
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled nanoparticles based on poly(ethylene glycol)–oleanolic acid conjugates for co-delivery of anticancer drugs
Yingsa Wanga,
Yajie Tiana,
Pengbo Zhua,
Yunyun Maa,
Jing He*ab and
Jiandu Lei *ab
*ab
aMOE Engineering Research Center of Forestry Biomass Materials and Bioenergy, Beijing Forestry University, Beijing, 100083, China. E-mail: hejing2008@sina.com; Fax: +86-10-62336349; Tel: +86-10-62336349
bBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing, 100083, China
First published on 7th June 2017
Abstract
Oleanolic acid (OA) has shown promising antitumor activity. However, it is difficult to formulate because of its poor water solubility and low bioavailability. In this study, self-assembled nanoparticles based on poly(ethylene glycol)–oleanolic acid conjugates (mPEG–OA) for co-delivery of anticancer drugs were developed to enhance the properties of OA. The mPEG–OA was synthesized by conjugating oleanolic acid (OA) to methoxy poly(ethylene glycol) carboxylic acid (mPEG–COOH). Then hydroxycamptothecin (HCPT), another anticancer drug was encapsulated into the mPEG–OA conjugates by the nanoprecipitation method. The mPEG–OA/HCPT nanoparticles (NPs) had an appropriate size, high drug loading of OA and HCPT, high stability, slow drug release and low cytotoxicity. In vivo studies revealed that the nanoparticles significantly improved the antitumor activity compared with free drugs. As no systemic toxicity was observed, the mPEG–OA NPs can serve as a promising drug delivery system for cancer therapy.
1. Introduction
Cancer is one of the most prevalent and deadly diseases worldwide.1 Existing chemotherapy is inadequate, with problems such as the increasing number of multidrug-resistant cancers and side effects of anticancer drugs.2 Drug delivery systems have been attracting considerable interest for a long time to improve the efficacy, reduce the toxicity and reduce the frequency of doses.3 Oleanolic acid (OA), (3β)-3-hydroxyolean-12-en-28-oic acid, is widely present in many Asian herbs.4 Although OA has been shown to have many significant biological actions including hepatoprotective, anti-inflammatory, antioxidant and antitumor effects, the clinical application of OA has been limited by its poor aqueous solubility and extremely low dissolution rate in the gastrointestinal tract.5–7Drug conjugation, as a prodrug strategy, provides the possibility to overcome these shortcomings, especially when combined with nanotechnology.8 Nanotechnology has evident merits in tumor therapy because of the improved accumulation of anticancer drugs in tumor tissues via the enhanced permeability and retention (EPR) effect.9–11 EPR is a passive targeting strategy for solid tumors mainly based on particle size. Angiogenic tumors tend to develop a permeable vasculature and can selectively recruit circulating small particles. The hydrophobic drugs are usually modified by hydrophilic agents, such as polyethylene glycol (PEG),12 which serves as a versatile prodrug platform. Linear PEG is most widely used in drug modification due to its simple synthetic steps.13
Although PEG-drug conjugation has made significant improvements, such as improved water solubility, prolonged drug release and low toxicity, it suffers from other problems such as large dosage and drug resistance. Hence polytherapy is often used in the treatment of cancer. Self-assembled nanoparticles for drug co-delivery have gained considerable attention.14–16 The synergistic combination of two or more drugs can overcome undesirable toxicity and other side effects and consequently reduces the dosage of each agent.17
In this study, we designed a self-assembled nanoparticle based on poly(ethylene glycol)–oleanolic acid conjugates (mPEG–OA) as a drug delivery system. On the one hand, the anticancer drug OA interacted with methoxy poly(ethylene glycol) carboxylic acid (mPEG–COOH) via ester linkages and mPEG–OA was an amphiphilic structure which was able to self-assemble into micelles. On the other hand, hydroxycamptothecin (HCPT) was entrapped into mPEG–OA to achieve a combination therapy (Scheme 1). Finally, the biophysical properties of the mPEG–OA/HCPT nanoparticles (NPs), including particle size, loading efficiency, in vitro drug release kinetics, cellular uptake, in vivo antitumor activities and side effects were investigated.
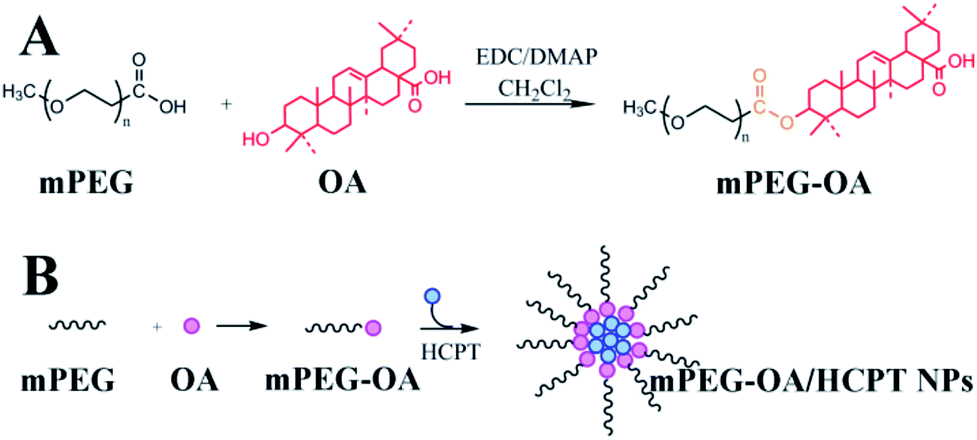 | ||
| Scheme 1 Synthesis of mPEG–OA (A). Illustration of mPEG–OA co-delivery of HCPT (B). | ||
2. Materials and methods
2.1. Materials
Methoxy poly(ethylene glycol) carboxylic acid (mPEG–COOH, Mw = 5000) was purchased from Jenkem Technology Co., Ltd (Beijing, China). It is an FDA and EU approved food grade material. OA and HCPT were purchased from Chengdu Preferred Biotechnology Co., Ltd (Sichuan, China). 4-Dimethylaminopyridin (DMAP), dimethyl sulfoxide (DMSO) and 1-ethyl-3-(3-dimethy-laminopropyl)-carbodiimide hydrogen chloride (EDC HCl) were purchased from J&K Chemical Reagent Co., Ltd (Beijing, China).Penicillin and streptomycin, Gibco Dulbecco's Phosphate-Buffered Saline (DPBS), and Gibco Dulbecco's Modified Eagle's Medium (DMEM) were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from HyClone. Cell-Counting Kit-8 (CCK-8) was supplied by Dojindo Laboratories. B16 murine melanoma (B16) cells were obtained from the Peking University Health Science Center (Beijing, China). The cells were cultured in the listed medium: DMEM with 10% FBS and 1% streptomycin–penicillin and maintained in an incubator supplied with a 5% CO2–95% air humidified atmosphere at 37 °C.
Female C57BL/6 mice, 6–7 weeks of age, were purchased from the National Institute for the Control of Pharmaceutical and Biological Products. All of the animal experiments were in accordance with the guidelines set by the National Institutes of Health (NIH Publication No. 85-23, revised 1985) and were approved by the Experimental Animal Ethics Committee, Beijing.
2.2. Synthesis of mPEG–OA conjugate
The mPEG–OA conjugate was synthesized via an esterification reaction by using DMAP as a catalyst and EDC as a coupling reagent in dichloromethane. Briefly, mPEG–COOH (2.0 g, 0.4 mmol) and OA (1.5 g, 3.2 mmol) were dissolved in 20 mL of dichloromethane. The solution was cooled to 0 °C and EDC (0.6 g, 1.8 mmol) and DMAP (0.4 g, 3.2 mmol) were added to the mixture. The mixture was stirred at 0 °C for 1 h and at room temperature overnight. Then the solvent was evaporated under vacuum. The residue was dissolved in 10 mL of dichloromethane and the crude product was precipitated with ethyl ether (100 mL). After filtration the solids were washed with ethyl ether (2 × 100 mL), and dried under vacuum to give mPEG–OA.2.3. Preparation of mPEG–OA/HCPT NPs
Nanoparticles were prepared by a precipitation method according to previous reports.18 mPEG–OA conjugate (10.0 mg) and free HCPT (5.0 mg) dissolved in DMSO (0.5 mL) were added dropwise to aqueous media (3.5 mL), which was then vortexed for 10 min at room temperature. The resulting solutions were transferred to a MWCO 3500 cartridge, and dialyzed against normal saline (100 mL) for 3 h, with two exchanges of dialysate.2.4. Determination of drug loading
The drug loading efficiencies (DLE) of OA and HCPT were analyzed by a high performance liquid chromatography (HPLC) method with a UV detector set at 210 nm and 254 nm, respectively.19 mPEG–OA/HCPT NPs (1.0 mg) were dissolved in acetonitrile (1.0 mL). The drug content in the mPEG–OA/HCPT NPs was determined using HPLC (OA: 210 nm, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mixture (v/v) of acetonitrile–water as a mobile phase, flow rate of 1.0 mL min−1; HCPT: 254 nm, 30:70 mixture (v/v) of acetonitrile–water as a mobile phase, flow rate of 1.0 mL min−1) and using a reverse phase column (C18). The column temperature was 25 °C. The DLE of OA and HCPT were calculated according to the following formula: DLE (wt%) = (weight of loaded drug/weight of nanoparticles) × 100%.
:5 mixture (v/v) of acetonitrile–water as a mobile phase, flow rate of 1.0 mL min−1; HCPT: 254 nm, 30:70 mixture (v/v) of acetonitrile–water as a mobile phase, flow rate of 1.0 mL min−1) and using a reverse phase column (C18). The column temperature was 25 °C. The DLE of OA and HCPT were calculated according to the following formula: DLE (wt%) = (weight of loaded drug/weight of nanoparticles) × 100%.
2.5. In vitro drug release
The release of OA and HCPT from mPEG–OA/HCPT NPs was analyzed by a dialysis method.20 Firstly, mPEG–OA/HCPT NPs were diluted to 1.0 mg mL−1, and then 5.0 mL of the solution was transferred to dialysis tubing (MWCO 3500 Da). The dialysis tubing was immersed into 100 mL of PBS (pH 7.4 or 5.5). Subsequently, the solution was shaken in a shaking water bath at 37 °C. At predetermined intervals, 2.0 mL of solution outside the dialysis bag was taken out and replaced by 2.0 mL of fresh PBS solution. The drug concentration in the medium was determined by HPLC. The release amount was then calculated. Each stability profile represents the average of three independent runs with the same sampling schedules. The standard deviation of each point is typically 2% or less. After 2 weeks, the solution inside the dialysis tubing was taken out and lyophilized, producing a white powder. 1H-NMR was used to demonstrate the hydrolysis.2.6. Characterization of mPEG–OA NPs and mPEG–OA/HC-PT NPs
2.7. Solubility studies
The solubility of mPEG–OA was analyzed. Briefly, excess amounts of mPEG–OA were added to screw-capped scintillation vials containing 10 mL of purified water. An aliquot of the sample (5 mL) was taken at 1 h intervals. The samples were analyzed directly by the HPLC method.2.8. In vitro cell cytotoxicity
The cytotoxicity of free OA, free HCPT, mPEG–OA NPs and mPEG–OA/HCPT NPs was evaluated with a CCK-8 assay.21 B16 cells were used as a model of a tumor cell line. Briefly, the cell lines were seeded in 96-well plates (Corning, USA) at an initial density of 3 × 103 cells per well in 180 μL of culture medium and incubated for 24 h. Then, the cells were treated with various samples (OA, HCPT, mPEG–OA NPs and mPEG–OA/HCPT NPs) at 37 °C in a humidified incubator with 5% CO2 for 48 h and 72 h. The samples were prepared as follows. The samples of mPEG–OA NPs or mPEG–OA/HCPT NPs were dissolved in DMSO (Merck, Darmstadt, Germany) and then diluted in tissue culture medium. Free OA or HCPT of equivalent concentrations were added in parallel for comparison. The cells were treated with various concentrations of those drugs. 20 μL of CCK-8 solution was added to each well and incubated for 1 h at 37 °C. The percentage cell viability can be calculated by measuring the absence of absorption at 450 nm. IC50 was calculated as polymer concentrations which inhibited the growth of 50% of the B16 cells. IC50 was calculated using the Boltzmann sigmoidal function from Origin 8.6 (OriginLab, Northampton, USA). Data are representative of three independent experiments.We evaluate the synergistic effects between OA and HCPT in the mPEG–OA/HCPT NPs by applying the combination index (CI) = OAcombined/OAsingle + HCPTcombined/HCPTsingle, whereby OA combined and HCPTcombined represent the IC50 of drugs used in the combination treatment, and OAsingle and HCPTsingle represent single drug IC50. An index lower than 1 denotes drug synergism while one larger than 1 shows an antagonistic effect.22
2.9. Cellular uptake study
The cellular uptake of HCPT from the mPEG–OA/HCPT NPs was evaluated by flow cytometry (CaAn ADP9, Beckman Coulter, America). B16 cells were seeded in a 12-well tissue culture plate (2 mL medium) at a density of 2 × 105 cells per well. After being cultured for 24 h, the original medium was replaced by HCPT and mPEG–OA/HCPT NPs (IC50) and incubated for 4 h at 37 °C. After the incubation, the cells were washed with PBS (pH 7.4) twice and treated with trypsin (0.5 mL) for 2 min. Then the cells were resuspended in PBS (2 mL) and subjected to the flow cytometry analysis.2.10. In vivo antitumor efficiency
A tumor xenograft model was established in the right axillary flank region of female C57BL/6 mice (6–7 weeks) after injection of 3 × 106 B16 cells. Treatments were initiated when tumors reached an average volume of 100 to 150 mm3, and this day was designated as day 0. On day 0, these mice were randomly divided into 6 groups (n = 6) and administered intravenous injection with PBS (control), free OA (10 mg kg−1), free HCPT (10 mg kg−1), OA/HCPT (OA: 4 mg kg−1, HCPT: 10 mg kg−1), mPEG–OA NPs (10 mg kg−1, OA equivalent), or mPEG–OA/HCPT NPs (10 mg kg−1, HCPT equivalent) every other day until day 8. All groups were intravenously injected via the tail vein. In the observation phase, the tumor sizes and body weights were monitored every other day. The tumor volume was calculated using the formula: (L × W2)/2, where L is the longest and W is the shortest tumor diameter (millimeter). For efficacy studies, the percentage of tumor growth inhibition (% TGI) was calculated using the following formula: [(C − T)/C] × 100%, where C is the mean tumor volume of the control group at a specified time and T is the mean tumor volume of the treatment group at the same time. At the end of the experiment (>6 weeks) or when the implanted tumor volume reached 5000 mm3, the mice were sacrificed for humane reasons. To further evaluate the hematological toxicity of the nanoparticles, 200 μL of blood was sampled from each mouse after final administration to test the white blood cell number (WBC) using a hematology analyzer (MEK-7222K, Nihon Kohden Celltac E).2.11. Side effects
Toxic side-effects of current chemotherapeutical drugs often lead to a severe reduction in the quality of life, so the detection of allergic reactions is very important. Type I hypersensitivity is the most common type of hypersensitivity reaction and IgE antibodies play an important role in mediating type I hypersensitivity responses.23,24 Thus we selected IgE levels as the parameter for rapid evaluation of type I hypersensitivity reactions. Six groups of tumor-bearing mice (26–28 g, n = 6) were used in allergy testing studies (control, OA, HCPT, OA/HCPT, mPEG–OA NPs, mPEG–OA/HCPT NPs). The samples were administrated via tail intravenous injection every other day at the OA and HCPT doses of 10 mg kg−1 body weight. After administration with different samples for 10 days, the orbit blood of mice in different groups was collected and centrifuged. Serum samples were analyzed according to the procedure of mouse IgE ELISA.2.12. Statistical analysis
All experiments in this study were performed three times, and the data were expressed as the means ± standard deviation (SD). Analysis of variance (ANOVA) was utilized to determine statistical significance between different groups. In all the analyses, p < 0.05 was considered to be of statistical significance.3. Results and discussion
3.1. Synthesis of mPEG–OA conjugate
A conjugate made of hydrophilic mPEG and hydrophobic drug OA was successfully synthesized. Fig. 1 shows the 1H-NMR spectra of mPEG–OA and OA. In Fig. 1A, typical peaks at 5.3 ppm (peak a) and 0.7–2.0 ppm were assigned to the protons of OA. After modification of OA, Fig. 1B, additional chemical signals at 2.8–3.9 ppm were detected and ascribed to the protons of mPEG, indicating a chemical linkage between OA and mPEG. Due to the formation of ester bonds, the proton peak δ 3.2 (1H, t) of OA moved to δ 4.2 (1H, t). The signal of OA was significantly weakened and that of mPEG was detected, further indicating the successful synthesis of mPEG–OA.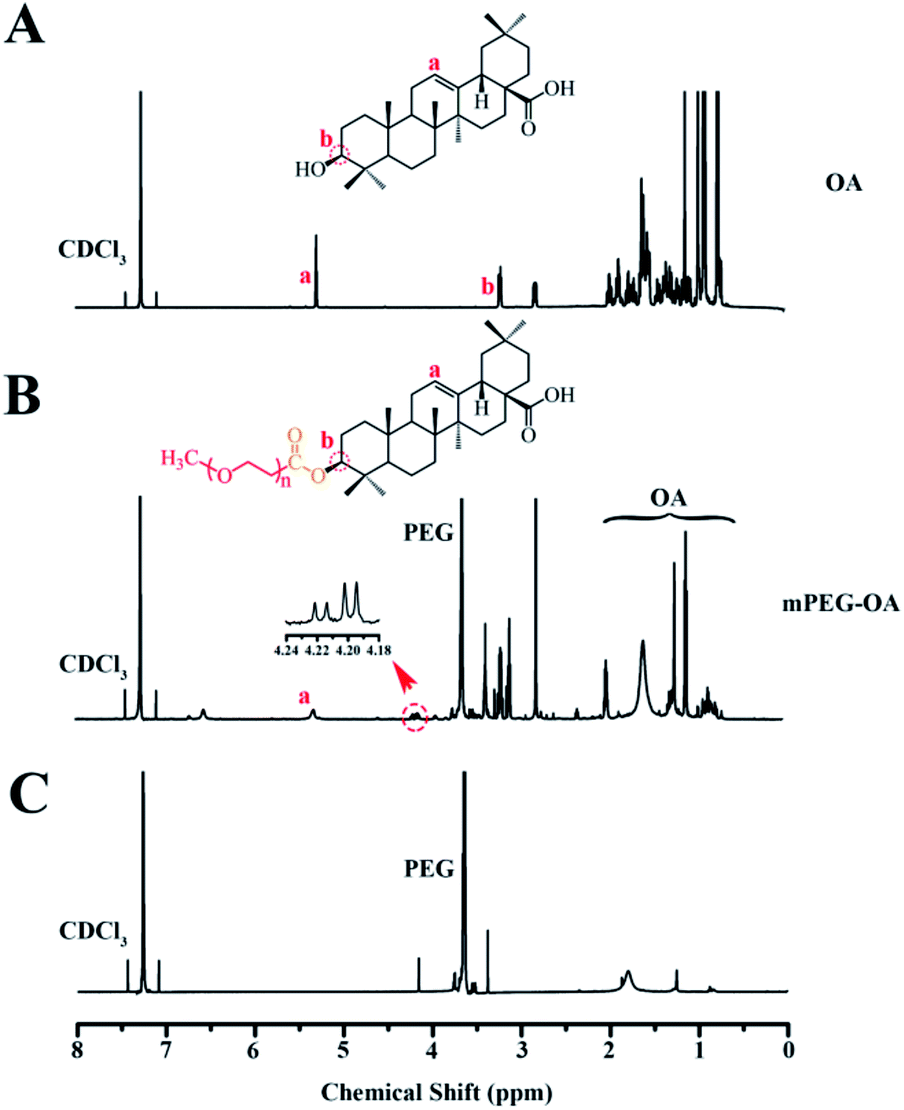 | ||
| Fig. 1 1H-NMR spectra of OA in CDCl3 (A), mPEG–OA in CDCl3 (B). 1H-NMR spectra of the hydrolysis result of the mPEG–OA (C). | ||
3.2. Characterization of the nanoparticles
Nanoparticles were prepared by the nanoprecipitation method according to our previous report.18 The mPEG–OA conjugate and HCPT were dissolved in 0.5 mL of DMSO solution. Then the mixture was added dropwise into aqueous media. The loading efficiency on nanoparticles was calculated and is shown in Table 1. The mPEG–OA/HCPT NPs contained 6.18 ± 1.20 wt% OA and 14.12 ± 0.73 wt% HCPT, suggesting the successful loading of HCPT into the mPEG–OA NPs.The desired diameters of the self-assembled nanoparticles were between 20 and 200 nm.25 The diameters of the nanoparticles were measured by dynamic light scattering (DLS). Fig. 2 presents the size distributions of the mPEG–OA NPs and mPEG–OA/HCPT NPs. The surface zeta potential is a key factor for better blood compatibility and prolonged circulation time of the nanoparticles for reduced clearance by the reticuloendothelial system (RES).26 The zeta potential of the nanoparticles was determined by a Nano-ZS Zeta Sizer. The size and the zeta potential of the nanoparticles are summarized in Table 1. It was observed that the average particle size of the mPEG–OA/HCPT NPs was larger than that of mPEG–OA NPs, which was attributed to the loaded drug. The final mPEG–OA/HCPT NPs had a diameter of 164.18 ± 40.12 nm and a negative surface charge of −5.13 ± 0.28 mV with a low polydispersity index. The negatively charged surface might be due to the carboxylate groups that resided on the outer surface of the nanoparticles after self-assembly in an aqueous solution. The carboxylate groups dominated the surface charge of the self-assembled nanoparticles.
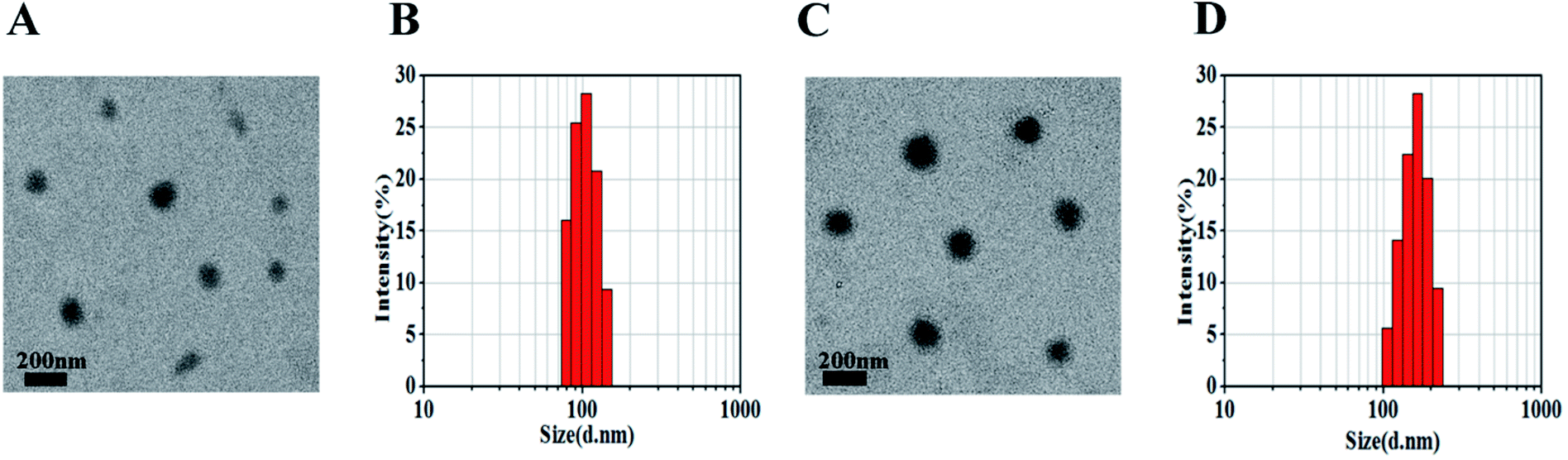 | ||
| Fig. 2 mPEG–OA NPs (A and B) and mPEG–OA/HCPT NPs (C and D) observed by TEM (scale bar = 200 nm) and DLS. | ||
3.3. Solubility studies
Solubility studies were carried out to explore the solubility of OA and mPEG–OA in purified water. The results indicated that PEGylation significantly increased the solubility of OA. The solubility of OA was about 0.018 mg mL−1. The mPEG–OA had 12.30 mg mL−1 solubility, which was equivalent to 1.03 mg mL−1 of OA (Table 1). The increased solubility makes it possible to systemically evaluate the therapeutic efficacy of OA in vivo.3.4. In vitro drug release
The release profiles of the nanoparticles at two different pH were studied in PBS (pH 5.5 and 7.4) at 37 °C. The nanoparticles were dialyzed against PBS through a membrane with a cutoff size of 3500 Da. This particular size was selected as the membrane pores were large enough to allow the diffusion of free drug but not nanoparticles. The results indicated that the mPEG–OA NPs and the mPEG–OA/HCPT NPs slowly hydrolyzed and released OA in PBS (pH 5.5 and 7.4) without burst release (Fig. 3A and C). As shown in Fig. 1C, the peaks at δ 3.3–4.2 were assigned to the methylene protons of the mPEG. And the OA typical peaks at 5.3 ppm and 0.7–2.0 ppm disappeared from the original position. The results indicated that the OA were successfully released from the mPEG–OA. | ||
| Fig. 3 Drug release curves in PBS at 37 °C from the mPEG–OA/HCPT NPs (A: OA and B: HCPT) and the mPEG–OA NPs (C: OA). The size distribution of nanoparticles after 14 days of storage at 4 °C (D). | ||
As for HCPT, there was an initial burst followed by a sustained drug release (Fig. 3B). The slight initial burst release of HCPT indicated that HCPT was dispersed on and near the surface of the mPEG. It might also be attributed to balancing the equilibrium between the inside and outside release settings.
The pH of tumor tissue is much lower than that of normal tissue.27 Thus, the drug release profile of the mPEG–OA/HCPT NPs was also investigated at pH 5.5. It was clearly observed that the drug released from the mPEG–OA/HCPT NPs was higher at pH 5.5 than at pH 7.4 over a 150 h period. The result was due to the re-protonation of the hydroxyl group of OA. As the external pH decreased, the nanoparticles quickly hydrolyzed and released the OA and HCPT which could increase the toxicity to the tumor. Therefore, these findings enable the development of a new type of nanoparticle for drug delivery applications.
As shown in Fig. 3D, the average particle size of the nanoparticles hardly changed during the investigation period. The results indicated that the mPEG–OA NPs and the mPEG–OA/HCPT NPs displayed a good redispersion stability.
3.5. In vitro cell cytotoxicity
The cell cytotoxicity was measured by cancer cell line viability. Free OA, free HCPT, mPEG–OA NPs and mPEG–OA/HCPT NPs were tested for their cytotoxic effects on B16 cells by CCK-8 assay. The cytotoxic effect of NPs was concentration-dependent. As shown in Fig. 4 and Table 2, IC50 was evaluated by survival curves. The results demonstrated that the IC50 values of the samples were ranked as OA > HCPT > mPEG–OA NPs > mPEG–OA/HCPT NPs. mPEG–OA/HCPT NPs provided more efficient delivery of the drug compared to the others. Nanoparticles had a higher efficacy than the free drug because of the continuous exposure of the drug into the cells. Entrapment of HCPT into mPEG–OA NPs reduced the IC50 comparable of mPEG–OA NPs, which confirmed the synergy between OA and HCPT. The mPEG–OA/HCPT NPs (6.18 wt% OA, 14.12 wt% HCPT) IC50 value of B16 cells was 1.37 μg mL−1 around 18-fold lower than that of OA. The CI value of OA and HCPT in the mPEG–OA/HCPT NPs was 0.14, which is smaller than 1. This result suggested that the mPEG–OA/HCPT NPs achieve a synergistic effect by co-delivery of two different anticancer drugs OA and HCPT. | ||
| Fig. 4 Cellular cytotoxicity of control, OA, HCPT, mPEG–OA NPs and mPEG–OA/HCPT NPs in B16 cells. CCK-8 assay of OA, HCPT and nanoparticles with different concentrations (equivalent to native OA and HCPT) in B16 cell lines (n = 3, error bars represent standard deviation). | ||
| Compound | B16 (μg mL−1) |
|---|---|
| OA | 25.04 |
| HCPT | 16.78 |
| mPEG–OA NPs | 2.82 |
| mPEG–OA/HCPT NPs | 1.37 |
3.6. Cellular uptake study
To investigate the cellular uptake, B16 cells were incubated with free HCPT and mPEG–OA/HCPT NPs for 4 h. It was found that the HCPT fluorescence intensity in B16 cells treated with the mPEG–OA/HCPT NPs was stronger than that treated with free HCPT (Fig. 5A and B). The results indicated that the mPEG–OA/HCPTNPs were efficiently endocytosed by B16 cells.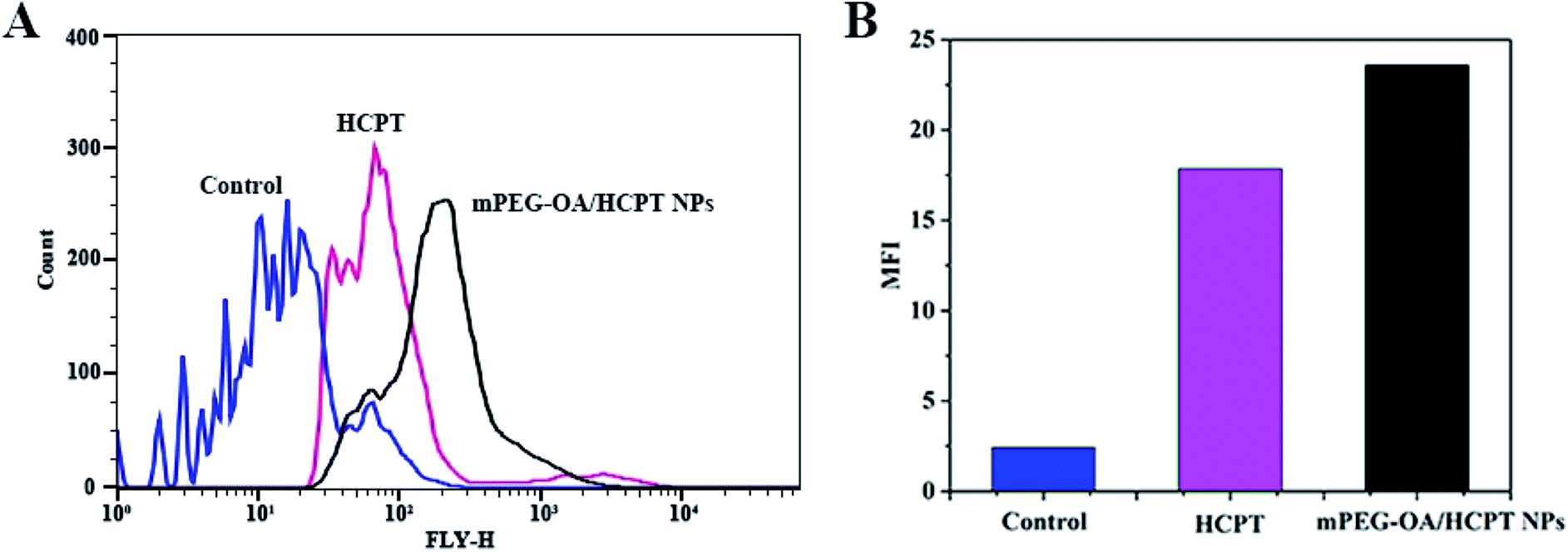 | ||
| Fig. 5 Flow cytometry analysis (A) and MFI of B16 cells incubated with free HCPT and the mPEG–OA/HCPT NPs at 37 °C for 4 h (B). The untreated cells are used as a control. | ||
3.7. In vivo antitumor efficiency
On the basis of the above results, the antitumor effect of mPEG–OA/HCPT NPs was explored in a mouse tumor model. The in vivo antitumor efficiency of the drug delivery nanoparticles was compared with free OA, free HCPT and OA/HCPT on a multiple-dose schedule in xenograft models of B16 murine melanoma. Tumor-bearing mice treated with the nanoparticles showed a prominent survival advantage compared with the control treated mice (Fig. 6B). The groups treated with free OA, free HCPT, OA/HCPT, mPEG–OA NPs and mPEG–OA/HCPT NPs showed varied levels of antitumor effects and they were in order mPEG–OA/HCPT NPs > mPEG–OA NPs > OA/HCPT > HCPT > OA (Fig. 6C). mPEG–OA/HCPT NPs resulted in 82.7% TGI (day 20) and 50% survival of animals (day 30). mPEG–OA NPs resulted in 73.4% TGI (day 20) and 33.3% survival of animals (day 30). Free OA resulted in 49.1% TGI (day 20) and 0% survival of animals (day 30), free HCPT resulted in 46.7% TGI (day 20) and 16.7% survival of animals (day 30) and OA/HCPT resulted in 58.2% TGI (day 20) and 16.7% survival of animals (day 30) (Fig. 6A and B and Table 3). | ||
| Fig. 6 Antitumor efficacy of OA, HCPT, OA/HCPT, mPEG–OA NPs, mPEG–OA/HCPT NPs in B16-bearing mice model. Tumor volumes of mice with different drug formulations; dashed line: stop chemotherapy (A). Survival percentage of different treatments (B). Tumor photograph from each treatment group excised on day 20 ((i): control, (ii): OA, (iii): HCPT, (iv): OA/HCPT, (v): mPEG–OA NPs, (vi): mPEG–OA/HCPT NPs) (C). Body weight variation of mice with different treatments (D). IgE levels of mice treated with different drug formulations for 30 min (E). WBC change during different administrations in normal mice. Blood samples were collected from mice on day 2 after the last treatment. Data are shown as means ± SD; n = 6 (F). | ||
| Compound | Mean TV ± SDa (mm3) | RTVa | TGIa (%) | Curesb (%) |
|---|---|---|---|---|
| a Mean tumor volume (TV), RTV, and % TGI data were taken at day 20 (by day 20, a significant percentage of control animals were euthanized due to excess tumor burden).b % cures were taken at day 30. | ||||
| Control | 4623 ± 1783 | 25.4 ± 13.6 | 0 | 0 |
| OA | 2354 ± 1252 | 13.3 ± 9.3 | 49.1 | 0 |
| HCPT | 2468 ± 1291 | 15.9 ± 8.7 | 46.7 | 16.7 |
| OA/HCPT | 1932 ± 864 | 11.4 ± 7.1 | 58.2 | 16.7 |
| mPEG–OA NPs | 1230 ± 575 | 7.4 ± 4.8 | 73.4 | 33.3 |
| mPEG–OA/HCPT NPs | 801 ± 486 | 4.3 ± 2.5 | 82.7 | 50.0 |
The systemic toxicity was evaluated by monitoring the body weight. After drug administration every other day, the body weight of free OA, free HCPT, OA/HCPT, mPEG–OA NPs and mPEG–OA/HCPT NPs exhibited no statistically significant changes compared to the PBS control (Fig. 6D). The results indicated that no signs of systemic toxicity were observed. These studies demonstrated that mPEG–OA/HCPT NPs have obviously mitigated the toxicities of the drug treatment while maintaining significant therapeutic efficacy.
3.8. Evaluation of the side effects
Many drug formulations, such as paclitaxel and docetaxel are always associated with serious side effects, including hematological toxicity and hypersensitivity reactions (usually type I hypersensitivity). The IgE antibodies play an important role in mediating type I hypersensitivity responses. Herein, we selected IgE levels as the parameter for rapid evaluation of type I hypersensitivity reactions. As shown in Fig. 6E, the blood IgE levels of mice in different groups (PBS, OA, HCPT, OA/HCPT, mPEG–OA NPs, mPEG–OA/HCPT NPs) were evaluated. Mice treated with OA, HCPT and OA/HCPT showed higher IgE levels than the PBS group, which might be due to the poor water solubility. The IgE levels of mPEG–OA NPs and mPEG–OA/HCPT NPs were observed to be without significant change. This explored the idea that the use of these nanoparticles could distinctly reduce the risk of hypersensitivity reactions. The blood of mice was also collected to test the WBC count after treatment with different OA formulations. The WBC count is often used as an indicator of hematological toxicity. The results demonstrated that the WBC count of mice treated with OA displayed a small decrease over the normal group (Fig. 6F). No obvious decrease in the WBC number of the mice treated with mPEG–OA NPs and mPEG–OA/HCPT NPs was observed, indicating that the nanoparticles could avoid severe hematotoxicity.4. Conclusions
In our study, mPEG–OA nanoparticles with a hydrophobic core of OA and a hydrophilic PEG shell have been developed and successfully synthesized which can also encapsulate another anticancer drug HCPT to achieve a synergistic effect. By evaluating the anticancer effects of the nanoparticles, mPEG–OA/HCPT NPs have shown significantly enhanced anticancer efficacy compared with free drug formulations. These nanoparticles for co-delivery of anticancer drugs also displayed a series of desirable properties, such as good water solubility, appropriate size and low side effects. It is believed that our study is a helpful and meaningful guide for future research.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21576029), and the China State Forestry Administration 948 Project (No. 2014-4-35).Notes and references
- F. Bray, A. Jemal, N. Grey, J. Ferlay and D. Forman, Lancet Oncol., 2012, 13, 790–801 CrossRef PubMed.
- T. Minko, L. Rodriguez-Rodriguez and V. Pozharov, Adv. Drug Delivery Rev., 2013, 65, 1880–1895 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. Hao, J. Liu, X. A. Wen and H. B. Sun, Bioorg. Med. Chem. Lett., 2013, 23, 2074–2077 CrossRef CAS PubMed.
- R. Martin, C. Cordova, J. A. San Roman, B. Gutierrez, V. Cachofeiro and M. L. Nieto, J. Mol. Cell. Cardiol., 2014, 72, 250–262 CrossRef CAS PubMed.
- T. Tian, X. Liu, E. Lee, J. Sun, Z. Feng, L. Zhao and C. Zhao, Arch. Pharmacal Res., 2017, 40, 458–468 CrossRef CAS PubMed.
- J. L. Maia, R. C. P. Lima-Junior, C. M. Melo, J. P. David, J. M. David, A. R. Campos, F. A. Santos and V. S. N. Rao, Pharmacol. Res., 2006, 54, 282–286 CrossRef CAS PubMed.
- X. Chuan, Q. Song, J. Lin, X. Chen, H. Zhang, W. Dai, B. He, X. Wang and Q. Zhang, Mol. Pharmaceutics, 2014, 11, 3656–3670 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- D. Lee, J. H. Na, S. Lee, C. M. Kang, H. N. Kim, S. J. Han, H. Kim, Y. S. Choe, K. Jung, K. C. Lee, K. Choi, I. C. Kwon, S. Y. Jeong, K. Lee and K. Kim, Mol. Pharmaceutics, 2013, 10, 2190–2198 CrossRef CAS PubMed.
- R. Ngoune, A. Peters, D. von Elverfeldt, K. Winkler and G. Puetz, J. Controlled Release, 2016, 238, 58–70 CrossRef CAS PubMed.
- C. Barrera, A. P. Herrera and C. Rinaldi, J. Colloid Interface Sci., 2009, 329, 107–113 CrossRef CAS PubMed.
- Z. Ahmad, S. Lv, Z. Tang, A. Shah and X. Chen, J. Biomater. Sci., Polym. Ed., 2016, 27, 40–54 CrossRef CAS PubMed.
- Y. Li, J. Lin, X. Yang, Y. Li, S. Wu, Y. Huang, S. Ye, L. Xie, L. Dai and Z. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 17573–17581 CAS.
- Y. Li, J. Lin, G. Liu, Y. Li, L. Song, Z. Fan, X. Zhu, G. Su and Z. Hou, RSC Adv., 2016, 6, 86717–86727 RSC.
- X. Wang, X. Gu, H. Wang, Y. Sun, H. Wu and S. Mao, Eur. J. Pharm. Sci., 2017, 96, 255–262 CrossRef CAS PubMed.
- R. K. Thapa, H. T. Nguyen, J. Jeong, B. S. Shin, S. K. Ku, H. Choi, C. S. Yong and J. O. Kim, Nanomedicine, 2016, 13, 885–896 CrossRef PubMed.
- M. J. Ernsting, W. L. Tang, N. MacCallum and S. D. Li, Bioconjugate Chem., 2011, 22, 2474–2486 CrossRef CAS PubMed.
- H. H. Y. Tong, H. B. Wu, Y. Zheng, J. Xi, A. H. L. Chow and C. K. Chan, Int. J. Pharm., 2008, 355, 195–202 CrossRef CAS PubMed.
- Y. X. Wang and Y. B. Tan, J. Drug Delivery Sci. Technol., 2016, 36, 183–191 CrossRef CAS.
- M. Lou, L. N. Zhang, P. G. Jia, F. Q. Feng, J. H. Liu, C. Yang, B. F. Li and L. Wang, Biomed. Pharmacother., 2016, 84, 1–9 CrossRef CAS PubMed.
- Y. Han, Z. He, A. Schulz, T. K. Bronich, R. Jordan, R. Luxenhofer and A. V. Kabanov, Mol. Pharmaceutics, 2012, 9, 2302–2313 CAS.
- A. Ariza, M. I. Montanez, T. D. Fernandez, J. R. Perkins and C. Mayorga, Curr. Pharm. Des., 2016, 22, 6773–6783 CrossRef CAS PubMed.
- S. M. Atkinson and A. Nansen, Basic Clin. Pharmacol. Toxicol., 2017, 120, 108–114 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- J. Du, T. Sun, W. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 3621–3626 CrossRef CAS PubMed.
- M. Karimi, M. Eslami, P. Sahandi-Zangabad, F. Mirab, N. Farajisafiloo, Z. Shafaei, D. Ghosh, M. Bozorgomid, F. Dashkhaneh and M. R. Hamblin, WIREs Nanomedicine and Nanobiotechnology, 2016, 8, 696–716 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |