
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dehalogenation of diatrizoate using nanoscale zero-valent iron: impacts of various parameters and assessment of aerobic biological post-treatment†
Pan-Pan He,
Chuan-Shu He,
Qi Liu and
Yang Mu *
*
CAS Key Laboratory of Urban Pollutant Conversion, Collaborative Innovation Centre of Suzhou Nano Science and Technology, Department of Chemistry, University of Science and Technology of China, Hefei, China. E-mail: yangmu@ustc.edu.cn; Fax: +86 551 63607907
First published on 23rd May 2017
Abstract
This study investigated the feasibility of nanoscale zero-valent iron (nZVI) for reductive dehalogenation of iodinated contrast medium diatrizoate (DTA). The impacts of various parameters, including nZVI dosage, DTA concentration, solution pH, aging time of nZVI, the presence of natural organic matter, and the type of competitive anions, on the dehalogenation of DTA as well as the formation of its reductive product 5-diacetamidobenzoate (DABA) using nZVI were evaluated. Furthermore, an aerobic biological post-treatment was conducted to study the biodegradability of reductive products of DTA dehalogenation using nZVI. The results showed that dosing with 0.5 g L−1 nZVI particles resulted in a rapid decrease in DTA concentration and a corresponding rise of the concentrations of DABA and I−. DTA dehalogenation in terms of its removal and DABA formation was enhanced with the increase in nZVI dosage but deteriorated when increasing solution pH. nZVI aging time had a negative impact on DTA dehalogenation. Natural organic matter at much low level could improve DTA dehalogenation, while had a negative influence at high concentrations. Contrary to sulfate, the presence of nitrate and phosphate strongly inhibited DTA removal using nZVI. The results also showed that the reductive product DABA could be degraded by aerobic biological post-treatment, suggesting DTA dehalogenation with nZVI may be a vital procedure for its biodegradability improvement and consequently complete removal.
1. Introduction
Iodinated contrast media (ICM) are widely used in hospitals for urinary tract imaging. A large number of ICM are used in tests for patients (>100 g per dose), and the annual world-wide consumption of ICM was reported as approximately 3.5 × 106 kg.1 Due to their heavy use and high biochemical stability, increasing concentrations of ICM have been detected in hospital and domestic wastewater, treated wastewater effluent, surface water, groundwater, and even finished drinking water, whose concentration is up to µg L−1.2–6 Although ICM are believed to be harmless, subtle effects of mixtures of ICM with their metabolites and other micropollutants are unknown and may exert unanticipated ecological or human health risks.7Due to high oxidation resistance of the tri-iodinated aromatic ring structure, ICM have been reported to only lose their alkyl side chains by biological and chemical oxidation.7 Alternatively, some reductive strategies have been developed for the removal of ICM from water, such as anaerobic biological transformation,8 dehalogenation through hydrogen with noble metal as the catalyst, and (bio)elelctrochemical reduction.7,9 However, there are some shortcomings in those methods. Anaerobic biodegradation needs high activity microbial species, and long processing time. Other chemical methods suffer from high cost, which inhibits their wide application. Therefore, it's urgent to find an efficient and low-cost technique for the treatment of ICM from water.
Zero-valent iron (ZVI) is an active metal whose electric potential is −0.440 V. Iron releases electrons when it is oxidized, making it be able to remove a variety of pollutants.10 It is noted that micro-scale ZVI powder has been successfully applied to treat iodine organic matters.11–13 Compared to micro-scale ZVI, the nano-scale ZVI (nZVI) possesses greater surface activity to remove water pollutants effectively,14 resulting from small particle size and sharp increase of specific surface area. Therefore, nZVI has been widely adopted to reductive removal of different pollutants such as chlorinated organic compounds,15,16 nitro-aromatic compounds,17,18 organic dyes19,20 and heavy metals.21,22 However, the utilization of nZVI to degrade ICM under anoxic conditions has seldom been investigated.
This study aimed at investigating the feasibility of nZVI for reductive dehalogenation of ICM from water, in which diatrizoate (DTA) was selected as the model ICM. The impacts of various parameters, including nZVI dosage, diatrizoate concentration, solution pH, aging time of nZVI, the presence of natural organic matter (NOM), and type of competitive anions, on DTA dehalogenation using nZVI were evaluated. Furthermore, an aerobic post-treatment was conducted to study the biodegradability of reductive products of DTA dehalogenation using nZVI.
2. Materials and methods
2.1 Materials
Sodium diatrizoate (≥98% purity) and its reductive product 5-diacetamidobenzoate (DABA, ≥98% purity) were purchased from Aladdin Industry Company (China) and Chem-Bridge Company (China), respectively. Humic acid was obtained from Sigma-Aldrich Company (Switzerland), and other chemicals including NaBH4, FeCl3, NaCl, NaNO3, Na2SO4, HCl, NaOH, ethanol and riboflavin were purchased from Sinopharm Chemical Reagent Ltd (China). High purity of compressed N2 was obtained from Shangyuan Gas Products Co. (Nanjing, China). All the chemicals were used as receipt except for that humic acid was purified according to a previous method.232.2 Synthesis and characterization of nZVI
The nZVI particles were synthesized by using the liquid-phase reduction method.24 Briefly, 19.32 g FeCl3·6H2O was dissolved into 200 mL 30% ethanol solution, following with N2 purge for 20 min. The reducing solution was prepared with 24.37 g NaBH4 dissolved into 400 mL deionized water. After that, the reducing solution was added into the ferric solution on a mechanical stirrer with the speed of 300 rpm. The drop rate of reducing solution was controlled at 60–70 drops per min. The mixture was left to stir for another 30 min after the addition of NaBH4 solution was completed. Centrifugation was used to separate the liquid phase solution and the solid black particles. The solid black particles were washed three times with deionized water and ethanol, respectively, and then dried in a vacuum oven overnight at 65 °C. The nZVI products were stored in an anaerobic operation box (Shellab, Bactron EZ, USA) containing 5% H2, 5% CO2 and 90% N2 to avoid oxygen exposure.The morphology of the synthesized nZVI nanoparticles (NPs) was examined using a transmission electron microscope (TEM) with a JED-2300T (JEM-2011, JEOL Ltd., Japan) and a ZEISS GEMINISEM 500 scan electron microscope (SEM) (EHT = 3.00 kV, Mag = 30.00k×, WD = 6.00 mm, JSM-6700F, JEOL Ltd., Japan). The freshly prepared nZVI samples were dispersed in ethanol by ultrasonic before TEM and SEM tests. X-ray diffraction (XRD) analysis of nZVI particles was performed by using a Japan Rigaku TTR-III X-ray diffractometer at 40 kV and 100 mA with a Cu Kα (λ = 1.54178) radiation source. Iron nanoparticles were scanned from 10° to 80° at 0.020° per step and 8° min−1. Also, Fourier Transform Infrared (FTIR) spectrometer was used to examine the nZVI before and after the dehalogenation reaction (Nicolet 8700, Thermo Nicolet, USA).
2.3 DTA dehalogenation experiments
In the typical degradation experiments, 0.5 mL of 6 mM DTA solution was added into a 150 mL serum bottle containing 100 mL deionized water with pH at 7.0, then was purged with nitrogen for 15 min. Further, 0.05 g nZVI was added into the solution in the anaerobic operation box, and then the bottle was immediately sealed tightly with rubber stoppers and aluminum caps following with continuous shaking with 150 rpm at 25 °C for 120 h by using a bath shaker.The effects of various parameters on DTA dehalogenation were investigated, including nZVI dosage, DTA concentration, pH, nZVI aging time, the presence of NOM (riboflavin and humic acid), and competitive anions (SO42−, PO43− and NO3−). The experimental conditions are summarized in Table 1. The initial solution pH was adjusted using 2 M HCl or NaOH. Each experiment was carried out in duplicate.
| Experiments | Operational conditions | |
|---|---|---|
| Variable item | Constant items | |
| Effect of nZVI dosage | Dosage of nZVI: 0.1, 0.2, 0.5, 1.0 g L−1 | DTA: 30 µM |
| pH: 7 | ||
| Aging time: fresh one | ||
| NOM: NA | ||
| Competitive anion: NA | ||
| Effect of DTA concentration | DTA: 5, 15, 30, 60 µM | nZVI dosage: 0.5 g L−1 |
| pH: 7 | ||
| Aging time: fresh one | ||
| NOM: NA | ||
| Competitive anion: NA | ||
| Effect of pH | pH: 3, 5, 7, 9 | nZVI dosage: 0.5 g L−1 |
| DTA: 30 µM | ||
| Aging time: fresh one | ||
| NOM: NA | ||
| Competitive anion: NA | ||
| Effect of nZVI aging time | Aging time: 0, 4, 8 weeks | nZVI dosage: 0.5 g L−1 |
| DTA: 30 µM | ||
| pH: 7 | ||
| NOM: NA | ||
| Competitive anion: NA | ||
| Effect of NOM | Riboflavin: 0, 0.1, 0.2, 0.5, 1, 5 µM | nZVI dosage: 0.5 g L−1 |
| DTA: 30 µM | ||
| Humic acid: 0, 0.1, 0.5, 1, 5, 10 mg L−1 | pH: 7 | |
| Aging time: fresh one | ||
| Competitive anion: NA | ||
| Effect of different anions | SO42−: 0, 2, 5, 10 mg L−1 | nZVI dosage: 0.5 g L−1 |
| DTA: 30 µM | ||
| PO43−: 0, 2, 5, 10 mg L−1 | pH: 7 | |
| NO3−: 0, 2, 5, 10 mg L−1 | NOM: NA | |
| Aging time: fresh one | ||
2.4 Aerobic post treatment
An aerobic post-treatment was conduct at room temperature to test the biodegradable ability of the product from DTA dehalogenation in this study. After 96 h dehalogenation by nZVI with initial DTA concentration of 30 µM, the solution was collected and then centrifuged to prepare basal medium for aerobic biological treatment. The composition of basal medium was as follows (mg L−1): glucose 1880, (NH4)2SO4 2830, KH2PO4 132, K2HPO4·3H2O 221, CaCl2·2H2O 250, FeSO4·7H2O 200, MgSO4·7H2O 200 and 1.0 mL trace element solution. The trace element solution contained (mg L−1): H3BO3 50, ZnCl2 50, CuCl2·2H2O 30, MnSO4·H2O 50, Na2Mo7O4·2H2O 60, AlCl3 50, CoCl2·6H2O 50, and NiCl2·6H2O 50.25 2 g L−1 NaHCO3 was added as buffer solution. 10 g VSS per L (VSS: volatile total solids) of aerobic sludge from Wangtang municipal wastewater treatment plant (Hefei, China) was inoculated into a 440 mL serum bottle with basal medium to a final working volume of 250 mL. To exclude the possibility of sludge adsorption, the control experiments were conducted without oxygen. A pump was used to purge air into the solution to guarantee adequate oxygen during experiments. The dissolved oxygen (DO) in the solution was monitored with LDO101 dissolved oxygen probe (HACH, USA). Additionally, experiments with 30 µM DTA in the basal medium were also conducted to investigate whether DTA could be degraded by the aerobic activated sludge. Each experiment was repeated twice.2.5 Analysis and calculations
Samples were collected at selected time intervals by syringes and then filtered through a 0.45 µm membrane for further analysis. The concentrations of DTA and DABA were analyzed using high performance liquid chromatography (Agilent 1260, USA) with an Agilent HC-C18 column (5 µm, 4.6 mm × 250 mm). The mobile phase was a mixture of 0.1% formic acid–methanol (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, v/v) with flow rate of 1.0 mL min−1 and the UV detector was set at 254 nm. Samples after 24 h dehalogenation reaction in the typical experiments were collected for the identification of intermediate products using liquid chromatography electrospray ionisation tandem mass spectrometry (LC-ESI-MS) (LTQ-Orbitrap XL, ThermoFisher Co., USA), in which the chromatographic conditions are as same as those for DTA and DABA analysis, and other parameters are summarized in Table S1 of the ESI.† The concentrations of SO42− and PO43− were determined by ion chromatography (DIONEX ICS-1100, USA) equipped with an AS19 column and an electrical conductivity detector. The KOH effluent was used as mobile phase with concentration gradient ranging from 10 to 40 mM and 25 to 65 mM at 1 mL min−1, respectively. I− concentration was analyzed by using UV-vis spectrophotometer (Agilent Cary 60, USA) at 226 nm,26 while Fe2+ concentration was determined with phenanthroline method at 510 nm.27 The concentrations of NO3− and NH4+ were measured with UV-vis spectrophotometer (Agilent Cary 60, USA) at 220 and 697 nm by coordinating with salicylic acid and hypochlorous acid, respectively.27
:25, v/v) with flow rate of 1.0 mL min−1 and the UV detector was set at 254 nm. Samples after 24 h dehalogenation reaction in the typical experiments were collected for the identification of intermediate products using liquid chromatography electrospray ionisation tandem mass spectrometry (LC-ESI-MS) (LTQ-Orbitrap XL, ThermoFisher Co., USA), in which the chromatographic conditions are as same as those for DTA and DABA analysis, and other parameters are summarized in Table S1 of the ESI.† The concentrations of SO42− and PO43− were determined by ion chromatography (DIONEX ICS-1100, USA) equipped with an AS19 column and an electrical conductivity detector. The KOH effluent was used as mobile phase with concentration gradient ranging from 10 to 40 mM and 25 to 65 mM at 1 mL min−1, respectively. I− concentration was analyzed by using UV-vis spectrophotometer (Agilent Cary 60, USA) at 226 nm,26 while Fe2+ concentration was determined with phenanthroline method at 510 nm.27 The concentrations of NO3− and NH4+ were measured with UV-vis spectrophotometer (Agilent Cary 60, USA) at 220 and 697 nm by coordinating with salicylic acid and hypochlorous acid, respectively.27
The DTA removal efficiency (DRE, %) and the DABA formation efficiency (DFE, %) at time t (h) were determined using the following equations:
| DRE = (CDTA,0 − CDTA,t)/CDTA,0 × 100 |
| DFE = CDABA,t/CDTA,0 × 100 |
3. Results and discussion
3.1 Typical DTA dehalogenation with nZVI
As shown in Fig. 1a, nZVI particles prepared in our study had a chain-like, core–shell structure and were nearly spherical with a size around 50 nm,28 which was also demonstrated by the SEM image (Fig. 1b). The peak at 44.67° in Fig. 1c confirmed that Fe0 was the majority of the particle component.28 Fig. 2a illustrates the trend in concentrations of DTA, DABA and I− over time in typical DTA dehalogenation experiments using nZVI particles. Dosing with 0.5 g L−1 nZVI particles resulted in a rapid decrease of DTA concentration and a corresponding rise of the concentrations of DABA and I−. The DTA removal efficiency reached to 100% within 24 h, while the DABA formation efficiency was only 39.6% in 24 and it reached to 84.0% after 120 h reaction. In addition, it should be mentioned that DABA concentration still slowly increased with reaction time after DTA was completely removed (Fig. 2a). Moreover, the adsorption of DABA on nZVI was negligible, as shown in Fig. S3.† These results indicate that DTA dehalogenation by nZVI was fast while the DABA formation was the limiting step reaction. Therefore, we speculate that all DTA could be completely reduced to DABA by nZVI but with much long reaction time at high pHs. | ||
| Fig. 1 Images of the synthesized nZVI particles: (a) TEM, (b) SEM and (c) XRD. | ||
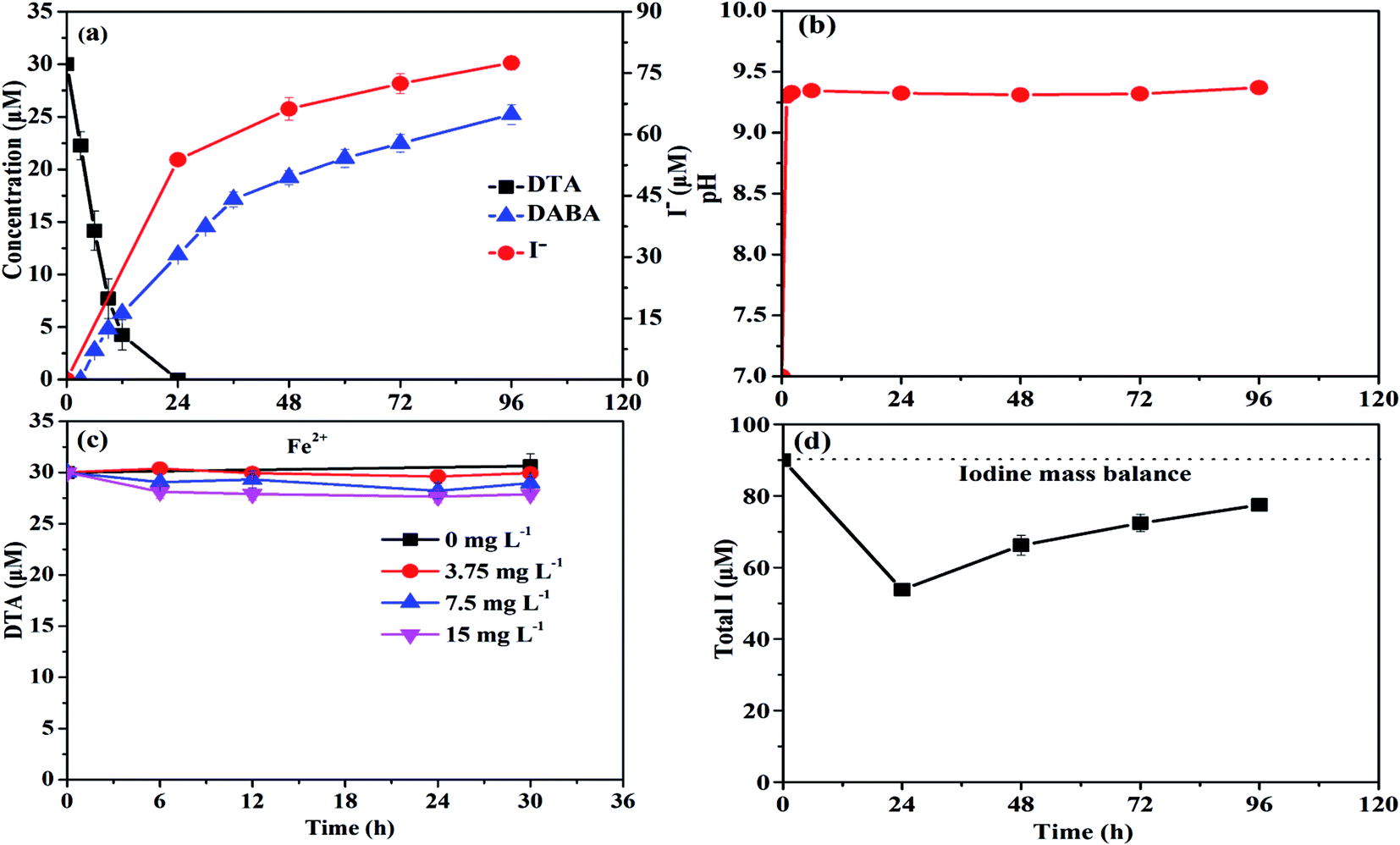 | ||
| Fig. 2 Variations of (a) DTA removal, DABA formation and I− release, (b) pH as a function of reaction time in typical dehalogenation experiments (30 µM DTA, 0.5 g L−1 nZVI, and pH at 7), and (c) DTA removal by Fe2+ at various concentrations, and (d) the total detectable I. | ||
As illustrated in Fig. 2b, the reaction pH increased rapidly from 7.0 to 9.4 within 2 h because of the reaction between nZVI and H2O or DTA with proton consumption.11 After that, the solution pH changed slightly, properly due to the equilibrium for the Fe(OH)2/H2O system (pH = 8.9).29 Fig. 1c shows that the crystal form of nZVI was totally destroyed after 96 h dehalogenation reaction. The nZVI surface passivation under high pH occurs easily, while DTA removal and DABA formation were continuous after 2 h in this study. Additionally, we also tested the reductive ability of Fe2+ and found that DTA concentration changed slightly regardless of Fe2+ concentration (Fig. 2c). Thus, the possible contribution to DTA removal and DABA formation in this study may partially result from surface-bound Fe2+ and/or other reactive corrosion products like green rust. Several studies have reported that the green rust could dechlorinate different pollutants effectively.30,31 Erbs et al. found that green rusts formed during corrosion of ZVI under nonacid conditions may considerably contribute to the total reduction of CCl4.30 Satapanajaru et al. reported that the green rusts provided a source of Fe(II) and produced magnetite (and other oxide surfaces) that coordinated Fe(II), which then facilitated dechlorination.31
As shown in Fig. 2a, I− concentration increased from 0 to 77.46 µM within 96 h due to the dehalogenation of DTA in the typical experiments. Based on the mass balance calculation, the total detectable iodine concentration (i.e., I− concentration + 3 × CDTA,t) decreased from 90 to 53.78 µM and then increased to 77.46 µM during reaction (Fig. 2d). The missing iodine was speculated to exist in the intermediate products with one or two iodine loss during DTA dehalogenation.8 The results from LC-ESI-MS analysis confirmed the formation of intermediate products (DTA-2I, DTA-I) during DTA dehalogenation process, as shown in Fig. S1 and S2.† Since each DTA dehalogenation experiment was carried out under anaerobic conditions, it could be concluded that reductive transformation was majorly happened during DTA dehalogenation by nZVI in this study. The oxidative Fenton like reaction for DTA degradation would be negligible because this reaction need an aerobic condition. Recently, Stieber et al. investigated ICM removal by granular ZVI with a particle size of 0.125–3.000 mm in the presence of oxygen at acidic pH conditions,11 and DTA only reduced by 60% in 8 h, indicating a slower removal rate compared to our study. Besides for reductive transformation by ZVI, they found that oxidative process were also responsible for the removal of ICM, which is due to the formation of hydrogen peroxide as an intermediate product for the oxidation of ZVI in aerobic water.
3.2 Effects of various parameters
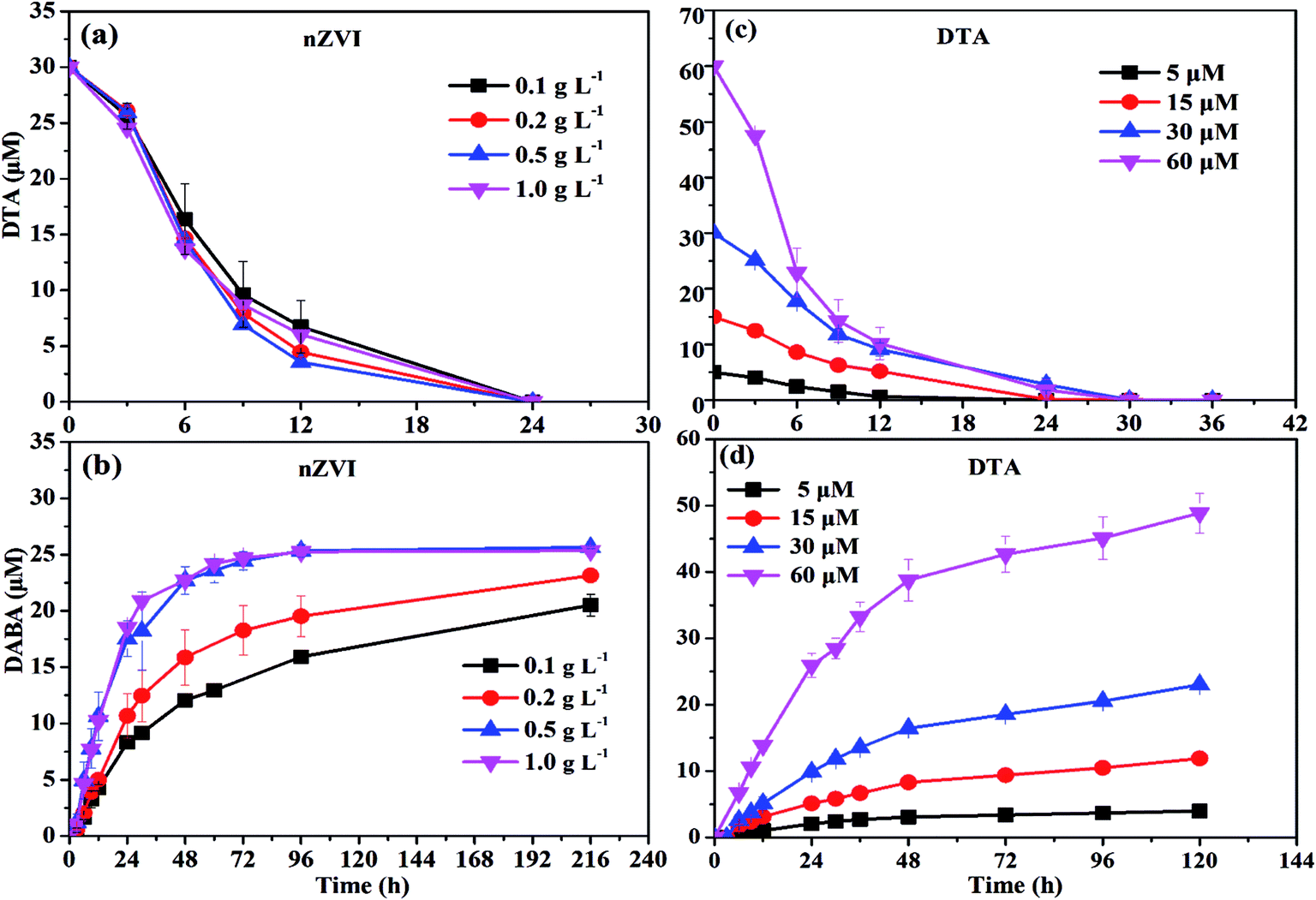 | ||
| Fig. 3 Effect of nZVI dosage on (a) DTA removal and (b) DABA formation, and effect of DTA concentration on (c) DTA removal and (d) DABA formation. | ||
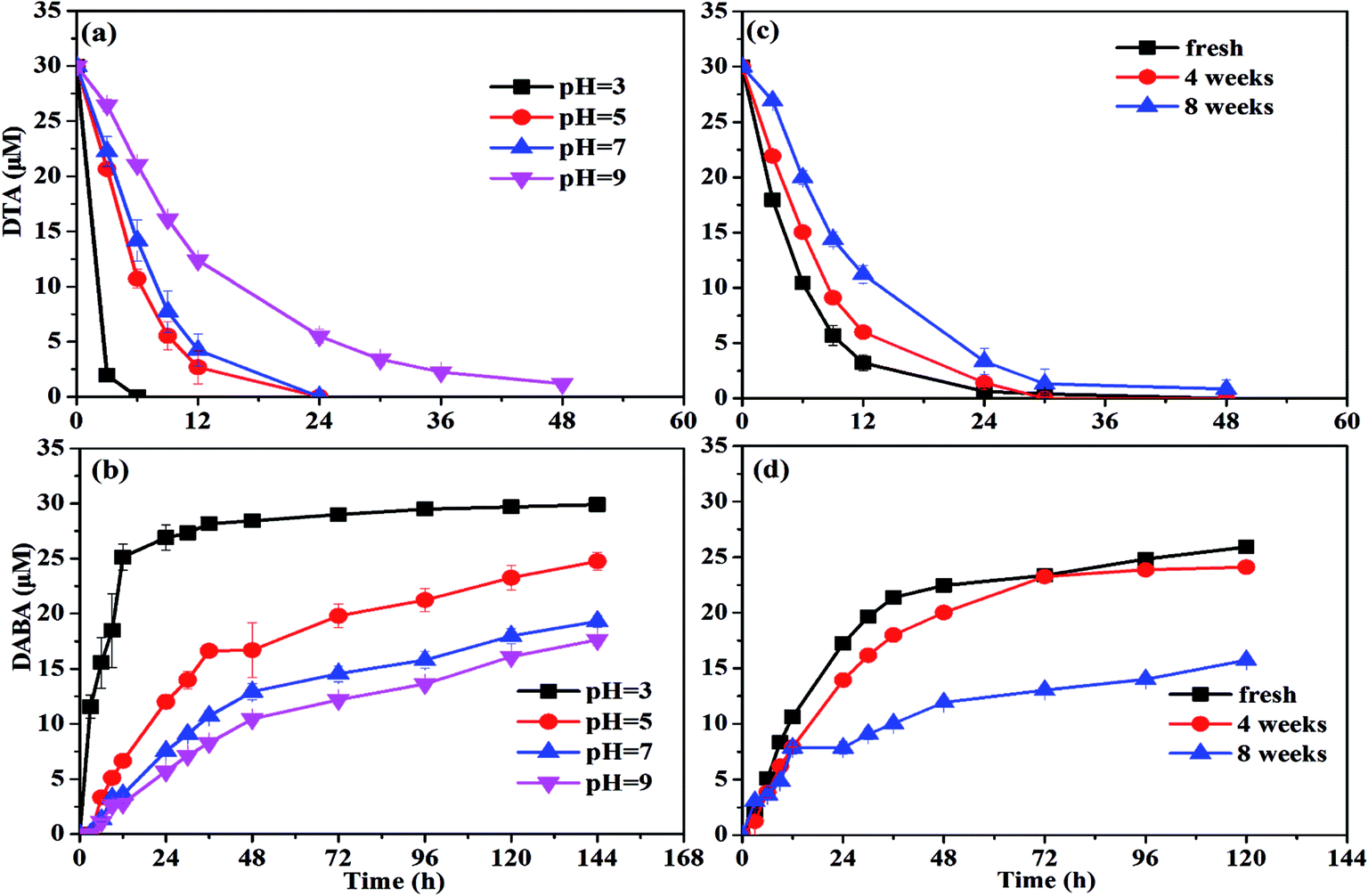 | ||
| Fig. 4 Effect of pH on (a) DTA removal and (b) DABA formation, and effect of nZVI aging time on (c) DTA removal and (d) DABA formation. | ||
37 to form the oxide shell on the surface of nZVI during aging and moreover the oxide shell increased with increasing aging time.29 This might prevent the contact between nZVI and DTA and thus result in the poor dehalogenation performance.To activate the aged nZVI and recover its reactivity, several methods can be effective, such as acid washing, H2-pretreatment and ultrasound. After acid washing, the oxide layer on the surface of nZVI is able to be dissolved into solution and transformed into Fe2+ or Fe3+. It involves the reconversion of oxidized iron compounds to iron chlorides, which can participate again as a precursor in a regenerating synthesis process with NaBH4 reductant to produce nZVI.38 The H2-pretreatment method offered several advantages over the acid-washing method practically without wastewater or sludge generation. Liou et al. employed hydrogen gas at 400 °C to activate the ZVI surface to improve nitrate removal by ZVI and they found that the lag period of nitrate removal by H2-pretreated ZVI disappeared and the first-order reaction rate constant was increased by 4.7 times compared with the non-pretreated ZVI.39 The ultrasound method serves to increase the active iron surface, improve reaction rates, and increase the concentration of terminal by-products. Geiger et al. showed the kinetic improvement caused by ultra-sound pretreatment and illustrated that ultrasound could clear the iron surfaces of debris and thus increase the surface area up to 169%.40
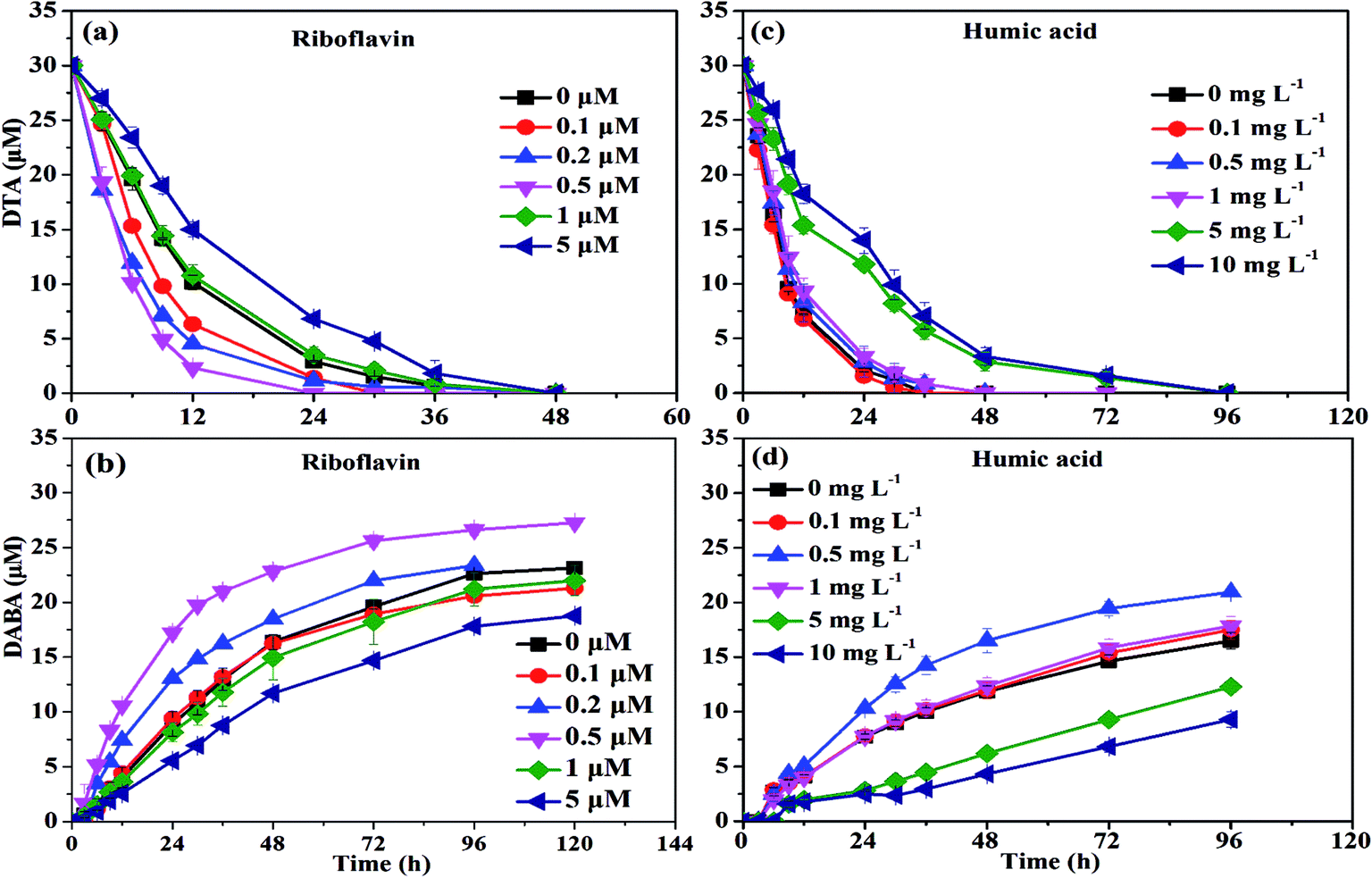 | ||
| Fig. 5 DTA dehalogenation with nZVI in the presence of two different NOMs. | ||
At low concentrations, several studies have found that NOM was able to act as the electron shuttle to rapidly transfer electrons, leading to enhanced reductive degradation of contaminants.42 For instance, Kim et al. observed that the degradation rate of NO3− by nZVI was accelerated in the presence of 6.25–100 mg L−1 humic acid, and they suggested that quinone moieties (e.g., p-hydroquinone, lawsone, and 9,10-anthraquinone-2,6-disulfonate (AQDS)) in humic acid played a pivotal role in enhancing degradation of NO3− by nZVI.42 On the contrary, in the presence of high concentration of NOM, adsorption of NOM onto reaction sites on the surface of iron phases would play a major role in inhibiting the reductive degradation of pollutants by nZVI. Adsorption of NOM can compete with contaminants in ways which can decelerate the kinetics of contaminant degradation.42,43 The FTIR images further proved this phenomenon. As shown in Fig. 6, a characteristic peak at 1425 cm−1, belongs to –CH deformation of –CH3 and –CH bending of CH2 (ref. 44), was found in the presence of 10 mg L−1 humic acid, strongly suggesting the adsorption of humic acid on the surface of nZVI.36
 | ||
| Fig. 6 FTIR images of nZVI before and after dehalogenation reaction in the presence or absence of humic acid. | ||
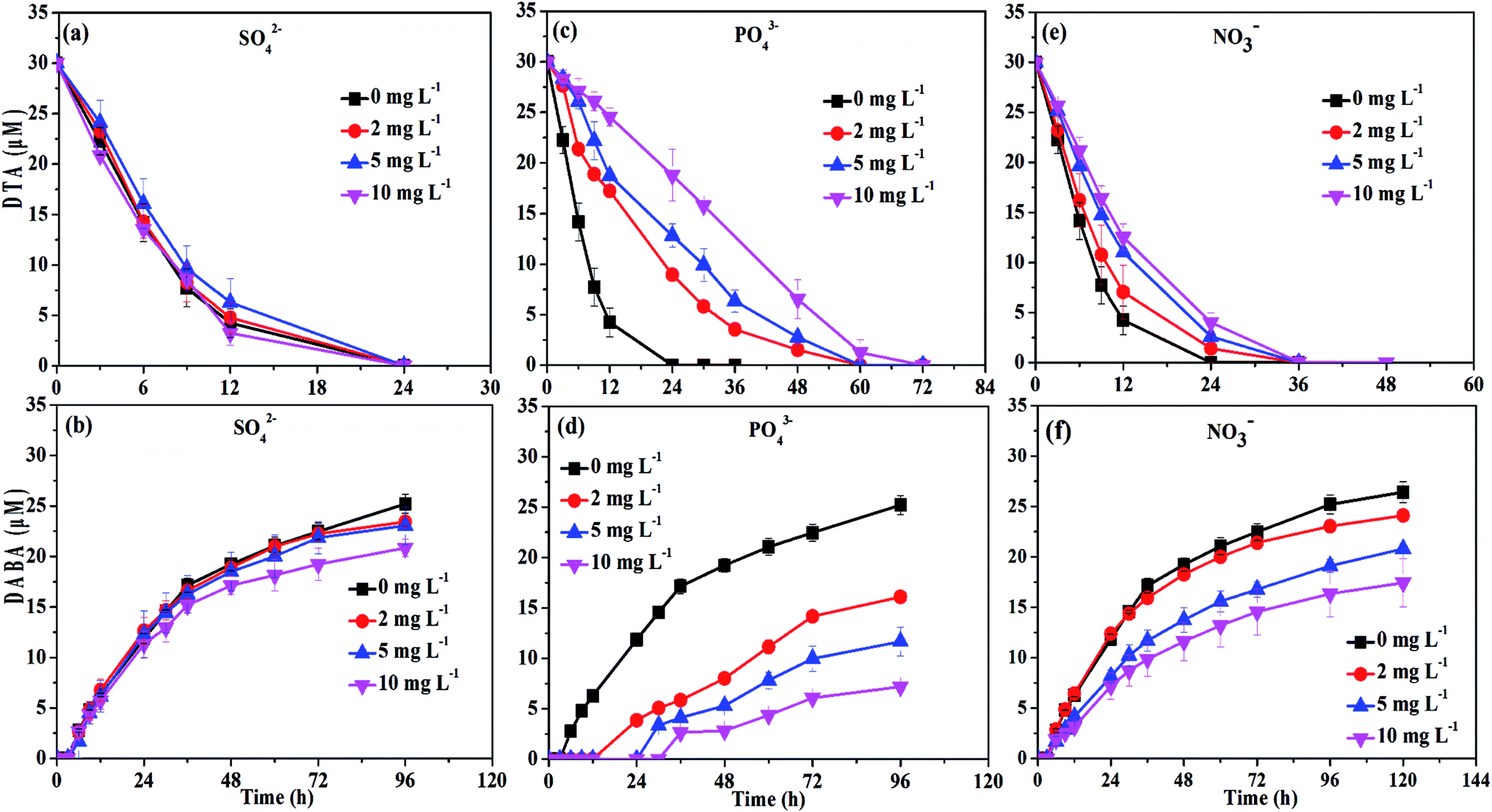 | ||
| Fig. 7 DTA dehalogenation with nZVI in the presence of different anions (SO42−, PO43−, and NO3−). | ||
The DTA removal efficiency at 24 h reduced from 100% to 37.4% when PO43− concentration was improved from 0 to 10 mg L−1, and at the same time the DABA formation efficiency at 48 h decreased from 64.1% to 9. 4% (Fig. 7c and d). The negative effect might be due to that PO43− is an inner-sphere complex-forming anion which could strongly sorb to mineral surfaces, thus hindering the sorption of DTA on the surface of nZVI particles. Indeed, we found that the PO43− in the solution completely disappeared after reaction. The disappearance of PO43− may result from precipitation with ferrous ion or co-precipitation with Fe(OH)2 due to high pH.45 Chen et al. also observed that PO43− had a negative effect on the removal of tetracycline with polyvinylpyrrolidone modified nZVI.33
Similarly, the rates of both DTA removal and DABA formation reduced when concentration of NO3− were increased from 0 to 10 mg L−1 (Fig. 7e and f). As shown in Fig. S4,† NO3− was completely transformed into NH4+ by nZVI after 24 h reaction with initial NO3− concentration of 10 mg L−1, and the removal efficiency at 12 h reached to 87.4%, suggesting that NO3− removal was faster than DTA reduction by nZVI. This implies that that NO3− was able to compete with DTA for reactive sites and electrons of the nZVI, resulting in inhibition of DTA dehalogenation. Meanwhile, the reaction between NO3− and nZVI could make the oxide-shell thicker, and the surface passivation become more seriously with the concentration of NO3− increased.46
3.3 Performance of aerobic biological post-treatment
To test the biodegradability of reductive products of DTA dehalogenation using nZVI, aerobic biological post-treatment was conducted in this study. We first excluded the volatilization possibility of DABA even with O2 supply (data not shown). As shown in Fig. 8, DABA concentration was unchanged without O2 supply, suggesting insignificant adsorption of DABA onto sludge. In the presence of oxygen, the DABA removal efficiency was 58.8% after 96 h, indicating that aerobic sludge had a good degradation ability on DABA. The other transformation products including DTA-2I and DTA-I, which are present with much low concentrations, were not examined during aerobic degradation experiments. Meanwhile, the DO concentration declined in first 8 h and then increased to around 8 mg L−1, majorly due to the consumption of glucose in the solution. In addition, we also found that DTA couldn't be degraded by aerobic biological treatment (Fig. 8b). These results illustrated that DTA dehalogenation with nZVI would be a vital procedure for its biodegradability improvement and consequently complete removal.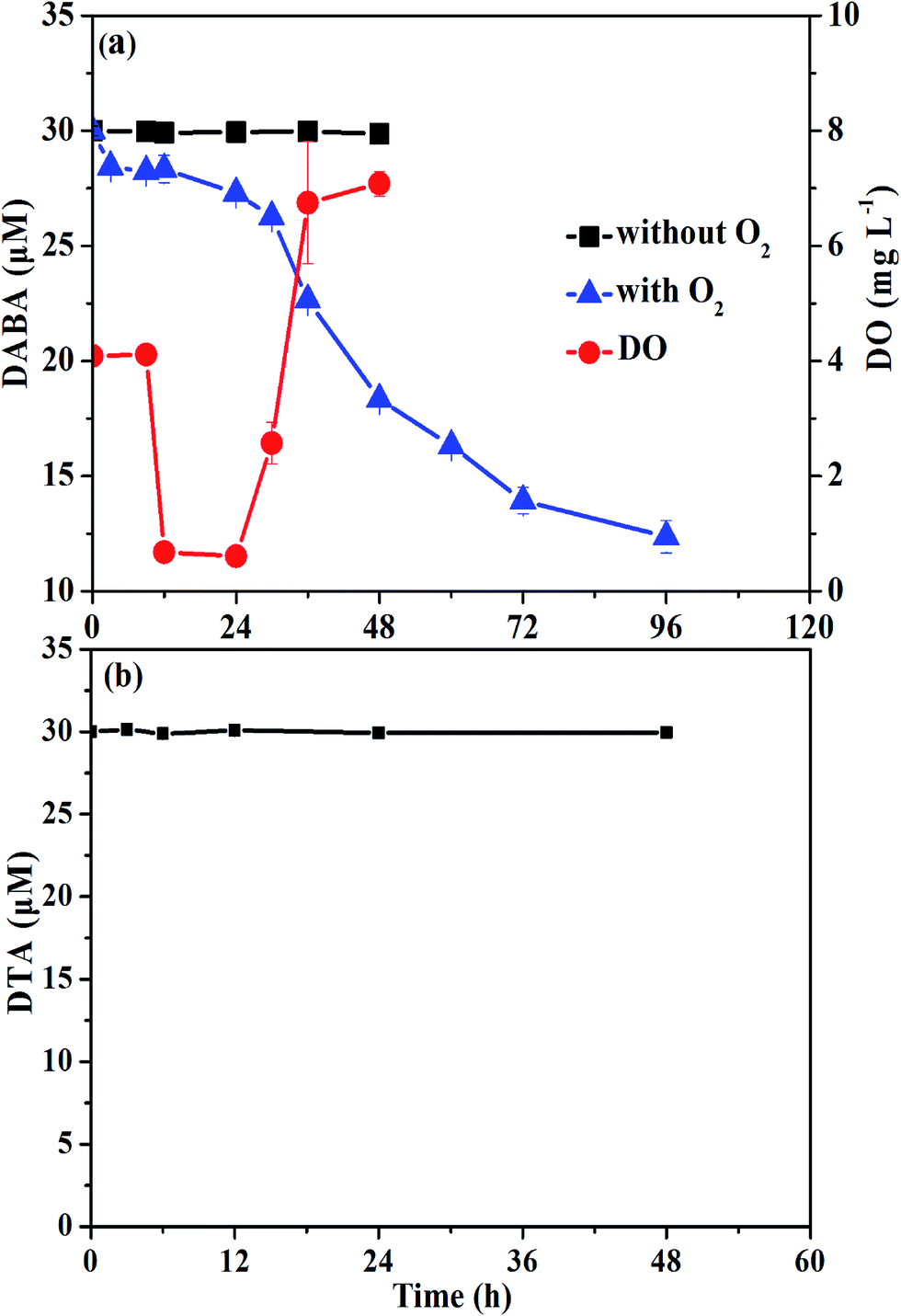 | ||
| Fig. 8 Concentration changes of DTA, DABA and DO during aerobic biological post-treatment. | ||
4. Conclusions
The present study demonstrated that the nZVI could be effectively adopted for the dehalogenation of DTA. The DTA removal efficiency reached to 100% within 24 h in typical experiments with 0.5 g L−1 nZVI particles. DTA dehalogenation in terms of its removal and DABA formation was enhanced with the increase in nZVI dosage but deteriorated when increasing solution pH. nZVI aging time had a negative impact on DTA dehalogenation. The presence of NOM at much low level could improve DTA dehalogenation, while negatively affected DTA removal at high concentrations. Contrary to sulfate, both nitrate and phosphate ions strongly inhibited DTA removal with nZVI. The results also showed that DABA could be degraded by aerobic biological post-treatment, suggesting DTA dehalogenation with nZVI may be a vital procedure for its biodegradability improvement and consequently complete removal.Acknowledgements
The authors wish to thank the Natural Science Foundation of China (51538012 and 51478446), the Recruitment Program of Global Experts, and the Fundamental Research Funds for the Central Universities for financially supporting this study.References
- S. Perez and D. Barcelo, Anal. Bioanal. Chem., 2007, 387, 1235–1246 CrossRef CAS PubMed.
- T. A. Ternes and R. Hirsch, Environ. Sci. Technol., 2000, 34, 2741–2748 CrossRef CAS.
- S. Echeverria, F. Borrull, N. Fontanals and E. Pocurull, Talanta, 2013, 116, 931–936 CrossRef CAS PubMed.
- A. Putschew, S. Schittko and M. Jekel, J. Chromatogr. A, 2001, 930, 127–134 CrossRef CAS PubMed.
- A. Putschew, S. Wischnack and M. Jekel, Sci. Total Environ., 2000, 255, 129–134 CrossRef CAS PubMed.
- F. Sacher, B. Raue and H. H. Brauch, J. Chromatogr. A, 2005, 1085, 117–123 CrossRef CAS PubMed.
- Y. Mu, J. Radjenovic, J. Y. Shen, R. A. Rozendal, K. Rabaey and J. Keller, Environ. Sci. Technol., 2011, 45, 782–788 CrossRef CAS PubMed.
- M. Redeker, A. Wick, B. Meermann and T. A. Ternes, Environ. Sci. Technol., 2014, 48, 10145–10154 CrossRef CAS PubMed.
- C. Zwiener, T. Glauner, J. Sturm, M. Worner and F. H. Frimmel, Anal. Bioanal. Chem., 2009, 395, 1885–1892 CrossRef CAS PubMed.
- F. L. Fu, D. D. Dionysiou and H. Liu, J. Hazard. Mater., 2014, 267, 194–205 CrossRef CAS PubMed.
- M. Stieber, A. Putschew and M. Jekel, Environ. Sci. Technol., 2011, 45, 4944–4950 CrossRef CAS PubMed.
- M. Stieber, A. Putschew and M. Jekel, Water Sci. Technol., 2008, 57, 1969–1975 CrossRef CAS PubMed.
- A. Putschew, U. Mlehe, A. S. Tellez and M. Jekel, Water Sci. Technol., 2007, 56, 159–165 CrossRef CAS PubMed.
- T. Masciangioli and W. X. Zhang, Environ. Sci. Technol., 2003, 37, 102a–108a CrossRef CAS PubMed.
- S. R. C. Rajajayavel and S. Ghoshal, Water Res., 2015, 78, 144–153 CrossRef CAS PubMed.
- M. Danish, X. G. Gu, S. G. Lu and M. Naqvi, Environ. Sci. Pollut. Res., 2016, 23, 13298–13307 CrossRef CAS PubMed.
- J. Dong, C. Y. Wen, D. F. Liu, W. J. Zhang, J. T. Li, H. J. Jiang, C. W. Qin and M. Hong, J. Nanopart. Res., 2015, 17 CAS.
- C. J. Ou, S. Zhang, J. G. Liu, J. Y. Shen, Y. Liu, X. Y. Sun, J. S. Li and L. J. Wang, Phys. Chem. Chem. Phys., 2015, 17, 22072–22078 RSC.
- S. Luo, P. F. Qin, J. H. Shao, L. Peng, Q. R. Zeng and J. D. Gu, Chem. Eng. J., 2013, 223, 1–7 CrossRef CAS.
- M. Khosravi and S. Arabi, Water Sci. Technol., 2016, 74, 343–352 CrossRef PubMed.
- P. P. Huang, Z. F. Ye, W. M. Xie, Q. Chen, J. Li, Z. C. Xu and M. S. Yao, Water Res., 2013, 47, 4050–4058 CrossRef CAS PubMed.
- T. Y. Liu, X. Yang, Z. L. Wang and X. X. Yan, Water Res., 2013, 47, 6691–6700 CrossRef CAS PubMed.
- J. Hur and M. A. Schlautman, Environ. Sci. Technol., 2003, 37, 4020 CrossRef CAS.
- C. B. Wang and W. X. Zhang, Environ. Sci. Technol., 1997, 31, 2154–2156 CrossRef CAS.
- Y. S. Li, J. S. Cao, B. B. Li, W. W. Li, F. Fang, Z. H. Tong and H. Q. Yu, Environ. Sci. Technol. Lett., 2016, 3, 36–40 CrossRef CAS.
- T. S. Yan, B. Y. Zhou, X. S. Xue and J. P. Cheng, J. Org. Chem., 2016, 81, 9006–9011 CrossRef CAS PubMed.
- X. Wang, F. Wei and W. Qi, Water and wastewater monitoring and analysis methods, China Environmental Science Press, Beijing, China, 2002 Search PubMed.
- X. Y. Wang, P. Wang, J. Ma, H. L. Liu and P. Ning, Appl. Surf. Sci., 2015, 345, 57–66 CrossRef CAS.
- Y. Q. Liu and G. V. Lowry, Environ. Sci. Technol., 2006, 40, 6085–6090 CrossRef CAS PubMed.
- M. Erbs, H. C. B. Hansen and C. E. Olsen, Environ. Sci. Technol., 1999, 33, 307–311 CrossRef CAS.
- T. Satapanajaru, P. J. Shea, S. D. Comfort and Y. Roh, Environ. Sci. Technol., 2003, 37, 5219–5227 CrossRef CAS PubMed.
- X. Q. Li, J. S. Cao and W. X. Zhang, Ind. Eng. Chem. Res., 2008, 47, 2131–2139 CrossRef CAS.
- H. Chen, H. J. Luo, Y. C. Lan, T. T. Dong, B. J. Hu and Y. P. Wang, J. Hazard. Mater., 2011, 192, 44–53 CAS.
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045–2053 CrossRef CAS PubMed.
- Y. P. Sun, X. Q. Li, J. S. Cao, W. X. Zhang and H. P. Wang, Adv. Colloid Interface Sci., 2006, 120, 47–56 CrossRef CAS PubMed.
- A. S. Adeleye, A. A. Keller, R. J. Miller and H. S. Lenihan, J. Nanopart. Res., 2013, 15, 1418–1435 CrossRef.
- X. Q. Li, D. W. Elliott and W. X. Zhang, Crit. Rev. Solid State Mater. Sci., 2006, 31, 111–122 CrossRef CAS.
- A. M. Khalil, O. Eljamal, R. Eljamal, Y. Sugihara and N. Matsunaga, Synthesis, 2017, 6, 1 Search PubMed.
- Y. H. Liou, S. L. Lo, C. J. Lin, W. H. Kuan and S. C. Weng, J. Hazard. Mater., 2005, 126, 189–194 CrossRef CAS PubMed.
- C. L. Geiger, N. E. Ruiz, C. A. Clausen, D. R. Reinhart and J. W. Quinn, Water Res., 2002, 36, 1342–1350 CrossRef CAS PubMed.
- A. B. M. Giasuddin, S. R. Kanel and H. Choi, Environ. Sci. Technol., 2007, 41, 2022–2027 CrossRef CAS PubMed.
- D. G. Kim, Y. H. Hwang, H. S. Shin and S. O. Ko, Desalin. Water Treat., 2012, 49, 147–156 CrossRef CAS.
- P. G. Tratnyek, M. M. Scherer, B. L. Deng and S. D. Hu, Water Res., 2001, 35, 4435–4443 CrossRef CAS PubMed.
- J. Niemeyer, Y. Chen and J. M. Bollag, Soil Sci. Soc. Am. J., 1992, 56, 135–140 CrossRef CAS.
- Z. P. Wen, Y. L. Zhang and C. M. Dai, Colloids Surf., A, 2014, 457, 433–440 CrossRef CAS.
- Y. Liu, T. Phenrat and G. V. Lowry, Environ. Sci. Technol., 2007, 41, 7881–7887 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03750c |
| This journal is © The Royal Society of Chemistry 2017 |