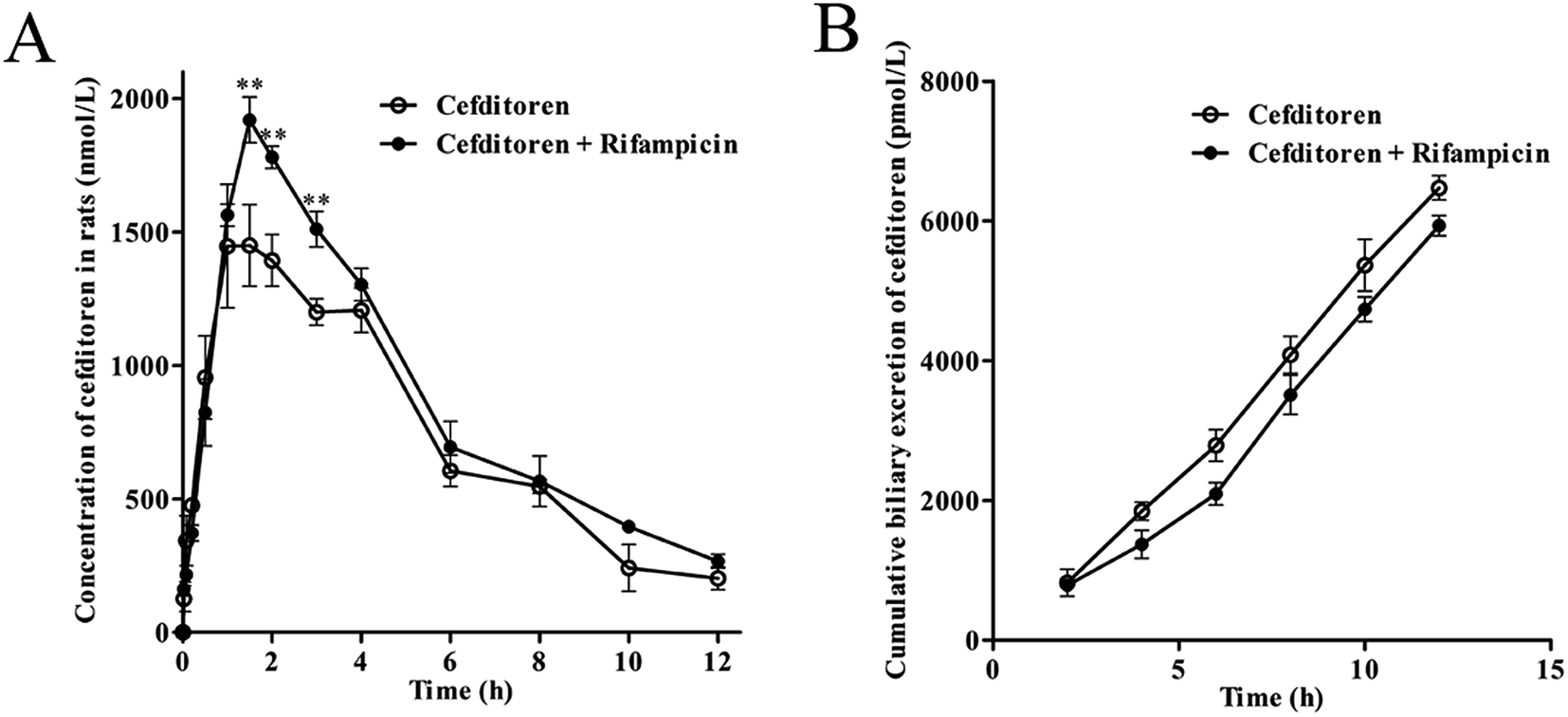 | ||
| Fig. 2 Mean plasma concentration–time curves (A) and cumulative biliary excretion (B) of cefditoren by oral co-administration with rifampicin in rats. Statistical differences between each set of points were compared with those for the control group by a two-tailed unpaired t-test, with P < 0.05 as the limit of significance (*P < 0.05, **P < 0.01; mean ± SD, n = 5). | ||
3.2 Hepatic uptake of cefditoren by perfused rat liver
To clarify the mechanism of the hepatic uptake of cefditoren, in situ perfused rat liver studies were implemented. The concentrations of cefditoren in the effluent perfusate were examined in the presence or absence of rifampicin (10 μmol L−1). Based on the value for the hepatic extraction ratio calculated by eqn (1) and (2), the concentrations of cefditoren reached a plateau after 5 min and the hepatic extraction ratio remained at a constant value of 34.70% ± 4.61% until the perfusion terminated (Fig. 3A). When cefditoren was administered simultaneously with rifampicin, the hepatic extraction ratio of cefditoren was reduced to 18.30% ± 2.89% (P = 0.009) (Fig. 3A). Furthermore, approximately 10% reduction (P = 0.009) of the hepatic accumulation of cefditoren was calculated based on the (attenuated) disappearance in the perfusate liver samples (Fig. 3B).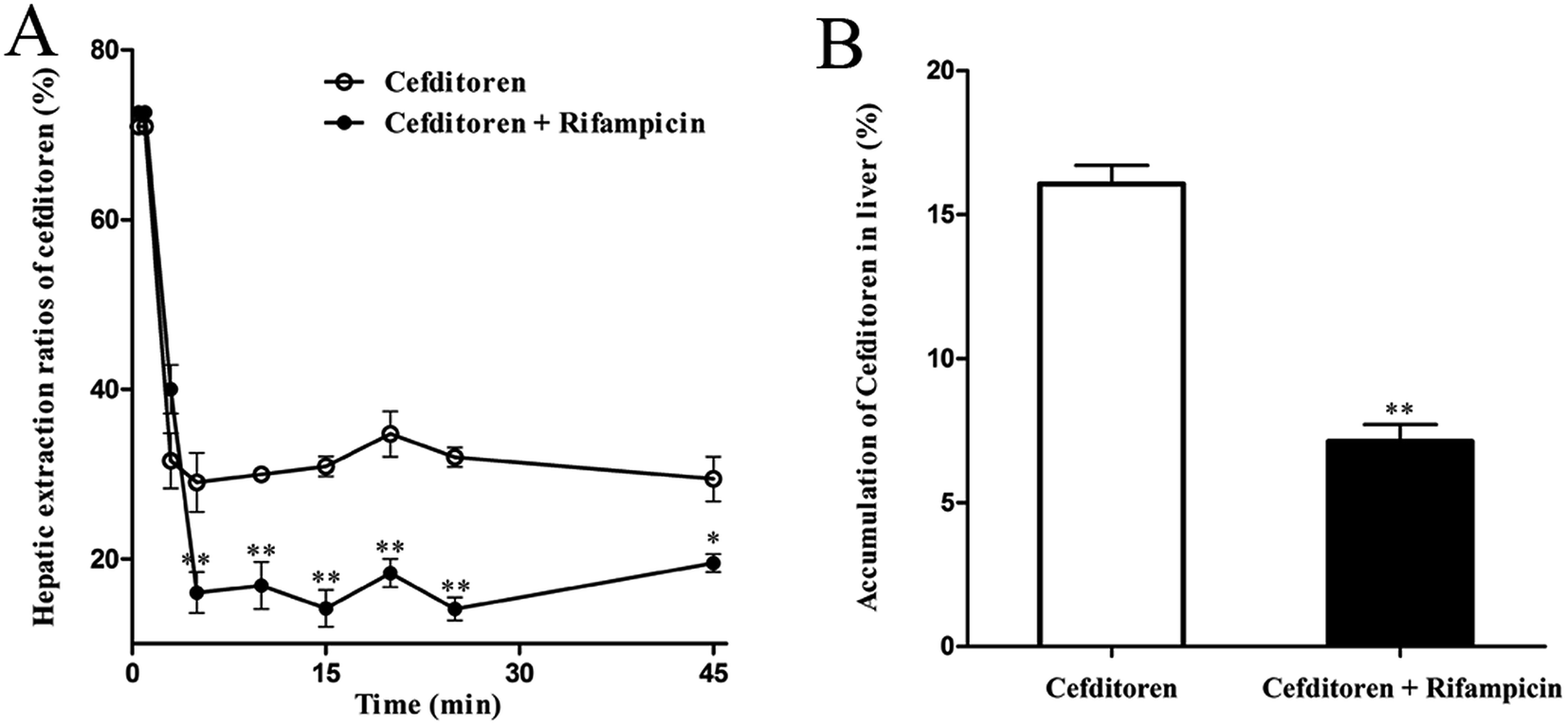 | ||
| Fig. 3 The hepatic extraction ratio of cefditoren in the presence or absence of rifampicin in the perfused rat liver experiment (A) and the accumulation of cefditoren in the presence or absence of rifampicin in the perfused rat liver experiment (B). (A) Rat livers were perfused with 1 μmol L−1 cefditoren, and the hepatic extraction ratio was calculated from the concentration of cefditoren in the effluent and the influent perfusates. (B) Calculation of the accumulation of cefditoren in liver. A significant difference from the control group in the uptake of cefditoren or digoxin (*P < 0.05, **P < 0.01; mean ± SD, n = 5). | ||
