DOI:
10.1039/C7RA04201A
(Paper)
RSC Adv., 2017,
7, 30870-30878
L-Glutamine-assisted synthesis of flower-like NiO and ball-flower-like NiO/Ag as an electrochemical sensor for lead(II) detection
Received
13th April 2017
, Accepted 1st June 2017
First published on 14th June 2017
Abstract
Flower-like NiO (F-NiO) and ball-flower-like NiO/Ag (BF-NiO–Ag) were synthesized in the presence of L-glutamine as an electrochemical sensor for lead(II) detection. The structure and morphology of the F-NiO and BF-NiO–Ag were studied by X-ray diffraction (XRD) and field emission scanning electron microscopy (FESEM). Energy dispersive X-ray confirmed the presence of Ag on the surface of the BF-NiO–Ag. The results confirmed that Ag+ was reduced to Ag metal in the presence of L-glutamine. The FESEM images confirmed the effect of L-glutamine and Ag on the morphology and the available surface area. Electrochemical impedance spectroscopy results confirmed the effect of the loaded Ag on the conductivity of the BF-NiO–Ag. Differential pulse voltammetry results gave a linear working range for the concentration of lead(II) between 1.5 and 10 nM with a LOD of 0.06 nM (S/N = 3). The sensitivity of this linear segment is 0.3 μA nM−1. The average recoveries, recovery percentage and relative standard deviation obtained from four determinations (n = 4) confirmed the feasibility of the proposed method for the quantitative detection of certain concentration ranges of Pb2+.
1 Introduction
Heavy metal ion contamination of drinking water is widely known to cause serious health effects to the human body through the food chain since they are scarcely biodegradable.1,2 Among the heavy metal ions, lead inflicts serious damage to the hematopoietic system, functionality of the central nervous system and kidneys even at low concentrations.3 Due to the dangerous health effects from exposure to lead ions, the fabrication and development of different methods for the detection of lead(II) ions has recently attracted huge attention from researchers. Some of these methods are: colorimetric spectrometry,4 inductively coupled plasma mass spectrometry,5 atomic absorption/emission spectrometry,6 fluorescence spectrometry7 and electrochemical methods.8 Among these methods, electrochemical detection has attracted the greatest attention due to the following reasons: short analytical time, low cost, low power consumption, acceptable sensitivity and ease of application.9,10 The selectivity and sensitivity are key elements for the successful fabrication and development of electrochemical sensors for lead(II) detection. Therefore, the synthesis of different types of materials with highly available surface area and appropriate functional groups for the adsorption and oxidation/reduction of the analyte are crucial steps in the development of electrochemical sensors.
In recent years, Kirowa-Eisner et al. fabricated different type of electrodes using gold11,12 and silver13,14 for the detection of cadmium, lead and copper. They concluded that gold was inappropriate for the selective determination of lead in a mixture of lead and cadmium ions due to the overlapping of peaks of both ions, while silver showed acceptable selectivity for the detection of lead and cadmium ions. They suggested that the sensitivity could be improved further by using advanced procedures or increasing the available surface area of the electrode. Graphene is one of the few materials that have attracted huge attention for the fabrication of different types of sensors due to its amazing properties. Different types of nanocomposites containing graphene were recently fabricated for the detection of Pb2+ and Cd2+.15 The detection of a heavy metal ion such as Cd2+ is also very important due to the dangerous effects on human health. Recent reports have shown that electrochemical sensors were successfully fabricated for the simultaneous detection of heavy metals such as Pb2+ and Cd2+.16–18 Zhang et al.19 recently reported the fabrication of an electrochemical sensor utilizing glassy carbon electrode modified with NiO for the detection of Pb(II) and Cd(II). They reported that among the different morphologies of NiO, the GCE modified with ball-shaped NiO gave better selectivity and sensitivity for the detection of Pb(II) and Cd(II). The improved detection of Pb(II) and Cd(II) was due to the faster charge transfer rate and greater available surface area of the ball shaped NiO. Therefore, the increase of surface area and decrease of charge transfer resistance are two important factors in the fabrication of electrochemical sensors for the detection of Pb(II) and Cd(II). Based on this, the synthesis of NiO/Ag in the presence of amino acids is an interesting phenomenon to be investigated because: Ag nanoparticles and NiO are sensitive materials for the detection of lead(II) and amino acids play a special role as the reducing agent,20 which could influence the morphology of the fabricated NiO/Ag. In this paper, we report the synthesis of NiO/Ag in the presence of L-glutamine as an electrochemical sensor for the detection of lead(II) ions.
2 Experimental methods
2.1 Green synthesis of F-NiO and BF-NiO–Ag
All chemicals such as nickel(II) nitrate hexahydrate, L-glutamine, silver nitrate, sodium acetate and acetic acid were procured from Sigma-Aldrich (St. Louis, Mo, USA). In a typical process, 1.164 g nickel(II) nitrate hexahydrate and 0.07 g silver nitrate were dissolved in 80 mL of ethanol and distilled water with a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v to form a homogeneous solution. Then, 0.073 g L-glutamine was added to the solution. After stirring for 30 min, 0.6 g urea was added to the solution with stirring. The suspension was transferred to a Teflon-lined stainless steel autoclave, and heated at 150 °C for 15 h. The solid product was centrifuged at 4000 rpm for 10 minutes to separate the product from the solution, followed by drying in a vacuum oven at 50 °C for 12 h. Finally, the product was annealed at 400 °C for 3 h to obtain the BF-NiO–Ag. The same synthetic procedures were carried out in the preparation of NiO but without the presence of silver nitrate, as the reference. The effect of the L-glutamine and reaction time on the morphology of NiO/Ag and the determination of the optimum amount of L-glutamine for the synthesis of the BF-NiO–Ag was also investigated.
:1 v/v to form a homogeneous solution. Then, 0.073 g L-glutamine was added to the solution. After stirring for 30 min, 0.6 g urea was added to the solution with stirring. The suspension was transferred to a Teflon-lined stainless steel autoclave, and heated at 150 °C for 15 h. The solid product was centrifuged at 4000 rpm for 10 minutes to separate the product from the solution, followed by drying in a vacuum oven at 50 °C for 12 h. Finally, the product was annealed at 400 °C for 3 h to obtain the BF-NiO–Ag. The same synthetic procedures were carried out in the preparation of NiO but without the presence of silver nitrate, as the reference. The effect of the L-glutamine and reaction time on the morphology of NiO/Ag and the determination of the optimum amount of L-glutamine for the synthesis of the BF-NiO–Ag was also investigated.
2.2 Electrode fabrication
1 mg of the synthesized BF-NiO–Ag and F-NiO were dispersed in 1 mL dimethylformamide (DMF) separately, with ultra-sonication for 10 minutes to obtain a homogenous green suspension. Then, 10 μL of the homogenous suspension was drop-casted onto the surface of a polished glassy carbon electrode (GCE) and dried at room temperature.
2.3 Instruments and characterizations
The BF-NiO–Ag and F-NiO were characterized by X-ray diffraction (Siemens D5000) with Cu Kα radiation, field emission scanning electron microscopy (FESEM, Quanta 200F), FT-IR spectrum (Spectrum 400 (FT-IR/FT-FIR spectrometer)) and energy dispersive X-ray (EDX) spectroscopy. Electrochemical impedance spectroscopy (EIS) measurements were performed in 100 μM Pb(NO3)2 in 0.1 M acetate buffer solution (ABS) at pH 6. The determination of the charge transfer resistance of the BF-NiO–Ag and F-NiO were performed between 100 kHz and 0.1 Hz, with an acquisition of 10 points per decade. A saturated Ag/AgCl and a platinum foil were the reference and the counter electrodes, respectively.
3 Results and discussions
3.1 Physicochemical characterizations
3.1.1 FESEM and EDX. The FESEM images of the F-NiO and BF-NiO–Ag in different magnifications synthesized in the presence of L-glutamine are given in Fig. 1(a–c) and (d–f), respectively. There are significant changes in the FESEM images of the NiO synthesized in the presence and absence of Ag. Fig. 1(a) shows a flower-like morphology of the NiO while Fig. 1(d) shows a ball-flower-like morphology of the NiO/Ag, both synthesized in the presence of L-glutamine. Moreover, the Ni/Ag oxidizes to a spongy morphology which leads to an increase in the surface area of NiO/Ag compared to NiO. The high magnification comparisons of the FESEM images of the F-NiO (Fig. 1(b and c)) with the BF-NiO–Ag synthesized in the presence of L-glutamine confirms the increase in the porosity, surface area and change of morphology. The increase of the surface area and detection sensitivity of NiO with Ag loading synthesized in the presence of L-glutamine is one of the important objectives of this work. The effect of L-glutamine on the morphology of the NiO/Ag can be seen from the comparison of the FESEM images of BF-NiO–Ag (Fig. 1(d)) and the NiO/Ag (Fig. 1(h)) synthesized in the presence and absence of L-glutamine, respectively. From the comparison of the FESEM images, it can be confirmed that L-glutamine plays a specific role on the morphology of the NiO/Ag composite, which will be discussed in the next section. The EDX results of the BF-NiO–Ag (Fig. 1(g)) together with the weight percentages of each element are given in Table 1. The EDX results clearly confirm the loading of Ag in NiO of the BF-NiO–Ag synthesized in the presence of L-glutamine. The growth mechanism of the BF-NiO–Ag was studied together with the effect of L-glutamine concentration on the morphology of the NiO/Ag, in concentrations of 0.00025, 0.0005 and 0.00075 M. During the synthesis of NiO/Ag in the presence of 0.00025 M and 0.0005 M (0.073 g) L-glutamine shown in Fig. 2(a) and (b) respectively, the nanowalls are closer to each other with the decrease of the L-glutamine concentration. In addition, with the increase of the L-glutamine concentration to 0.00075 M, the insoluble L-glutamine was observed even with stirring after 10 minutes. Therefore, 0.0005 M of L-glutamine is the optimum amount for the synthesis of NiO/Ag. Fig. 3(a) to (c) are the FESEM images which shows the various morphology of the BF-NiO–Ag synthesized in the presence of L-glutamine after 5, 10 and 15 hours. The mechanism growth can be explained based on four steps. In the first step, the Ni (L-glutamine)n or Ag (L-glutamine)n complexes21 are formed in solution predominantly through the coordination of the amide and carboxyl groups. It is reasonable that some primary crystals of Ni(OH)2 and AgOH are formed initially, suspended in the solution or precipitated at the bottom of the container during the process. It is possible that the Ni(OH)2 crystals forms the nucleation site of the nanoflower. In the second growth step, the Ni (L-glutamine)n complexes attack the Ni(OH)2 core by an ion exchange process and forms the coordination core. The growth of the coordination core is accelerated by the intermolecular interactions between the Ni (L-glutamine)n and the Ni(OH)2 core. This interaction enhances the agglomeration process where the growth of nanoflowers is observed in Fig. 3(a) (after 5 h). In the final step, the full-grown nanoflower can be observed in Fig. 3(b) (after 10 h). The ligand–ligand interactions in the Ni (L-glutamine)n complexes and the steric effect of the side chains could promote the anisotropic growth and formation of a branched flower-like structure as seen in Fig. 3(c) (after 15 h). Fig. 3(d) shows the synthesized Ag in the presence of L-glutamine. The results confirmed the nanoflower morphology of the synthesized Ag in the presence of L-glutamine. The proposed mechanism discussed previously is an explanation for the formation and growth of the Ag nanoflower morphology. The effect of Ag on the structure and morphology of the BF-NiO–Ag can be observed by the comparison of the FESEM images in Fig. 1(d)–(f) and 2(b) which are related to the BF-NiO–Ag, with Fig. 1(a)–(c) which are related to the F-NiO. These results clearly show the effect of Ag on the morphology of the BF-NiO–Ag, where Ag could decrease the size of the ball shape NiO.
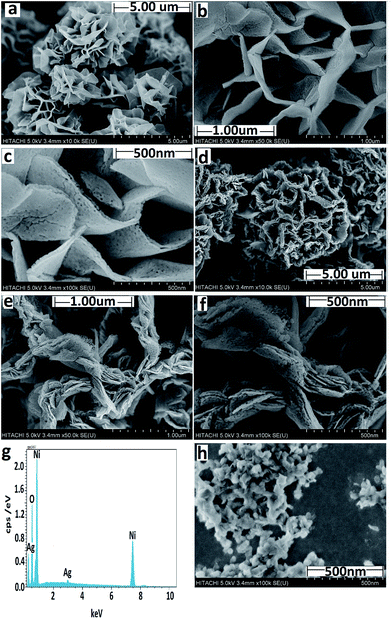 |
| | Fig. 1 (a) and (b, c) Low and high magnification FESEM images of the F-NiO synthesized in the presence of L-glutamine. (d) and (e, f) Low and high magnification FESEM images of the BF-NiO–Ag synthesized in the presence of L-glutamine. (g) EDX result of BF-NiO–Ag (h) FESEM image of the NiO/Ag prepared in the absence of L-glutamine. | |
Table 1 EDX result of BF-NiO–Ag
| Elements |
O |
Ni |
Ag |
| W% |
19.83 |
79.31 |
0.86 |
 |
| | Fig. 2 FESEM results of: the BF-NiO–Ag synthesized in the presence of 0.00025 M (a) and 0.0005 M (b) of L-glutamine. | |
 |
| | Fig. 3 FESEM results of the variation of morphology of the synthesized BF-NiO–Ag in the presence of L-glutamine after 5 (a), 10 (b) and 15 (c) hours formation process time. (d) FESEM result of synthesized Ag in the presence of L-glutamine. | |
3.1.2 XRD. Fig. 4(a) and (b) show the XRD pattern of the F-NiO and the BF-NiO–Ag synthesized in the presence of L-glutamine, respectively. The XRD pattern of the F-NiO shows the intensities for the (111), (200), (220), (311) and (222) peaks of NiO (ref. code: 00-002-1216, nickel oxide, cubic). The comparison of XRD patterns of the F-NiO and BF-NiO–Ag show the presence of peaks attributed to the Ag metal (the intensity for the (111), (200) and (220) (ref. code: 00-001-1167)) (Fig. 4(b)). The EDX and XRD results not only confirm the loading of Ag on NiO but also the reduction of Ag+ to Ag. Moreover, the presence of impurity peaks is undetected in the XRD patterns. Based on previous reports, the L-glutamate anion creates a coordination bond with Ag+ and the carbonyl groups when glutamine reduces the Ag ions.21 With the addition of urea, the pH of the mixture increases due to the hydrolysis of urea. With the increase of pH, Ni2+ converts to Ni(OH)2 and later converts to NiO during the annealing process. It is notable that CO2 gas are formed in two steps, i.e. during the reduction of Ag+ by L-glutamine (Scheme 1) and during the hydrolysis of urea, which results in the formation one mole of CO2. The release of CO2 gas during the synthesis of NiO/Ag generates pores in the structure of the product. The release of CO2 during the NiO synthesis is due to the hydrolysis of the urea, therefore the NiO/Ag is highly porous compared to NiO. The increase of the porosity in the structure of the BF-NiO–Ag increases the surface area that is necessary for improving the efficiency of the sensor. It can be concluded that during the synthesis of the BF-NiO–Ag, the L-glutamine plays a role as: (i) a surfactant, and (ii) a reducing agent. Indeed L-glutamine has an effect on the loading process of Ag into the NiO.
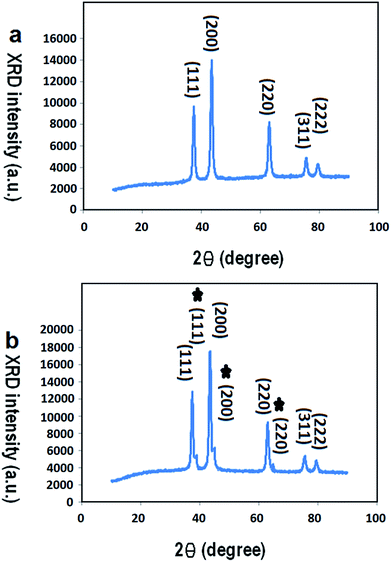 |
| | Fig. 4 XRD patterns of: (a) F-NiO prepared in the presence of L-glutamine, (b) BF-NiO–Ag prepared in the presence of L-glutamine. | |
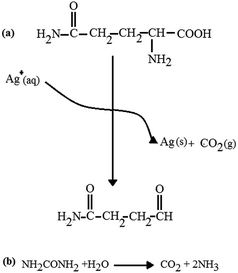 |
| | Scheme 1 The reaction between Ag+ and L-glutamine. | |
3.2 Electrochemical characterizations
The interfacial properties of the GCE, F-NiO/GCE and BF-NiO–Ag/GCE were investigated by EIS. The Nyquist plots of bare GCE (1), F-NiO/GCE (2) and BF-NiO–Ag/GCE (3) in 10 μM Pb2+ with 0.1 M (ABS) at pH 6 are shown in Fig. 5(a). The Nyquist plot clearly shows that the semicircle diameter of the BF-NiO–Ag/GCE significantly decreases compared to the bare GCE and F-NiO/GCE, with the loading of Ag on the surface of NiO. The reason of this is due the increase of the conductivity of NiO loaded with Ag. The increase of conductivity affects the sensitivity of the BF-NiO–Ag/GCE. These results are confirmed by the electrochemical parameters (Table 2) that were obtained by the simulation of the EIS measurements in Fig. 5(b) which shows the equivalents circuits of the electrodes. The R1 is related to the electrolyte resistance while R2 is correlated to the first resistance against ionic mobility which occurs from the solution to the coating layer in the equivalent circuits. The Q1 and Q2 parameters are the constant phase elements which were used instead of the pure capacitor in the equivalents circuits. The pure capacitor was substituted by the constant phase element Q in the equivalents circuits to present a closer agreement between the simulated and experimental data. An overview of the total resistance of each electrode confirms the effect of Ag loading on the resistance. On the other hand, the amount of total Q also shows that the porosity and the surface area of the BF-NiO–Ag are higher than the F-NiO. Previous reports also confirm that the variation of the “n” factor is between 0 and 1.22 The decrease of the “n” factor is the evidence for the increase in the surface roughness. Therefore from the EIS results, it can be concluded that the conductivity and the surface roughness of NiO were enhanced by Ag loading on the surface, which is the main objective of this work.
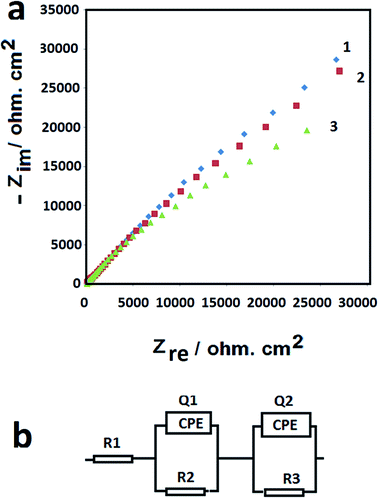 |
| | Fig. 5 Nyquist plots of: bare GCE (1), F-NiO/GCE (2) and BF-NiO–Ag/GCE (3) and their equivalents circuits in 10 μM Pb2+ with 0.1 M (ABS) at pH 6. | |
Table 2 Electrochemical parameters obtained from the simulation of the EIS results: the bare GCE, F-NiO/GCE and BF-NiO–Ag/GCE; in 10 μM Pb2+ with 0.1 M (ABS) at pH 6
| Electrode |
R1(Rs) ohm cm2 |
R2 kohm cm2 |
R3 ohm cm2 |
Q1 Y (mohm−1 sn cm−2) |
n1 |
Q2 Y (mohm−1 sn cm−2) |
n2 |
| GCE |
8.22 |
173.3 |
86.6 |
0.1349 |
0.6274 |
0.03320 |
0.8813 |
| NiO flower like/GCE |
9.51 |
81.1 |
442 |
0.1534 |
0.7021 |
0.1044 |
0.7985 |
| NiO/Ag ball-flower like/GCE |
9.43 |
52.3 |
252.5 |
0.1625 |
0.6048 |
0.1102 |
0.6484 |
3.3 Electrochemical behavior of lead(II) at different modified electrodes
Cyclic voltammetry (CV) were performed in 10 μM Pb2+ with 0.1 M (ABS) at pH 6 at 100 mV s−1 to investigate the electrochemical behavior of lead(II) on the bare GCE (Fig. 6(a1)), F-NiO/GCE (Fig. 6(a2)) and BF-NiO–Ag/GCE (Fig. 6(a3)). The result clearly shows the current attributed to the reduction of Pb2+ to Pb+ and Pb+ to Pb on the GCE electrode modified with the BF-NiO–Ag, which is higher than the F-NiO/GCE and bare GCE. The comparison between the BF-NiO–Ag/GCE and the F-NiO/GCE voltammograms shows two important phenomena. First, the reduction peak potential shifts to more positive region and second, the reduction of Pb2+ on the surface of BF-NiO–Ag/GCE occurs in two steps, while the oxidation of Pb occurs in a single step. The anodic peak at −0.55 V is attributed to the oxidation of Pb to Pb2+. From our previous report,23 a possible mechanism for the electrochemical reduction of Pb(II) on the BF-NiO–Ag/GCE surface is as follows:| | |
Pb2+ (aq) + BF-NiO–Ag/GCE → BF-NiO–Ag/GCE/Pb2+ (adsorption)
| (1) |
| | |
BF-NiO–Ag/GCE/Pb2+ + e− → Pb + BF-NiO–Ag/GCE/Pb+ (reduction)
| (2) |
| | |
BF-NiO–Ag/GCE/Pb+ + e− → Pb + BF-NiO–Ag/GCE (reduction)
| (3) |
| | |
Pb → Pb2+ + 2e− (oxidation)
| (4) |
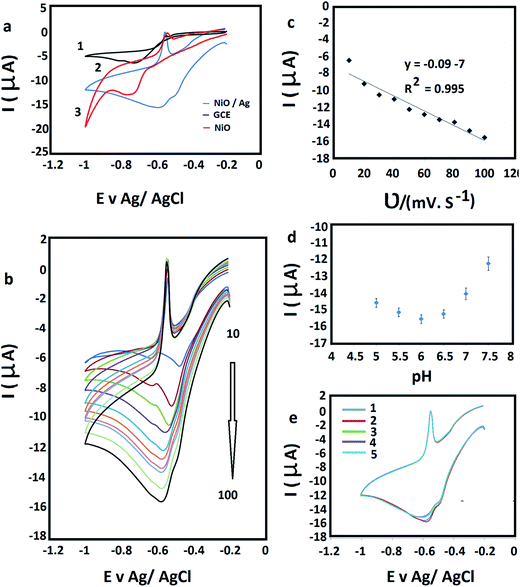 |
| | Fig. 6 (a) Cyclic voltammograms (CVs) of: bare GCE (1), F-NiO/GCE (2) and BF-NiO–Ag/GCE (3) prepared in the presence of L-glutamine, in 10 μM Pb2+ with 0.1 M (ABS) at pH 6 at 100 mV s−1. (b) CVs of BF-NiO–Ag/GCE at different scan rates from 10 to 100 mV s−1, in 10 μM Pb2+ with 0.1 M (ABS) at pH 6 at 100 mV s−1. (c) Linear plots of the reduction currents versus scan rate. (d) The effect of pH on the electrochemical response of the BF-NiO–Ag/GCE in 10 μM Pb2+. (e) The stability and repeatability of the BF-NiO–Ag/GCE for the detection of lead(II) by CV in 10 μM Pb2+ with 0.1 M (ABS) at pH 6, after keeping the electrode in air and at ambient temperature for 10 days. | |
Based on the CV and EIS results of the BF-NiO–Ag/GCE, it can be concluded that the increase of conductivity of the BF-NiO–Ag is due to the loading of Ag on the NiO surface and the increase of the total surface area compared to the F-NiO. Therefore, the adsorption and reduction of Pb2+ on the surface of BF-NiO–Ag/Ag significantly increases compared to the F-NiO. Moreover, the behavior of BF-NiO–Ag/GCE with the variation of scan rate in 10 μM Pb2+ solution with 0.1 M (ABS) (pH 6) was studied at different scan rates (from 10 to 100 mV s−1) (Fig. 6(b)). The linear increase of the second reduction peak current with the increase of scan rate shows that the electrode reaction is a surface-confined electrochemical process.24 The linear regression equations are articulated as Ipc (μA) = −0.09υ (mV s−1) − 7 (R2 = 0.995) (Fig. 6(c)). The effect of pH of the acetate buffer solution (ABS) on the reduction current on the surface of BF-NiO–Ag/GCE was studied at 100 mV s−1 in 10 μM lead(II) solution (Fig. 6(d)). The result indicates that the current increases with the increase of pH until pH 6, but decreases in alkaline solution. The stability and repeatability of the modified BF-NiO–Ag for the detection of lead(II) was also investigated by CV. The experiment was performed in 10 μM Pb2+ with 0.1 M (ABS) at pH 6, after keeping the electrode in air and at ambient temperature for 10 days. After four consecutive determinations of lead(II) concentration, the current response decreased only 2.61%, which confirms that the BF-NiO–Ag had an acceptable repeatability and stability for the detection of lead(II) (Fig. 6(e)).
The typical amperometric response of the BF-NiO–Ag/GCE to consecutive concentration changes of Pb(II) was investigated with differential pulse voltammetry (DPV) in 0.1 M ABS (pH 6.0) (Fig. 7(a)). The results clearly shows the dependence of the reduction peak current (Ipc) on the Pb2+ concentration. Based on the Ipc, the sensor calibration was performed three times and the calibration curve with the error bars is plotted in Fig. 7(b). A linear relation of the peak current and the Pb(II) concentration with a correlation coefficient (R2) of 0.991 is obtained. The range of the Pb(II) concentration is between 1.5 nM and 10 nM, with a sensitivity of −0.3 μA nM−1. The linear equation is Ipc (μA) = −0.3c (nM−1) − 0.2. The limit of detection (LOD) and the limit of quantification (LOQ) of the BF-NiO–Ag composite were calculated using the following equations:25
| |
 | (5) |
| |
 | (6) |
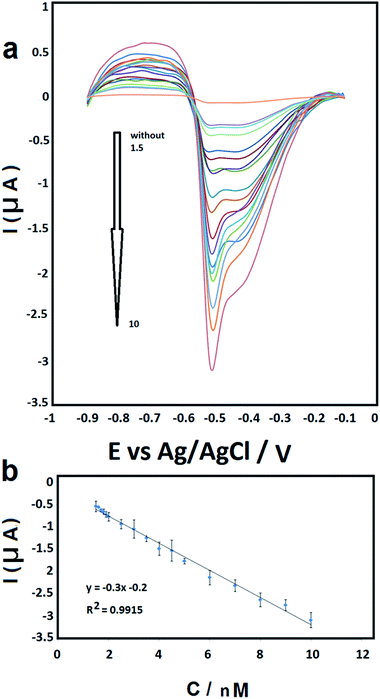 |
| | Fig. 7 (a) DPV curves of different concentration of Pb2+ in 0.1 M (ABS) (1.5, 1.6, 1.7, 1.8, 1.9, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 7, 8, 9 and 10 nM) at pH 6. (b) The calibration curve. Error bars represent ± standard deviation. | |
The LOD and LOQ (S/N = 3) are estimated as 0.062 nM and 0.207 nM for the linear segments, respectively.
The recovery experiments for examining the analytical application was also performed in different concentrations of Pb2+ (0.5, 5 and 10 nmol L−1) in 0.1 M ABS (pH 6.0). The average recoveries, recovery percentage and relative standard deviation (RSD) obtained from four determinations (n = 4) are given in Table 3a. The results confirm the feasibility of the proposed method for the quantitative detection of Pb2+ at certain concentration ranges.
Table 3 The detection of Pb2+ concentration in test samples (results based on four replicate determinations per sample) and (b) analysis of real samples
| a |
| Sample |
Added (nM) |
Found (nM) |
RSD% |
Recovery% |
| 1 |
0.5 |
0.49 |
3.36 |
99.85 |
| 2 |
5 |
4.94 |
2.13 |
98.87 |
| 3 |
10 |
9.95 |
1.32 |
99.57 |
| b |
| Added (nm) |
Found (nM) (mean) |
Recovery% |
RSD% |
LOD (nM) |
| Pipe water |
| 5 |
4.988 |
99.765 |
2.587 |
1.125 |
| 6 |
5.707 |
98.458 |
5.925 |
| 7 |
6.992 |
99.892 |
2.394 |
| 8 |
7.877 |
98.468 |
2.518 |
| 9 |
8.852 |
98.361 |
3.554 |
| 10 |
9.970 |
99.7 |
1.943 |
|
| Ground water |
| 5 |
4.917 |
98.350 |
4.056 |
1.825 |
| 6 |
5.885 |
98.083 |
4.918 |
| 7 |
6.919 |
98.846 |
2.572 |
| 8 |
7.903 |
98.787 |
3.664 |
| 9 |
8.935 |
99.286 |
3.061 |
| 10 |
9.853 |
98.535 |
3.282 |
3.4 Analysis of real samples
The analytical application of the BF-NiO–Ag/GCE was investigated with ground water and pipe water in University of Malaya, Kuala Lumpur. The peak current and the corresponding concentration of Pb2+ was determined by using prepared samples with ground water and pipe water with a concentration range between 5 and 10 nM Pb2+. The RSD, LOD for Pb2+ and recovery percentage are given in Table 3b. A linear relation between the peak current and the Pb(II) concentration of the pipe water and ground water are obtained (Fig. 8(a) and (b) respectively). The results show an acceptable recovery percentage and RSD. The recovery percentage of the ground water also confirms the presence of Pb2+. The selectivity of the BF-NiO–Ag/GCE as an electrochemical sensor for the detection of Pb2+ was determined by examining it in the presence of transition metal ions (Cd2+, Zn2+, Co2+, Fe2+, Mn2+, Ni2+, Cu2+, Pd2+, Ag+) and alkaline earth metal ion (K+). The DPV result of Pb2+ in the presence of 100-fold individual metal ions in the solution with respect to Pb2+ is given in Fig. 9. The result clearly shows very low interference effect of the mixture of metal ions toward the detection of Pb2+. Table 4 shows that the LOD of lead(II) for the BF-NiO–Ag/GCE is lower compared to the shuttle-like Fe2O3 NPs/GCE,26 Nafion, carbon nanotubes/benzo-18-crown-6/GCE27 and antimony film/GCE,28 but is comparable to the Fe3O4/3% RGO/GCE.29
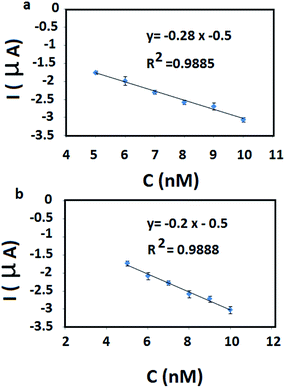 |
| | Fig. 8 Linear relation of the peak current and the Pb(II) concentration related pipe water and ground water (a) and (b) respectively. | |
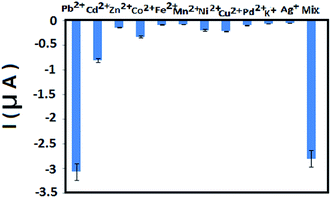 |
| | Fig. 9 The selectivity of the sensor by investigating it with interfering transition metal ions (Cd2+, Zn2+, Co2+, Fe2+, Mn2+, Ni2+, Cu2+, Pd2+, Ag+) and alkaline earth metal ion (K+). The electrochemical response of Pb2+ in the presence of 100-fold individual species in the solution with respect to Pb2+. The illustrated error bars represent the standard deviations of measurements taken from at least three independent experiments. | |
Table 4 A summary and comparison of the estimated LOD values from the present work with previous reports
| Type of electrode |
LOD (nM) |
Linear range (nM) |
Reference |
| Shuttle-like Fe2O3 NPs/GCE |
0.1 |
0.12–40 |
26 |
| Nafion, carbon nanotubes/benzo-18-crown-6/GCE |
1 |
1–30 |
27 |
| Fe3O4 nanospheres/multi-walled carbon NTs/GCE |
0.006 |
0.02–1.6 |
28 |
| Antimony film/GCE |
4.3 |
0.1–0.7 |
29 |
| Fe3O4/3% RGO/GCE |
0.082 |
0.05–1.5 |
30 |
| p-tert-Butylthiacalix[4]arene/GCE |
8 |
0.1–25 |
31 |
| NiO/Ag ball-flower like/GCE |
0.06 |
1.5–10 |
This work |
4 Conclusions
The BF-NiO–Ag was synthesized in the presence of L-glutamine and utilized as an electrochemical sensor for lead(II) detection. The XRD and EDX results confirmed that Ag+ was reduced to Ag metal and loaded on the F-NiO during the synthesis of the BF-NiO–Ag. The FESEM images clearly showed the effect of Ag loading and the presences of L-glutamine, on the morphology of the BF-NiO–Ag, which lead to a significant increase in the total surface area. The EIS result shows a larger value of the constant phase element of the BF-NiO–Ag/GCE compared to the other electrodes, and is a strong evidence for the presence of increased porosity, cracks and parapets. From the differential pulse voltammetry results, a linear working range for the concentration of lead(II) between 1.5 and 10 nM, with a LOD of 0.06 nM (S/N = 3) was obtained. The sensitivity of this linear segment is 0.3 μA nM−1.
Acknowledgements
The authors wish to thank Mojdeh Yeganeh for valuable discussion. This research is supported by High Impact Research MoE Grant UM.C/625/1/HIR/MoE/SC/04 from the Ministry of Education Malaysia, PRGS grant PR002-2014A, RP038C-15HTM, FP039 2016 and University Malaya Centre for Ionic Liquids (UMCiL).
References
- M. Tuzen, E. Melek and M. Soylak, J. Hazard. Mater., 2006, 136, 597 CrossRef CAS PubMed.
- M. Kobya, E. Demirbas, E. Senturk and M. Ince, Bioresour. Technol., 2006, 96, 518 Search PubMed.
- H. L. Needleman and P. J. Landrigan, Am. J. Public Health, 2004, 94, 8 CrossRef PubMed.
- Y. Zhang and S. B. Adeloju, Talanta, 2015, 137, 148 CrossRef CAS PubMed.
- P. Ugo, S. Zampieri, L. M. Moretto and D. Paolucci, Anal. Chim. Acta, 2001, 434, 291 CrossRef CAS.
- S. A. Asher, A. C. Sharma, A. V. Goponenko and M. M. Ward, Anal. Chem., 2003, 75, 1676 CrossRef CAS PubMed.
- D. Arunbabu, A. Sannigrahi and T. Jana, Soft Matter, 2011, 7, 2592 RSC.
- L. Cui, J. Wu and H. X. Ju, ACS Appl. Mater. Interfaces, 2014, 6, 16210 CAS.
- Z. Cai, J. T. Zhang, F. Xue, Z. Hong, D. Punihaole and S. A. Asher, Anal. Chem., 2014, 86, 4840 CrossRef CAS PubMed.
- E. A. Hutton, J. T. van Elteren, B. Ogorevc and M. R. Smyth, Talanta, 2004, 63, 849 CrossRef CAS PubMed.
- Y. Bonil, M. Brand and E. Kirowa-Eisner, Anal. Chim. Acta, 1999, 387, 85 CrossRef.
- Y. Bonfil, M. Brand and E. Kirowa-Eisner, Anal. Chim. Acta, 2002, 464, 99 CrossRef CAS.
- M. Brand, I. Eshkenazi and E. Kirowa-Eisner, Anal. Chem., 1997, 69, 4660 CrossRef CAS.
- V. K. Guptaa, M. L. Yolab, N. Atar, Z. Ustundag and A. O. Solak, Electrochim. Acta, 2013, 112, 541 CrossRef.
- M. L. Yola, T. Eren, H. İlkimen, N. Atar and C. Yenikaya, J. Mol. Liq., 2014, 197, 58 CrossRef CAS.
- M. L. Yola, N. Atar, M. S. Qureshi, Z. Ustunda and A. O. Solak, Sens. Actuators, B, 2012, 171–172, 1207 CrossRef CAS.
- V. K. Gupta, M. L. Yola, N. Atare, A. O. Solak, L. Uzunh and Z. Ustunda, Electrochim. Acta, 2013, 105, 149 CrossRef CAS.
- E. Kirowa-Eisner, M. Brand and D. Tzur, Anal. Chim. Acta, 1999, 385, 325 CrossRef CAS.
- X. Li, H. Wen, Q. Fu, D. Peng, J. Yu, Q. Zhang and X. Huang, Appl. Surf. Sci., 2016, 363, 7 CrossRef CAS.
- C. Blanco-Andujar, L. D. Tung and N. T. K. Thanh, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2010, 106, 553 RSC.
- O. Yamauchi, A. Odani and M. Takani, J. Chem. Soc., Dalton Trans., 2002, 18, 3411 RSC.
- L. C. Gruen, Biochim. Biophys. Acta, 1975, 386, 270 CrossRef CAS.
- W. H. Mulder, J. H. Sluyters, T. Pajkossy and I. Nyikos, J. Electroanal. Chem. Interfacial Electrochem., 1990, 285, 103 CrossRef CAS.
- G. Yang, X. Qu, M. Shen, C. Wang, Q. Qu and X. Hu, Microchim. Acta, 2008, 160, 275 CrossRef CAS.
- C. Xiang, Y. Zou, L. X. Sun and F. Xu, Talanta, 2007, 74, 206 CrossRef CAS PubMed.
- I. Krull and M. Swartz, LC-GC, 1998, 16, 922 CAS.
- P. Dai and Z. Yang, Microchim. Acta, 2012, 176, 109 CrossRef CAS.
- A. Sukeri and M. Jayaraman, Microchim. Acta, 2013, 180, 1065 CrossRef.
- S. B. Hocevar, I. Svancara, B. Ogorevc and K. Vytras, Anal. Chem., 2007, 79, 8639 CrossRef CAS PubMed.
- M. R. Mahmoudian, Y. Alias, W. J. Basirun, P. M. Woi, M. Sookhakian and F. Jamali-Sheini, Electrochim. Acta, 2015, 169, 126 CrossRef CAS.
- H. Zheng, Z. N. Yan, H. M. Dong and B. X. Ye, Sens. Actuators, B, 2007, 120, 603–609 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
W. J. Basiruna,
E. Zalnezhadc,
M. Ladana and
Y. Alias*a
*ab,
W. J. Basiruna,
E. Zalnezhadc,
M. Ladana and
Y. Alias*a











