
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple access toward 3-halo- and 3-nitro-pyrazolo[1,5-a]pyrimidines through a one-pot sequence†
Juan-Carlos Castillo,
Hernán-Alejandro Rosero and
Jaime Portilla *
*
Bioorganic Compounds Research Group, Department of Chemistry, Universidad de los Andes, Carrera 1 No. 18A-10, Bogotá 111711, Colombia. E-mail: jportill@uniandes.edu.co
First published on 31st May 2017
Abstract
Herein, a regioselective, time-efficient and one-pot route for the synthesis of diversely substituted 3-halo- and 3-nitropyrazolo[1,5-a]pyrimidines in good to excellent yields through a microwave-assisted process is provided. The reaction features a sequential cyclocondensation reaction of β-enaminones with NH-5-aminopyrazoles, followed by a regioselective electrophilic substitution with easily available electrophilic reagents. This methodology is distinguished by its short reaction times, high-yield, operational simplicity, broad substrate scope and pot-economy. Furthermore, these 3-functionalized heterocycles have been successfully used in the synthesis of 3-alkynyl and 3-aminopyrazolo[1,5-a]pyrimidines in yields up to 92%.
Introduction
Among the broad range of privileged structures, fused-pyrimidines represents one of the most prominent classes of heterocyclic scaffolds for novel synthetic drug discovery.1 Particularly, some synthetic purine analogues containing pyrazolo[1,5-a]pyrimidine nucleus have received augmented interest in recent years due to their broad range of applications in medicinal chemistry and drug design.2 For instance, the anxiolytic agent ocinaplon,3 and the hypnotic drugs indiplon,4 lorediplon,5 and zaleplon,6 among others potential drugs7 have this structural motif of pyrazolo[1,5-a]pyrimidine (Fig. 1). Too, some 3-halopyrazolo[1,5-a]pyrimidines have been reported as a nonbenzodiazepinoid class of antianxiety agents.8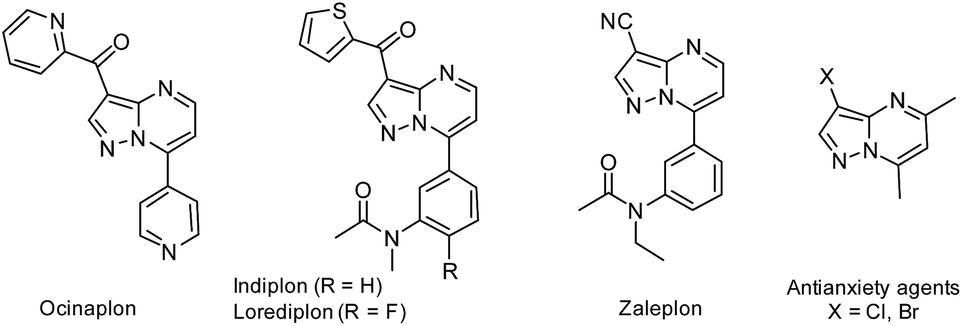 | ||
| Fig. 1 Examples of biologically active pyrazolo[1,5-a]pyrimidines. | ||
Besides medicinal applications, pyrazolo[1,5-a]pyrimidines and structural analogues have been found in the dyestuff industry (also used as fluorophores),9 organic light emitting diodes (OLEDs)10 and semiconductors materials.11 These promising biological and photophysical properties have prompted the development of various methods for the synthesis of functionalized pyrazolo[1,5-a]pyrimidines.12–16 The vast majority of these methods mostly involve the interaction between NH-5-aminopyrazoles with 1,3-bis-electrophilic reagents (e.g., β-dicarbonyl compounds,12 β-enaminones,13 β-haloenones,14 β-ketonitriles,15 among others16). Although such methodologies are generally reliable, many of them involved harsh reaction conditions, prolonged reaction times and tedious work-up, require use of additives, and often proceed with moderate yields. Particularly, ultrasound- and microwave-assisted reactions using β-enaminones as precursor have been successfully implemented (shorter reaction times and good yields). However, these methods suffer some limitations, including the use of potassium bisulphate as additive or Mg–Al hydrotalcite as solid base catalyst (Scheme 1a).17 Despite the success of different methods to obtain the interesting pyrazolo[1,5-a]pyrimidine core, we are surprised that its construction and subsequent functionalization in a single pot has remained largely unexplored. The introduced functional groups in this heterocyclic core are susceptible to undergoing successive post-functionalization reactions, which is important in drug discovery among other applications. For example, halogenation reactions have been conveniently explored (Scheme 1a)18 by obtaining suitable substrates for coupling reactions, however, the incorporation of others functional groups such as nitro or sulphonyl is less frequent. From the viewpoint of economy and environmental sustainability, developing operationally simple, high-yielding, and additive-free method for preparing 3-functionalized pyrazolo[1,5-a]pyrimidines in a one-pot manner is highly desirable.
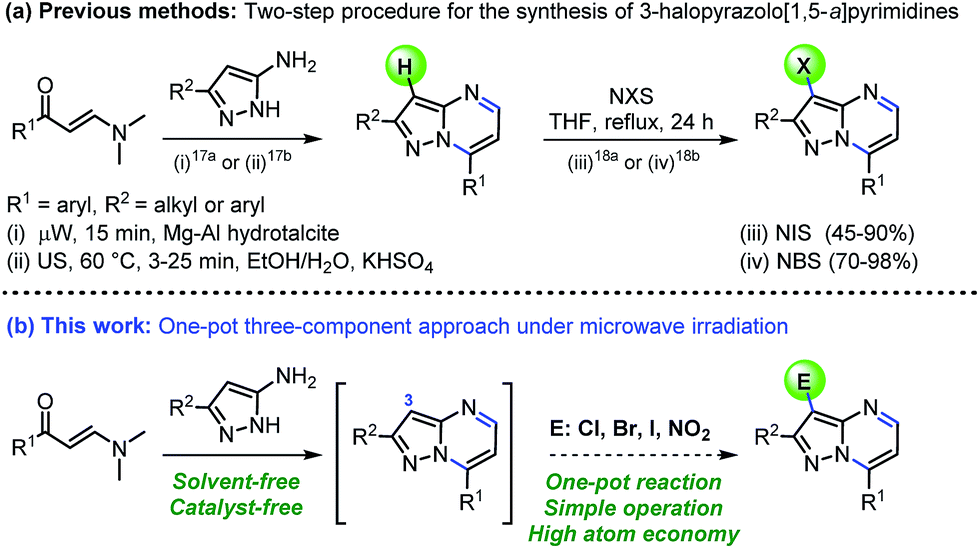 | ||
| Scheme 1 Synthesis of pyrazolo[1,5-a]pyrimidines under mild conditions. | ||
Recently, we reported a microwave-assisted synthesis of 6-aryldiazenylpyrazolo[1,5-a]pyrimidin-7-amines from α-arylhydrazinylidene-β-ketonitriles, and their use in the preparation of substituted pyrazolo[5,1-b]purines.15b Inspired by these results, and our continuing interest in the synthesis of biologically active N-heterocycles using green chemistry tools,19 we envisioned that microwave-assisted reactions of β-enaminones with NH-5-aminopyrazoles might generate the pyrazolo[1,5-a]pyrimidine core, which could be halogenated and nitrated at the C-3 position of the heterocycle. As a result, leading to the formation of 3-halo and 3-nitropyrazolo[1,5-a]pyrimidines through a catalyst- and additive-free one-pot approach (Scheme 1b). It is important to note that the cyclocondensation-electrophilic substitution sequence between β-enaminones, NH-5-aminopyrazoles and easily available electrophilic reagents would open a practical and eco-compatible synthetic approach to properly functionalized pyrazolo[1,5-a]pyrimidines through the formation of three new bonds in a single reaction vessel and the release of water as the unique byproduct. In this sense, the microwave-assisted one-pot synthesis has proved to be an extremely powerful tool because several synthetic transformations and bond-forming steps can be carried out in a single pot. Thereby, minimal chemical waste, time saving, and operational simplicity are achieve.20
Results and discussion
Considering the aforementioned aspects and prior to test the feasibility of the one-pot approach towards properly functionalized pyrazolo[1,5-a]pyrimidines, we started our investigation by examining a strategy to efficiently prepared β-enaminones under microwave irradiation. Although there are diverse protocols to obtain these 1,3-bis-electrophiles,21 the development of convenient, high-yielding and sustainable methods are still highly desirable due to their synthetic importance. As a test reaction, we submitted a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 mixture of acetophenone (1a) and N,N-dimethylformamide-dimethylacetal under microwave irradiation at 160 °C for 15 min in a sealed tube under solvent-free conditions. We were pleased to find that the desired β-enaminone 2a could be obtained with high purity and almost quantitative yield after simple evaporation of residues in the reaction (Scheme 2). Stimulated by this result, we examined the scope of this eco-compatible reaction with a variety of methylketones 1b–f, providing the β-enaminones 2b–f in nearly quantitative yields without further purification (Scheme 2).
:1.5 mixture of acetophenone (1a) and N,N-dimethylformamide-dimethylacetal under microwave irradiation at 160 °C for 15 min in a sealed tube under solvent-free conditions. We were pleased to find that the desired β-enaminone 2a could be obtained with high purity and almost quantitative yield after simple evaporation of residues in the reaction (Scheme 2). Stimulated by this result, we examined the scope of this eco-compatible reaction with a variety of methylketones 1b–f, providing the β-enaminones 2b–f in nearly quantitative yields without further purification (Scheme 2).
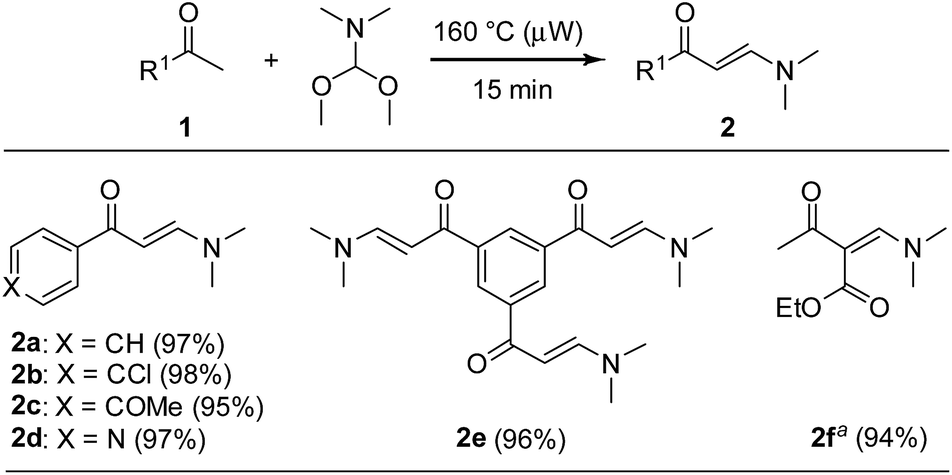 | ||
| Scheme 2 Solvent-free microwave-assisted synthesis of β-enaminones. Reaction conditions: methyl ketone 1 (8.0 mmol) and N,N-dimethylformamide-dimethylacetal (12.0 mmol); see ESI† for details. a 8.0 mmol of ethyl acetoacetate used. | ||
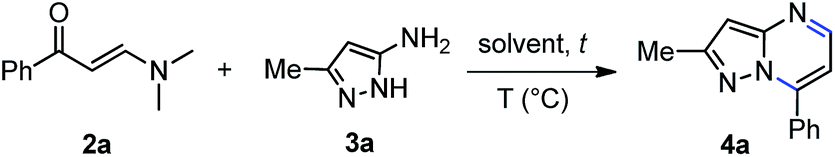
Subsequently, our exploratory study toward the synthesis of pyrazolo[1,5-a]pyrimidine core began with the model reaction between the phenylpropenone (2a) and 5-amino-3-methyl-1H-pyrazole (3a) (Table 1). We carried out the optimization by varying the temperature, the solvent and testing the effect of conventional heating versus microwave irradiation. Heating to reflux of an equimolar mixture of 2a and 3a in ethanol or toluene for 12 h did not lead to the pyrazolo[1,5-a]pyrimidine 4a (Table 1, entries 1 and 2, respectively). When we performed the reaction in DMF at higher temperature, provided compound 4a in poor yield (Table 1, entry 3). The structure of 4a was confirmed by 1H NMR, 13C NMR and mass spectroscopy. These preliminary results, showed that higher temperatures favor the formation of the pyrazolo[1,5-a]pyrimidine 4a. To further improve the reaction yield, we examined the influence of microwave irradiation on this reaction in different solvents such as ethanol, toluene, and DMF, and among these, ethanol gave the worst yield of compound 4a (10%) (Table 1, entries 4–6). In the reaction with ethanol as solvent, was not possible to increase the temperature above 150 °C due to system overpressure. In a modified protocol, we performed the solvent-free reaction of 2a and 3a at 180 °C for 1–2 min under microwave irradiation to afford the desired product 4a in high to nearly quantitative yield, after simple collection with mixtures of ethanol–water (Table 1, entries 7 and 8, respectively). Indeed, the reaction proceed in quantitative yield (Table 1, entry 9) after high vacuum removal of all volatiles of the reaction mixture. This result was evidenced by 1H NMR analysis of the crude product (ESI,† page S22). Therefore, we corroborated the feasibility of our hypothesis to carry out reliable functionalization reactions in the same reactor without further isolation of the pyrazolo[1,5-a]pyrimidine. In order to determine the specific microwave effect on accelerating the reaction vs. conventional heating, a pre-heated sand bath was used as a source of heat in comparative experiments (Table 1, entries 8 vs. 10). The low yield obtained with conventional heating at the same temperature for 30 min, indicated that the effect of microwave irradiation is not purely thermal.
|
||||
|---|---|---|---|---|
| Entry | Solvent | T (°C) | Time t | Yieldb (%) |
| a Reaction conditions: β-enaminone 2a (0.50 mmol) and NH-5-aminopyrazole 3a (0.50 mmol).b Isolated yield.c Conventional heating.d Run in 10 mL sealed tubes at a power of 300 W in anhydrous solvent (2.0 mL).e Run in 10 mL sealed tubes at a power of 300 W in the absence of solvent.f Conventional heating with a sand bath without solvent (fusion procedure).g Yield is given for the crude product. | ||||
| 1 | EtOH | Refluxc | 12 h | — |
| 2 | PhMe | Refluxc | 12 h | — |
| 3 | DMF | 150c | 12 h | 15 |
| 4 | EtOH | 150d | 3 min | 10 |
| 5 | PhMe | 180d | 3 min | 78 |
| 6 | DMF | 180d | 3 min | 80 |
| 7 | — | 180e | 1 min | 91 |
| 8 | — | 180e | 2 min | 95 |
| 9 | — | 180e | 2 min | Quantitativeg |
| 10 | — | 180f | 30 min | 51 |
With the optimized reaction conditions in hand (Table 1, entry 8), we then examined the scope of the reaction with a variety of β-enaminones 2a–f and NH-5-aminopyrazoles (3a–e). The results are summarized in Scheme 3. In general, the solvent-free microwave-assisted reaction of β-enaminones 2a–f with a wide range of NH-5-aminopyrazoles (3a–e) bearing both electron-donating and electron-withdrawing substituents regioselectively afforded pyrazolo[1,5-a]pyrimidines 4a–p in 85–97% yields, without tedious purification (Scheme 3). Almost no loss of efficiency was observed for the substrates tested, which indicated the low electronic influence of the substituents on the reactivity. This methodology also provided an interesting synthetic access to the valuable product 4q which could be used to synthesize pyrazolo[1,5-a]pyrimidine-based dendritic architectures with potential applications in pharmaceutical and medicinal chemistry, as well as in materials science.22 The structure of the tris-pyrazolo[1,5-a]pyrimidine 4q was solved by single-crystal X-ray diffraction analysis.23 Additionally, β-enaminone 2f exhibit two different types of carbonyl groups, which reacted chemo- and regioselectively with 3-methyl-1H-pyrazol-5-amine (3a) under optimized conditions, to give ethyl 2,7-dimethylpyrazolo[1,5-a]pyrimidine-6-carboxylate (4r) in 89% isolated yield. Also, aminopyrazol 3f substituted with an electron-withdrawing group (R: 4-NO2Ph) reacted efficiently with β-enaminone 2c (Ar: 4-MeOPh) under optimized conditions, giving the corresponding pyrazolo[1,5-a]pyrimidine 4s in 87% isolated yield (Scheme 3). Notably, this synthetic methodology to obtain pyrazolo[1,5-a]pyrimidines 4a–s was distinguished by its broad substrate scope, operational simplicity, no additives, no solvent, no excess of substrate is used, short reaction times, high-yielding without chromatographic purification, and eco-compatibility in terms of energy and waste.
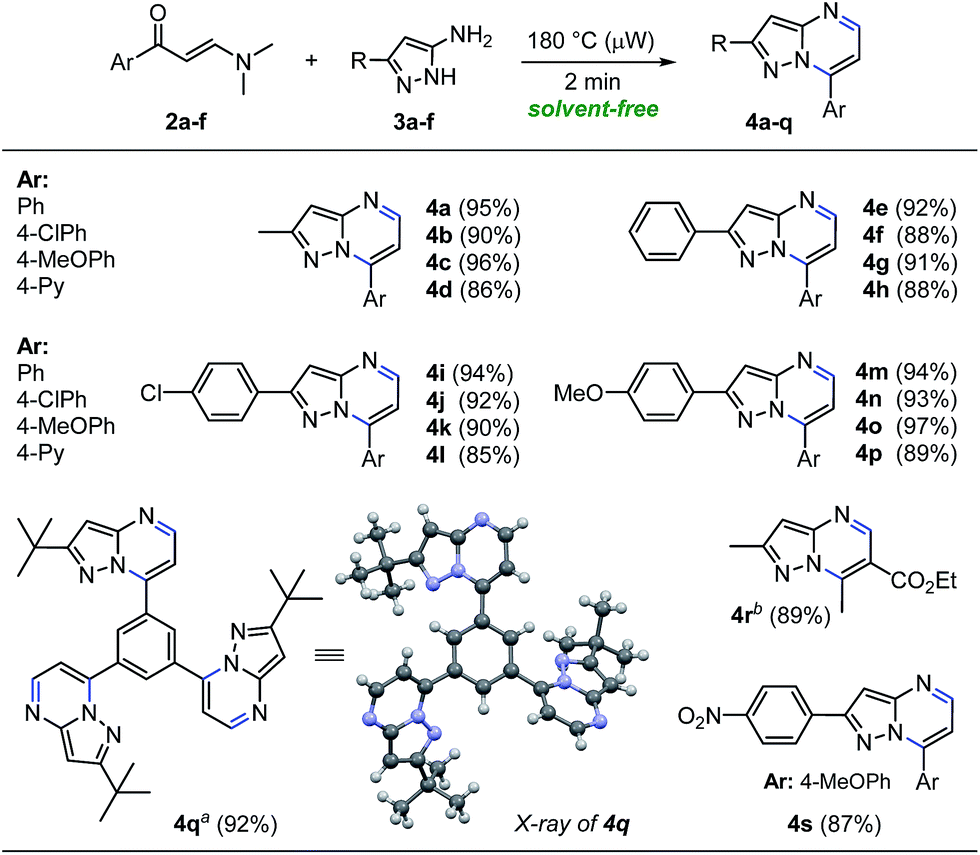 | ||
| Scheme 3 Microwave-assisted synthesis of β-enaminones and pyrazolo[1,5-a]pyrimidines. Reaction conditions: β-enaminone 2 (0.50 mmol) and NH-5-aminopyrazole 3 (0.50 mmol) at 180 °C for 2 min under solvent-free conditions. a 1.50 mmol of 3-(tert-butyl)-1H-pyrazol-5-amine used. b 0.50 mmol of β-enaminoester used. | ||
Halogenated pyrazolo[1,5-a]pyrimidines belong to a class of important building blocks and versatile synthons, which could be converted into more complex molecules by cross-coupling reactions.25 Nevertheless, existing methods to synthesize 3-halopyrazolo[1,5-a]pyrimidines are scarcely reported. Most of these involve the cyclocondensation reaction between 1,3-bis-electrophilic reagents and 5-amino-4-halopyrazoles25a or by multistep sequences that proceed with the preformation of pyrazolo[1,5-a]pyrimidines and subsequent halogenation reaction with diverse halogen sources such as N-halosuccinimides,18,24 iodine monochloride,25b bromine25c and 1-chlorobenzotriazole.25d The drawbacks of these strategies include: long reaction times, large volume of hazardous solvents, as well as difficult protocols. Therefore, and continuing with the implementation of greener and economic strategies, the development of an efficient and practical one-pot approach to halogenated pyrazolo[1,5-a]pyrimidines that allows use of readily available starting materials, short reaction times, and easier workup is highly desirable. Consequently, and according to the noteworthy results obtained in our former synthetic method (Scheme 3), we planned to study a consecutive approach toward 3-halopyrazolo[1,5-a]pyrimidine 5 in a single reaction vessel, starting from an equimolar mixture of β-enaminone 2 and NH-5-aminopyrazole (3), and finishing with the addition of 1 equiv. of N-iodosuccinimide (NIS). Usually, halogenation reactions have been carried out successfully in chlorinated solvents, thus we decided to use 1,2-dichloroethane (EDC) instead of dichloromethane (DCM) to evaluate the effect of temperature. In addition, the first step reaction was performed using conditions analogous to those previously optimized (Table 1, entry 8), while the second step was optimized as indicated below (Table 2).
|
|||
|---|---|---|---|
| Entry | T (°C) | Time t | Yieldb (%) |
| a Reaction conditions: β-enaminone 2a (0.25 mmol) and NH-5-aminopyrazole 3a (0.25 mmol) at 180 °C under microwave irradiation for 2 min; after cooling by airflow addition of NIS (0.25 mmol) and EDC (2.0 mL).b Isolated yield.c Run in 10 mL sealed tubes under microwave irradiation.d Run in a 10 mL round-bottom flask with 2.0 mL of EDC under reflux (not one-pot procedure).e Run in 10 mL sealed tubes at 25 °C. | |||
| 1 | 80c | 5 min | 96 |
| 2 | 60c | 5 min | 94 |
| 3 | 40c | 5 min | 95 |
| 4 | 30c | 5 min | 62 |
| 5 | 40c | 2 min | 70 |
| 6 | Refluxd | 20 min | 73 |
| 7 | 25e | 20 min | 94 |
| 8 | 25e | 10 min | 76 |
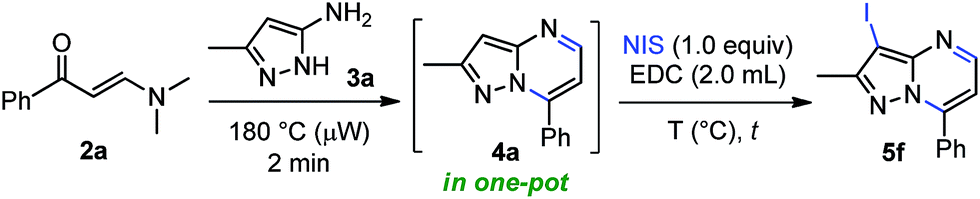
This approach is termed one-pot when the last reaction occurred in the same microwave tube. In this sense, we carried out the optimization by varying the temperature and testing the effect of microwave irradiation in contrast to conventional heating. The reaction of the crude product 4a with 1 equiv. of NIS in 2.0 mL of EDC using temperatures between 30–80 °C for 2–5 minutes under microwave irradiation afforded the desired product 5f in good to nearly quantitative yield (Table 2, entries 1–5). However, the reaction at reflux was not completely satisfactory due to loss of precursor mass during the transfer process from the microwave tube to the round-bottom flask (Table 2, entry 6). Pleasingly, this one-pot synthesis could be performed at room temperature without microwave heating, affording the 3-iodoheterocycle 5f in good to excellent yields and short reaction times (Table 2, entries 7 and 8, respectively). These results evidenced the high electron density of carbon atom at position 3 of the pyrazolo[1,5-a]pyrimidine ring (see 13C NMR spectra of compounds 4a–s in the ESI†). In consequence, the optimum conditions for the regioselective 3-iodination of the pyrazolo[1,5-a]pyrimidine 4a were achieved using a stoichiometric amount of N-iodosuccinimide by a simple and energy-saving protocol (Table 2, entry 7). With the optimized reaction conditions, we explored the scope of this one-pot synthesis with diverse N-halosuccinimides and pyrazolo[1,5-a]pyrimidines preformed. The microwave-assisted reaction between β-enaminones 2 and NH-5-aminopyrazoles (3) afforded the crude products 4, which were subsequently halogenated using diverse N-halosuccinimides in 1,2-dichloroethane as solvent at room temperature for 20 min. As shown in Scheme 4, the 3-halopyrazolo[1,5-a]pyrimidines 5a–i were successfully obtained in 89–96% yields. The structure of the 3-iodopyrazolo[1,5-a]pyrimidine 5g was solved by single-crystal X-ray diffraction analysis.26
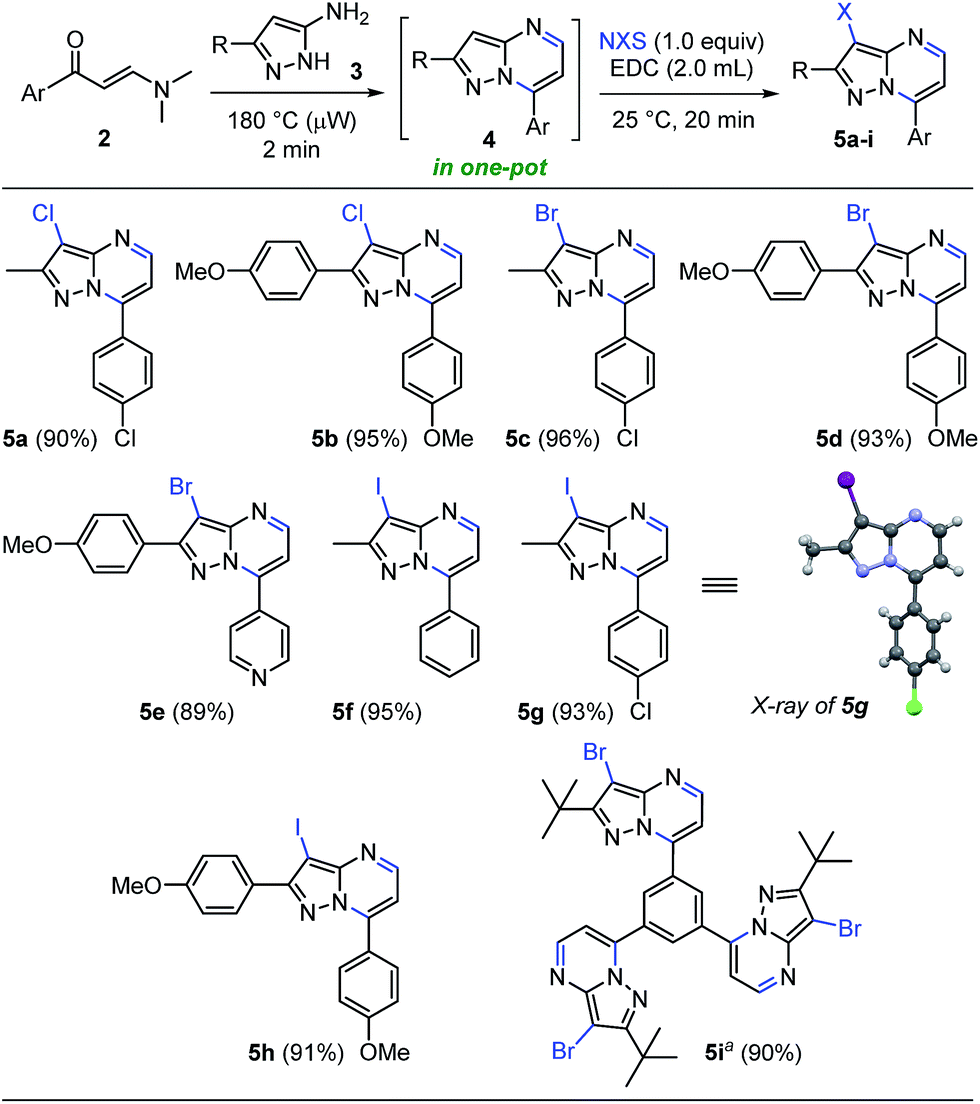 | ||
| Scheme 4 Synthesis of 3-halopyrazolo[1,5-a]pyrimidines. Reaction conditions: β-enaminone 2 (0.50 mmol) and NH-5-aminopyrazole 3 (0.50 mmol) at 180 °C under microwave irradiation for 2 min; after cooling by airflow addition of N-halosuccinimide (0.50 mmol) and EDC (2.0 mL) at 25 °C for 20 min. a From 4q, 1.50 mmol of NBS used. | ||
Although some publications are devoted to the linear synthesis of halogenated pyrazolo[1,5-a]pyrimidines, the regioselective incorporation of others functional groups such as nitroso, nitro, sulphonyl and amine have been poorly explored. In fact, most of the existing reports are patents. Thereby, we propose the introduction of diverse functional groups on this heterocyclic core through a one-pot approach according to our former results, starting from β-enaminone 2 (or another 1,3-bis-electrophilic reagents) and the appropriate NH-5-aminopyrazole (3). Particularly, nitroaromatic compounds have been employed to develop not only bioreductive prodrugs27a but also fluorescent probes for the detection of nitroreductase and tumor hypoxia.27b,c These results encouraged us to investigate the cyclocondensation-nitration sequence under microwave irradiation in order to afford 3-nitropyrazolo[1,5-a]pyrimidines 6 within a single reaction vessel (Scheme 5). Our initial testing of the nitration reaction proceeded under conventional heating, attempting to carry out the reaction in the same microwave tube. The crude product 4 with 4 equiv. of the nitrating mixture (HNO3:H2SO4, 2:1) in 2.0 mL of EDC or water as solvent, and with temperatures between 5–45 °C for 1–3 hours, did not work without microwave irradiation. However, microwave-assisted reactions at 60 °C for 10 min under solvent-free conditions, enabled the regioselective formation of the 3-nitrosubstituted products 6a–d in yields up to 89% (Scheme 5). Remarkably, these 3-nitropyrazolo[1,5-a]pyrimidines 6 could be used as potential colorimetric or fluorescent probes for the detection of nitroreductase and hypoxic tumor cell imaging, because the reduced systems have interesting photophysical properties, as well as their unsubstituted precursors 4 (Image 1 in the ESI†).
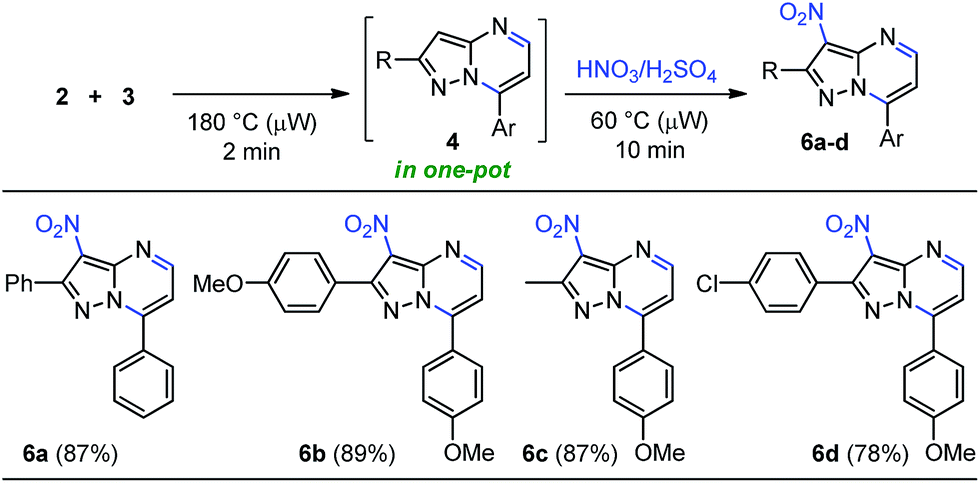 | ||
| Scheme 5 Synthesis of 3-nitropyrazolo[1,5-a]pyrimidines. Reaction conditions: β-enaminone 2 (0.50 mmol) and NH-5-aminopyrazole 3 (0.50 mmol) at 180 °C under microwave irradiation for 2 min; after cooling by airflow addition of nitric acid (2.0 mmol) and sulfuric acid (1.0 mmol) at 60 °C under solvent-free microwave irradiation for 10 min. | ||
Notably, the synergy between functionality and modularity is the key aspect of this consecutive one-pot approach towards the syntheses of properly functionalized pyrazolo[1,5-a]pyrimidines 5a–i and 6a–d. This is important because such structures are attractive scaffolds for medicinal chemistry and can be prepared by convenient and eco-compatible one-pot synthetic methods. For example, the chemically manipulating the halogen and nitro groups in the functionalized heterocycles 5 and 6 was briefly explored. Because of easy availability of water, we choose to use this green solvent and microwave heating in order to start our studies using some iodine-derivatives 5. Gratifyingly, the palladium-catalyzed Sonogashira cross-coupling reaction of phenylacetylene with 3-iodopyrazolo[1,5-a]pyrimidines 5f and 5g using 10% Pd/C–PPh3–CuI as a catalyst system, afforded the corresponding 3-phenylethynyl derivatives 7a and 7b in 75% and 60% yields, respectively (Scheme 6, right). Further applications of this methodology is under active investigation due to their interesting extended π-conjugation.27c Furthermore, the palladium-catalyzed reduction of representative 3-nitropyrazolo[1,5-a]pyrimidines 6a and 6b was explored. As expected, the reduction of the nitro group was achieved at 25 °C under H2 atmosphere at ambient pressure using Pd/C as catalyst in ethanol, giving the corresponding pyrazolo[1,5-a]pyrimidin-3-amines 8a and 8b in good yields, which have inherent synthetic utility (Scheme 6, left).
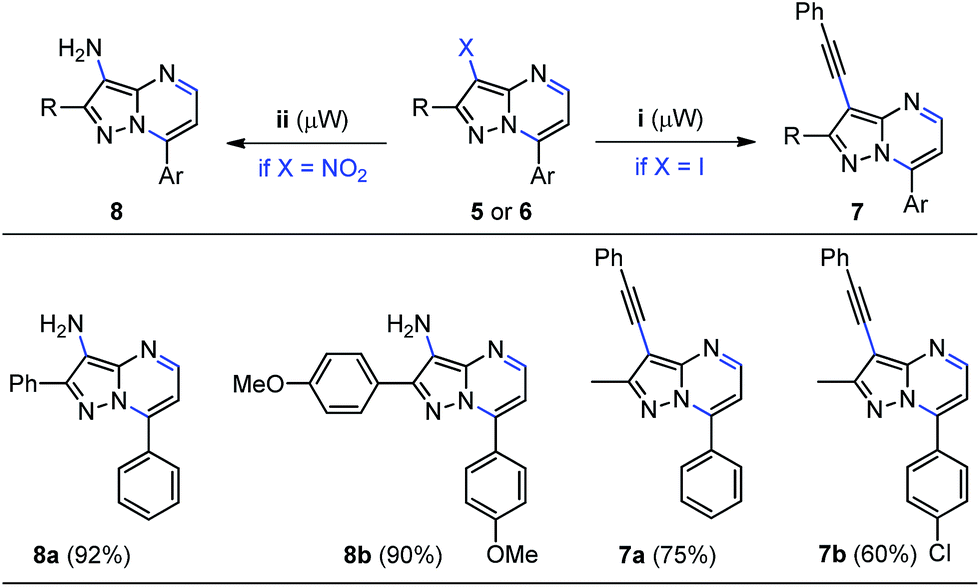 | ||
| Scheme 6 Post-functionalization of 3-halo- and 3-nitropyrazolo[1,5-a]pyrimidines. Reaction conditions: (i) substrate 5f or 5g (0.25 mmol), phenylacetylene (0.38 mmol), 1:4:2 ratio of 10% Pd/C–PPh3–CuI and Et3N (5.0 equiv.) in water (2.0 mL) at 80 °C under microwave irradiation for 1 h; (ii) substrate 6a or 6b (0.50 mmol) and 10% Pd/C (5 wt%) under an H2 atmosphere in EtOH (5.0 mL) at 25 °C for 3 h. | ||
Conclusions
In summary, β-enaminones and pyrazolo[1,5-a]pyrimidines can be easily and conveniently prepared in excellent yields from methylketones via microwave irradiation under catalyst- and solvent-free conditions, and easy purification. These protocols provide a better and practical alternative to existing procedures. Likewise, we have developed an efficient and expeditious microwave-assisted synthesis of functionalized pyrazolo[1,5-a]pyrimidines in good to excellent yields with the formation of three new bonds in a one-pot manner. Interestingly, the time-efficient construction of a pyrazolo[1,5-a]pyrimidine core and subsequent regioselective functionalization on the pyrazole moiety with functional groups such as halogen or nitro in a single reaction vessel has not been previously reported. This cyclocondensation-electrophilic substitution sequence offers marked improvements in terms of general applicability, efficacy, simplicity, and eco-compatibility. Also, this one-pot methodology could be used to introduce others electrophilic substituents on the pyrazolo[1,5-a]pyrimidine core. Finally, some representative post-functionalization reactions have also been developed, affording heteroamines and π-extended heterocycles of valuable synthetic and medicinal interest.Acknowledgements
The authors are grateful for the financial support from Universidad de los Andes and Colombian Institute for Science and Research (COLCIENCIAS). We also acknowledge Edwin Guevara for acquiring the high-resolution mass spectra, Dr Eduardo C. Escudero-Adan (X-ray Diffraction Unit of Institute of Chemical Research of Catalonia (ICIQ) in Tarragona-Spain) and Dr Mario Macias (Universidad de los Andes) for their help with the analysis of the X-ray crystallographic data.Notes and references
- (a) J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley-Blackwell, West Sussex, United Kingdom, 5th edn, 2010 Search PubMed; (b) S. Schenone, M. Radi, F. Musumeci, C. Brullo and M. Botta, Chem. Rev., 2014, 114, 7189 CrossRef CAS PubMed.
- (a) A. Damont, V. Médran-Navarrete, F. Cacheux, B. Kuhnast, G. Pottier, N. Bernards, F. Marguet, F. Puech, R. Boisgard and F. Dollé, J. Med. Chem., 2015, 58, 7449 CrossRef CAS PubMed; (b) S. Cherukupalli, R. Karpoormath, B. Chandrasekaran, G. A. Hampannavar, N. Thapliyal and V. N. Palakollu, Eur. J. Med. Chem., 2017, 126, 298 CrossRef CAS PubMed.
- A. Lippa, P. Czobor, J. Stark, B. Beer, E. Kostakis, M. Gravielle, S. Bandyopadhyay, S. J. Russek, T. T. Gibbs, D. H. Farb and P. Skolnick, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7380 CrossRef CAS PubMed.
- L. Xu, J. Wang, B. Xiao, J. Yang, Y. Liu and R. Huang, Bioorg. Med. Chem. Lett., 2012, 22, 963 CrossRef CAS PubMed.
- L. Horoszok, T. Baleeiro, F. D'Aniello, S. Gropper, B. Santos, A. Guglietta and T. Roth, Hum. Psychopharmacol Clin. Exp., 2014, 29, 266 CrossRef CAS PubMed.
- J. Jablan, G. Szalontai and M. Jug, J. Pharm. Biomed. Anal., 2012, 71, 35 CrossRef CAS PubMed.
- (a) M. Zhao, H. Ren, J. Chang, D. Zhang, Y. Yang, Y. He, C. Qi and H. Zhang, Eur. J. Med. Chem., 2016, 119, 183 CrossRef CAS PubMed; (b) C. Ravi, A. Qayum, D. Mohan, S. K. Singh and S. Adimurthy, Eur. J. Med. Chem., 2017, 126, 277 CrossRef CAS PubMed; (c) S. Cherukupalli, R. Karpoormath, B. Chandrasekaran, G. A. Hampannavar, N. Thapliyal and V. N. Palakollu, Eur. J. Med. Chem., 2017, 126, 298 CrossRef CAS PubMed.
- W. E. Kirkpatrick, T. Okabe, I. W. Hillyard, R. K. Robins, A. T. Dren and T. Novinson, J. Med. Chem., 1977, 20, 386 CrossRef CAS PubMed.
- (a) A. Z. Sayed, M. S. Aboul-Fetouh and H. S. Nassar, J. Mol. Struct., 2012, 1010, 146 CrossRef CAS; (b) H. Kim, A. Jo, Y. Lee, Y. S. Hwang and S. B. Park, Chem. Commun., 2016, 52, 7822 RSC.
- M. A. El-Gahami, A. E. M. Mekky, T. S. Saleh and A. S. Al-Bogami, Spectrochim. Acta, Part A, 2014, 129, 209 CrossRef CAS PubMed.
- A. G. Al-Sehemi, A. Irfan and A. M. Fouda, Spectrochim. Acta, Part A, 2013, 111, 223 CrossRef CAS PubMed.
- (a) A. V. Ivachtchenko, E. S. Golovina, M. G. Kadieva, V. M. Kysil, O. D. Mitkin, S. E. Tkachenko and I. M. Okun, J. Med. Chem., 2011, 54, 8161 CrossRef CAS PubMed; (b) J. Portilla, J. Quiroga, M. Nogueras and J. Cobo, Tetrahedron, 2012, 68, 988 CrossRef CAS.
- (a) H. A. Abdel-Aziz, T. S. Saleh and H. S. A. El-Zahabi, Arch. Pharm., 2010, 343, 24 CAS; (b) A. O. Abdelhamid, T. T. El-Idreesy, N. A. Abdelriheem and H. R. M. Dawoud, J. Heterocycl. Chem., 2016, 53, 710 CrossRef CAS.
- K. Shekarrao, P. P. Kaishap, V. Saddanapu, A. Addlagatta, S. Gogoi and R. C. Boruah, RSC Adv., 2014, 4, 24001 RSC.
- (a) J. Sun, J.-K. Qiu, B. Jiang, W.-J. Hao, C. Guo and S.-J. Tu, J. Org. Chem., 2016, 81, 3321 CrossRef CAS PubMed; (b) J. C. Castillo, D. Estupiñan, M. Nogueras, J. Cobo and J. Portilla, J. Org. Chem., 2016, 81, 12364 CrossRef CAS PubMed.
- (a) J. Quiroga, J. Portilla, R. Abonia, B. Insuasty, M. Nogueras and J. Cobo, Tetrahedron Lett., 2008, 49, 6254 CrossRef CAS; (b) H. Bel Abed, O. Mammoliti, G. Van Lommen and P. Herdewijn, Tetrahedron Lett., 2013, 54, 2612 CrossRef CAS; (c) X. Zhang, Y. Song, L. Gao, X. Guo and X. Fan, Org. Biomol. Chem., 2014, 12, 2099 RSC; (d) P. Saikia, S. Gogoi and R. Boruah, J. Org. Chem., 2015, 80, 6885 CrossRef CAS PubMed; (e) P. Golubev, E. A. Karpova, A. S. Pankova, M. Sorokina and M. A. Kuznetsov, J. Org. Chem., 2016, 81, 11268 CrossRef CAS PubMed.
- Examples of environmentally friendly conditions, see: (a) M. Mokhtar, T. S. Saleh and S. N. Basahel, J. Mol. Catal. A: Chem., 2012, 353, 122 CrossRef; (b) S. Kaping, U. Kalita, M. Sunn, L. I. Singha and J. N. Vishwakarma, Monatsh. Chem., 2016, 147, 1257 CrossRef CAS.
- (a) L. Yin and J. Liebscher, Synthesis, 2004, 14, 2329 Search PubMed; (b) M. A. P. Martins, E. Scapin, C. F. Frizzo, F. A. Rosa, H. G. Bonacorso and N. Zanatta, J. Braz. Chem. Soc., 2009, 20, 205 CrossRef CAS.
- (a) J. Galvez, J. C. Castillo, J. Quiroga, M. Rajzmann, J. Rodriguez and Y. Coquerel, Org. Lett., 2014, 16, 4126 CrossRef CAS PubMed; (b) J. Orrego-Hernández, J. Cobo and J. Portilla, Eur. J. Org. Chem., 2015, 5064 CrossRef; (c) J. Orrego-Hernández, N. Nuñez-Dallos and J. Portilla, Talanta, 2016, 152, 432 CrossRef PubMed; (d) J. C. Castillo, J. Orrego-Hernández and J. Portilla, Eur. J. Org. Chem., 2016, 3824 CrossRef CAS.
- (a) V. Polshettiwar and R. S. Varma, Acc. Chem. Res., 2008, 41, 629 CrossRef CAS PubMed; (b) Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC.
- For precedents, see: (a) N. D. Koduri, H. Scott, B. Hileman, J. D. Cox, M. Coffin, L. Glicksberg and S. R. Hussaini, Org. Lett., 2012, 14, 440 CrossRef CAS PubMed; (b) Y.-W. Kang, S. J. Han and H.-Y. Jang, Org. Lett., 2016, 18, 272 CrossRef CAS PubMed.
- (a) L.-L. Lai, C.-H. Lee, L.-Y. Wang, K.-L. Cheng and H.-F. Hsu, J. Org. Chem., 2008, 73, 485 CrossRef CAS PubMed; (b) M. Nasr-Esfahani, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, V. Mirkhani, S. Tangestaninejad and H. A. Rudbari, J. Org. Chem., 2014, 79, 1437 CrossRef CAS PubMed.
- CCDC 1542479 (4q) contains the supplementary crystallographic data for this paper.
- (a) D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993 CrossRef CAS PubMed; (b) L. Yin and J. Liebscher, Synthesis, 2005, 1, 131 Search PubMed.
- (a) R. A. Nugent, C. J. Dunn, N. D. Staite, M. J. Murphy, S. T. Schlachter, T. Stephen, D. G. Aspar, S. K. Shields and L. A. Galinet, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 109, 229 CrossRef; (b) R. K. Robins, D. E. Brien and T. Novinson, US. Pat., Appl. Publ., US 19784093617 A1, 1978; (c) Z. Luo, D. Slee, J. E. Tellew, J. Williams and X. Zhang, PCT Int. Appl, WO 2005063755 A1, 2005; (d) J. D. Albright and J. P. Dusza, US. Pat., Appl. Publ, US 19794178449 A1, 1979.
- CCDC 1542481 (5g) contains the supplementary crystallographic data for this paper.†.
- (a) L. Cui, Y. Zhong, W. Zhu, Y. Xu, Q. Du, X. Wang, X. Qian and Y. Xiao, Org. Lett., 2011, 13, 928 CrossRef CAS PubMed; (b) Z. Li, X. Li, X. Gao, Y. Zhang, W. Shi and H. Ma, Anal. Chem., 2013, 85, 3926 CrossRef CAS PubMed; (c) A. Chevalier, Y. Zhang, O. M. Khdour, J. B. Kaye and S. M. Hecht, J. Am. Chem. Soc., 2016, 138, 12009 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, characterization data, X-ray structures of 4q, 5g, and NMR spectra. CCDC 1542479 and 1542481. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04336h |
| This journal is © The Royal Society of Chemistry 2017 |