3.3 Hepatic uptake of cefditoren in rat liver slices
In the interest of immediately investigating the hepatic uptake of cefditoren on a tissue level, rat liver slices experiment was performed in vitro. The uptake of cefditoren (1 μmol L−1) at 37 °C was markedly increased within 30 minutes, compared with the uptake at 4 °C. In addition, a significant difference was observed between the concentration of cefditoren in the liver slices at 4 °C and 37 °C (approximately 2.3-fold greater than at 4 °C, P = 0.009) (Fig. 4A). The results (saturable and temperature-dependent manner) were similar to those of digoxin (5 μmol L−1, an Oatp1a4 substrate) (Fig. 4B). The method of measuring the concentration of digoxin by LC-MS/MS was similar to that of previous studies. Meanwhile, the effects of inhibitors on the cefditoren uptake were investigated in rat liver slices. Ibuprofen (an Oatp1a1 inhibitor), digoxin (an Oatp1a4 substrate), glycyrrhizic acid (an Oatp1b2 inhibitor) and cyclosporin A (Oatp1a1 and Oatp1a4 inhibitor) could inhibit the primary hepatic uptake of cefditoren in rat liver slices. In contrast, no significant changes were recognized in TEA (an OCT substrate) and PAH (an OAT substrate) groups (Fig. 4C).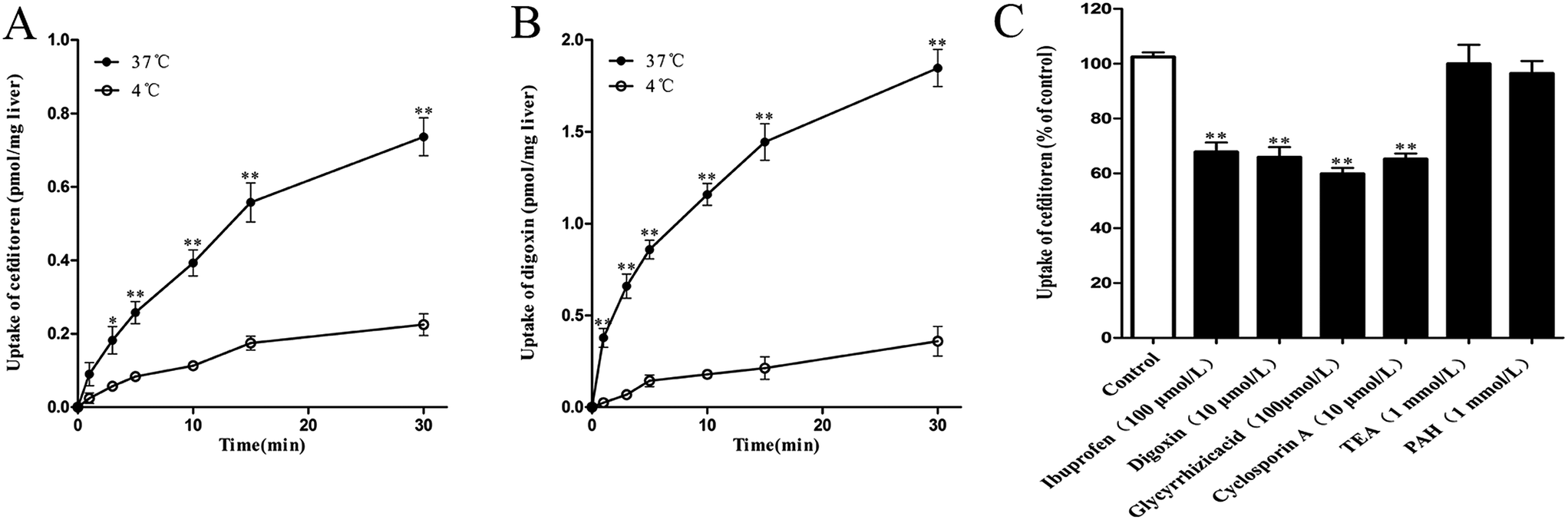 | ||
| Fig. 4 Time profile of cefditoren (A) and digoxin (B) uptake, and inhibitory effects of transporter modulators on uptake of cefditoren in rat liver slices (C). (A) Cefditoren concentration was 1 μmol L−1. (B) Digoxin concentration was 5 μmol L−1. (C) Results are shown as a percentage of cefditoren uptake (control) measured in the absence of inhibitor. A significant difference from the control group in the uptake of cefditoren or digoxin (*P < 0.05, **P < 0.01; mean ± SD, n = 5). | ||
3.4 Uptake of cefditoren in hOATP1B1-, hOATP2B1-, hOATP1B3-HEK293 cells
In order to clarify the target transporters involved in cefditoren hepatic transport in humans, the uptake of cefditoren by hOATP1B1-, hOATP2B1-, and hOATP1B3-HEK293 cells was measured. The uptake of cefditoren (5 μmol L−1) in hOATP1B1- and hOATP2B1-HEK293cells was visibly higher than that in vector-HEK293 cells. Meanwhile, an obvious reduction was observed in both hOATP1B1- and hOATP2B1-HEK293cells when combined with rifampicin (Fig. 5A–C). However, no significant difference was found in hOATP1B3-HEK293 cells. Eadie–Hofstee plot analysis indicated that the Km values of cefditoren were 189.7 ± 60.4 μmol L−1 and 122.7 ± 37.37 μmol L−1, respectively, in hOATP1B1- and hOATP2B1-HEK293 cells. Besides the Vmax values were 391.1 ± 53.15 pmol per mg protein and 277.6 ± 30.14 pmol per mg protein. All the pharmacokinetic parameters were calculated by eqn (3) and (4) (Fig. 5D and E). Furthermore, the inhibitors or substrates of OATP1B1 (rifampicin, cyclosporin A, E17β, pravastatin) (Fig. 6A) and OATP2B1 (rifampicin, cyclosporin A, E3S) (Fig. 6B) significantly inhibited the uptake of cefditoren in hOATP-HEK293 cells.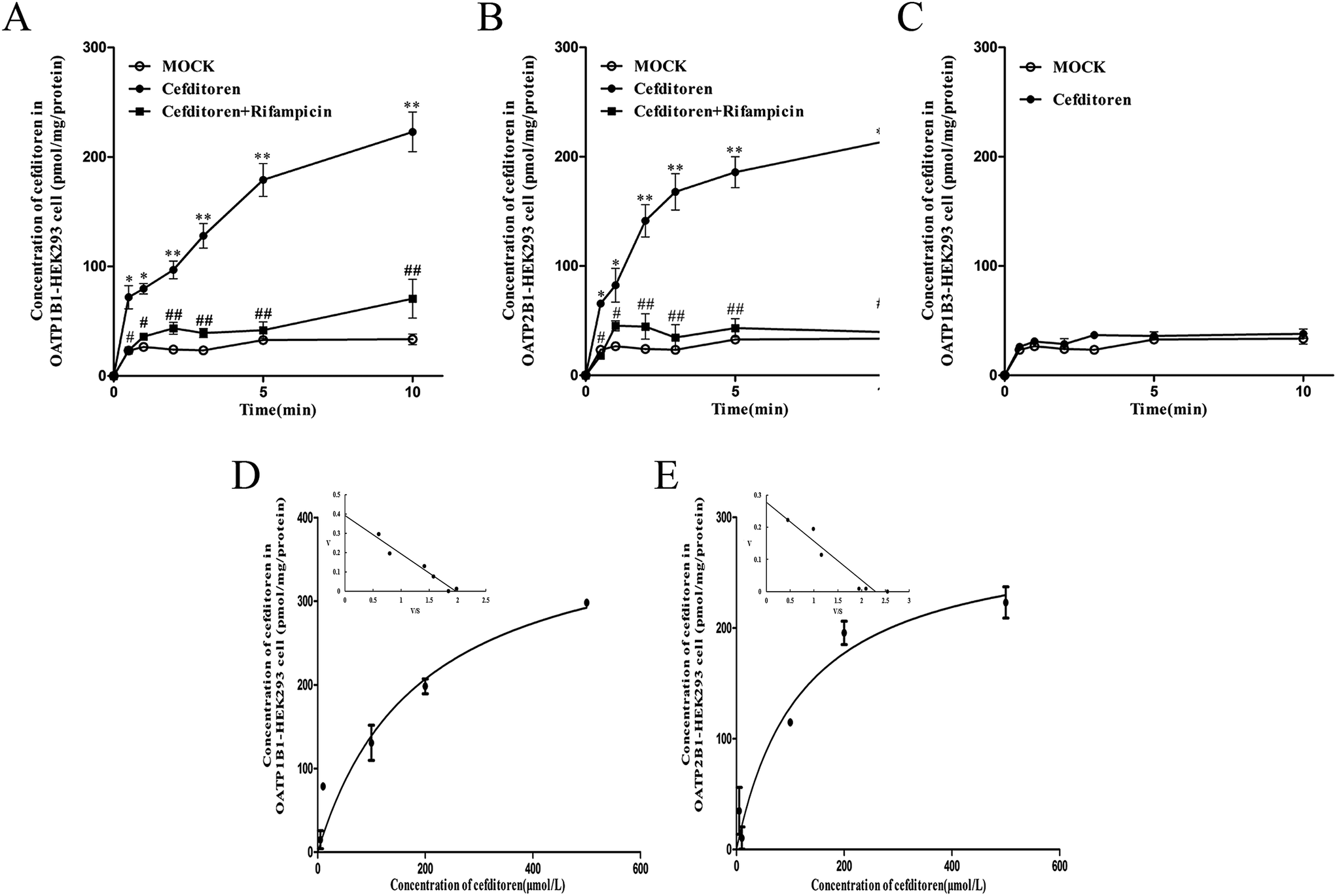 | ||
| Fig. 5 Time profiles and Eadie–Hofstee plot for the uptake of cefditoren in hOATP-HEK293 cells. The cefditoren concentration was 5 μmol L−1. (A) Uptake of cefditoren in hOATP1B1-HEK293 cells. (B) Uptake of cefditoren in hOATP2B1-HEK293 cells. (C) Uptake of cefditoren in hOATP1B3-HEK293 cells. The uptake of cefditoren by hOATP1B1- (D) and hOATP2B1-HEK293 cells (E) was measured at a concentration between 1 μmol L−1 and 500 μmol L−1. The specific OATP-mediated cefditoren transport was obtained by subtracting the uptake in vector-HEK293 cells from that in hOATPs-HEK293 cells for 1 minute. The continuous line represents the fitted curve (*P < 0.05, **P < 0.01; mean ± SD, n = 3). | ||
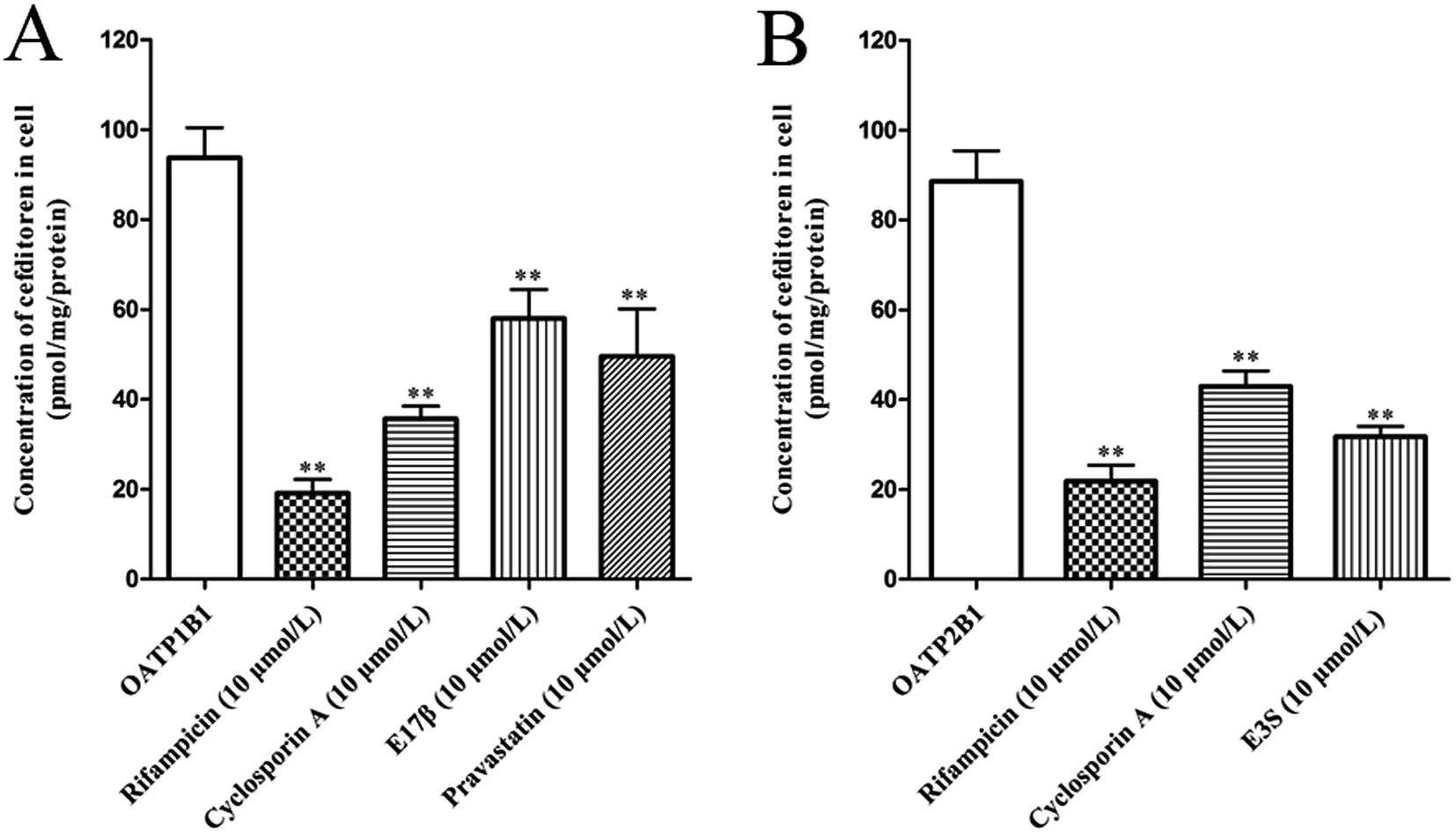 | ||
| Fig. 6 Inhibitory effects of inhibitors or substrates on the uptake of cefditoren in hOATP1B1-HEK293 cells (A) and hOATP2B1-HEK293 cells (B) for 1 minute. The cefditoren concentration was 5 μmol L−1. The absolute values for cefditoren uptake in control group were 93.25 and 88.38 pmol per mg protein, respectively, in hOATP1B1-HEK293 cells and hOATP2B1-HEK293 cells. A significant difference from the uptake in control group (*P < 0.05, **P < 0.01; mean ± SD, n = 3). | ||
3.5 Inhibitory effects of sartans on the uptake of cefditoren in hOATP1B1- and hOATP2B1-HEK293 cells
The effects of eprosartan, telmisartan, irbesartan and valsartan on the cellular uptake of cefditoren were investigated in hOATP1B1- and hOATP2B1-HEK293 cells (Fig. 7A and B). All potential inhibitors were used at a concentration of 1 μmol L−1, whereas cefditoren was used at a concentration of only 5 μmol L−1. All sartans which are frequently used in the clinic could inhibit the uptake of cefditoren significantly.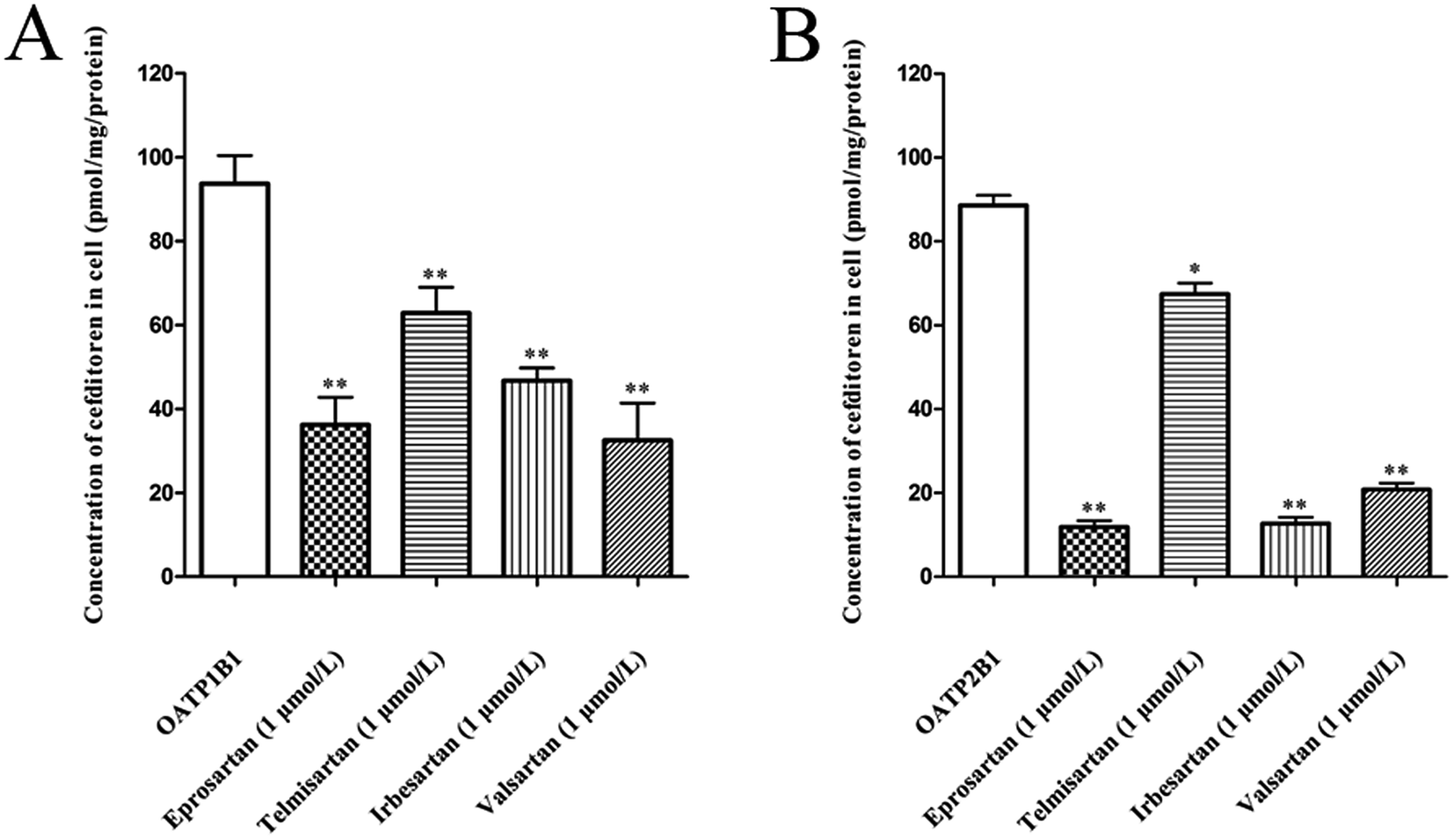 | ||
| Fig. 7 Inhibitory effects of sartans on the uptake of cefditoren in hOATP1B1-HEK293 cells (A) and hOATP2B1-HEK293 cells (B) for 1 minute. All sartans were used at a concentration of 1 μmol L−1, and cefditoren was at a concentration of 5 μmol L−1. The absolute values for cefditoren uptake in control group were 93.25 and 88.38 pmol per mg protein, respectively, in hOATP1B1-HEK293 cells and hOATP2B1-HEK293 cells. A significant difference from the uptake in control group (*P < 0.05, **P < 0.01; mean ± SD, n = 3). | ||
3.6 Effects of cefditoren on the mRNA and protein expression of Oatp1a1, Oatp1a4 and Oatp1b2 in rat liver
To clarify the molecular mechanism of cefditoren-facilitated hepatic uptake, the effects of cefditoren on Oatp1a1, Oatp1a4 and Oatp1b2 gene and protein expression were examined by qRT-PCR and western blotting in rat liver. After pretreating the rats with cefditoren for one week, Oatp1a1 mRNA expression was markedly increased 0.5-fold, while Oatp1b2 and Oatp1a4 mRNA expressions were unchanged (Fig. 8). In the same manner, the protein expression status of the above transporters (Fig. 9) was similar to the corresponding change in gene expression, the Oatp1a1 protein expression being 3-fold greater than control group (Fig. 8).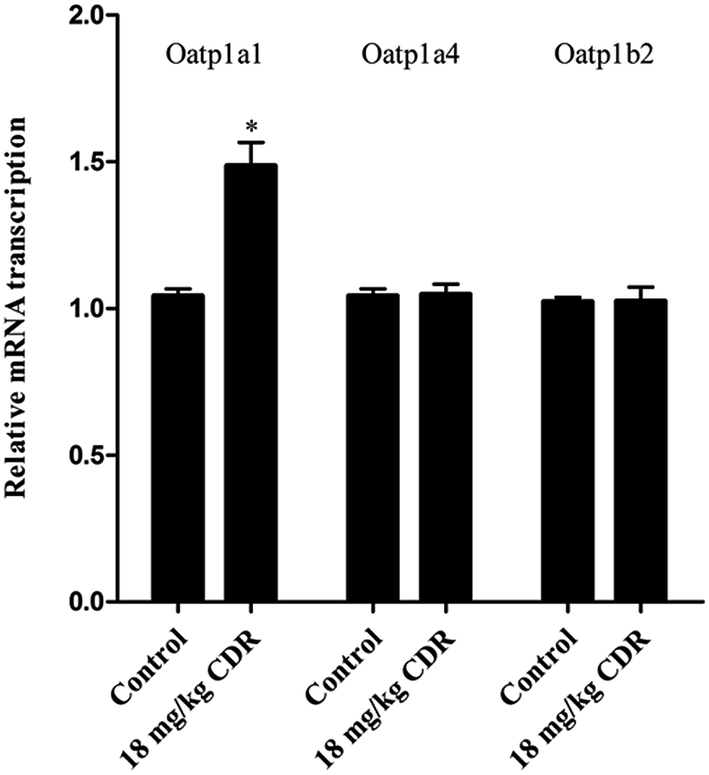 | ||
| Fig. 8 Effect of cefditoren (CDR) on the expression of Oatp1a1, Oatp1a4 and Oatp1b2 mRNA in rat liver. Data were expressed mean ± SD (*P < 0.05 vs. control; **P < 0.01 vs. control; n = 3). | ||
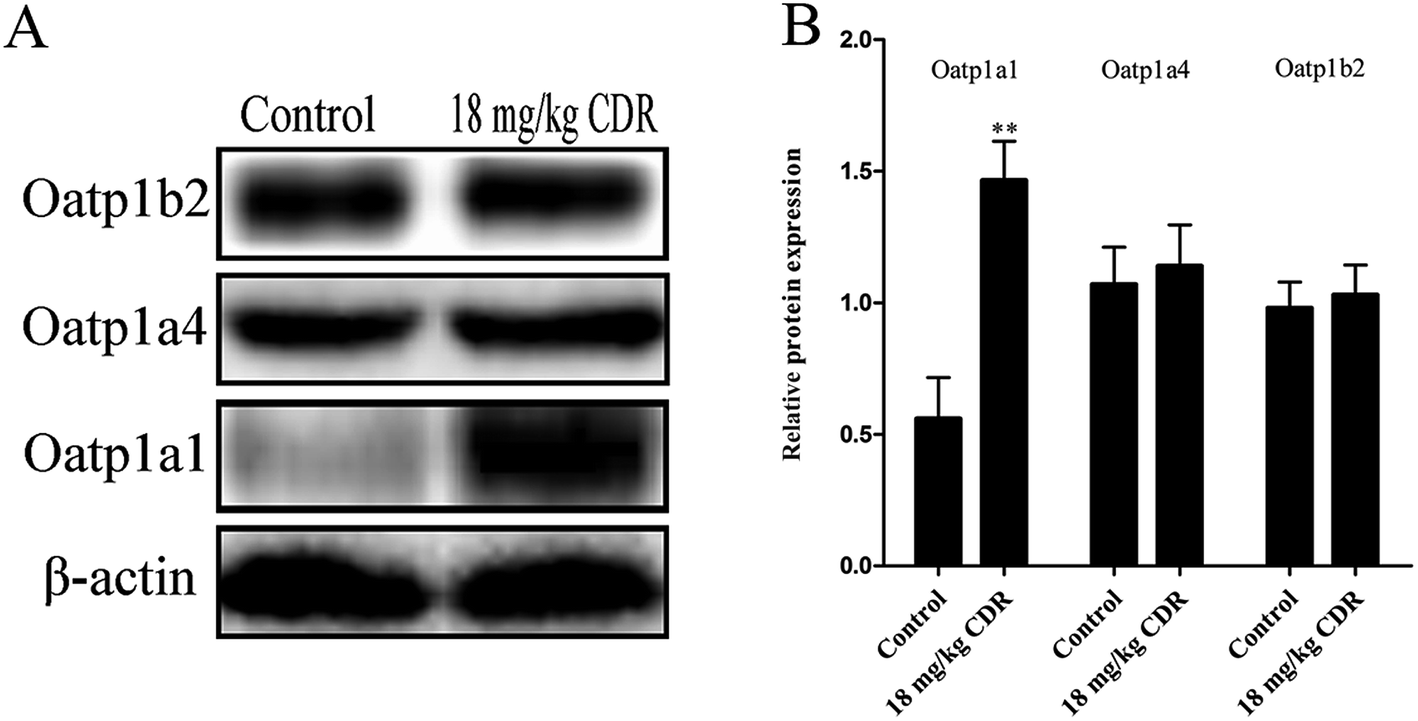 | ||
| Fig. 9 Effect of cefditoren (CDR) on the expression of Oatp1a1, Oatp1a4 and Oatp1b2 protein in rat liver. (A) Lane 1: control group; lane 2: cefditoren alone group (18 mg kg−1). (B) The bar graph shows the relative expression ratio of each protein calculated after normalization by β-actin protein. Data were expressed mean ± SD (*P < 0.05 vs. control; **P < 0.01 vs. control; n = 3). | ||
4. Discussion
The change in function of transporters directly affects the processes of drug absorption, distribution, metabolism and excretion.8,10,43–46 The transporters on cell membranes are the target sites of DDIs.8,10,45,47 Furthermore, transporter-mediated DDI is related to drug therapy, pharmacokinetics and safe medication in the clinic.43,45,48Cefditoren is freely soluble in water and the function of transmembrane transport is relatively poor. It was reported that cefditoren was a substrate of PEPT1,4 and both Mrp2 and Bcrp were involved in the biliary excretion of cefditoren.3 These phenomena indicate that the liver may play a non-negligible role in the absorption and elimination of cefditoren in rats. Considering this, we can infer that the process of entering the liver of cefditoren might be involved with hepatic uptake transporter. Therefore, a detailed investigation of hepatic uptake for cefditoren is crucial for understanding the pharmacokinetics of cefditoren in rats and humans.
The plasma concentration of cefditoren was increased significantly when orally co-administrated with rifampicin (an inhibitor of Oatps)42 in rat (Fig. 2A). And the Cmax of cefditoren (control group) was 0.80 ± 0.06 μg mL−1. According to this, the concentration of cefditoren was determined as 1 μmol L−1 (below the Cmax) in all rat experiments in this study. Besides, no obvious changes occurred in the cumulative biliary excretion of cefditoren (Fig. 2B). This phenomenon indicated that the increased plasma concentration of cefditoren was just on account of the inhibitory effect of rifampicin on the uptake of cefditoren in liver. Furthermore, the perfused rat liver experiment in situ supported this point of view. The hepatic extraction ratio of cefditoren was decreased from 34.7% ± 4.61% to 18.3% ± 2.89% in the presence of rifampicin (Fig. 3A), and approximately 10% reduction was found in the hepatic accumulation of cefditoren (Fig. 3B). These results indicated that cefditoren was transported by the hepatic transporters belonging to the Oatps family in rat liver. Therefore, the phenomena mentioned above indicated that the uptake of cefditoren might be mediated by Oatps in rats, and liver was the potential target organ for cefditoren uptake. Moreover, the uptake of cefditoren in rat liver slices was 2.3-fold greater at 37 °C than at 4 °C after incubation for 30 minutes (Fig. 4A). The uptake of cefditoren in the liver at 4 °C might be explained by nonspecific membrane binding or intracellular binding after passive diffusion into the cells.49 The difference in drug uptake at 4 °C and 37 °C indicated that the uptake of cefditoren in rat liver was mediated by the hepatic transporters. Furthermore, the specific inhibitors of different Oatp isoforms, such as cyclosporin A, ibuprofen, digoxin and glycyrrhizic acid, markedly decreased the uptake of cefditoren in rat liver slices at concentrations which are close to the respective Ki values.42,50 These phenomena indicated that multiple Oatp isoforms were involved in the rat liver extraction process of cefditoren. By comparison, high concentration of TEA and PAH did not inhibit the uptake of cefditoren (Fig. 4B). Therefore, it could be confirmed that the Oatps family might be involved in the uptake of cefditoren in rat liver slices. These series of experiments mentioned above clearly clarified a potential mechanism concerning the uptake of cefditoren in liver through multiple Oatp isoforms (Oatp1a1, Oatp1a4, and Oatp1b2) as the major underlying transporters in rats.
In the interest of clarifying the potential OATP isoforms for the transport of cefditoren, hOATP1B1-, hOATP2B1- and hOATP1B3-HEK293 cells were used to evaluate the transport properties of cefditoren in humans. In transfected cell studies, no significant time-dependent uptake of cefditoren was observed in hOATP1B3-HEK293 cells. On the contrary, in hOATP1B1- and hOATP2B1-HEK293 cells, significant uptake was observed. Meanwhile, an obvious reduction was found in both hOATP1B1- and hOATP2B1-HEK293 cells when cefditoren was combined with rifampicin (a potential inhibitor of hepatic OATPs)51–53 (Fig. 5A–C). Furthermore, concentration-dependent uptakes of cefditoren were significantly observed in hOATP1B1- and hOATP2B1-HEK293 cells (Fig. 5D and E). The Km values of cefditoren were 189.7 ± 60.4 μmol L−1 and 122.7 ± 37.37 μmol L−1, respectively. The results mentioned above suggested that cefditoren was transported into hepatocytes by OATP1B1 and OATP2B1 rather than OATP1B3 in humans. Therefore, the hepatic uptake of cefditoren might be influenced when cefditoren is administered simultaneously with drugs known as substrates or inhibitors of OATP1B1 and/or OATP2B1 (Fig. 6). Furthermore, to provide more useful information about the clinical relevance of interactions with co-administered drugs, in vitro uptake assays were conducted using human transporter expressing cells. According to the daily doses of cefditoren (200 mg d−1, Cmax = 3.44 ± 0.71 mg L−1) in the clinic, the concentration of cefditoren was identified as 5 μmol L−1 in all uptake experiments. Uptake studies using hOATP1B1- and hOATP2B1-HEK293 cells indicated that eprosartan, telmisartan, irbesartan and valsartan exhibited a normal clinical concentration inhibition on the uptake of cefditoren (Fig. 7). All the concentrations of sartans were at 1 μmol L−1 according to the normal doses in the clinic. Taking into account these phenomena, it could be speculated that possible clinical interactions may occur when cefditoren is co-administrated with drugs that are substrates or inhibitors of OATP1B1 and/or OATP2B1.
In this study, the effects of cefditoren on regulation of Oatp1a1, Oatp1b2 and Oatp1a4 expression were analyzed in rat liver. Rats were administrated orally with a corresponding dose of 18 mg kg−1 of cefditoren for one week, since the routine human dose of cefditoren is 200 mg twice a day. After this period, the gene expression levels of Oatp1a1 were increased by cefditoren, while those for Oatp1a4 and Oatp1b2 were unchanged (Fig. 8). However, the observation of increased Oatp1a1 mRNA levels was only indicated in rats. One cannot expect the same induction in humans. Further studies are needed to investigate the details of Oatp1a1 mRNA expression changes in humans. In the same manner, the protein expression status of the above transporters was similar to the corresponding change in gene expression (Fig. 9). The regulation effects of cefditoren on these drug transporters described here may have functional and clinical implications for clarifying the mechanisms of potential cefditoren-related DDIs. And the detailed mechanisms of transporter regulation by cefditoren need further examination.
In the clinic, β-lactam antibiotics are widely used to promote health, although the mechanisms of action are generally unknown. It has been reported that Oatp2 (Oatp1a4) is the predominant contributor to the hepatic uptake of nafcillin, a type of β-lactam antibiotic.54 Additionally, cefadroxil, cefazolin, cefmetazole, cefoperazone, cefsulodin, and cephalexin are also substrates of Oatp2.54 And OATP1B3 plays a major role in the hepatic uptake of β-lactam antibiotics in humans.55 However, in this study, the results indicated that cefditoren was a substrate of OATP1B1 and OATP2B1, but not a substrate of OATP1B3. Therefore, OATP1B1 and OATP2B1 also perform an indispensable role in the hepatic uptake of β-lactam antibiotics in humans. To date, only a very small proportion of currently available drugs have been investigated for their potential interactions with β-lactam antibiotics in humans. Thus, further clinical studies are required to gain knowledge of transporter-based β-lactam antibiotics–drug interactions.
5. Conclusion
In summary, our results demonstrate for the first time that the uptake of cefditoren by the liver is mediated by a hepatic transport system belonging to the OATP family, including OATP1B1 and OATP2B1 in humans and multiple Oatps (Oatp1a1, Oatp1a4 and Oatp1b2) in rats. In addition, cefditoren could increase the protein and gene expression of Oatp1a1. And the sartans could inhibit the hepatic uptake of cefditoren; more attention should be paid to the combination of these drugs in the clinic.Conflict of interest
The authors report no conflict of interest.Acknowledgements
We especially thank Dr Yuichi Sugiyama (Graduate School of Pharmaceutical Sciences, University of Tokyo, Tokyo, Japan) for helpfully providing OATP1B3-HEK293 cells and vector-HEK293 cells and Dr Xiulin Yi (State Key Laboratory of Drug Delivery Technology and Pharmacokinetics, Tianjin Institute of Pharmaceutical Research, Tianjin, China) for helpfully providing OATP1B1- and OATP2B1-HEK293 cells. This work was supported in part by a grant from the National Natural Science Foundation of China (no. 81302838, 81473280).References
- R. N. Jones, M. A. Pfaller, M. R. Jacobs, P. C. Appelbaum and P. C. Fuchs, Diagn. Microbiol. Infect. Dis., 2001, 41, 1–14 CrossRef CAS PubMed.
- D. M. Johnson, D. J. Biedenbach, M. L. Beach, M. A. Pfaller and R. N. Jones, Diagn. Microbiol. Infect. Dis., 2000, 37, 99–105 CrossRef CAS PubMed.
- Q. Meng, Q. Liu, C. Wang, H. Sun, T. Kaku, Y. Kato and K. Liu, Drug Metab. Pharmacokinet., 2010, 25, 320–327 CrossRef CAS PubMed.
- Q. Zhang, Q. Liu, J. Wu, C. Wang, J. Peng, X. Ma and K. Liu, Eur. J. Pharmacol., 2009, 612, 9–14 CrossRef CAS PubMed.
- D. R. Guay, Clin. Ther., 2001, 23, 1924–1937 CrossRef CAS PubMed ; discussion 1923.
- K. Wellington and M. P. Curran, Drugs, 2004, 64, 2597–2618 CrossRef CAS PubMed.
- S. K. Spangler, M. R. Jacobs and P. C. Appelbaum, J. Antimicrob. Chemother., 1997, 39, 141–148 CrossRef CAS PubMed.
- S. Shugarts and L. Z. Benet, Pharm. Res., 2009, 26, 2039–2054 CrossRef CAS PubMed.
- N. F. Smith, W. D. Figg and A. Sparreboom, Expert Opin. Drug Metab. Toxicol., 2005, 1, 429–445 CrossRef CAS PubMed.
- P. Sun, C. Wang, Q. Liu, Q. Meng, A. Zhang, X. Huo, H. Sun and K. Liu, Pharmacol. Rep., 2014, 66, 311–319 CrossRef CAS PubMed.
- T. Abe, M. Kakyo, T. Tokui, R. Nakagomi, T. Nishio, D. Nakai, H. Nomura, M. Unno, M. Suzuki, T. Naitoh, S. Matsuno and H. Yawo, J. Biol. Chem., 1999, 274, 17159–17163 CrossRef CAS PubMed.
- T. Abe, M. Unno, T. Onogawa, T. Tokui, T. N. Kondo, R. Nakagomi, H. Adachi, K. Fujiwara, M. Okabe, T. Suzuki, K. Nunoki, E. Sato, M. Kakyo, T. Nishio, J. Sugita, N. Asano, M. Tanemoto, M. Seki, F. Date, K. Ono, Y. Kondo, K. Shiiba, M. Suzuki, H. Ohtani, T. Shimosegawa, K. Iinuma, H. Nagura, S. Ito and S. Matsuno, Gastroenterology, 2001, 120, 1689–1699 CrossRef CAS.
- B. Hsiang, Y. Zhu, Z. Wang, Y. Wu, V. Sasseville, W. P. Yang and T. G. Kirchgessner, J. Biol. Chem., 1999, 274, 37161–37168 CrossRef CAS PubMed.
- J. Konig, Y. Cui, A. T. Nies and D. Keppler, J. Biol. Chem., 2000, 275, 23161–23168 CrossRef CAS PubMed.
- I. Tamai, J. Nezu, H. Uchino, Y. Sai, A. Oku, M. Shimane and A. Tsuji, Biochem. Biophys. Res. Commun., 2000, 273, 251–260 CrossRef CAS PubMed.
- K. N. Faber, M. Muller and P. L. Jansen, Adv. Drug Delivery Rev., 2003, 55, 107–124 CrossRef CAS PubMed.
- M. Hirano, K. Maeda, Y. Shitara and Y. Sugiyama, J. Pharmacol. Exp. Ther., 2004, 311, 139–146 CrossRef CAS PubMed.
- Y. Shitara, T. Horie and Y. Sugiyama, Eur. J. Pharm. Sci., 2006, 27, 425–446 CrossRef CAS PubMed.
- T. De Bruyn, Z. W. Ye, A. Peeters, J. Sahi, M. Baes, P. F. Augustijns and P. P. Annaert, Eur. J. Pharm. Sci., 2011, 43, 297–307 CrossRef CAS PubMed.
- A. Zhang, C. Wang, Q. Liu, Q. Meng, J. Peng, H. Sun, X. Ma, X. Huo and K. Liu, Drug Metab. Dispos., 2013, 41, 994–1003 CrossRef CAS PubMed.
- B. Hagenbuch and P. J. Meier, Pflugers Archiv : European journal of physiology, 2004, 447, 653–665 CrossRef CAS PubMed.
- B. Hagenbuch and P. J. Meier, Biochim. Biophys. Acta, 2003, 1609, 1–18 CrossRef CAS.
- N. Ishiguro, K. Maeda, W. Kishimoto, A. Saito, A. Harada, T. Ebner, W. Roth, T. Igarashi and Y. Sugiyama, Drug Metab. Dispos., 2006, 34, 1109–1115 CrossRef CAS PubMed.
- R. Nakagomi-Hagihara, D. Nakai, K. Kawai, Y. Yoshigae, T. Tokui, T. Abe and T. Ikeda, Drug Metab. Dispos., 2006, 34, 862–869 CrossRef CAS PubMed.
- W. Yamashiro, K. Maeda, M. Hirouchi, Y. Adachi, Z. Hu and Y. Sugiyama, Drug Metab. Dispos., 2006, 34, 1247–1254 CrossRef CAS PubMed.
- M. Hirano, K. Maeda, Y. Shitara and Y. Sugiyama, Drug Metab. Dispos., 2006, 34, 1229–1236 CrossRef CAS PubMed.
- S. Kitamura, K. Maeda, Y. Wang and Y. Sugiyama, Drug Metab. Dispos., 2008, 36, 2014–2023 CrossRef CAS PubMed.
- M. Shimizu, K. Fuse, K. Okudaira, R. Nishigaki, K. Maeda, H. Kusuhara and Y. Sugiyama, Drug Metab. Dispos., 2005, 33, 1477–1481 CrossRef CAS PubMed.
- M. Trauner and J. L. Boyer, Physiol. Rev., 2003, 83, 633–671 CrossRef CAS PubMed.
- X. Bossuyt, M. Muller, B. Hagenbuch and P. J. Meier, J. Pharmacol. Exp. Ther., 1996, 276, 891–896 CAS.
- J. E. van Montfoort, B. Hagenbuch, K. E. Fattinger, M. Muller, G. M. Groothuis, D. K. Meijer and P. J. Meier, J. Pharmacol. Exp. Ther., 1999, 291, 147–152 CAS.
- B. Hagenbuch and C. Gui, Xenobiotica, 2008, 38, 778–801 CrossRef CAS PubMed.
- J. Kindla, M. F. Fromm and J. Konig, Expert Opin. Drug Metab. Toxicol., 2009, 5, 489–500 CrossRef CAS PubMed.
- Y. Jia, Z. Liu, X. Huo, C. Wang, Q. Meng, Q. Liu, H. Sun, P. Sun, X. Yang, X. Shu and K. Liu, Int. J. Pharm., 2015, 495, 588–598 CrossRef CAS PubMed.
- Y. Shitara, H. Sato and Y. Sugiyama, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 689–723 CrossRef CAS PubMed.
- X. Huo, Q. Liu, C. Wang, Q. Meng, H. Sun, J. Peng, X. Ma and K. Liu, Eur. J. Pharm. Sci., 2013, 50, 420–428 CrossRef CAS PubMed.
- A. Zhang, Y. Jia, Q. Xu, C. Wang, Q. Liu, Q. Meng, J. Peng, H. Sun, P. Sun, X. Huo and K. Liu, Toxicol. Appl. Pharmacol., 2016, 305, 127–135 CrossRef CAS PubMed.
- K. X. Liu, Y. Kato, M. Yamazaki, O. Higuchi, T. Nakamura and Y. Sugiyama, Hepatology, 1993, 17, 651–660 CrossRef CAS PubMed.
- Q. Miao, Q. Liu, C. Wang, Q. Meng, X. Guo, J. Peng, T. Kaku and K. Liu, Drug Metab. Pharmacokinet., 2011, 26, 494–502 CrossRef CAS PubMed.
- L. Wang, Q. Meng, C. Wang, Q. Liu, J. Peng, X. Huo, H. Sun, X. Ma and K. Liu, J. Nat. Prod., 2013, 76, 909–914 CrossRef CAS PubMed.
- X. Guo, Q. Meng, Q. Liu, C. Wang, H. Sun, J. Peng, X. Ma, T. Kaku and K. Liu, Toxicol. Appl. Pharmacol., 2013, 271, 285–295 CrossRef CAS PubMed.
- K. Fattinger, V. Cattori, B. Hagenbuch, P. J. Meier and B. Stieger, Hepatology, 2000, 32, 82–86 CrossRef CAS PubMed.
- X. Huo, Q. Liu, C. Wang, Q. Meng, H. Sun, J. Peng, X. Ma, P. Sun and K. Liu, J. Pharm. Sci., 2014, 103, 719–729 CrossRef CAS PubMed.
- K. M. Morrissey, S. L. Stocker, M. B. Wittwer, L. Xu and K. M. Giacomini, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 503–529 CrossRef CAS PubMed.
- L. Wang, C. Wang, Q. Liu, Q. Meng, X. Huo, P. Sun, X. Yang, H. Sun, Y. Zhen, J. Peng, X. Ma and K. Liu, Eur. J. Pharm. Sci., 2014, 63, 77–86 CrossRef CAS PubMed.
- F. Muller and M. F. Fromm, Pharmacogenomics, 2011, 12, 1017–1037 CrossRef PubMed.
- G. Ciarimboli, Anticancer Res., 2014, 34, 547–550 CAS.
- K. M. Giacomini, S. M. Huang, D. J. Tweedie, L. Z. Benet, K. L. Brouwer, X. Chu, A. Dahlin, R. Evers, V. Fischer, K. M. Hillgren, K. A. Hoffmaster, T. Ishikawa, D. Keppler, R. B. Kim, C. A. Lee, M. Niemi, J. W. Polli, Y. Sugiyama, P. W. Swaan, J. A. Ware, S. H. Wright, S. W. Yee, M. J. Zamek-Gliszczynski and L. Zhang, Nat. Rev. Drug Discovery, 2010, 9, 215–236 CrossRef CAS PubMed.
- J. H. Proost, L. Beljaars, P. Olinga, P. J. Swart, M. E. Kuipers, C. Reker-Smit, G. M. Groothuis and D. K. Meijer, Eur. J. Pharm. Sci., 2006, 27, 123–132 CrossRef CAS PubMed.
- Y. Shitara, D. Sugiyama, H. Kusuhara, Y. Kato, T. Abe, P. J. Meier, T. Itoh and Y. Sugiyama, Pharm. Res., 2002, 19, 147–153 CrossRef CAS.
- I. Tamai and T. Nakanishi, Curr. Opin. Pharmacol., 2013, 13, 859–863 CrossRef CAS PubMed.
- M. K. Choi, Q. R. Jin, Y. L. Choi, S. H. Ahn, M. A. Bae and I. S. Song, Biopharm. Drug Dispos., 2011, 32, 175–184 CrossRef CAS PubMed.
- S. R. Vavricka, J. Van Montfoort, H. R. Ha, P. J. Meier and K. Fattinger, Hepatology, 2002, 36, 164–172 CrossRef CAS PubMed.
- M. Nakakariya, T. Shimada, M. Irokawa, H. Koibuchi, T. Iwanaga, H. Yabuuchi, T. Maeda and I. Tamai, Pharm. Res., 2008, 25, 578–585 CrossRef CAS PubMed.
- M. Nakakariya, T. Shimada, M. Irokawa, T. Maeda and I. Tamai, Drug Metab. Pharmacokinet., 2008, 23, 347–355 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |