
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enabling the facile conversion of acyl hydrazides into N-acyl carbamates via metal-free ionic-based rupture of the N–N linkage†
André Shamsabadi a,
Jack Rena and
Vijay Chudasama*ab
a,
Jack Rena and
Vijay Chudasama*ab
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk
bResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal
First published on 23rd May 2017
Abstract
Herein we report the one-pot transformation of readily synthesised/easily accessed acyl hydrazides into N-acyl carbamates via an alkylation-elimination reaction sequence. A range of N-acyl carbamates are prepared in consistent yields, including those featuring protecting groups and having alkyl & aryl N-acyl functionality. The reaction conditions also tolerate a wide variety of sensitive functional groups.
Acyl hydrazides have been prepared using many methods through the years with a substantial increase in the number of reported methodologies in the past decade.1 Their synthesis can now be considered to be largely optimal in view of the plethora of distinct routes by which they can be formed in good to excellent yields.1 They represent synthetically versatile scaffolds that have been exploited for the creation of various moieties such as 1,3,4-oxadiazoles,2–5 thioesters,6 esters,6 amides6 and ketones,7 as well as building blocks for the creation of bioactive molecules such as hydroxamic acids8 and macrocylic enamides9 (Fig. 1). However, to date, all approaches have focused on substituting the hydrazide moiety or alkylating at the acidic N–H position, with the latter being used to good effect by exploiting the conformation of the N–N bond in ring-closing metathesis to form medium-sized macrocycles.9
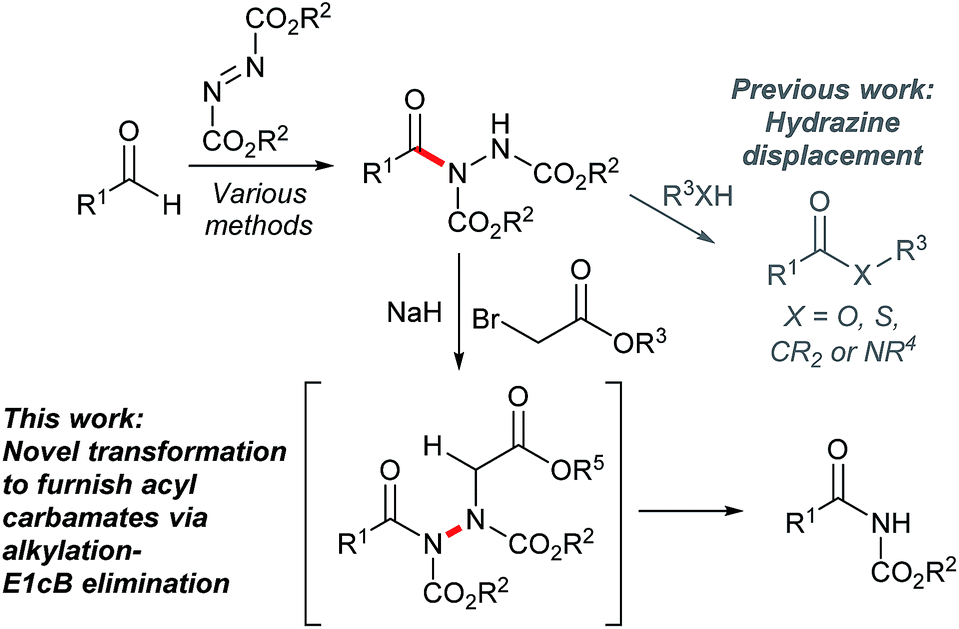 | ||
| Fig. 1 General method for the synthesis of acyl hydrazides via hydroacylation, how acyl hydrazides have commonly been used and how we propose to use them in this article. | ||
In this article, we focus on developing a new method to form acyl carbamates from acyl hydrazides via rupture of the N–N bond in a one-pot alkylation-E1cB elimination procedure (Fig. 1). Owing to the importance of N-acyl carbamates in various bioactive compounds,10–13 e.g. anti-malassezia agents,10 insecticides,11 antibiotics,12 and pro-drugs,13 its use as a directing a group in synthesis,14,15 as well as phototriggers,16 there was plenty of incentive to explore this chemistry. Further to this, the synthetic strategy proposed in this article is complementary to existing traditional approaches for the synthesis of N-acyl carbamates, which tend to react acyl halides with carbamates or primary/secondary amides with chloroformates.17,18 Furthermore, the ability to cleave the N–N bond without the use of hydrogenation, expensive catalysts and/or undesirable metals was seen as a key aspect in itself;19–24 this is particularly well described in related work by Magnus et al. where conversion of hydrazines to amines was achieved using an elegant alkylation-elimination sequence, albeit in two distinct steps and requiring relatively high temperatures (ca. 80 °C).23
Our study began with the reaction of acyl hydrazide 1a with low-cost bromoacteate 2a with the aim of forming acyl carbamate 3a in a two-step one-pot procedure. Initially, the use of 1.5 eq. of ethyl bromoacetate and 3 eq. of NaH was trialled at room temperature (Table 1, entry 1). These conditions resulted in the formation of several entities: desired acyl carbamate 3a; alkylated-hydrazine intermediate 4a; trans-esterified acyl carbamate 3b; and over-alkylated product 5. Rationally, to limit over-alkylation, the reaction was repeated with 1.1 eq. of ethyl bromoacetate (Table 1, entry 2); this completely suppressed formation of over-alkylated product 5. This also lowered the yield of trans-esterified acyl carbamate 3b; suggesting that the ethoxide, which is likely to facilitate the trans-esterification process, may have been liberated from the ethyl bromoacetate starting material under the basic conditions. Whilst the formation of by-products 3b and 5 was suppressed by lowering the equivalents of alkylating agent, it was coincident with inadequate conversion of the singly alkylated intermediate 4a to the desired acyl carbamate 3a. Most pleasingly, however, simply increasing the equivalents of base from 3 to 5 resulted in full conversion of the intermediate to afford acyl carbamate 3a in 69% yield (Table 1, entry 3). Whilst the reaction was improved, yields were still limited by the formation of trans-esterified by-product 3b. To circumvent the issue, we utilised tert-butyl bromoacetate 2b as the α-bromo acetate source; it was thought that liberation of any tert-butoxide under basic conditions would not result in trans-esterification. To our delight this was the case, with complete suppression of the formation of the trans-esterification product being observed (Table 1, entry 4); this resulted in an 83% yield of the desired product. It is also important to note that tert-butyl bromoacetate 2b is more readily obtained and even more economical than its ethyl variant. Lowering the equivalence of base to 3 once again showed incomplete conversion of intermediate, 4b in this case, to acyl carbamate 3a (Table 1, entry 5).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ra | Baseb | Base/eq. | 3a/% | 4/% | 3b/% | 5/% |
| a 1.1 eq. of alkylating agent utilised unless otherwise stated in parenthesis.b NaH utilised as a 60% dispersion in mineral oil unless otherwise stated in parenthesis.c tBuOH utilised as solvent. | |||||||
| 1 | Et (1.5 eq.) | NaH | 3 | 24 | 38 | 16 | 11 |
| 2 | Et | NaH | 3 | 52 | 30 | 6 | 0 |
| 3 | Et | NaH | 5 | 69 | 0 | 8 | 0 |
| 4 | tBu | NaH | 5 | 83 | 0 | 0 | 0 |
| 5 | tBu | NaH | 3 | 58 | 25 | 0 | 0 |
| 6 | tBu | Cs2CO3 | 5 | 0 | 86 | 0 | 0 |
| 7c | tBu | tBuOK | 5 | 0 | 82 | 0 | 0 |
| 8 | tBu | NaH (95%) | 5 | 78 | 0 | 0 | 0 |
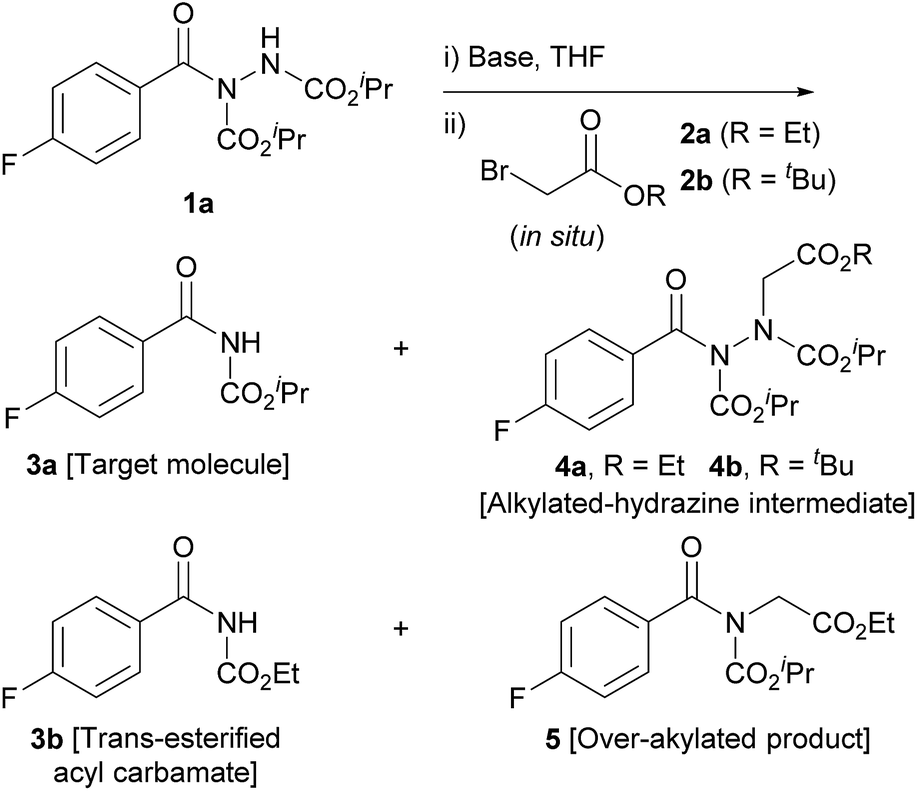
Having established a procedure which led to sole formation of the desired product, the suitability of other bases was appraised (Table 1, entries 6–8). Using Cs2CO3 (pKa = 10.3), or tBuOK (pKa = 17, reaction in tBuOH) only resulted in formation of the alkylated species 4b, suggesting that at 25 °C for 16 h, these bases were unable to facilitate E1cb elimination. Finally, trialling of 95% NaH resulted in a slightly lower yield, 78%, than when using a 60% dispersion in mineral oil.
With optimised conditions in hand we took the opportunity to investigate the applicability of our protocol for the formation of various acyl carbamates (Table 2). A range of acyl hydrazides were trialled (1a, 1c–m) under the developed reaction conditions. All starting hydrazides were prepared using our previously reported procedure for the hydroacylation of azodicarboxylates in good yields.25,26
|
||
|---|---|---|
| Entry | R1 = | Yield/% |
| 1 |  |
83 |
| 2 |  |
85 |
| 3 | 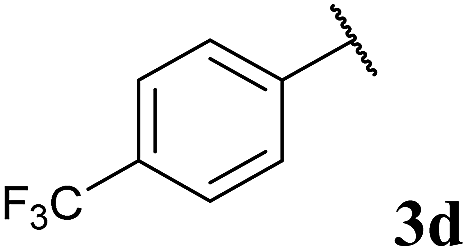 |
71 |
| 4 | 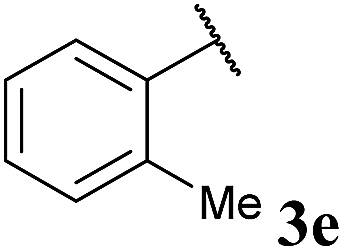 |
70 |
| 5 | 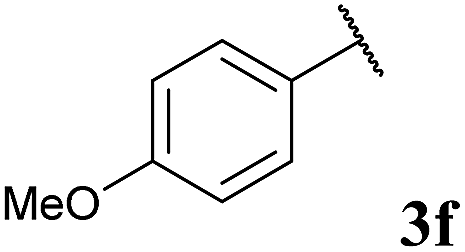 |
86 |
| 6 | 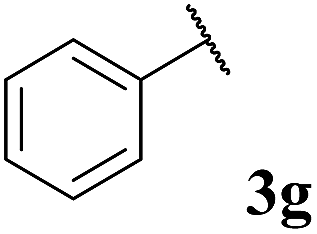 |
76 |
| 7 |  |
87 |
| 8 |  |
72 |
| 9 |  |
68 |
| 10 |  |
74 |
| 11 | 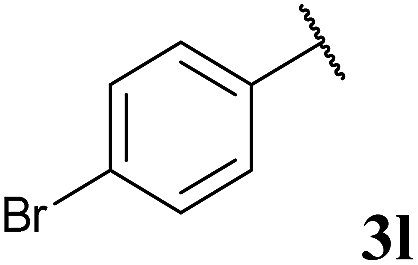 |
80 |
| 12 | 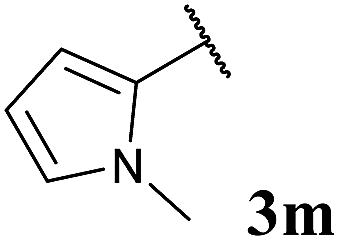 |
61 |
To our delight, the reaction was tolerant of various functional groups on the aromatic acyl hydrazide motif, e.g. halo, trifluoromethyl, methoxy, ester and methyl functionalities (Table 2, entries 1–5, 7–11) were tolerated with good and consistent yields being observed throughout the series. The reaction proved to be compatible with electron rich (Table 2, entry 5, 9), electron neutral (Table 2, entry 6) and electron poor (Table 2, entry 1, 3, 10) moieties, irrespective of the functional groups existing in ortho-, meta- or para-positions on the aryl ring. Gratifyingly, even a sensitive methyl ester functionality was tolerated in the reaction conditions (Table 2, entry 8). The only moderate yield that was observed was when using a rather sensitive N-methylpyrrole acyl hydrazide (Table 2, entry 12).
We also appraised the tolerance of the reaction with respect to the use of acyl hydrazides that were synthesised from the hydroacylation of azodicarboxylates other than DIAD (see ESI for details on synthesis†). The optimised reaction conditions allowed for the formation of aryl acyl carbamates with common protecting groups, e.g. tert-butoxy carbamate (Boc), benzyloxy carbamate (CBz) and ethyl carbamate, in high yields (Scheme 1). These acyl carbamates can undergo facile conversion to various species by reaction at the carbamate nitrogen atom before cleavage of the protecting group.27,28
 | ||
| Scheme 1 One-pot conversion of acyl hydrazide 1b, 1n–o to protected acyl carbamates 3b, 3n–o. | ||
Despite the majority of potent bioactive N-acyl carbamates being aryl in their acyl character, we decided to test our protocol on alkyl acyl hydrazides also. A selection of appropriate acyl hydrazides (i.e. primary, secondary and tertiary alkyl examples) were prepared using our previously described aerobic C–H activation protocol.25,26,29–31 Whilst reaction in our one-pot protocol only achieved minimal yields of carbamates for the primary and secondary alkyl acyl hydrazides 3p and 3q, a step-wise protocol provided good yields (Scheme 2). The major limitation on yield in all cases was elimination of the hydrazine via deprotonation of the α-H (highlighted in red), which was confirmed by isolation of the by-product 6 in a yield that accounted for the mass balance. In fitting with this, the absence of an α-H acidic proton in tertiary alkyl acyl hydrazide 1r resulted in a very good yield of carbamate 3r.
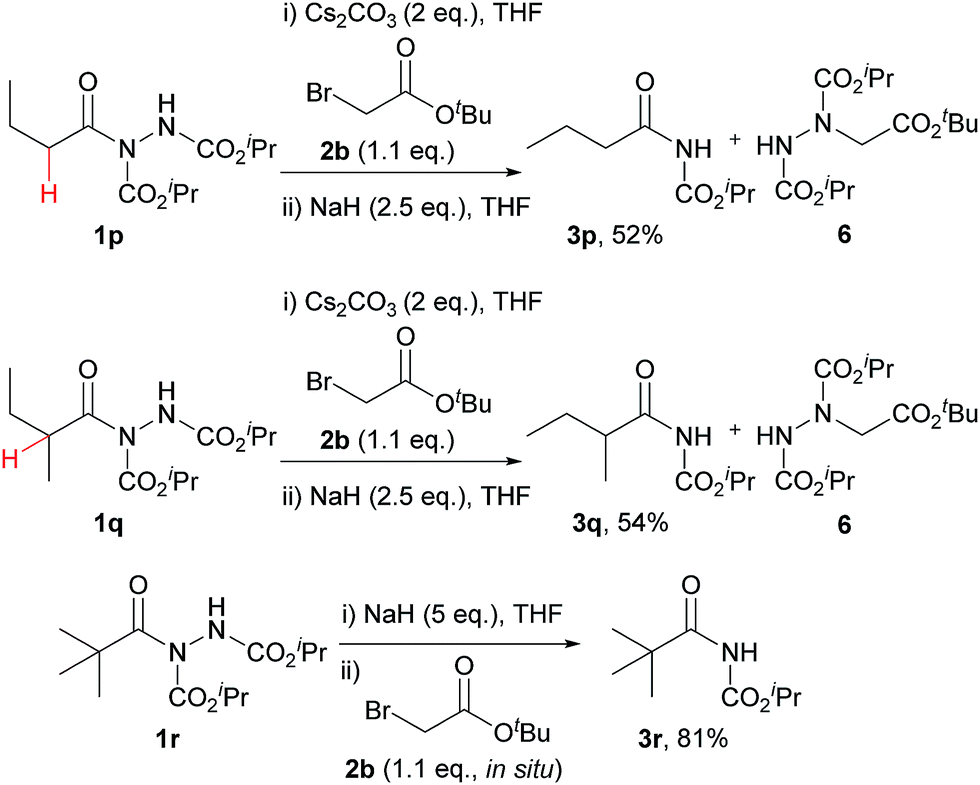 | ||
| Scheme 2 Conversion of alkyl acyl hydrazide 1p–r to alkyl acyl carbamates 3p–r. | ||
Conclusions
In conclusion, we have shown readily accessed acyl hydrazides to be excellent candidates for the synthesis of N-acyl carbamates, which have several applications in terms of their synthetic utility and are often inherently biologically useful (as exemplified by the range of bioactive compounds that bear such a motif). A robust one-pot procedure that is tolerant of a large number of functional groups has been developed; including the synthesis of acyl carbamates with a protecting group on the nitrogen atom – often a key synthetic building block – and adaptability to alkyl & aryl N-acyl functionalities.Acknowledgements
The authors gratefully acknowledge Prof. Alwyn Davies for productive discussions. We also acknowledge UCL (UCL Graduate Research Scholarship) for funding AS.Notes and references
- A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2017, 15, 17–33 CAS.
- K. Tokumaru and J. N. Johnston, Chem. Sci., 2017, 8, 3187–3191 RSC.
- J. Boström, A. Hogner, A. Llinàs, E. Wellner and A. T. Plowright, J. Med. Chem., 2012, 55, 1817–1830 CrossRef PubMed.
- K. D. Patel, S. M. Prajapati, S. N. Panchal and H. D. Patel, Synth. Commun., 2014, 44, 1859–1875 CrossRef CAS.
- H. A. Rajapakse, H. Zhu, M. B. Young and B. T. Mott, Tetrahedron Lett., 2006, 47, 4827–4830 CrossRef CAS.
- A. Maruani, M. T. W. Lee, G. Watkins, A. R. Akhbar, H. Baggs, A. Shamsabadi, D. A. Richards and V. Chudasama, RSC Adv., 2016, 6, 3372–3376 RSC.
- A. R. Akhbar, V. Chudasama, R. J. Fitzmaurice, L. Powell and S. Caddick, Chem. Commun., 2014, 50, 743–746 RSC.
- G. N. Papadopoulos and C. G. Kokotos, Chem.–Eur. J., 2016, 22, 6964–6967 CrossRef CAS PubMed.
- Y. J. Kim and D. Lee, Org. Lett., 2004, 6, 4351–4353 CrossRef CAS PubMed.
- V. K. Thulam, S. C. B. Kotte, H. Sanjay Kumar, P. M. Murali, K. Mukkanti and P. S. Mainker, J. Pharm. Res., 2013, 7, 195–199 CAS.
- J. Fraser, P. G. Clinch and R. C. Reay, J. Sci. Food Agric., 1965, 16, 615–618 CrossRef CAS PubMed.
- C. Davidovich, A. Bashan, T. Auerbach-Nevo, R. D. Yaggie, R. R. Gontarek and A. Yonath, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4291–4296 CrossRef CAS PubMed.
- A. H. Kahns and H. Bundgaard, Int. J. Pharm., 1991, 71, 31–43 CrossRef CAS.
- S. Shaw-Ponter, G. Mills, M. Robertson, R. D. Bostwick, G. W. Hardy and R. J. Young, Tetrahedron Lett., 1996, 37, 1867–1870 CrossRef CAS.
- R. J. Young, S. Shaw-Ponter, G. W. Hardy and G. Mills, Tetrahedron Lett., 1994, 35, 8687–8690 CrossRef CAS.
- P. Bourbon, Q. Peng, G. Ferraudi, C. Stauffacher, O. Wiest and P. Helquist, Bioorg. Med. Chem. Lett., 2013, 23, 6321–6324 CrossRef CAS PubMed.
- A. Bach, N. Stuhr-Hansen, T. S. Thorsen, N. Bork, I. S. Moreira, K. Frydenvang, S. Padrah, S. B. Christensen, K. L. Madsen, H. Weinstein, U. Gether and K. Strømgaard, Org. Biomol. Chem., 2010, 8, 4281–4288 CAS.
- K. Li, X. Cheng and K. K. Hii, Eur. J. Org. Chem., 2004, 959–964 CrossRef CAS.
- P. Sinha, C. C. Kofink and P. Knochel, Org. Lett., 2006, 8, 3741–3744 CrossRef CAS PubMed.
- P. A. Jacobi and M. J. Martinelli, J. Am. Chem. Soc., 1984, 106, 5594–5598 CrossRef CAS.
- M. A. Brimble and C. H. Heathcock, J. Org. Chem., 1993, 58, 5261–5263 CrossRef CAS.
- H. Ding and G. K. Friestad, Tetrahedron, 2004, 6, 637–640 CAS.
- P. Magnus, N. Garizi, K. A. Seibert and A. Ornholt, Org. Lett., 2009, 11, 5646–5648 CrossRef CAS PubMed.
- P. Magnus and A. J. Brozell, Org. Lett., 2012, 14, 3952–3954 CrossRef CAS PubMed.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 CAS.
- U. Jacquemard, V. Bénéteau, M. Lefoix, S. Routier, J.-Y. Mérour and G. Coudert, Tetrahedron, 2004, 60, 10039–10047 CrossRef CAS.
- S. Routier, L. Saugé, N. Ayerbe, G. Coudert and J.-Y. Mérour, Tetrahedron Lett., 2002, 43, 589–591 CrossRef CAS.
- V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133–135 RSC.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS PubMed.
- V. Chudasama, J. M. Ahern, R. J. Fitzmaurice and S. Caddick, Tetrahedron Lett., 2011, 52, 1067–1069 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all small molecules. See DOI: 10.1039/c7ra04178k |
| This journal is © The Royal Society of Chemistry 2017 |