
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderlying mechanism for the modulation of apoptosis induced by a new benzoindole derivative on HT-29 colon cancer cells
Fatemeh Hajiaghaalipour *ab,
Elham Bagheric,
Fadhil Lafta Farajde,
Mahmood Ameen Abdullab and
Nazia Abdul Majid*a
*ab,
Elham Bagheric,
Fadhil Lafta Farajde,
Mahmood Ameen Abdullab and
Nazia Abdul Majid*a
aInstitute of Biological Sciences, Faculty of Science, University of Malaya, 50603, Kuala Lumpur, Malaysia. E-mail: sara.h.alipour@um.edu.my; nazia@um.edu.my
bDepartment of Biomedical Sciences, Faculty of Medicine, University of Malaya, 50603, Kuala Lumpur, Malaysia
cDepartment of Pharmacy, Faculty of Medicine, University of Malaya, 50603, Kuala Lumpur, Malaysia
dDepartment of Chemistry, Faculty of Science, University of Malaya, 50603, Kuala Lumpur, Malaysia
eDepartment of Chemistry, Faculty of Science, University of Diyala, Diyala Governorate, Iraq
First published on 3rd August 2017
Abstract
Colorectal cancer is the third most common form of cancer affecting both men and women around the world. The chemical and biological studies of heterocyclic compounds have been an interesting area in pharmaceutical and medicinal chemistry. A new synthetic compound namely 2-(1,1-dimethyl-1H-benzo[e]indol-2-yl)-3-((2-hydroxyphenyl)amino)acrylaldehyde, abbreviated as DBID was screened for the antiproliferation effects against the colorectal cancer cell line, HT-29 and its possible mechanism of action was elucidated. To determine the IC50 value, MTT assay was employed and further verified by the LDH release assay and apoptosis-inducing effect. DBID inhibited the proliferation of HT-29 cells and significantly increased the levels of caspase -8, -9 and -3/7 in the treated cells compared to untreated cells. Apoptosis features in HT-29 cells were detected in treated cells by using the AO/PI staining and flow cytometric analysis of Annexin V. The changes in expression of some apoptotic genes were confirmed by gene quantification using RT-PCR. The current study showed that the DBID compound exhibited chemotherapeutic activity, which was evident by significant increases in the expression and activation of caspase, up-regulation of the expression of specific apoptotic genes and exploitation of the apoptotic signaling pathways to trigger cancer cell death.
Introduction
The American Cancer Society reported colorectal cancer as the third most common form of cancer in both genders around the world. Their report indicates that, for 2017, colorectal cancer would be the third leading cause of cancer deaths in women, and the second leading cause of death in men estimating about 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 260 deaths as a result from the disease. Recently, there has been a drastic increase in the number of colon cancer cases. This surge has highlighted this cancer as a health issue of serious concern, with 135430 cases of colorectal cancer being estimated and expected to be reported in 2017.1
260 deaths as a result from the disease. Recently, there has been a drastic increase in the number of colon cancer cases. This surge has highlighted this cancer as a health issue of serious concern, with 135430 cases of colorectal cancer being estimated and expected to be reported in 2017.1
Age is the one of the risk factors for colorectal cancer as 91% of patients are diagnosed at 50 years of age or older. The other risk factors are personal or family history of colorectal cancer and/or polyps, known to have chronic inflammatory bowel disease, and some inherited genetic manifestations. Modifiable risks factors include alcohol consumption, long-term smoking, obesity, less physical activity and high amounts of red or processed meat in diet. The standard treatment for colon and rectal cancer is surgical removal, followed by adjuvant chemotherapy or radiotherapy. For localized cancers, surgical resection may be curative. However, this therapy is just moderately successful especially for late stage cancers; therefore, new approaches to the treatment of colorectal cancer are required.1–3
The chemistry and biological study of heterocyclic compounds have been an interesting area for a long time in pharmaceutical and medicinal chemistry. Medicinally many indole derivatives are acknowledged to have a wide range of medicinal properties.4–6
In the present study, new Schiff base 2-(1,1-dimethyl-1H-benzo[e]indol-2-yl)-3-((2-hydroxyphenyl)amino)acrylaldehyde (DBID) has been synthesized and the potential chemotherapeutic properties of DBID, as a novel synthetic compound and its possible mechanism of action was investigated on colorectal cancer cell line, HT-29.
Experimental
Synthetic compound
The new compound 2-(1,1-dimethyl-1H-benzo[e]indol-2-yl)-3-((2-hydroxyphenyl)amino)acrylaldehyde (DBID) was synthesized, characterized and confirmed by spectroscopic techniques. These data have already been published by the same authors.7 The compound was provided by the Department of Chemistry, University of Malaya.Cell culture
To evaluate the antiproliferative activity of DBID, a human colonic carcinoma cell line, HT-29 (ATCC® HTB-38™) was purchased from the American Type Culture Collection (ATCC, VA, USA). Normal human colon cell CCD 841 CoN (ATCC® CRL-1790™) was provided from American Type Culture Collection, VA, USA. The HT-29 and CCD 841 CoN cells were cultured in ATCC-formulated McCoy's 5A Medium (ATCC® 30-2007™) and Eagle's Minimum Essential Medium (ATCC® 30-2003™), respectively. The cells were supplemented with 10% FBS (fetal bovine serum), and 1% penicillin–streptomycin (iDNA, South America), incubated at 37 °C in a 5% CO2 incubator.Antiproliferative effects of DBID
The inhibitory effect of DBID was investigated using a human colorectal carcinoma cell, HT-29 while the antiproliferative effect of DBID was evaluated by using the MTT (3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay.8 Normal colon cell line, CCD 841 CoN was used to identify any cytotoxic effect of the compound. The cells were treated with various concentrations of the compound (0–50 μg ml−1) for 48 h. All the experiments were performed in triplicate. In this study 5-fluorouracil (5-FU) was used as a standard drug. The IC50 known as the concentration of the compound that caused 50% growth inhibition was determined based on the following formula;| Inhibition (%) = [(OD untreated − OD treated)/(OD untreated)] × 100 |
Determination of lactate dehydrogenase (LDH) release
The release of lactate dehydrogenase (LDH) into the culture medium was assessed using a CytoTox-ONE™ Homogeneous Membrane Integrity Assay kit (Promega, Madison, WI, USA) according to the manufacturer protocol. In this experiment, the cells were treated with different concentration of DBID for 24 h. The lysis solution (9% weight/volume solution of Triton® X-100 in water) was used to determine the maximum amount of LDH present. The percentage of cytotoxicity was calculated by using the following formula:| Cytotoxicity (%) = 100 × [(sample LDH release − culture medium background)/(maximum LDH release − culture medium background)] |
Reactive oxygen species generation
The intracellular reactive oxygen species (ROS) generation was measured using Cellular Reactive Oxygen Species Detection Assay Kit (Abcam, Cambridge, UK; ab113851) according to the manufacturer protocol. The procedure was based on oxidation of DCFH-DA to form the fluorescent compound, 2′,7′-dichlorofluorescein (DCF) by intracellular ROS and other peroxides. The cells were treated with DBID at the different concentrations for 24 h.Fluorescence microscopic examination
To investigate the morphological characterization of HT-29 cells, propidium iodide and acridine orange double staining was performed where the cells were then observed under a fluorescence microscope according to the method described previously.9,10 The HT-29 cells were treated with two different concentrations of DBID for 24 h. The harvested cells were stained with an equal volume of fluorescent dye (AOPI) and observed under a fluorescence microscope.Flow cytometric Annexin V
The Annexin V assay was carried out using FITC Annexin V Apoptosis Detection Kit supplied from BD Pharmingen™ (ApoAlert Annexin V, Clontech, California, USA). The cultured cells in a 25 cm flask were treated with the IC50 concentration of DBID compound determined by MTT assay for 24 h. The cells were then subjected with the FITC Annexin V staining protocol to measure apoptosis.Caspase activity assays
The caspase -3/7, -8 and -9 activities was evaluated by using commercial kits purchased from Promega Company (Madison, WI, USA) according to the manufacturer's protocol. The cells were treated with different concentrations of DBID for 24 h. Cells without any treatment, cell treated with respected caspase inhibitor and media alone without cells were used as the controls and blank.Gene expression analysis by RT-PCR
HT-29 cells were treated with the IC50 concentration of DBID and incubated for 24 h in 25 cm2 culture flasks. Total RNA was extracted with RNeasy® plus mini kit (Cat. no. 74134, QIAGEN, Germany), and assessed for quality and quantity using absorption measurements (Thermo Scientific Nanodrop 2000 Spectrophotometer, Massachusetts, USA). Reverse transcription into cDNA was performed by using Two-Step qRT-PCR kit, high capacity RNA to cDNA by Applied Biosystems, USA. The relative quantification of the genes was performed using the TaqMan® Gene Expression Master Mix by following the manufacturer's protocol. The expression of two endogenous controls ACTB (Hs99999903_m1) and GAPDH (Hs03929097_g1) were used in this study. Apoptosis-related cysteine peptidase specific primers; caspase 3 (CASP3, Hs00234387_m1), caspase 8 (CASP8, Hs01018151_m1), caspase 9 (CASP9, Hs00609647_m1), apoptosis regulator; BCL2 (BCL2, Hs00608023_m1), BCL2 associated X (BAX, Hs00180269_m1), tumor protein p53 (TP53, Hs01034249_m1) and BH3 interacting domain death agonist (BID Hs00609632_m1) were purchased from TaqMan® (MGB probes, FAM™ dye-labeled). All experimental samples were performed in biological triplicates. The ΔΔCt method was used to determine the relative expression and comparative Ct values analysis via differences between the treated and untreated groups were performed.Statistical analysis
All experiments were carried out in triplicate, and the results were analyzed and presented as means ± standard deviation (SD). Statistical analysis was done by using SPSS V.18.0 statistical software, Student's t-test. P-value less than 0.05 was considered statistically significant.Results and discussion
In vitro antiproliferative effect
The colonic carcinoma cell line HT-29 was selected to determine the antiproliferative activity of the compound. In addition, CCD 841 CoN cells was included to investigate the cytotoxicity of the compound on normal colon cells. As shown in Table 1, DBID exhibited the potent antiproliferative effect against HT-29 cells (IC50 = 11.85 ± 2.7) whereas the IC50 of the compound on normal colon cells (IC50 > 150 μg ml−1) was significantly higher than HT-29 (p < 0.05). The 5-fluorouracil (5-FU) acting as the reference control showed high anti-proliferative activities against tumorigenic HT-29 cells. The findings of this study showed strong inhibitory effect of the DBID compound against the tumorigenic cell line; HT-29 without no cytotoxic effect on normal cells. The MTT assay showed that DBID at concentration of 11.86 μg ml−1 can inhibit 50% of HT-29 proliferation in vitro.| Compound | HT-29 | CCD 841 CoN |
|---|---|---|
| a Data are presented as IC50. IC50 is the effective concentration of the compound which was used to prevent the 50% of the cells' growth (μg ml−1). Results were expressed as means ± SD (n = 3). | ||
| DBID | 11.85 ± 2.7 | 189.73 ± 4.1 |
| 5-Fluorouracil | 3.9 ± 1.26 | 43.8 ± 6.3 |
The leakage of LDH
The cytotoxic effect of DBID compound against HT-29 cells was further verified by the LDH release assay. The measurement of the LDH release is commonly used for testing cytotoxicity based on the integrity of the cell membrane. The loss of integrity of the cell membrane leads to the leakage of LDH from the cytoplasm into the surrounding culture medium in non-viable cells.11 DBID compound induced LDH leakage in HT-29 cells when compared to untreated cells (Fig. 1). The LDH leakage from HT-29 cells treated with DBID compound was significantly different at concentration as low as 10 μg ml−1 compared to the untreated cells (p < 0.05).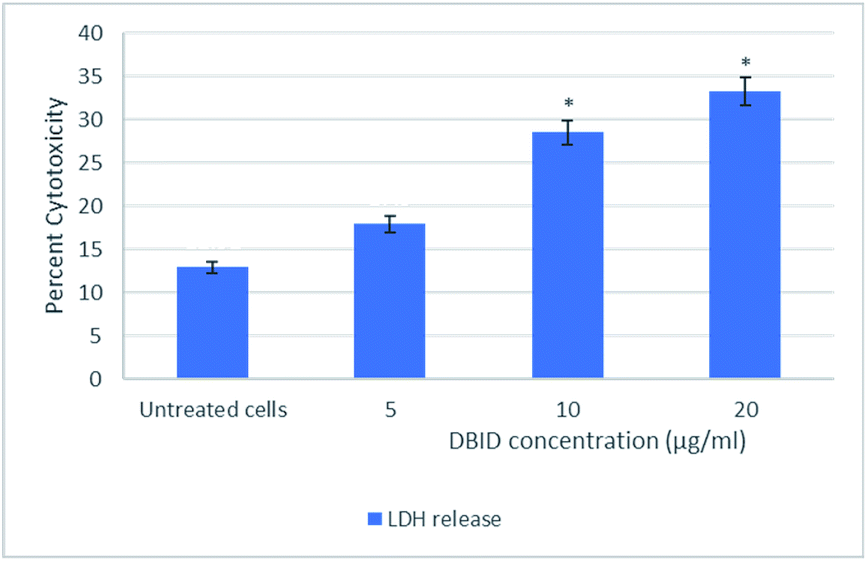 | ||
| Fig. 1 Lactate dehydrogenase (LDH) release in HT-29 cells treated with different concentration of DBID compound for 24 h. Results were expressed at means ± SD (n = 3). *Indicates significant differences compared to the control (untreated cells) verified by the Student's t-test (p < 0.05). | ||
In determining the release of lactate dehydrogenase (LDH), the significant percent of cytotoxicity of DBID against HT-29 cells was shown at concentration 10 μg ml−1, which was compatible with the IC50 value of DBID (11.85 ± 2.7 μg ml−1) determined in the MTT assay. Based on the presented results, DBID compound was toxic to HT-29 cells, which resulted in a loss of membrane integrity, release of LDH into the culture medium, and generated a greater fluorescent signal in comparison with untreated cells. The anticancer effect of heterocyclic compounds has been proposed in many studies.12–16
Intracellular ROS formation
The reactive oxygen species are produced continuously in living cells, as a result of metabolic and other biochemical reactions. Under oxidative stress conditions, excessive ROS leads to various biochemical and physiological impairments and promotes cell death. Fig. 2 illustrates the intracellular reactive oxygen species (ROS) generation in treated cells with different concentration of DBID for 24 h compared with untreated cells. The formation of ROS in HT-29 cells treated with the DBID compound increased in a dose-dependent manner and the strong significant production of ROS in treated cells was generated at concentration of 10 and 20 μg ml−1 upon treatment. At 5 μg ml−1 of DBID concentration treatment, a slight increase of ROS was observed.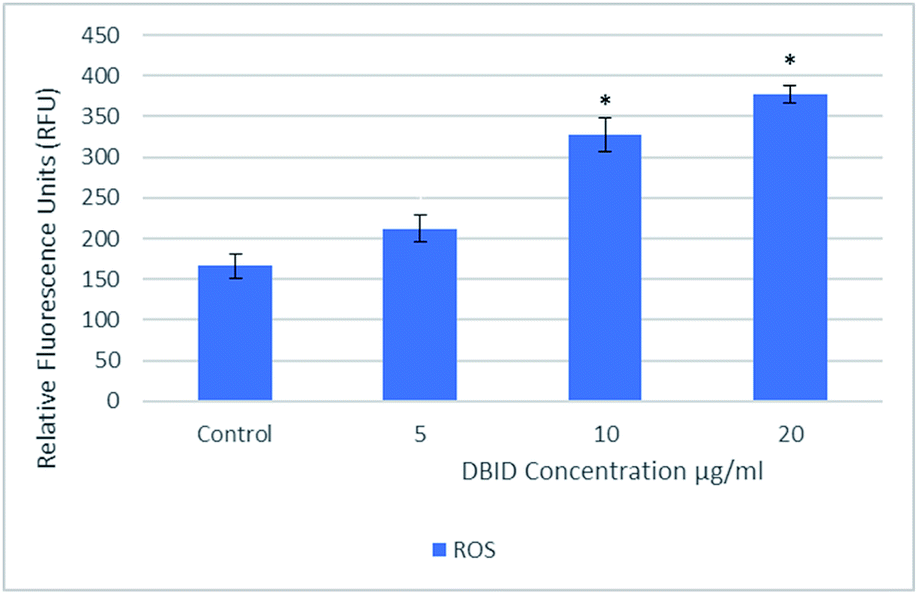 | ||
| Fig. 2 Intracellular ROS generated in HT-29 cells treated with different concentration of DBID compound for 24 h. Results are expressed as mean ± standard deviation. *p < 0.05 versus control (untreated cells) by Student's t-test. | ||
DBID was able to elevate the production of ROS in the treated cells significantly compared with untreated cells. The ROS concentration is vital in cells and tissue. Under oxidative stress conditions, excessive ROS leads to a various biochemical and physiological impairments and promotes cell death. The important role of intracellular ROS in apoptosis induction has been reviewed previously.17 Previous studies have suggested that the reactive oxygen species are responsible as part of the signaling process for activating an important mechanism for eliminating cancer cells such as apoptosis. It has been proposed that the disproportional increase in intracellular ROS leads to release cytochrome c from mitochondria which trigger caspase activation.18–20 There are several anticancer agents depend on this form of cell death for their efficacy.19,21 The present results propose the apoptosis inductions through the production of intracellular ROS formation in cells treated with DBID compound.
Fluorescence microscopic examination
The occurrence of apoptosis was qualitatively investigated. This involved the use of the two dyes of AO/PI and fluorescence microscopy. AO penetrates the plasma membrane of viable and early apoptotic cells where stained nucleated cells would generate a green-coloured fluorescence while PI would only enter the cells with compromised membranes and later produce an orange/red fluorescence intensity in dying, dead, and necrotic nucleated cells stained with PI.22,23As shown in Fig. 3, a normal morphology with green nuclei and also an intact structure was observed in untreated cells while the cells treated with DBID showed bright green nuclei indicating the condensation of nucleic chromatin and interpolated propidium iodide (orange fluorescence) amongst the fragmented DNA and nuclear fragmentation. Apoptosis characteristics e.g. cell shrinkage, membrane blebbing and chromatin condensation were observed in DBID treated cell. Fig. 3 shows remarkable morphological changes of the HT-29 cells treated with DBID in both concentrations (Fig. 3B and C) in apoptotic bodies. The results of this study proved that the activation of the caspase pathway may trigger apoptosis in HT-29 cells that had been treated with DBID.
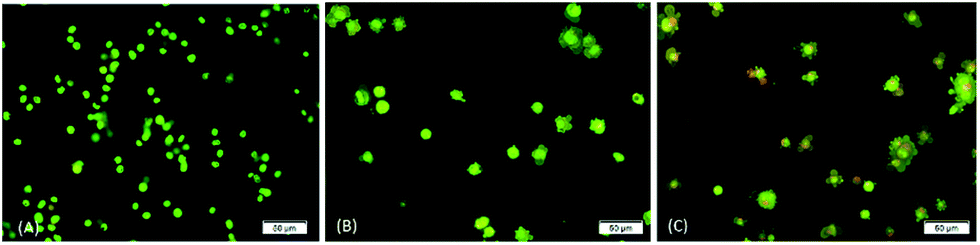 | ||
| Fig. 3 Morphological charges of untreated and DBID-treated HT-29 cells. Fluorescence microscopic examination by using double fluorescent dye (AO/PI) staining method on cells treated with DBID after 24 h incubation. From left to right; (A) untreated HT-29 cells, (B) 10 μg ml−1 DBID-treated, (C) 20 μg ml−1 DBID-treated. | ||
Annexin V assay
Annexin V/propidium iodide (PI) flow cytometry was performed to confirm the presence of apoptosis. Annexin V is used to detect apoptosis by targeting the loss of plasma membrane asymmetry by specific interaction with phosphatidylserine (PS) on the cell surface. The intact cell membrane of viable cells is not permeable to PI and represent AV−/PI− staining. The loss of plasma membrane asymmetry and strong affinity of AV-FITC with PS leads to AV+/PI− staining in the early apoptotic cells, whereas AV+/PI+ represents late apoptotic. Necrotic stage is representing by AV−/PI+ as a result of PI penetration through the membranes and intercalate into nucleic acid due to loss of plasma and nuclear membrane integrity. The flow cytometry of Annexin V results and analysis are presented in Fig. 4(A and B). The cells treated with DBID significantly underwent apoptosis after 24 h post treatment compared with untreated cells (p < 0.05). More than 40% of treated cells were at the early and late stages of apoptosis after 24 h (Fig. 4(B)). In addition, treatment with DBID showed no concurrent increase in necrotic cells. Consistent with AOPI stating results, flow cytometric Annexin V analysis of DBID treated cells clearly demonstrated that antiproliferation and apoptosis in HT-29 treated cells are closely related.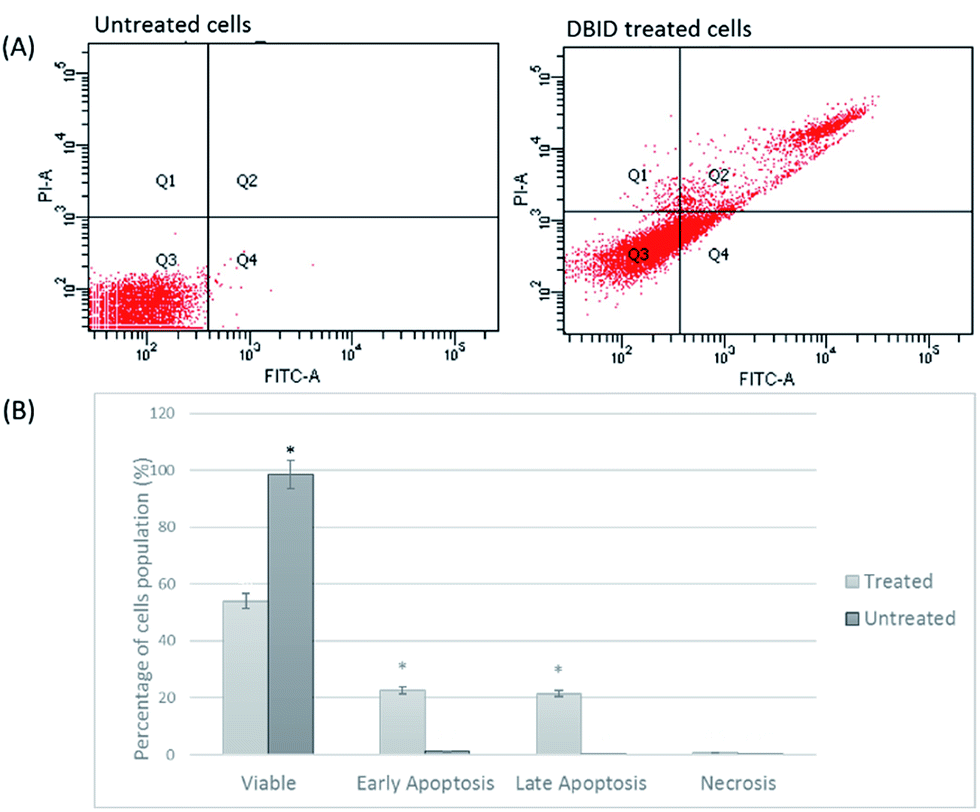 | ||
| Fig. 4 Flow cytometric Annexin V/PI. (A) HT-29 cells (untreated and DBID treated) were stained with Annexin V/PI (B) flow cytometric analysis of the percentage level of the four quadrants representing viable cells (Annexin V−PI−), early apoptosis (Annexin V+PI−), late apoptosis (Annexin V+PI+) or necrotic (Annexin V−PI+) stages in treated and untreated cells. Values shown are percentages of each quadrant *p < 0.05. | ||
Cellular caspase activities (caspase -9, -8 and -3/7)
The activation of caspases -9, -8 and -3/7 in HT-29 cells treated with different concentration of DBID compound were determined. Each of these assays were linear with respect to the number of cells undergoing apoptosis in the experiment. As shown in Fig. 5(A–C), the compound (DBID) significantly increased the activities of caspases -9, -8 and -3/7 in HT-29 cells, compared with the control in a dose-dependent manner (p < 0.05). Fig. 6 represents the caspase activity fold changes over control cells. The caspase-8 activity was significant only at the concentration of 10 and 20 μg ml−1 of the compound (p < 0.05). The activity of caspase -3/7 and -9 in treated cells was steadily increased in a dose-dependent manner and statistically significant compared to the untreated cells (p < 0.05). Apoptosis is typically accompanied by the activation of caspase proteins. Caspases are cysteine–aspartic proteases or cysteine-dependent aspartate-directed proteases, a family of cysteine proteases that play essential roles in apoptosis. Caspase-9 is known as an indicator for the activation of a branch caspase cascade, namely, the intrinsic (mitochondria mediated) apoptotic pathway and caspase-8 is a marker for the extrinsic (receptor) pathway of apoptosis. Caspase-3 is activated in apoptotic cells by both the extrinsic and intrinsic pathways.24 The results of caspase activity suggested that the intrinsic or mitochondrial pathway were mainly mediated by the activation of caspase-9, and also the extrinsic or death receptor pathway mediated by the activation of caspase-8, triggered the apoptosis induction in treated cells. These results revealed that the caspase -9 and -8 as initiator caspases and caspase-3/7 as executioner caspases were involved in the mechanism of apoptosis induced by the compound on HT-29 cells. Consistent with this result, the cytotoxic effect and antiproliferative activity of heterocyclic compounds synthetic compounds, against colon cancer cells by activating caspases and inducing apoptosis have also been shown in a previous study.25–27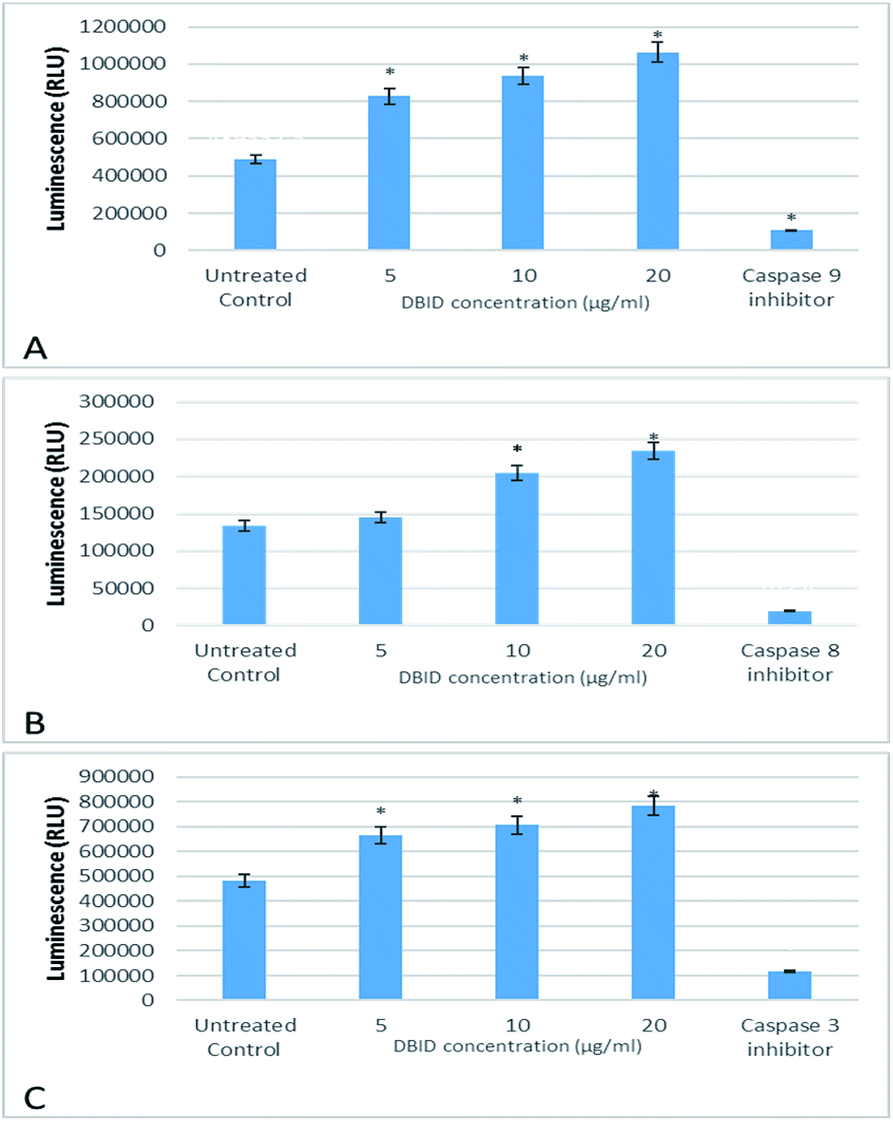 | ||
| Fig. 5 The effect of DBID on caspase activity in HT-29 colon cancer cells. The activity of (A) caspase-9 (B) caspase-8 and (C) caspase-3/7. Data shown are the mean values of the triplicates. *Indicates significant differences from untreated cells (p < 0.05). | ||
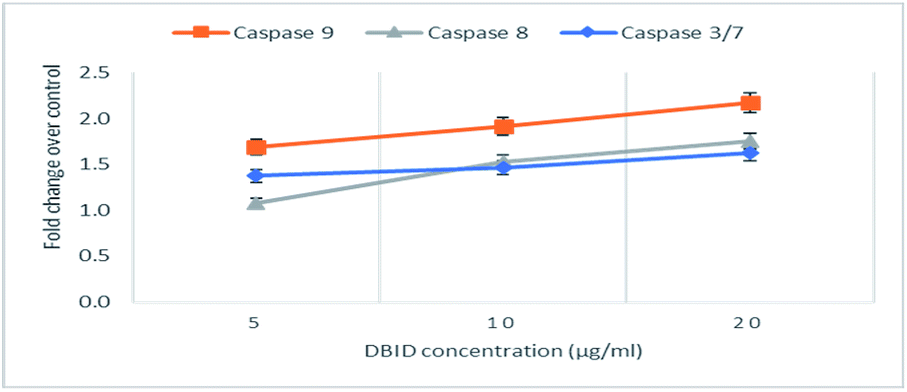 | ||
| Fig. 6 The fold change in caspase activity of HT-29 cells treated with DBID at different concentrations. | ||
Gene expression
The HT-29 cells were treated with the IC50 concentration of the DBID compound to further investigate the possible pathways involved in antiproliferative effect of the compound at gene expression level using real time-PCR technique.The gene expression study of some of these genes involved in apoptosis showed the overexpression of TP53, BID and BAX in HT-29 cells. Furthermore, the genes encode the cysteine–aspartic acid protease (caspase) family e.g. CASP9, CASP8 and CASP3 were also up-regulated in this study. Fig. 7 shows that CASP3, CASP9, CASP8, TP53, BAX and BID were significantly overexpressed in treated HT-29 cells by a mean factor of 2.48, 2.79, 1.67, 3.7, 4.1 and 1.47, respectively. The apoptosis regulator gene, BCL2 was slightly downregulated in DBID treated cells by a mean factor of 0.89.
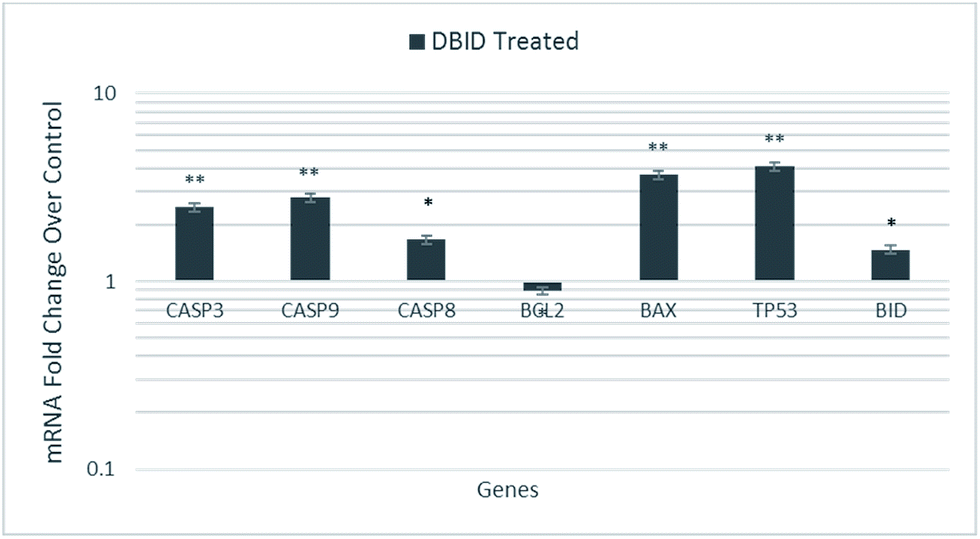 | ||
| Fig. 7 Gene expression analysis of HT-29 cells treated with the IC50 concentration of DBID for 24 h. Results are expressed as mean ± standard deviation of three replicates *p < 0.05, **p < 0.01. | ||
The activation of caspases is known as a hallmark of apoptosis which proposals new therapeutic agents for the treatment of cancer cells.28–31 The p53 tumor suppressor gene (TP53) encodes the tumor protein p53, a transcriptional factor that binds to DNA and activates the expression of downstream genes, which mediates cellular stress responses i.e. cell-cycle arrest, senescence and apoptosis.32 BID, a pro-apoptotic member of the Bcl-2 family, mediates the activation of caspase-9 by caspase-8 through the mitochondrial pathway. The ability of DBID to activate BAX can be inhibited by the anti-apoptotic Bcl-2 proteins resulting in apoptosis inhibition by sequestering DBID, leading to reduced Bax activation.33,34 Moreover, the intrinsic or mitochondrial pathway is mediated by the alteration of the pro-apoptotic (Bax) and anti-apoptotic (Bcl2) ratio. Bcl2L (Bcl2-Like), BclXL (Bcl2 related protein long isoform) and other anti-apoptotic Bcl2 family members reside in the outer mitochondrial membrane and prevent cytochrome c release.35 These results revealed that the treatment with DBID promotes apoptosis by alerting the Bax/Bcl2 ratio by increasing the expression of Bax protein and down-regulating the expression of Bcl-2 as well as upregulating the genes involved in apoptosis. These findings of the current study are consistent with previous studies reported by several researchers.28–31,36,37
Conclusions
The present study demonstrates the antiproliferative activity of a new benzoindole derivative (DBID) against colon cancer cell line, HT-29, without being toxic to the normal colon cell (CCD 841 CoN). The quantitatively and qualitatively investigation of caspase activities by using luminescence spectrophotometry, fluorescence microscopy (AO/PI), flow cytometric Annexin V and gene expression provided in the present study, proposed that the antiproliferative effect of DBID on colon cancer cell line could be due to the occurrence of apoptosis through the generation of intracellular ROS, induction of cellular caspase activities as demonstrated by, initiation of both the intrinsic and extrinsic apoptosis pathways and upregulation of apoptotic genes. In conclusion, DBID which is a novel synthetic compound shows potential and promising chemotherapeutic properties.Acknowledgements
This study was supported by University of Malaya Research Grants, PG073-2015B and RP043A-15HTM.References
- American Cancer Society, Cancer Facts & Figures 2017, American Cancer Society, Atlanta, 2017 Search PubMed.
- B. Levin, D. A. Lieberman, B. McFarland, K. S. Andrews, D. Brooks, J. Bond, C. Dash, F. M. Giardiello, S. Glick, D. Johnson, C. D. Johnson, T. R. Levin, P. J. Pickhardt, D. K. Rex, R. A. Smith, A. Thorson and S. J. Winawer, Gastroenterology, 2008, 134, 1570–1595 CrossRef CAS PubMed.
- A. B. Benson, D. Schrag, M. R. Somerfield, A. M. Cohen, A. T. Figueredo, P. J. Flynn, M. K. Krzyzanowska, J. Maroun, P. McAllister and E. Van Cutsem, J. Clin. Oncol., 2004, 22, 3408–3419 CrossRef PubMed.
- H. Patel, N. Darji, J. Pillai and B. Patel, Int. J. Drug Dev. Res., 2012, 2, 225–230 Search PubMed.
- N. Pahari, D. Saha, V. K. Jain, B. Jain and D. Mridha, Int. J. Pharm. Sci. Res., 2010, 1, 399–408 CAS.
- M. M. Ibrahim, H. M. Ali, M. A. Abdullah and P. Hassandarvish, Molecules, 2012, 17, 12449–12459 CrossRef CAS PubMed.
- F. Hajiaghaalipou, F. Faraj, E. Bagheri, H. Khaledi, H. Ali, N. Majid and M. Abdulla, Curr. Pharm. Des., 2017, 1–10 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- K.-B. Ng, A. Bustamam, M. A. Sukari, S. I. Abdelwahab, S. Mohan, M. J. C. Buckle, B. Kamalidehghan, N. M. Nadzri, T. Anasamy and A. H. A. Hadi, BMC Complementary Altern. Med., 2013, 13, 1 CrossRef PubMed.
- F. Hajiaghaalipour, M. Kanthimathi, J. Sanusi and J. Rajarajeswaran, Food Chem., 2015, 169, 401–410 CrossRef CAS PubMed.
- A. Jarosz, M. Skoda, I. Dudek and D. Szukiewicz, Oxid. Med. Cell. Longevity, 2016, 2016, 1–14 CrossRef PubMed.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem. Lett., 2010, 20, 7205–7211 CrossRef CAS PubMed.
- M. Hajrezaie, M. Paydar, S. Zorofchian Moghadamtousi, P. Hassandarvish, N. S. Gwaram, M. Zahedifard, E. Rouhollahi, H. Karimian, C. Y. Looi and H. M. Ali, Sci. World J., 2014, 2014, 1–12 CrossRef PubMed.
- M. Zahedifard, F. L. Faraj, M. Paydar, C. Y. Looi, M. Hajrezaei, M. Hasanpourghadi, B. Kamalidehghan, N. A. Majid, H. M. Ali and M. A. Abdulla, Sci. Rep., 2015, 5, 11544 CrossRef PubMed.
- W. M. Cholody, T. Kosakowska-Cholody, M. G. Hollingshead, H. K. Hariprakasha and C. J. Michejda, J. Med. Chem., 2005, 48, 4474–4481 CrossRef CAS PubMed.
- J. N. Weinstein, T. G. Myers, P. M. O'Connor, S. H. Friend, A. J. Fornace, K. W. Kohn, T. Fojo, S. E. Bates, L. V. Rubinstein and N. L. Anderson, Science, 1997, 275, 343–349 CrossRef CAS PubMed.
- H. U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 5, 415–418 CrossRef CAS PubMed.
- Y. M. Chung, Y. S. Bae and S. Y. Lee, Free Radical Biol. Med., 2003, 34, 434–442 CrossRef CAS PubMed.
- G.-Y. Liou and P. Storz, Free Radical Res., 2010, 44, 479–496 CrossRef CAS PubMed.
- J.-E. Ricci, R. A. Gottlieb and D. R. Green, J. Cell Biol., 2003, 160, 65–75 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- E. V. Mironova, A. A. Evstratova and S. M. Antonov, J. Neurosci. Methods, 2007, 163, 1–8 CrossRef PubMed.
- L. von Bertalanffy, Protoplasma, 1963, 57, 51–83 CrossRef CAS.
- S. Fulda and K. M. Debatin, Oncogene, 2006, 25, 4798–4811 CrossRef CAS PubMed.
- M. E. l. Juan, U. Wenzel, H. Daniel and J. M. Planas, J. Agric. Food Chem., 2008, 56, 4813–4818 CrossRef CAS PubMed.
- C. Sanmartín, D. Plano, A. K. Sharma and J. A. Palop, Int. J. Mol. Sci., 2012, 13, 9649–9672 CrossRef PubMed.
- P. J. Barnard and S. J. Berners-Price, Coord. Chem. Rev., 2007, 251, 1889–1902 CrossRef CAS.
- P. Beauparlant and G. Shore, Curr. Opin. Drug Discovery Dev., 2003, 6, 179–187 CAS.
- C. Jiang, Z. Wang, H. Ganther and J. Lu, Cancer Res., 2001, 61, 3062–3070 CAS.
- P. Hensley, M. Mishra and N. Kyprianou, Biol. Chem., 2013, 394, 831–843 CrossRef CAS PubMed.
- A. Philchenkov, M. Zavelevich, T. J. Kroczak and M. J. Los, Exp. Oncol., 2004, 26, 82–97 CAS.
- A. Vazquez, E. E. Bond, A. J. Levine and G. L. Bond, Nat. Rev. Drug Discovery, 2008, 7, 979–987 CrossRef CAS PubMed.
- H. Li, H. Zhu, C.-j. Xu and J. Yuan, Cell, 1998, 94, 491–501 CrossRef CAS PubMed.
- S. Cory, D. C. Huang and J. M. Adams, Oncogene, 2003, 22, 8590–8607 CrossRef CAS PubMed.
- A. B. Parrish, C. D. Freel and S. Kornbluth, Cold Spring Harbor Perspect. Biol., 2013, 5, 1–24 Search PubMed.
- E. Asselin, G. B. Mills and B. K. Tsang, Cancer Res., 2001, 61, 1862–1868 CAS.
- M. Olsson and B. Zhivotovsky, Cell Death Differ., 2011, 18, 1441–1449 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |