DOI:
10.1039/C7RA03885B
(Paper)
RSC Adv., 2017,
7, 32327-32335
Y-doped V2O5 with enhanced lithium storage performance
Received
5th April 2017
, Accepted 20th June 2017
First published on 23rd June 2017
Abstract
Y-doped V2O5 was prepared by the sol–gel method combined with the freeze drying technique followed by annealing in air. The microstructure of the Y-doped V2O5 was analyzed by XRD, SEM, XPS, and first-principles calculations. The electrochemical performance of the Y-doped V2O5 was characterized by CV, EIS, and charge–discharge tests. The results demonstrated that Y doping had great influence on the morphology and crystalline orientation of the prepared V2O5 sample. In particular, Y formed covalent bonds with the adjacent O atoms within the V2O5 layers and decreased the bond orders of the neighboring V–O bonds. Compared with undoped V2O5, Y-doped V2O5 showed much improved rate capability, higher lithium ion diffusion coefficient, lower electrochemical reaction resistance, and greatly enhanced cycling stability. Y-doped V2O5 could be a promising cathode material for lithium ion batteries.
1. Introduction
Lithium-ion batteries (LIBs) have been widely used as power sources for portable electronic devices, and are being developed for high-power applications, such as electric vehicles and hybrid electric vehicles.1,2 In this context, further improvements in the energy and power densities, safety, and lifetime of LIBs are urgently required. The development of new and improved electrode materials is very crucial for the production of high performance LIBs.3 There has been much progress in the exploration of different anode materials for LIBs.4,5 In contrast, the capacity of the cathode is usually far below that of the anode, which limits the development of high power and energy density LIBs.6,7 Among the potential cathode materials for the next generation LIBs, orthorhombic vanadium pentoxide (V2O5) has been attracting great attention due to its very high theoretical capacity (294 mA h g−1 when storing two Li+), good safety, low cost, and abundant sources.8–11 However, drawbacks such as poor structural stability, low electronic and ionic conductivity, and slow electrochemical kinetics drastically reduce its cycling stability and rate capability. To overcome these drawbacks, numerous efforts have been carried out. Some groups prepared V2O5 with various nanostructures, such as nanorods,12,13 nanoribbons,14,15 nanowires,16–18 nanosheets,19–22 porous/hollow spheres,23–25 and even hierarchical/heterogonous nanostructures.26,27 These nanostructured materials showed enhanced electrochemical kinetics, specific capacity, and rate performance than the pristine V2O5 due to their small size and large surface area, which could greatly increase the contact area between active materials and electrolyte, shorten the diffusion pathways of Li+, and relax the mechanical stress associated Li+ intercalation/deintercalation.28–30 However, it should be noted that these nanostructured V2O5 still suffered from poor cycling stability because of the intrinsic poor electronic conductivity and/or crystal structural fracture during Li+ intercalation/deintercalation process. In order to improve the capacity retention of V2O5, some guest cations, such as Cu2+,31 Mn2+,32 Fe3+,33 Cr3+,34 Al3+,35 and Sn4+,36,37 has been introduced into the material matrix. It has been demonstrated the dopant cations increase the structural stability of V2O5, resulting in improved electrochemical performance. Rare earth elements, which have been regarded as an ‘industrial vitamin’ are widely applied in modern industries. The chemistry of rare earth differs from transition metals because of the nature of the 4f orbitals, which are ‘buried’ inside the atom and are shielded from the atom's environment by the 4d and 5p electrons.38 These unique properties can be exploited to accomplish new types of applications that are not possible with transition and main group metals.39–41 However, there are few reports about the influence of rare earth element doping on the electrochemical performance of V2O5. In particular, the action mechanism of dopant cations has not been well understood till now. Herein, we prepared Y-doped V2O5 material by a facile sol–gel method combined with freeze drying technique followed with annealing in air. The effect of Y doping on the microstructure of V2O5 was analyzed by physical characterizations combined with first-principles calculations, and the electrochemical properties of the Y-doped V2O5 were evaluated as potential cathode materials for LIBs.
2. Materials and methods
2.1 Synthesis of Y-doped V2O5
All the chemical reagents were of analytical purity and used without further purification. The synthesis of Y-doped V2O5 follows three main processes. Firstly, V2O5 gel was prepared using the method derived from the one initially developed by Fontenot et al.42 In brief, 0.25 g V2O5 powder was added into 3.85 mL de-ionized (DI) water and 1.15 mL H2O2 (30 wt% in H2O) to form a yellow suspension. The above suspension was stirred for 15 min and sonicated for 15 min respectively while kept in water bath at room temperature for reactions to form a transparent red solution. Then 20 mL DI water was added in the red solution and sonicated for about 1 h to form a brownish red V2O5 gel. Secondly, Y(NO3)3·6H2O was added into the V2O5 gel, dispersed and diluted by adding DI water to form a homogenous solution with CV2O5 = 0.03 M and the n(Y)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(V) of 3%. This solution was pre-frozen in a refrigerator for 1 day and then the solvent (water) in the frozen sample was removed using a freeze dryer (FD-1A-50, Boyikang Corp., Beijing, China) under a vacuum at −50 °C for 2 days to get the Y-doped V2O5 precursor. Thirdly, the Y-doped V2O5 precursor was annealed in air at 400 °C for 1 h to form Y-doped V2O5. For comparison, pure V2O5 was also prepared under the same condition described above but without adding Y(NO3)3·6H2O.
:n(V) of 3%. This solution was pre-frozen in a refrigerator for 1 day and then the solvent (water) in the frozen sample was removed using a freeze dryer (FD-1A-50, Boyikang Corp., Beijing, China) under a vacuum at −50 °C for 2 days to get the Y-doped V2O5 precursor. Thirdly, the Y-doped V2O5 precursor was annealed in air at 400 °C for 1 h to form Y-doped V2O5. For comparison, pure V2O5 was also prepared under the same condition described above but without adding Y(NO3)3·6H2O.
2.2 Computational method
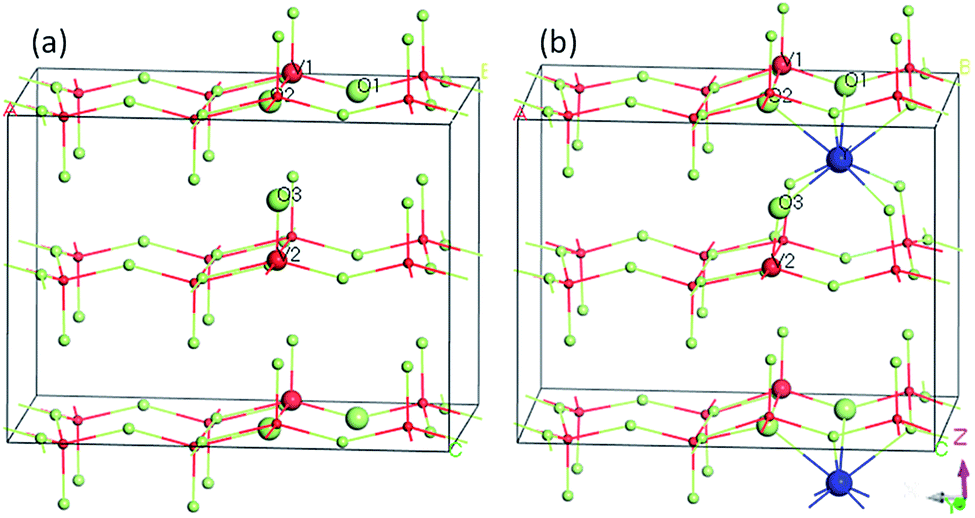
The geometric and electronic structures of the samples were calculated using plane-wave based density functional theory with a CASTEP program package.43 The super cell (V16O40) was used to model pure V2O5 (Fig. 1a) and Y-doped V2O5 (V16O40Y) was constructed by inserting one Y atom in between two adjacent V2O5 layers (Fig. 1b). The two models are shown in Fig. 1. The exchange–correlation function is PBEsol of the generalized gradient approximation (GGA).44 The interaction between valence electrons and the ionic core was described by the ultrasoft pseudopotential.45 A plane-wave cutoff energy of 400 eV and a k-point mesh of 1 × 2 × 2 were used for the geometry optimizations. The threshold for self-consistent field iterations is 5 × 10−7 eV per atom. The convergence tolerance parameters of optimized calculation were the energy of 5 × 10−6 eV per atom, the maximum force of 0.01 eV Å, the maximum inner stress of 0.02 GPa and the maximum displacement of 5 × 10−5 nm.
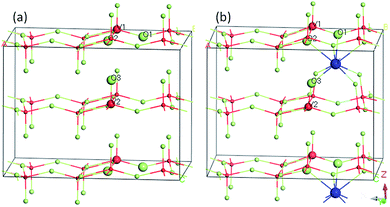 |
| | Fig. 1 Computational models of (a) pure V2O5 supercell (V16O40) and (b) Y-doped V2O5 (V16O40Y). | |
2.3 Material characterization
The crystal structure of the prepared samples was determined on X'Pert3 diffractometer (PANalytical, Netherlands) with a Cu-Kα radiation source (λ = 1.54056 Å). The morphology of the prepared samples was characterized on a field-emission scanning electron microscopy (FE-SEM) (Hitachi, SU-5000). The content of Y and the valent state of V in the prepared samples were analyzed by XPS spectrometer (ESCALAB 250Xi) using monochromic AlKα excitation. The pressure in the analytical chamber during spectral acquisition was 10 × 10−10 mbar. To evaluate the exact amount of Y actually present in the synthesized material, inductively coupled plasma atomic emission spectroscopy (ICP-AES, Perkin-Elmer Optima 2000 DV) was also carried out in addition to the XPS analysis.
2.4 Electrochemical measurements
The electrochemical performances of the samples were tested using CR 2016 coin-type cell with metallic lithium as the anode and polypropylene (PP) film as the separator. The cathodes were fabricated by mixing Y-doped V2O5, super P carbon black, and poly(vinyldifluoride) (PVDF) with a weight ratio of 70:20:10 in n-methyl-2-pyrrolidone (NMP) solvent. The resulting slurry was then uniformly spread on an aluminium foil current collector. The cathodes were dried at 80 °C for 12 h in an oven and then punched into small disks with a diameter of 15 mm. The thickness of the electrode was about 15 μm and the mass loading of the active material was about 1.0 mg cm−2. The electrolyte solution was made of 1 M LiPF6 in EC/DMC/DEC (1:1:1 by weight). The cells were galvanostatically charged and discharged between 2.0 V and 4.0 V (vs. Li/Li+) using LAND-CT2001A battery tester at room temperature. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were carried out on an electrochemical workstation (CHI 760). The CV test was performed between 2.0 and 4.0 V at scan rates ranging from 0.1 to 0.5 mV s−1. The EIS was measured in the frequency range from 0.01 Hz to 100 kHz at the open circuit voltage (OCV) after given discharge/charge cycles with 5 mV voltage amplitude.
3. Results and discussion
3.1 Structure characterizations
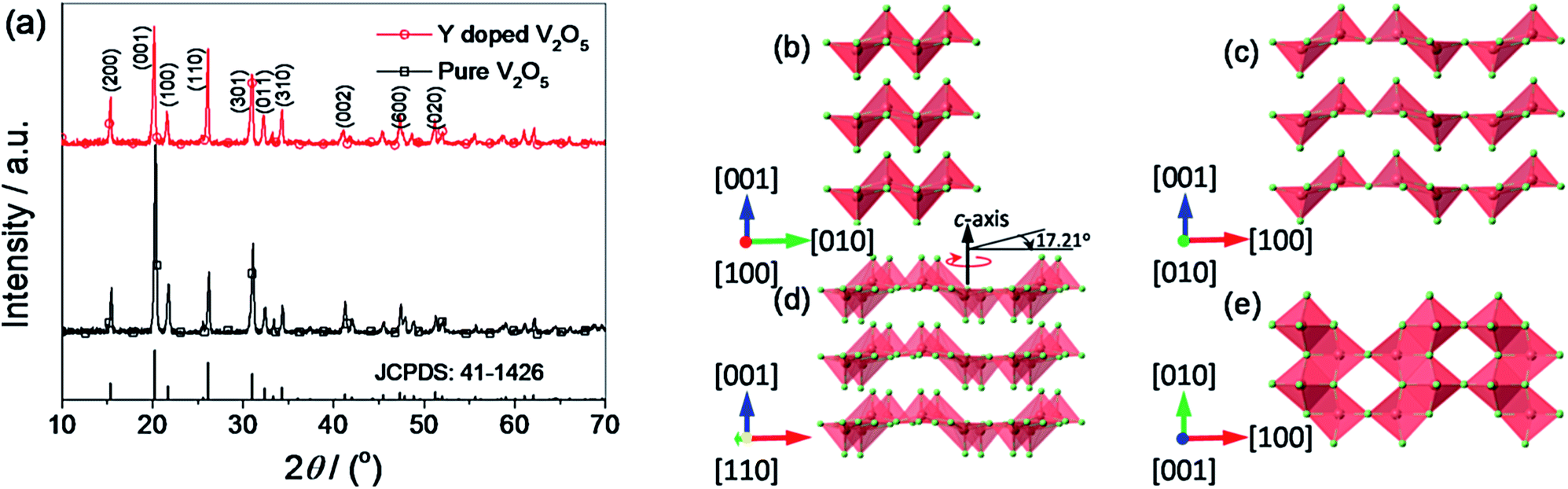
Fig. 2 presents the XRD patterns of the pure V2O5 and Y-doped V2O5 samples. Both pure V2O5 and Y-doped V2O5 samples show a single orthorhombic V2O5 phase (JCPDS card no. 41-1426) without detectable secondary phase. The lattice constants of the samples were calculated based on the Pmmn space group, which were a = 11.493 Å, b = 3.562 Å, c = 4.369 Å for pure V2O5 and a = 11.515 Å, b = 3.566 Å, c = 4.373 Å for Y-doped V2O5. The results suggest that the Y doping causes slight lattice expansions in V2O5. From this changes of lattice constants, it can be deduced that the doped Y is likely to locate between the VO5 slabs and forming the [YO6] octahedral with oxygen atoms in the V2O5 structure.46 By careful observation, it can be found that there is obvious difference of the relative intensities of (001) and (110) diffraction peaks for the two samples. For Y-doped V2O5, the intensity ratio between (110) and (001) diffractions is 0.81, which is significantly larger than that (0.31) of pure V2O5. This indicates that the Y-doped V2O5 exposed more facets than the pure V2O5. As illustrated in Fig. 2c and d, the (010) crystal plane that has the two-dimensional diffusion path is presented when the (110) crystal plane is turned 17.21° along the c axis. Clearly, this unique crystal orientation of the Y-doped V2O5 can facilitate lithium ion intercalation and deintercalation.
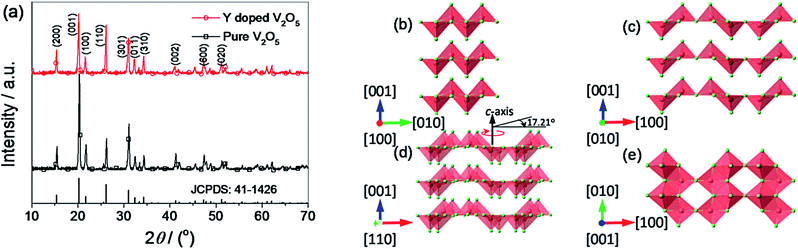 |
| | Fig. 2 (a) XRD patterns of the pure V2O5 and Y-doped V2O5. The vertical lines on the x-axis correspond to the standard XRD reflections of orthorhombic V2O5. (b–e) The refined structural model of the orthorhombic V2O5 viewed along the [100] (b), [010] (c), [110] (d), and [001] (e) directions. The V atoms and O atoms are colored by light red and light green, respectively. | |
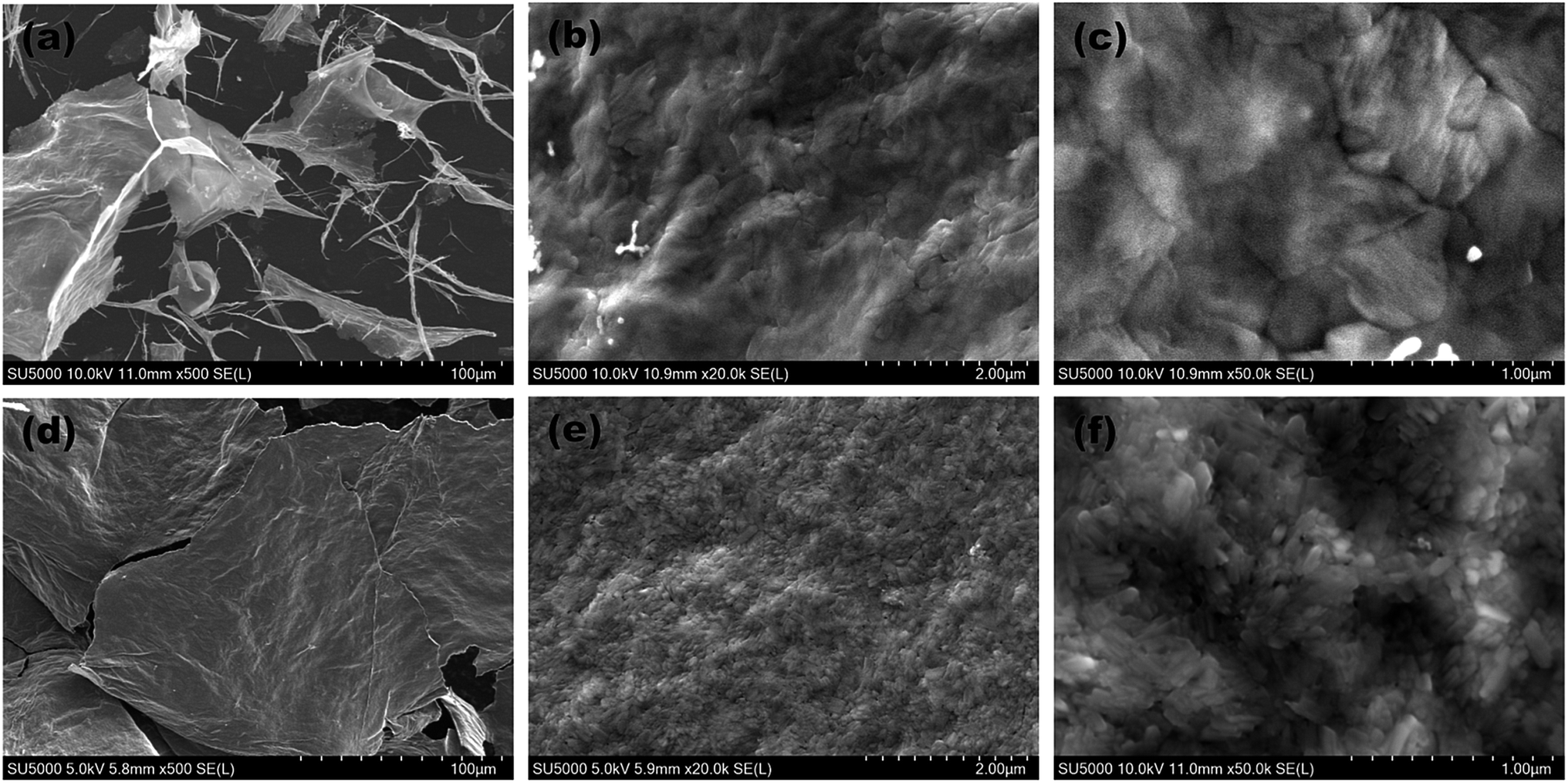
Fig. 3 gives the SEM images of the pure V2O5 and Y-doped V2O5 samples. There is significant difference between the morphologies of the two samples. The pure V2O5 shows mixed sheet-like and fibrous morphologies. The lateral size of the sheets in pure V2O5 ranges from several to several ten microns. Moreover, these sheets have a rather smooth surface. The diameter of the fibers in pure V2O5 is a few tens to a few hundreds of nanometers. In contrast, the Y-doped V2O5 presents only sheet-like morphology and the lateral size (few hundreds of microns) of these V2O5 sheets are much larger than that of the pure V2O5. In addition, the V2O5 sheets consist of interconnected nanoparticles and show a rather coarse and porous surface. This indicates that Y dopant could greatly modify the morphology of V2O5. For the Y-doped V2O5, this nano-sized building blocks (nanoparticles) and porous 2D sheet-like structure are anticipated to offer fast transport of Li-ions and electrons, large material-electrolyte contact area, and easy penetration and diffusion of electrolyte, which are crucial for the improvement of rate performance of electrode materials. Moreover, the pores could also effectively relax the mechanical strain generated upon the lithium ion intercalation/deintercalation cycles.
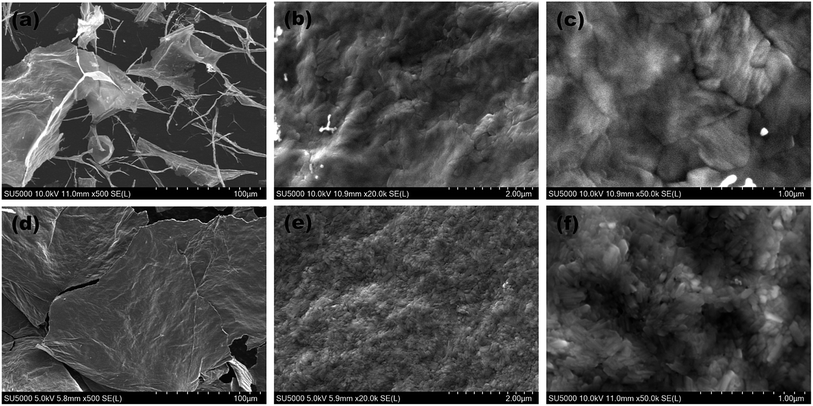 |
| | Fig. 3 SEM images of the (a–c) pure V2O5 and (d–f) Y-doped V2O5. | |
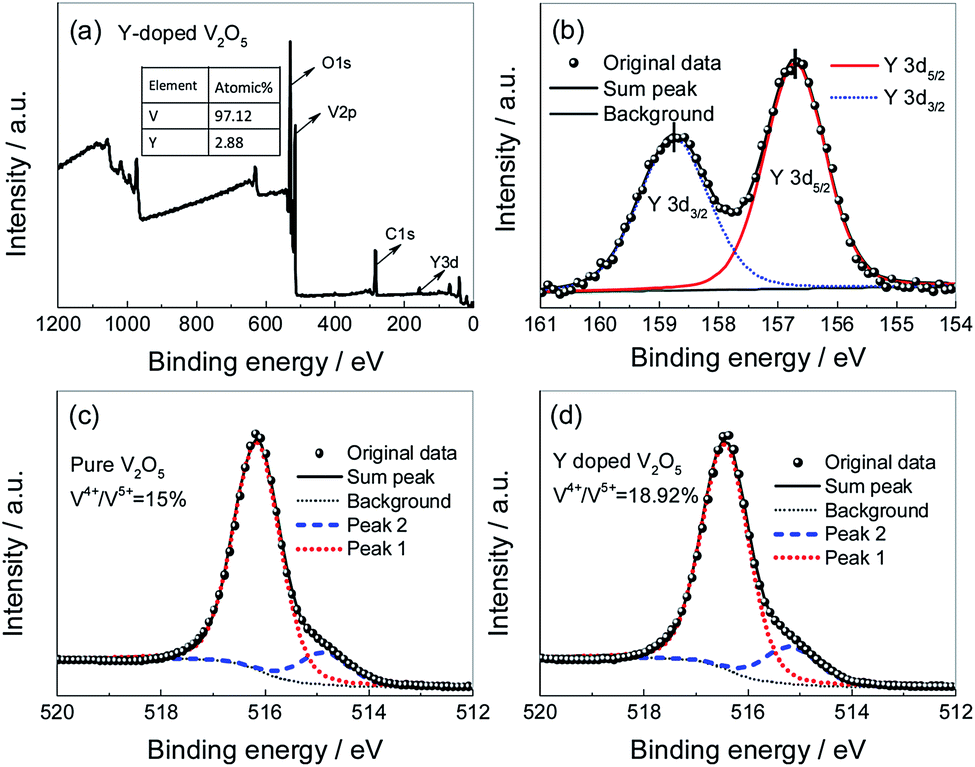
To investigate the oxidation state of vanadium, XPS measurements were carried out on the pure V2O5 and Y-doped V2O5. Fig. 4a shows the XPS survey spectrum of Y-doped V2O5. The line belongs to Y is observed in the spectra and the calculated atomic ratio of n(Y):n(V) is of 2.96%. Furthermore, ICP-AES measurement revealed that the n(Y):n(V) in the Y-doped V2O5 sample is 2.99%. The results from XPS and ICP-AES measurements were consistent and almost equal to the theoretical value of 3%, suggesting that Y was successfully doped in V2O5. Fig. 4b depicts the XPS spectra take from the Y 3d regions. Two intense peaks at 156.7 and 158.8 eV correspond to the binding energy of Y 3d5/2 and Y 3d3/2, respectively. The energy difference between Y 3d5/2 and Y 3d3/2 is 2.1 eV, which is identical to the standard energy difference value for Y2O3,47 indicating that the oxidation state of Y ions in Y-doped V2O5 is mainly trivalent. Fig. 4c and d present the V 2p3/2 core peak spectra of the pure V2O5 and Y-doped V2O5, respectively. Both of them composes of two components located at 516.4 eV and 515.2 eV, which can be associated with two formal oxidation degree, V5+ and V4+, respectively. The calculated molar ratios of V4+/(V4+ + V5+) in the pure V2O5 and Y-doped V2O5 are 15% and 19%, respectively. These increased low valence state V4+ in the Y-doped V2O5 are necessary to balance the net charge in the crystal structure of V2O5 matrix due to the accommodation of Y3+ ions. The low valence state V4+ could enhance the conductivity of V2O5 and accordingly facilitate the Li+ intercalation/deintercalation process in V2O5.48
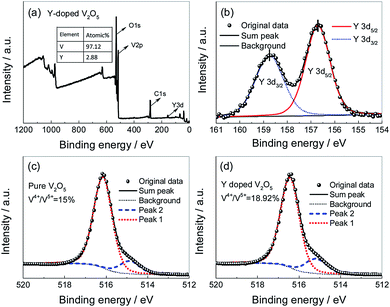 |
| | Fig. 4 (a) XPS survey spectrum of the Y-doped V2O5; (b) high-resolution spectra for Y 3d performed on the Y-doped V2O5; (c) high-resolution spectra for V 2p3/2 performed on the pure V2O5; (d) high-resolution spectra for V 2p3/2 performed on the Y-doped V2O5. | |
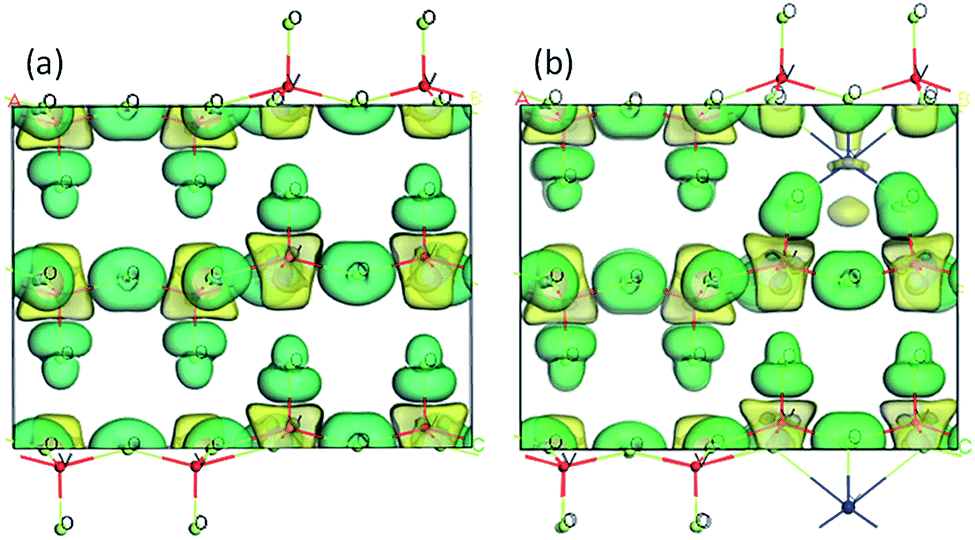
To further understand the effect of Y doping on the geometric and electronic structures of V2O5, first-principles calculations were performed on both the pure V2O5 and Y-doped V2O5. Table 1 presents the selected bond orders and bond lengths of the two model systems. For pure V2O5, the calculated V–O bond lengths are in very good agreement with the available experimental values.49 After Y doping, the length of these V–O bonds (V1–O1, V1–O2, V1–O3) adjacent to Y atom was elongated obviously and their corresponding bond orders decrease, suggesting the covalent character of these V–O bonds were weakened.50 Moreover, the Y dopant shows noticeable interaction with the adjacent O atoms in the VO5 slabs. The bond orders of the Y–O1 and Y–O2 are 0.20 and 0.18, respectively; the bond order of the Y–O3 is up to 0.33, implying that these Y–O bonds have a covalent character. Fig. 5 presents the isosurface of the electron density difference of the pure V2O5 and Y-doped V2O5. In this plot a loss of electrons is indicated in yellow, while electron enrichment is indicated in green. As shown in Fig. 5, the bonding O atoms are surrounded by enriched electrons, while the corresponding V atoms are surrounded by fewer electrons. Notably, there is an obvious redistribution of electrons near the doped Y atoms. The Y atom loses electrons and the adjacent O atoms gain electrons. The interaction between Y atom and O atoms is also supported by the distortion of the V–O bond near Y atoms, where the four apical vanadyl oxygen atoms (O3 as labeled in Fig. 1) move closer to the Y atoms.
Table 1 The bond lengths and corresponding bond orders of selected V–O and Y–O bonds for the pure V2O5 and Y-doped V2O5 calculated by first-principles calculations
| System |
Selected bond |
Bond order |
Length/Å |
| Experimental values from ref. 49. |
| Pure V2O5 |
V1–O1 |
0.46 |
1.776 |
| 1.780a |
| V1–O2 |
0.34 |
1.871 |
| 1.881a |
| V2–O3 |
0.81 |
1.588 |
| 1.581a |
| Y-doped V2O5 |
V1–O1 |
0.39 |
1.834 |
| V1–O2 |
0.30 |
1.936 |
| V2–O3 |
0.70 |
1.641 |
| Y–O1 |
0.20 |
2.436 |
| Y–O2 |
0.18 |
2.520 |
| Y–O3 |
0.33 |
2.420 |
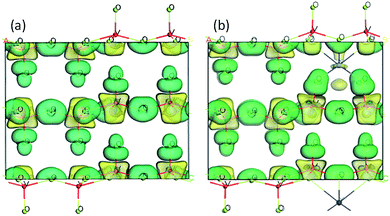 |
| | Fig. 5 Electronic density difference isosurfaces for the (a) pure V2O5 and (b) Y-doped V2O5. The green region shows the electron accumulation, while the yellow region shows the electron loss. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.) | |
It is known that the LixV2O5 will undergo various phase transitions (α, ε, δ, and γ) depending on the degree of lithium ion intercalation.51 In particular, when the lithium intercalation increases up to the range of 1 < x < 2, a structural modification to γ-phase LixV2O5 occurs, in which the layer puckering becomes more pronounced than in δ-phase LixV2O5 and the VO5 pyramids alternate up and down individually, easily resulting collapse of the layer structure and poor cycling stability of V2O5. Based on the above theoretical calculation results, it can be deduced that the Y dopant could have two favorable impacts for improving the cycling stability of V2O5. First, the chemical interaction between Y atom and the adjacent O atoms in the VO5 slabs could stabilize the layer structure of V2O5 phase during electrochemical interaction of Li+, allowing better structure stability during electrochemical cycling. Second, since the V–O bonds adjacent to Y atom were weakened, these local structural defects caused by Y dopant may serve as possible nucleation centers for phase transformation during Li+ intercalation/deintercalation process, which could also enhance the cycling performance of V2O5.
3.2 Electrochemical studies
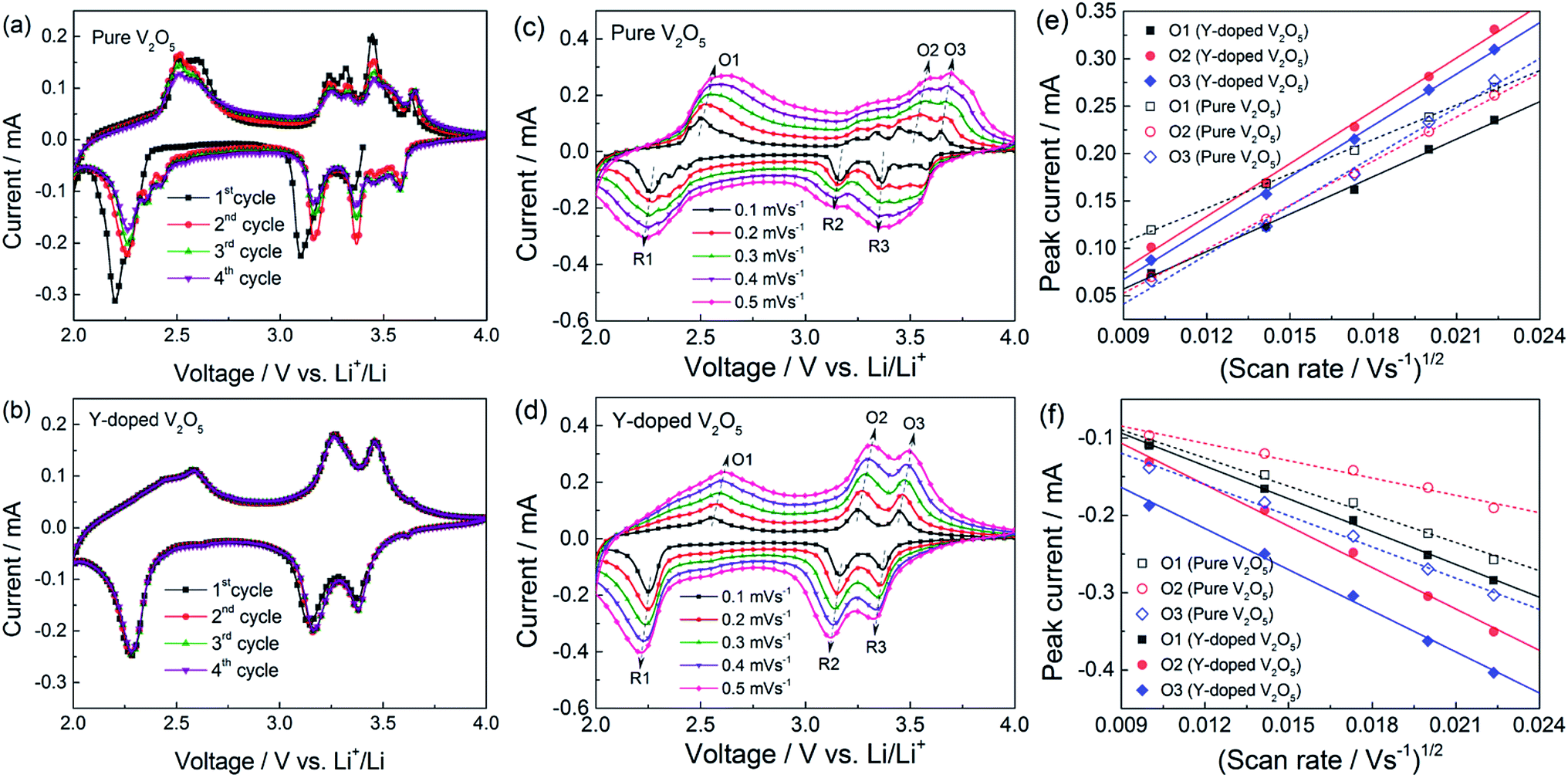
Fig. 6a and b show the cyclic voltammetry (CV) profiles of the first four cycles at a scan rate of 0.1 mV s−1 for the pure V2O5 and Y-doped V2O5 electrodes, respectively. For the pure V2O5, three reduction peaks at 3.35 V, 3.10 V, and 2.20 V are observed, which correspond to the α/ε, ε/δ, and δ/γ phase transitions, respectively.21,52 However, after this first intercalation process, the electrochemical behavior of the pure V2O5 is not completely reversible as indicated by the emergence of new redox systems. For example, two additional weak reduction peaks (at 2.35 V and 2.42 V) appear in the low potential region and a new redox couples (3.58/3.64 V) appear in the high potential region. This suggests that the pure V2O5 undergoes a permanent structural change during the first cycle as reported previously.21,34,53–56 In contrast, the Y-doped V2O5 exhibited three well reversible cathodic/anodic redox couples (3.38/3.46 V, 3.16/3/27 V, and 2.28/2.59 V) in the whole scanning process as shown in Fig. 6b, implying that the irreversible phase transitions of V2O5 are greatly depressed by Y doping. Similar results have also been observed for the Al doped V2O5 (ref. 57) and Cr doped V2O5.34,55,58 Due to the less number of phase transition during the redox process, this Y-doped V2O5 is expected to show better cycling performance than the pure V2O5. Moreover, the CV profiles of Y-doped V2O5 show good repeatability with cycling, implying that the material has good electrochemical and structural reversibility. To understand the kinetics of Li+ diffusion in the pure V2O5 and Y-doped V2O5 electrodes, CV measurements were performed at various scan rates ranging from 0.1 to 0.5 mV s−1 and the results are presented in Fig. 6c and d. The oxidation peaks are labeled as O1 to O3, while the reduction peaks as R1 to R3. The good linear relationship (Fig. 6e and f, all the correlation coefficients of linear fit are higher than 0.99) between the peak current (ip) and the square root of the scan rate (v1/2) indicates a diffusion-controlled process. In this case, the apparent Li+ diffusion coefficient (DLi) can be evaluated base on the Randles–Sevcik equation:33,36| | |
ip = (2.06 × 105)n3/2ADLi1/2CLiv1/2
| (1) |
where ip is the peak current (A), n is number of electron involved in the redox process, A is the electrode area (cm2), CLi is the concentration of Li+ in the cathode (mol cm−3), and v is the scan rate (V s−1). Table 2 summarized the calculated DLi values for the pure V2O5 and Y-doped V2O5 electrodes. Obviously, all the DLi values of Y-doped V2O5 are larger than those of pure V2O5. Similar results were also observed in Sn-doped V2O5 and Cu-doped V2O5.31,36 The larger Li+ diffusion coefficient of the Y-doped V2O5 can be attributed to the unique (110) crystal orientation, which could provide two-dimensional diffusion paths for Li+ intercalation as illustrated in Fig. 2d.
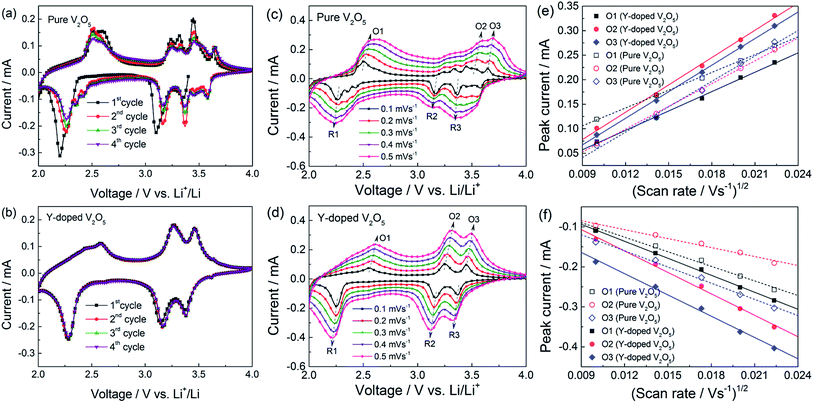 |
| | Fig. 6 (a, b) Cyclic voltammetry (CV) profiles of the pure V2O5 and Y-doped V2O5 electrodes as a scan rate of 0.1 mV s−1. (c, d) CV profiles of the pure V2O5 and Y-doped V2O5 electrodes at various scan rates. (e, f) Relationship of the peak current (ip) and the square root of scan rate (v1/2) for the pure V2O5 and Y-doped V2O5 electrodes. Symbols and lines represent the experimental data and fitted linear lines, respectively. | |
Table 2 Comparison of the Li+ diffusion coefficients (DLi) in the pure V2O5 and Y-doped V2O5 electrodes
| System |
Li+ diffusion coefficients, DLi × 10−12/cm2 s−1 |
| Peak O1 |
Peak O2 |
Peak O3 |
Peak R1 |
Peak R2 |
Peak R3 |
| Y-doped V2O5 |
2.227 |
4.430 |
4.190 |
2.567 |
4.086 |
4.025 |
| Pure V2O5 |
1.188 |
3.082 |
3.827 |
1.889 |
0.713 |
2.326 |
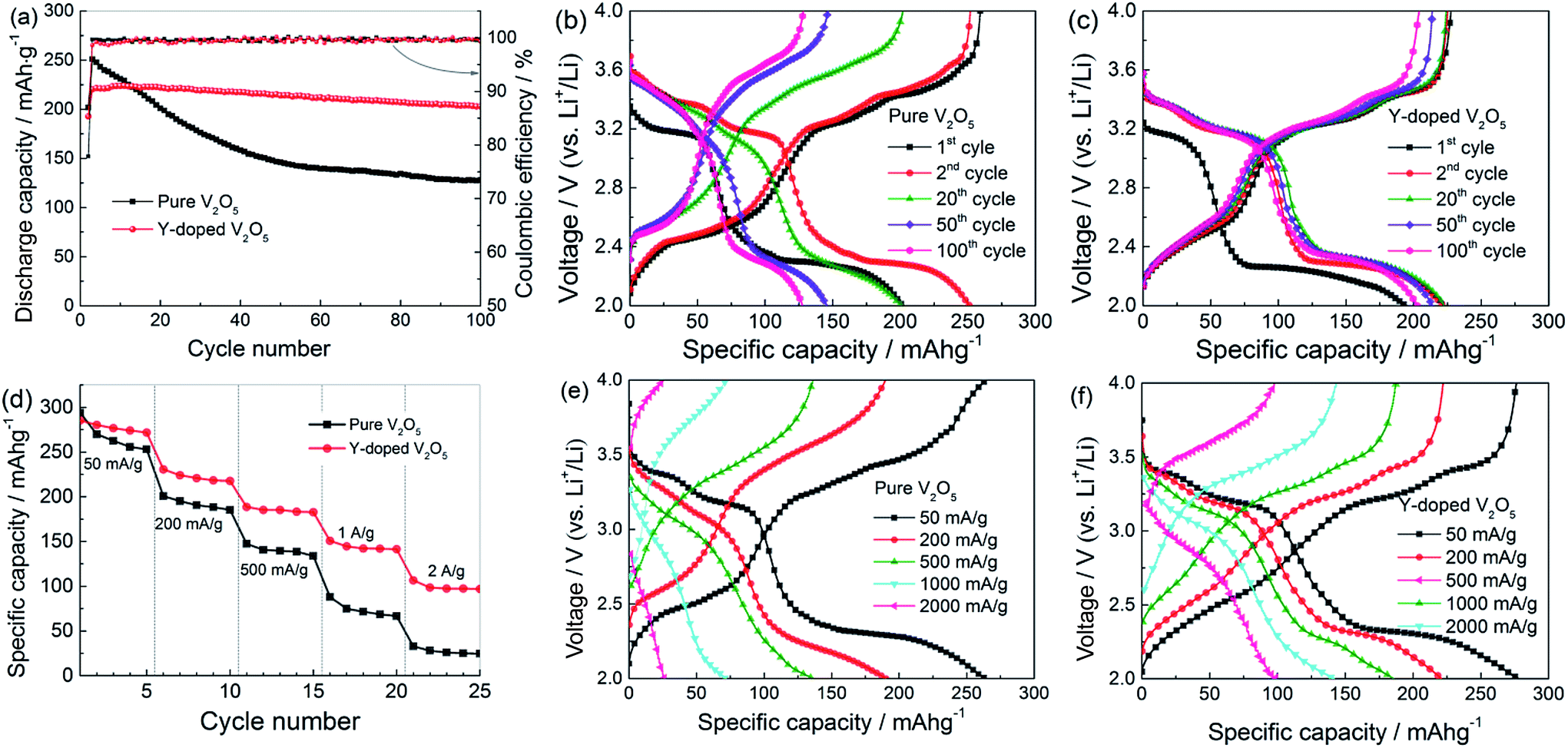
Fig. 7a gives the cycling response of the pure V2O5 and Y-doped V2O5 electrodes at a current density of 200 mA g−1. Obviously, the Y-doped V2O5 electrode exhibits much better cycling stability than the pure V2O5 electrode. In the case of the pure V2O5 electrode, the discharge capacity reaches a maximum of 251 mA h g−1 at the 2nd cycle, which is 31 mA h g−1 higher than that (220 mA h g−1) of the Y-doped V2O5 electrode; but with the increase in cycle number, the discharge capacity of the pure V2O5 electrode declines rapidly and at 100th cycle the discharge capacity decreases to 128 mA h g−1. This change of discharge capacity is in accordance with the CV results (Fig. 6a). Compared to the 2nd cycle, the capacity retention of the pure V2O5 electrode at 100th cycle is only 51.0%. While for the Y-doped V2O5 electrode, the discharge capacity reaches a maximum of 224 at the 10th cycle and then decreases very slowly with the increase in cycle number; at 100th cycle, the Y-doped V2O5 electrode can still deliver a specific discharge capacity of 203 mA h g−1, which is 75 mA h g−1 higher than that (128 mA h g−1) of the pure V2O5 electrode. Compared to the maximum capacity in 10th cycle, the capacity retention of the Y-doped V2O5 electrode in 100th cycle is 90.6%. The capacity retention capability of this Y-doped V2O5 is superior to those of the previously reported Cu-doped V2O5, Fe-doped V2O5, Cr-doped V2O5, Al-doped V2O5, and Sn-doped V2O5 as summarized in Table 3. Fig. 7b and c show the corresponding discharge/charge voltage profiles of the pure V2O5 and Y-doped V2O5 electrodes at different cycles. Both electrodes show a typical multi-stage discharge/charge profile, which agrees well with the redox peaks shown in the CV curves (Fig. 6a and b). With the cycle number increasing from 2 to 100, the pure V2O5 electrode shows a significant increase (decrease) of charge (discharge) voltage and the capacity gets reduced sharply. It is noticeable that the capacity loss with the plateau of about 3.2 V (corresponding to ε/δ transition) is much larger than those of others. Therefore, it can be inferred that the pure V2O5 processes relatively poorer reversibility for Li intercalation with the voltage plateau of about 3.2 V (ε/δ transition), which is considered a main reason for the fast capacity fading. In contrast, for the Y-doped V2O5 electrode, the discharge/charge voltage profile was well maintained since the second cycle, indicating an excellent capacity retention capability. Fig. 7d compares the rate performance of the pure V2O5 and Y-doped V2O5 electrodes with discharge/charge rates from 50 to 2000 mA g−1. As low rates, for example 50 mA g−1, the pure V2O5 displays a stable discharge capacity of 256 mA g−1, while the Y-doped V2O5 electrode displays a stable discharge capacity of 274 mA h g−1, indicating the specific capacity was improved by Y-doping. At higher rates, the superior capacity characteristics of the Y-doped V2O5 electrode are even clearer. For example, at 1000 mA g−1, the discharge capacity of the Y-doped V2O5 electrode retains a capacity of 142 mA h g−1 in contrast to only 68 mA h g−1 for the pure V2O5 electrode. Fig. 7e and f present the selected discharge/charge profiles of the pure V2O5 and Y-doped V2O5 electrodes are various current densities. With the increase of current density, the discharge voltage decreases and the charge voltage increases due to the increasing polarization effect. For the Y-doped V2O5 electrode, good reversible plateau regions can be observed at all the current densities. However, for the pure V2O5 electrodes, reversible plateau regions almost disappear when the current density is higher than 500 mA g−1.
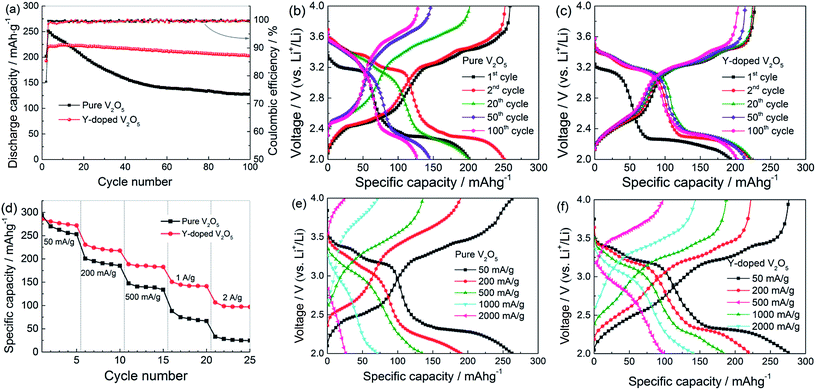 |
| | Fig. 7 (a) Cycling performance and (b, c) selected discharge/charge profiles at different cycles for the pure V2O5 and Y-doped V2O5 electrodes at a current density of 200 mA g−1. (d) Rate performance, and (e, f) the corresponding discharge/charge profiles at various current densities for the pure V2O5 and Y-doped V2O5 electrodes. | |
Table 3 The summarization of electrochemical performance of the Y-doped V2O5 in this work and other doped V2O5 materials reported previously in the literatures
| V2O5 materials |
Voltage range/V |
Current density/mA g−1 |
Maximum capacity/mA h g−1 |
Capacity retention/% |
Ref. |
| Y-doped V2O5 |
2.0–4.0 |
200 |
224 |
91.6 (after 100 cycles) |
This work |
| Cu-doped V2O5 |
2.0–4.0 |
58.8 |
242 |
69.0 (after 50 cycles) |
31 |
| Fe-doped V2O5 |
2.0–4.0 |
58.8 |
244 |
80.0 (after 48 cycles) |
33 |
| Cr-doped V2O5 |
2.0–4.0 |
150 |
196 |
86.7 (after 50 cycles) |
34 |
| 300 |
159 |
76.0 (after 50 cycles) |
34 |
| Al-doped V2O5 |
1.75–4.0 |
35 |
350 |
85.0 (after 20 cycles) |
35 |
| 350 |
208 |
70.0 (after 50 cycles) |
35 |
| Sn-doped V2O5 |
2.0–4.0 |
259 |
200 |
82.0 (after 50 cycles) |
37 |
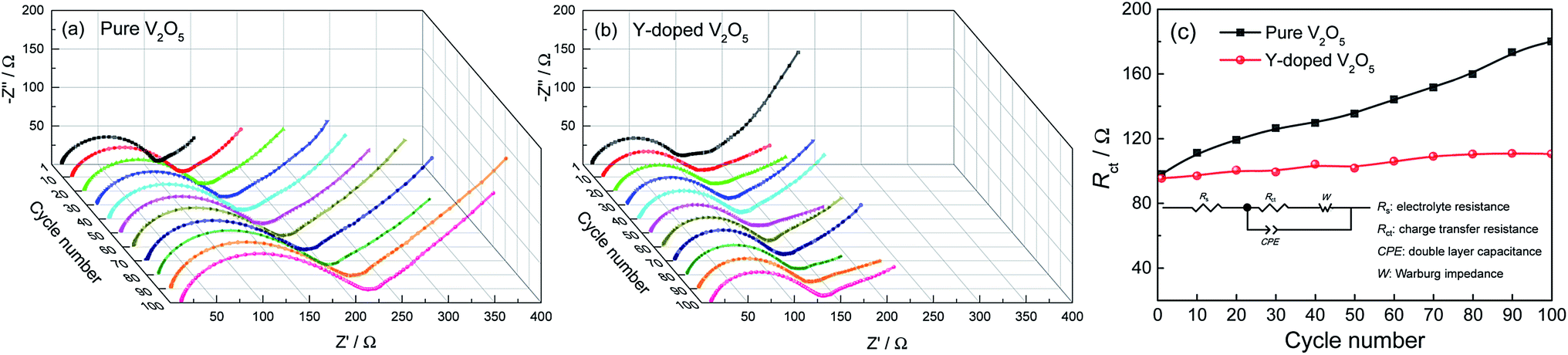
Fig. 8a and b give the Nyquist plots of the pure V2O5 and Y-doped V2O5 electrodes after every ten discharge/charge cycles at the current density of 200 mA g−1, respectively. The depressed semicircle observed at high frequency region is due to the charge transfer resistance (Rct); the inclined line at low frequency region relates to the Warburg impedance.59 The Rct values were determined by plot fitting with the equivalent circuit depicted in the inset in Fig. 8c. A constant phase element (CPE) is used in the equivalent circuit instead of a pure capacitance due to the inhomogeneous surface of the working electrode. Fig. 8c compares the evolutions of Rct of the pure V2O5 and Y-doped V2O5 electrodes with cycling. Clearly, the Rct of the pure V2O5 electrode increases rapidly with cycling, while that of the Y-doped electrode increases very slowly. After one discharge/charge cycle, the Rct values for the pure V2O5 electrode (98.0 Ω) is slightly higher than that (95.3) for the Y-doped V2O5 electrode. The Rct value for the pure V2O5 electrode enlarges drastically from 98.0 Ω at the first cycle to 180.1 Ω at the 100th cycle, whereas the Rct value for Y-doped V2O5 increase slowly from 95.3 Ω at the first cycle to 110.5 Ω at the 100th cycle, indicating that Y-doping has a positive effect on the depressing the charge transfer resistance during the process of discharge/charge cycles. The lower increase of impedance during cycling means smaller polarization and faster kinetics, which indicates good cycling stability and rate capability. These results are consistent with the superior electrochemical performance of Y-doped V2O5.
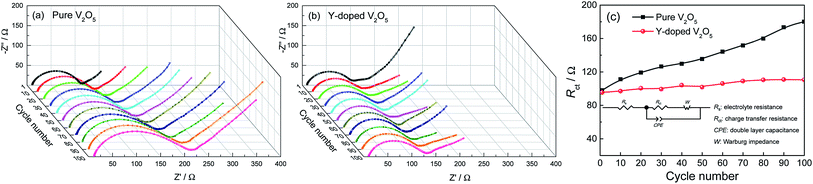 |
| | Fig. 8 Nyquist plots of the (a) pure V2O5 and (b) Y-doped V2O5 electrodes after various discharge/charge cycles. (c) Variations of Rct as a function of discharge/charge cycle number of the pure V2O5 and Y-doped V2O5 electrodes at a current density of 200 mA g−1. The electrical equivalent circuit is shown as an inset. | |
4. Conclusions
Y-doped V2O5 was prepared by a facile sol–gel method combined with freeze drying technique followed with annealing in air. FE-SEM and XRD analysis revealed that this Y-doped V2O5 has a sheet-like morphology and consists of small nanoparticles with the (110) preferred orientation. XPS tests demonstrated that Y doping induced more low valence state V4+ in V2O5. First-principles simulation indicated that Y dopant forms covalent bond with the adjacent O atoms within V2O5 layers and decreases the strength of the neighbored V–O bonds. When used as cathode material for Li-ion batteries, the Y-doped V2O5 exhibited much enhanced rate capability and cycling stability as compared to the pure V2O5 counterpart. The superior lithium storage performance of the Y-doped V2O5 could be ascribed to the following reasons: the nano-sized primary particles in Y-doped V2O5 reduce the diffusion path of both Li+ and electrons, which benefits the improvement of rate capability; furthermore, the predominantly exposed (110) crystal planes of Y-doped V2O5 provide channels for facile Li+ intercalation and deintercalation, which also contributes to the enhanced rate capability; the increased low valence state V4+ may improve the conductivity of Y-doped V2O5 and decrease electrochemical reaction resistance; the chemical interaction between Y atom and the adjacent O atoms in the VO5 slabs could stabilize the layer structure of V2O5 phase during electrochemical interaction of Li+, allowing better structure stability during electrochemical cycling; the local structural defects caused by Y dopant may serve as possible nucleation centers for phase transformation during Li+ intercalation/deintercalation process and accordingly improve the kinetics of Li+ intercalation/deintercalation in Y-doped V2O5 as well as its cycling performance.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51664012, 51464009, and 51562006), Guangxi Natural Science Foundation of China (2015GXNSFGA139006), Postdoctoral Science Foundation of Central South University, and China Postdoctoral Science Foundation (2016M590754).
Notes and references
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926–5940 RSC.
- N. Mahmood, T. Tang and Y. Hou, Adv. Energy Mater., 2016, 6, 1600374 CrossRef.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 CAS.
- J. W. Fergus, J. Power Sources, 2010, 195, 939–954 CrossRef CAS.
- J. Xu, S. Dou, H. Liu and L. Dai, Nano Energy, 2013, 2, 439–442 CrossRef CAS.
- H. T. Tan, X. Rui, W. Sun, Q. Yan and T. M. Lim, Nanoscale, 2015, 7, 14595–14607 RSC.
- D. McNulty, D. N. Buckley and C. O'Dwyer, J. Power Sources, 2014, 267, 831–873 CrossRef CAS.
- X. Huang, X. Rui, H. H. Hng and Q. Yan, Part. Part. Syst. Charact., 2015, 32, 276–294 CrossRef CAS.
- Y. Wang and G. Cao, Chem. Mater., 2006, 18, 2787–2804 CrossRef CAS.
- S. Tian, A. Xing, H. Tang, Z. Bao and G. Wu, J. Mater. Chem. A, 2014, 2, 2896–2900 CAS.
- A. Q. Pan, J. G. Zhang, Z. M. Nie, G. Z. Cao, B. W. Arey, G. S. Li, S. Q. Liang and J. Liu, J. Mater. Chem., 2010, 20, 9193–9199 RSC.
- C. K. Chan, H. Peng, R. D. Twesten, K. Jarausch, X. F. Zhang and Y. Cui, Nano Lett., 2007, 7, 490–495 CrossRef CAS PubMed.
- S.-L. Chou, J.-Z. Wang, J.-Z. Sun, D. Wexler, M. Forsyth, H.-K. Liu, D. R. MacFarlane and S.-X. Dou, Chem. Mater., 2008, 20, 7044–7051 CrossRef CAS.
- D. Yu, C. Chen, S. Xie, Y. Liu, K. Park, X. Zhou, Q. Zhang, J. Li and G. Cao, Energy Environ. Sci., 2011, 4, 858–861 CAS.
- H.-g. Wang, D.-l. Ma, Y. Huang and X.-b. Zhang, Chem. - Eur. J., 2012, 18, 8987–8993 CrossRef CAS PubMed.
- L. Mai, L. Xu, C. Han, X. Xu, Y. Luo, S. Zhao and Y. Zhao, Nano Lett., 2010, 10, 4750–4755 CrossRef CAS PubMed.
- X. Rui, Z. Lu, H. Yu, D. Yang, H. H. Hng, T. M. Lim and Q. Yan, Nanoscale, 2013, 5, 556–560 RSC.
- Z. l. Wang, D. Xu, L. m. Wang and X. b. Zhang, ChemPlusChem, 2012, 77, 124–128 CrossRef CAS.
- Y. Li, J. Yao, E. Uchaker, J. Yang, Y. Huang, M. Zhang and G. Cao, Adv. Energy Mater., 2013, 3, 1171–1175 CrossRef CAS.
- D.-J. Yan, X.-D. Zhu, K.-X. Wang, X.-T. Gao, Y.-J. Feng, K.-N. Sun and Y.-T. Liu, J. Mater. Chem. A, 2016, 4, 4900–4907 CAS.
- J. Liu, Y. Zhou, J. Wang, Y. Pan and D. Xue, Chem. Commun., 2011, 47, 10380–10382 RSC.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- E. Uchaker, N. Zhou, Y. Li and G. Cao, J. Phys. Chem. C, 2013, 117, 1621–1626 CAS.
- G. Li, Y. Qiu, Y. Hou, H. Li, L. Zhou, H. Deng and Y. Zhang, J. Mater. Chem. A, 2015, 3, 1103–1109 CAS.
- S. Zhou, X. Yang, Y. Lin, J. Xie and D. Wang, ACS Nano, 2012, 6, 919–924 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. Wen Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- M. Wagemaker and F. M. Mulder, Acc. Chem. Res., 2013, 46, 1206–1215 CrossRef CAS PubMed.
- H. Yu, X. Rui, H. Tan, J. Chen, X. Huang, C. Xu, W. Liu, Y. Denis, H. H. Hng and H. E. Hoster, Nanoscale, 2013, 5, 4937–4943 RSC.
- D. Yu, S. Zhang, D. Liu, X. Zhou, S. Xie, Q. Zhang, Y. Liu and G. Cao, J. Mater. Chem., 2010, 20, 10841–10846 RSC.
- S.-R. Li, S.-Y. Ge, Y. Qiao, Y.-M. Chen, X.-Y. Feng, J.-F. Zhu and C.-H. Chen, Electrochim. Acta, 2012, 64, 81–86 CrossRef CAS.
- S. Zhan, C. Wang, K. Nikolowski, H. Ehrenberg, G. Chen and Y. Wei, Solid State Ionics, 2009, 180, 1198–1203 CrossRef CAS.
- Y. L. Cheah, V. Aravindan and S. Madhavi, Electrochim. Acta, 2012, 4, 3270–3277 CAS.
- Y. Li, J. Yao, E. Uchaker, M. Zhang, J. Tian, X. Liu and G. Cao, J. Phys. Chem. C, 2013, 117, 23507–23514 CAS.
- Z. Li, C. Zhang, C. Liu, H. Fu, X. Nan, K. Wang, X. Li, W. Ma, X. Lu and G. Cao, Electrochim. Acta, 2016, 222, 1831–1838 CrossRef CAS.
- C. M. Carbonaro, D. Chiriu and P. C. Ricci, Phys. Status Solidi C, 2016, 13, 1017–1022 CrossRef CAS.
- N. Zhao, Y. Li, X. Zhi, L. Wang, X. Zhao, Y. Wang and G. Liang, J. Rare Earths, 2016, 34, 174–180 CrossRef CAS.
- L. Wang, C. Jiao, G. Liang, N. Zhao, Y. Wang and L. Li, J. Rare Earths, 2014, 32, 895–899 CrossRef CAS.
- Z. Lan, J. Li, B. Wei, R. Zhu and J. Guo, J. Rare Earths, 2016, 34, 401–406 CrossRef CAS.
- C. J. Fontenot, J. W. Wiench, M. Pruski and G. L. Schrader, J. Phys. Chem. B, 2000, 104, 11622–11631 CrossRef CAS.
- M. D. Segall, J. D. L. Philip, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. Laasonen, R. Car, C. Lee and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 6796–6799 CrossRef CAS.
- M. Giorgetti, M. Berrettoni and W. H. Smyrl, Chem. Mater., 2007, 19, 5991–6000 CrossRef CAS.
- J. H. Zheng, J. L. Song, Q. Jiang and J. S. Lian, Appl. Surf. Sci., 2012, 258, 6735–6738 CrossRef CAS.
- D. Liu, Y. Liu, A. Pan, K. P. Nagle, G. T. Seidler, Y.-H. Jeong and G. Cao, J. Phys. Chem. C, 2011, 115, 4959–4965 CAS.
- R. Enjalbert and J. Galy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1467–1469 CrossRef.
- M. D. Segall, R. Shah, C. J. Pickard and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16317–16320 CrossRef CAS.
- C. Delmas, H. Cognac-Auradou, J. M. Cocciantelli, M. Ménétrier and J. P. Doumerc, Solid State Ionics, 1994, 69, 257–264 CrossRef CAS.
- X. Li, C. Liu, C. Zhang, H. Fu, X. Nan, W. Ma, Z. Li, K. Wang, H. Wu and G. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 24629–24637 CAS.
- R. Baddour-Hadjean, A. Marzouk and J. P. Pereira-Ramos, J. Raman Spectrosc., 2012, 43, 153–160 CrossRef CAS.
- H. Zhao, L. Pan, S. Xing, J. Luo and J. Xu, J. Power Sources, 2013, 222, 21–31 CrossRef CAS.
- J. P. Pereira-Ramos, P. Soudan, R. Baddour-Hadjean and S. Bach, J. Power Sources, 2011, 196, 1392–1398 CrossRef CAS.
- W. Ma, C. Zhang, C. Liu, X. Nan, H. Fu and G. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 19542–19549 CAS.
- S. Zhan, C. Wang, G. Chen, F. Du and Y. Wei, Ionics, 2010, 16, 209–213 CrossRef CAS.
- S. Zhan, G. Chen, D. Liu, A. Li, C. Wang and Y. Wei, J. Alloys Compd., 2009, 479, 652–656 CrossRef CAS.
- Y. W. Li, J. H. Yao, C. J. Liu, W. M. Zhao, W. X. Deng and S. K. Zhong, Int. J. Hydrogen Energy, 2010, 35, 2539–2545 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b