
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication and interfacial characteristics of surface modified Ag nanoparticle based conductive composites†
Yingsi Wu a,
Lun-De Liaob,
Han-Chi Panb,
Leng Hebc,
Chin-Teng Lind and
Mei Chee Tan*a
a,
Lun-De Liaob,
Han-Chi Panb,
Leng Hebc,
Chin-Teng Lind and
Mei Chee Tan*a
aEngineering Product Development, Singapore University of Technology and Design, 8 Somapah Rd, Singapore 487372, Singapore. E-mail: meichee.tan@sutd.edu.sg
bInstitute of Biomedical Engineering and Nanomedicine, National Health Research Institutes, 35 Keyan Rd., Zhunan Town, Miaoli County 35053, Taiwan, Republic of China
cInstitute of Biomedical Engineering, National Chiao Tung University, 1001 Ta-Hsueh Road, Hsinchu 300, Taiwan, Republic of China
dFaculty of Engineering and Information Technology, University of Technology Sydney, City Campus 15 Broadway, Sydney 2007, Australia
First published on 7th June 2017
Abstract
The recent emergence of wearable electronics has driven the advancements of flexible and elastic conductive metal–polymer composites as electrodes and sensors. Surface modification of the conductive metal fillers are required to achieve a good dispersion within the matrix to obtain suitable conductivity and sensing properties. Additionally, it would be critical to ensure that the inclusion of these fillers does not affect the curing of the pre-polymers so as to ensure sufficient filler loading to form functional composites. In this work, a one-step approach is used to modify Ag–PAA nanoparticles via hydrogen bonds to form PAA–PVP complex modified Ag nanoparticles. The interfacial characteristics and thermal stability of these surface-modified Ag nanoparticles were studied to elucidate the underlying chemistries that governed the surface modification process. After surface modification, we successfully improved the dispersion of Ag nanoparticles and enabled curing of PDMS to higher Ag loadings of ∼25 vol%, leading to much lower electrical resistivity of ∼6 Ω cm. Our studies also showed that Ag nanoparticles modified at a PAA/PVP molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 resulted in a minimal particle aggregation. In a preliminary testing of our conductive composites as electrodes, clear electrocardiography signals were obtained. The facile surface modification method introduced here can be adapted for other systems to modify the particle interfacial behavior and improve the filler dispersion and loading without adversely affecting the polymer curing chemistry.
:10 resulted in a minimal particle aggregation. In a preliminary testing of our conductive composites as electrodes, clear electrocardiography signals were obtained. The facile surface modification method introduced here can be adapted for other systems to modify the particle interfacial behavior and improve the filler dispersion and loading without adversely affecting the polymer curing chemistry.
1. Introduction
Flexible and elastic conductive electrodes have a diverse range of applications from electronic skin sensors, diagnostic devices, light-emitting diodes to electrochemical devices.1–4 Amongst these various applications, conductive electrodes are most pervasively used for health monitoring via electrocardiography (ECG), electrooptigraphy (EOG) and electroencephalography (EEG) systems to measure the electricity activity of heart, eyes and brain, respectively.5–8 Most of these conductive electrodes are fabricated by embedding conductive fillers within elastomers. The conductivity of these composites depends on the intrinsic properties of the fillers and matrix, filler loading and filler–matrix interaction. A continuous electrical pathway is needed for these conductive filler–polymer networks so as to achieve a sufficiently low resistivity. To create the continuous electrical pathway, the volume fraction of fillers used would need to exceed the percolation threshold concentration. The resistivity (or conductivity) would dictate the sensing performance of these composites as electrodes. In order to obtain a good signal, the resistivity of these electrodes should be less than ∼150 Ω cm.9 To date, the commonly used conductive fillers are metals (e.g., silver (Ag) and copper (Cu)), carbon nanomaterials (e.g., carbon nanotubes and carbon black), or conducting polymers (e.g., PEDOT:PSS). Amongst these materials, Ag continues to be the most popular filler in conductive composites due to its low resistivity.10,11The ability of elastomers to adhere and conform to a given surface or shape enables a good contact between the sensor and sensing surface. Having a good contact would ensure clear signals to be obtained. Furthermore, using elastomers would also avoid the use of gel electrolytes that are needed for most conventional electrodes to ensure low interfacial resistance and consistent electrode performance. Subsequently, the poly(dimethylsiloxane) (PDMS) elastomer is one of the most commonly used elastomer. In addition, the liquid uncured PDMS can be molded easily and could be used as direct printing inks.12 The surface of cured PDMS can also be further modified using plasma treatment to control its physiochemical properties. Consequently, we can integrate both adhesion and conducting functions by creating a conductive PDMS composite that are loaded with Ag nanoparticles. To ensure uniform performance of these composites in both its stretched and upstretched states, the Ag nanoparticles loading must be sufficiently high and above the percolation threshold such that the percolation pathways are not affected and the composites' resistivity remain unchanged.
In this work, a facile approach (see Fig. 1) to modify the surfaces of Ag nanoparticles via inter-polymer complexation is reported. The interfacial characteristics and thermal stability of these surface-modified Ag nanoparticles were studied to elucidate the underlying chemistries that dictated the surface modification process. Polyacrylic acid (PAA) was selected as the particle stabilizer in this work for its wide utilization in producing and stabilizing Ag nanoparticles as inks for direct writing and printing.13–15 Compared to small molecules such as citrate, PAA prevents the aggregation of Ag nanoparticles due to the larger steric hindrance from its longer polymer chains.16 However, since PAA is a carboxylic acid-based polymer, the PAA-modified Ag nanoparticles were not well disperse within PDMS due to incompatible interfacial chemistries. What's worse, the Ag–PAA/PDMS mixture could not be cured when Ag loading exceeded 7 vol% in our experiments. The poor curing of the mixture at such low Ag loadings was consistent with our earlier observation of incompatible interfacial chemistries between Ag–PAA and PDMS. Thus, the modification of the Ag–PAA particles surface chemistry was needed to improve its dispersion and extend the range of Ag loading in PDMS such that it can exceed the percolation threshold. Since carboxylic acid and pyridine chemical functional groups are good hydrogen bonding partners, the complexation between PAA and PVP can be achieved under a mild and facile condition.17 Therefore, PVP was used to modify the PAA–Ag nanoparticles by a one-step polymer complexation process to improve the dispersion of the particles in PDMS, as shown in Fig. 1. Together with a demonstration of the use of our Ag–PDMS composite as an ECG electrode, the dispersion characteristics, electrical percolation threshold limits and electrical properties of our conductive composites will also be discussed.
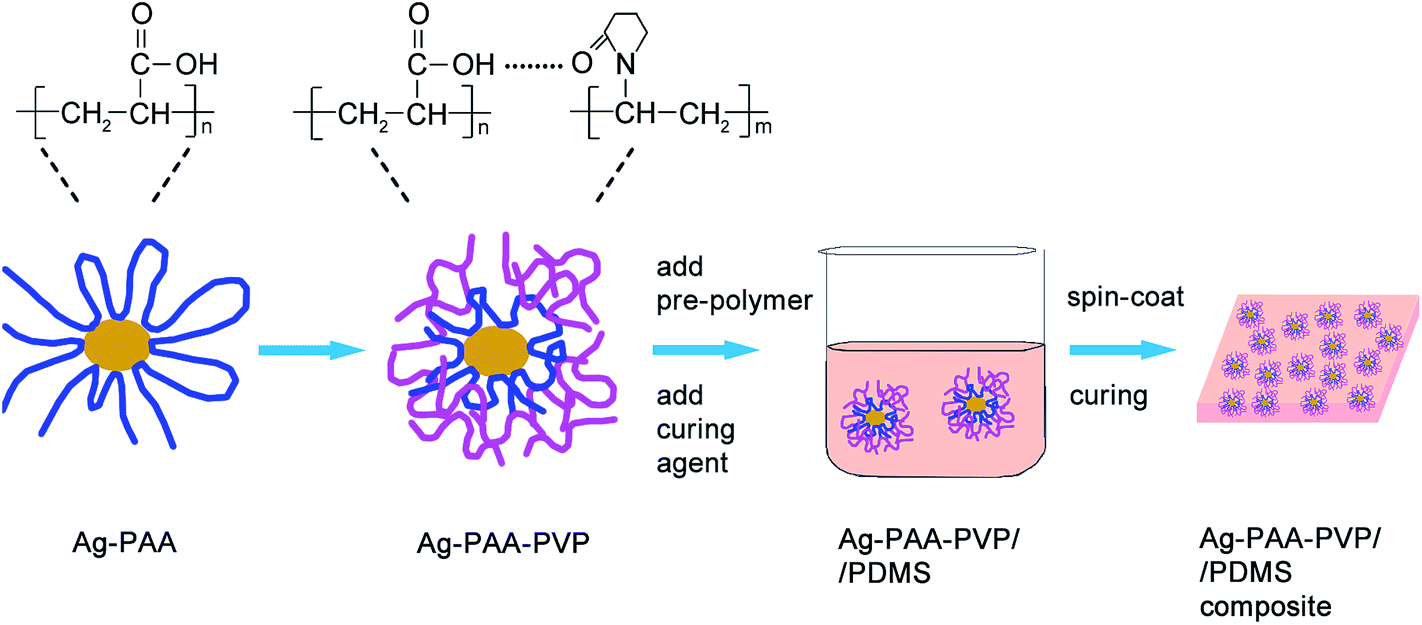 | ||
| Fig. 1 Schematic illustration of the surface modification process for Ag nanoparticles and the fabrication of Ag/PDMS conductive composite. | ||
2. Results and discussion
2.1. Morphology and size distribution of surface-modified Ag nanoparticles
Ag nanoparticles with different surface characteristics were synthesized as described detailed in the Methods section. PAA was selected as the particle stabilizer in this work for its wide utilization in producing and stabilizing Ag nanoparticles as inks for direct writing and printing. In our proposed method, PVP was used to modify the PAA-coated Ag nanoparticles via a complexation approach, where the molar ratios of PAA to PVP tested were 1:1 and 1:10. The theoretical molar ratios of the PAA and PVP monomer units that were added and used during the surface adsorption process, were [PAA]/[PVP] = 1:1 and [PAA]/[PVP] = 1:10. We have chosen to represent the relative amount of polymer used based on the molar ratio of monomeric units so as to deduce the extent of intermolecular interactions regardless of polymer chain length. PVP-Coated Ag nanoparticles were also prepared to allow us to make a comparison of the surface dispersion characteristics of these PVP-modified particles in solution and within PDMS. The typical morphologies and size distribution of our surface-modified Ag nanoparticles were characterized using the SEM and DLS (see Fig. 2 and S1 and Table S1†). Ag–PVP nanoparticles of 40 ± 5.5 nm with a spherical morphology and Ag–PAA particles of 158 ± 52 nm with irregular morphologies were synthesized. The Ag–PAA nanoparticles were composed of both large (∼80–200 nm) and small (∼10 nm) particles, where the smaller particles were located mostly on the surfaces of the larger particles. This suggests that the Ag–PAA particles were formed via the agglomeration and coalescence of smaller Ag nanoparticles at the elevated temperature (65 °C) used during synthesis. The broader size distribution of filler particles will be beneficial towards achieving a high conductivity for our composites, since the smaller particles can easily fill the voids leading to improved particle network connectivity.12,18 Fig. 2c and d shows that after PVP modification of the Ag–PAA nanoparticles at different molar ratios, the PAA–PVP modified nanoparticles exhibited similar size distributions, which are 138 ± 46 nm and 149 ± 50 nm for Ag–PAA–PVP (1:1) and Ag–PAA–PVP (1:10), respectively. The differences in particle sizes were mostly due to the difference in the effectiveness of either PVP or PAA as a particle stabilizer or surface capping agent during synthesis. In addition, since different reaction chemistries and solvents were used to synthesize Ag–PAA and Ag–PVP, the differences in the resulting particle sizes was also attributed to the differing nuclei sizes and varying particle growth rates in solution.
 | ||
| Fig. 2 Irregular shape and broad distributions of PAA- and PAA–PVP complexes-modified Ag nanoparticles were observed. Morphology of the as-synthesized (a) Ag–PAA, (b) Ag–PVP, (c) Ag–PAA–PVP (1:1) and (d) Ag–PAA–PVP (1:10) nanoparticles shown in the SEM images. | ||
2.2. PAA–PVP-modified particle characteristics
Using the DLS, we observed that the hydrodynamic radii of the particles were ∼15–18.5 nm larger than that of SEM (Fig. S1 and Table S1†). Since the hydrodynamic particle radius measured by DLS includes the thickness of the surfactant and solvent layers, we could attribute the size differences to mainly that of the polymer coating surrounding our Ag particles, especially considering the large molecular weights of our polymer coatings (PVP ∼ 10000 g mol−1; PAA ∼ 5000 or 50000 g mol−1). The thickness of PAA–PVP surfactants on the Ag nanoparticles were next analyzed to elucidate the molecular conformation of the polymer coatings on the particle surfaces. The calculated polymer thickness in Table 1 was estimated based on the weight percentages of polymer coatings measured using the TGA, the SEM average particle diameter (assuming the hard core sphere model), as well as the densities of Ag (10.5 g cm−3) and polymer (PAA ∼ 1.15 g cm−3 and PVP 1.2 g cm−3). From our calculations, the PVP-coating was the thinnest at ∼1.5 nm, whilst the thicknesses of PAA and PAA–PVP were ∼3.3 to 4.7 nm. We observed that the coating thickness was reduced from 4.1 to 3.3 nm once PVP was complexed with the PAA-coated Ag nanoparticles to form the Ag–PAA–PVP (1:1) nanoparticles. The coating thickness further increased to ∼4.7 nm upon further addition of more PVP to form the Ag–PAA–PVP (1:10) nanoparticles. This observed trend where the coating thickness follows this order of PAA–PVP (1:10) > PAA > PAA–PVP (1:1) > PVP, was also consistent with that of our data from SEM (dry state) and DLS (aqueous state) (Table S1†). However, we observed that the calculated thickness based on polymer weight loss was always smaller than the estimated thickness based on the difference between the SEM and DLS sizes (Table S1†). The difference in magnitude for coating thickness was attributed to the existence of the polymer coating in the two possible states: (1) collapsed, and (2) swollen configuration which will depend on the polymers' interfacial interaction with the solvent as well as with each other (in this case, water). The observed trend in varying coating thickness is also attributed to the differences in polymer–particles interactions and polymer chain lengths. Considering the molecular weights of PVP to PAA (PVP ∼ 10000 g mol−1; PAA ∼ 5000 or 50000 g mol−1), the PVP coated particle would have the thinnest coating since it has the lowest possible chain length (even when we assume that all polymer chains were fully extended). In contrast, with the use of the much higher molecular weight PAA, a thicker PAA coating (4.1 nm) would be expected compared to the PVP coated samples. The observed slight reduction in coating thickness from 4.1 to 3.3 nm once PVP was complexed with the PAA-coated Ag nanoparticles to form the Ag–PAA–PVP (1:1) nanoparticles was most likely due to the increased interactions between PAA–PVP polymer chains that led to an overall slight reduction in coating thickness. The addition of excess PVP to complex with PAA to coat the particles to form the Ag–PAA–PVP (1:10) nanoparticles was most likely due to the formation of multi-layered polymer structures in the coating or polymer swelling due to repulsive interactions or additional solvent interactions.
| Samples | TGA (wt%) | Calculated thickness (nm) |
|---|---|---|
| Ag–PAA | 1.8 | 4.1 |
| Ag–PVP | 2.8 | 1.5 |
| Ag–PAA–PVP (1:1) |
1.7 | 3.3 |
| Ag–PAA–PVP (1:10) |
2.3 | 4.7 |
In addition, based on the weights percentages of the polymer coatings, we could also calculate the number of polymer chains that were coated on the particle. From our calculations, it shows that we estimated ∼12000 to 24000 polymer chains of PAA or PAA–PVP per particle. Considering this number with that of the theoretical estimate of a single polymer monolayer in the extended configuration (∼30000 to 40000 polymer chains per particle, assuming a diameter of ∼0.5 nm), the molecular conformation of these polymer coatings will most likely follow the loop-train-tail configuration that is also commonly observed amongst high molecular macromolecules.
The electric double layer characteristic of our polymers were also measured to verify the PVP–PAA chemical complexation and coating properties (Fig. 3). For the Ag–PAA nanoparticles, the isoelectric point was at pH ∼ 2.4, whilst all the PVP-based systems showed an isoelectric point at pH ∼ 0.9. The observed shift in the isoelectric point of the Ag–PAA nanoparticles towards that of the Ag–PVP nanoparticles upon PAA–PVP complexation suggested that the PVP polymers had uniformly intercalated or coated with the surface bound PAA. As a consequence of the PAA–PVP complexation, the surface charge characteristics of the Ag–PAA–PVP followed more closely with that of the Ag–PVP nanoparticle system. The relatively high negative zeta potential of ∼−40 to −60 mV within a wide pH range suggests that the Ag–PAA–PVP nanoparticle suspension would remain stable over a large range of solvents due to the high electrostatic repulsion between the nanoparticles.
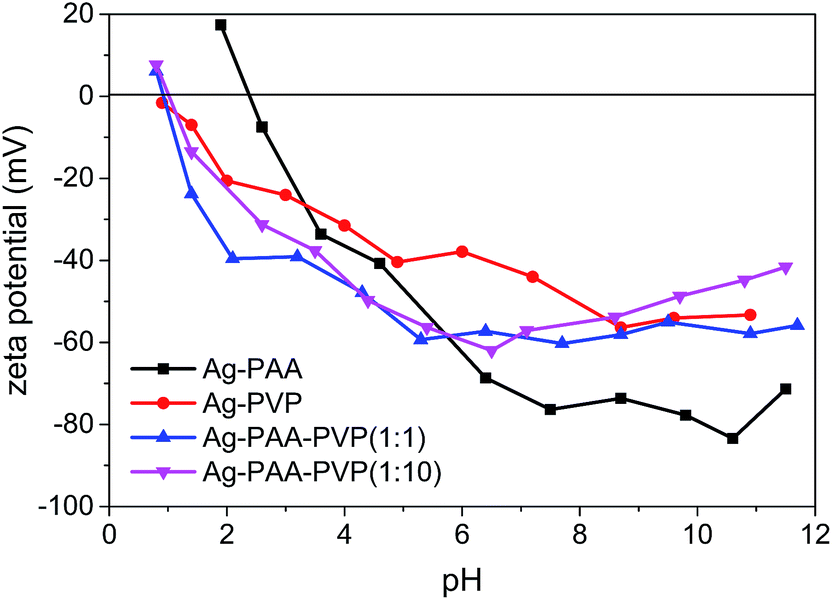 | ||
| Fig. 3 Surface charge characteristics of the aqueous Ag nanoparticle suspension (∼20 mg L−1) as a function of pH. Isoelectric point of the Ag–PAA nanoparticles was at pH ∼ 2.4, and the isoelectric point of Ag–PVP, Ag–PAA–PVP (1:1) and Ag–PAA–PVP (1:10) nanoparticles was at pH ∼ 0.9. This suggests that the PVP-based Ag nanoparticle system would be more stable over a wider range of solvents. | ||
2.3. Complexation of PAA and PVP polymers
The underlying chemical mechanisms that govern polymer complexation on Ag nanoparticles was studied by investigating the interaction between the 2 pure polymers (PVP or PAA only) under 2 separate conditions: (1) physical blend and (2) chemical complex formation. In this way, the effects of Ag particle sizes would not interfere with our evaluation of the polymer complexation process. We have hypothesized that a complex of PAA–PVP is formed via hydrogen bonding in a weakly acidic aqueous solution, where the carboxyl groups on PAA are protonated (i.e. pH of solution was tuned to ∼pKa of PAA). The hydrogen bonds between PAA is hypothesized to occur between the hydroxyl group on the carboxyl of PAA and the lone pair on the oxygen of the carbonyl group of PVP (see Fig. 4d). The pH of 5.8 was used to allow some of the carboxyl groups to be protonated while also avoiding Ag nanoparticles sintering which would lead to severe particle agglomeration and increase in the secondary particle sizes. In contrast, we did not expect any hydrogen bond interactions between PAA and PVP (i.e. the physical blends) prepared via the dissolution of the two polymers in ethanol (i.e. non-acidic environment), since most of the carboxyl groups of PAA would not be protonated in this environment.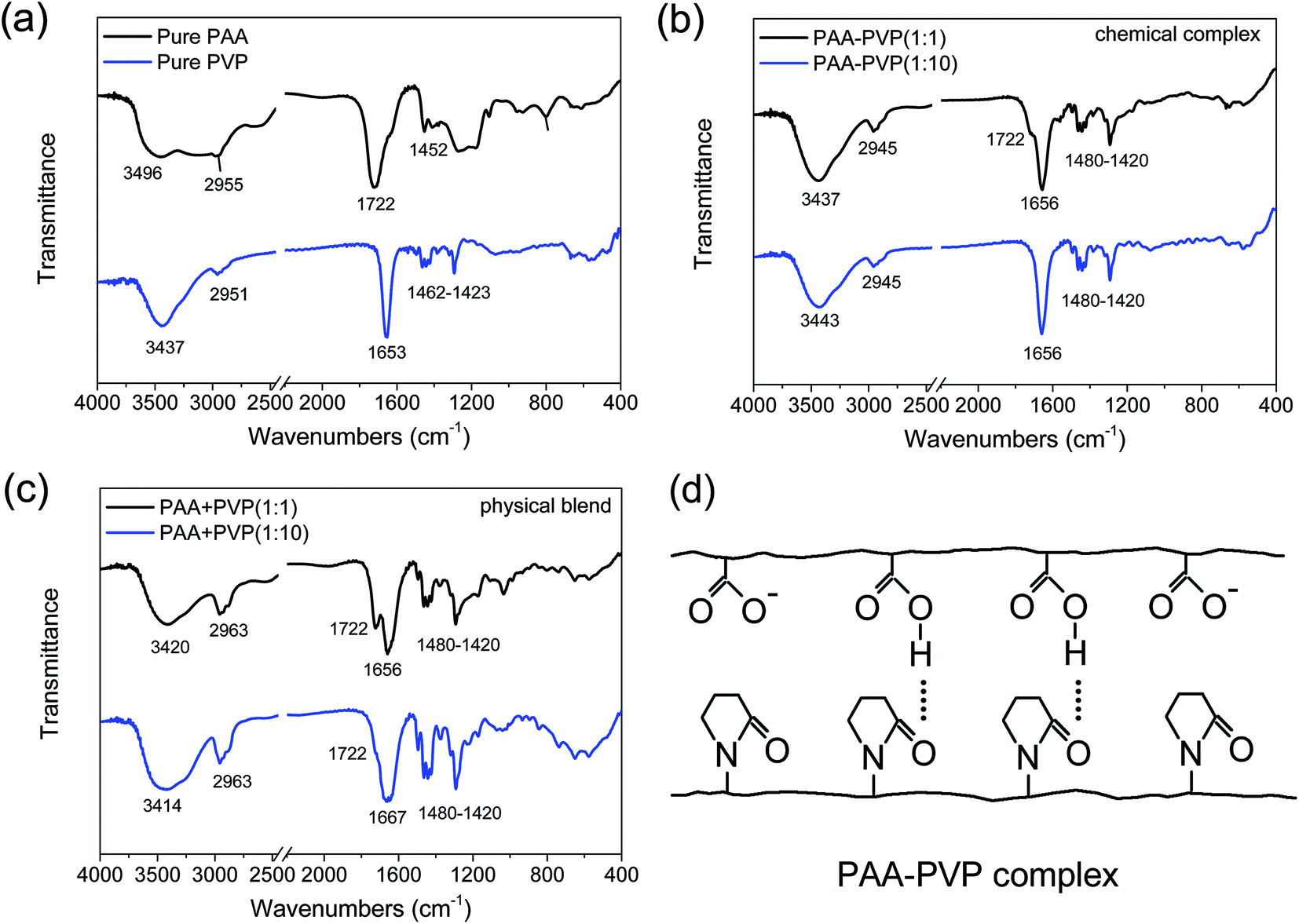 | ||
| Fig. 4 Interactions between PAA, PVP and PAA–PVP polymers using FTIR. FTIR spectra of (a) pure PAA and PVP polymers, the (b) chemical complexes and (c) physical blends of PAA–PVP at different molar ratios. (d) Schematic of the PAA–PVP intermolecular complexation. | ||
The chemical complexation and physical blending of PAA and PVP polymers were firstly studied by FTIR (see Fig. 4). The FTIR spectra show the characteristic chemical functional groups of the pure PAA and PVP samples (see Fig. 4a). For the pure PAA samples, in addition to the typical alkyl chain (CH2) bands observed at 2840–3000 cm−1 and 1340–1485 cm−1, we observed the characteristics signatures of dimer formations often associated with the carboxyl groups of PAA. The presence of PAA dimer formation is evident by the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption at ∼1710 cm−1 and broad OH absorption band at 3000 cm−1. In contrast, in the absence of dimer formation, one would have expected the CO and OH absorption bands to be positioned at 1760 and ∼3550 cm−1. The weak shoulder peak at ∼1628 cm−1 that is associated with the charged carboxyl group (COO−) was also observed, which suggested the incomplete protonation of these carboxyl groups in PAA. Since our OH absorption profile for PAA shows a broad absorption from 3200 to 3600 cm−1, it suggests that both dimeric and monomeric OH groups were present in our pure PAA. In the right pH conditions, these monomeric OH groups would facilitate hydrogen bond interactions with other functional groups (e.g., CO). For the pure PVP samples, the FTIR bands associated with alkyl chain (CH2) was again observed at 2840–3000 cm−1 and 1340–1485 cm−1. The band at ∼3437 cm−1 is attributed to the NH stretching which is associated with the amide functional group of PVP, and is also generally less affected by the hydrogen bonding. In addition, due to the lower electronegativity of N compared to O, the carbonyl band (CO) of PVP is observed at a lower frequency of ∼1653 cm−1, compared to that of the carboxylic acid (COOH, 1780–1700 cm−1 and COO−, 1650–1550 cm−1) and ester (COO, 1750–1700 cm−1) functional groups. It should be noted that the 1653 cm−1 absorption is a combined mode with contribution from both (CO and C–N) of PVP, where the absorption from CO of PVP would be much stronger.19–21
O absorption at ∼1710 cm−1 and broad OH absorption band at 3000 cm−1. In contrast, in the absence of dimer formation, one would have expected the CO and OH absorption bands to be positioned at 1760 and ∼3550 cm−1. The weak shoulder peak at ∼1628 cm−1 that is associated with the charged carboxyl group (COO−) was also observed, which suggested the incomplete protonation of these carboxyl groups in PAA. Since our OH absorption profile for PAA shows a broad absorption from 3200 to 3600 cm−1, it suggests that both dimeric and monomeric OH groups were present in our pure PAA. In the right pH conditions, these monomeric OH groups would facilitate hydrogen bond interactions with other functional groups (e.g., CO). For the pure PVP samples, the FTIR bands associated with alkyl chain (CH2) was again observed at 2840–3000 cm−1 and 1340–1485 cm−1. The band at ∼3437 cm−1 is attributed to the NH stretching which is associated with the amide functional group of PVP, and is also generally less affected by the hydrogen bonding. In addition, due to the lower electronegativity of N compared to O, the carbonyl band (CO) of PVP is observed at a lower frequency of ∼1653 cm−1, compared to that of the carboxylic acid (COOH, 1780–1700 cm−1 and COO−, 1650–1550 cm−1) and ester (COO, 1750–1700 cm−1) functional groups. It should be noted that the 1653 cm−1 absorption is a combined mode with contribution from both (CO and C–N) of PVP, where the absorption from CO of PVP would be much stronger.19–21
Chemical complexes of PAA–PVP at different molar ratios were formed by controlling the pH to form a weakly acidic environment (pH ∼ pKa), where the carboxyl groups on PAA are protonated. In these PAA–PVP chemical complexes, hydrogen bonds between the PAA and PVP chains are formed between the hydroxyl group on the carboxyl of PAA (COOH) and the lone pair on the oxygen of the carbonyl group of PVP (CO) (see Fig. 4d). For the chemical complexes of PAA–PVP at molar ratios of 1:1 and 1:10, we observed that their FTIR spectra was identical to that of pure PVP, where a broadening of the peak at 1653 cm−1 was observed when compared to that of pure PVP. These observed spectra similarities suggest that PVP was successfully complexed with the PAA chains. Only a weak shoulder peak at 1722 cm−1 was observed for the sample of PAA–PVP (1:1). The general absence of a distinct 1722 cm−1 band shows that the hydrogen bonded dimers between carboxyl groups of PAA were disrupted, whilst the broadening of the 1653 cm−1 band was attributed to the hydrogen bonds formed between COOH of PAA and CO of PVP (see Fig. 4d). The presence of deprotonated carboxyl functional group of PAA (COO−) would also lead to a similar further broadening of the 1653 cm−1 band.19–21
In contrast, minimal hydrogen bond interactions between PAA and PVP are likely when the two polymers are physically mixed after dissolution in ethanol (i.e. non-acidic environment), since most of the carboxyl groups of PAA would not be protonated (i.e. COO−). The FTIR spectra of our physical mixtures in Fig. 4c shows a much broader peak at ∼3423 cm−1 compared to that of pure PVP (Fig. 4a) and the chemical complexes (Fig. 4b), which suggested that a more diverse range of intermolecular interactions (e.g., hydrogen bonds, electrostatic dipole–dipole interactions) between the NH groups of PVP was present. In addition, a much more distinct peak at 1722 cm−1 was observed together with the broadening of the peak at 1653 cm−1 for the physical blends of PAA + PVP at both molar ratios of 1:1 and 1:10, especially when compared to that of the chemical complexes. The presence of the observed peak at 1722 cm−1 suggested that the dimers formed between the COOH groups of PAA chains were not disrupted. Also, the difference in absorption intensity at 1722 cm−1 for the different concentrations (1:1 vs. 1:10) suggested that these dimers were formed between PAA chains (i.e., intermolecular interactions) rather than within the PAA chain (i.e., intramolecular interactions). The broadening and shifting of the peak from 1653 to 1667 cm−1 at increased PAA:PVP ratios was most likely due to the presence of the deprotonated carboxyl groups of PAA (COO−) and its associated electrostatic interactions. Comparing this 1650–1670 cm−1 band of the chemical complexes and physical blends (Fig. 4b vs. 4c for 1:10), the much broader peak for the physical blends suggested that the distances for the intermolecular interactions occurred over a wider range of distances leading to a broader range of vibrational frequencies. The shifting of the peak centers from 1653 to 1667 cm−1 suggested that much higher energy intermolecular interactions (likely arising from the electrostatic interactions) were formed in our physical blends. Therefore, our FTIR data for our physical blends suggested that the addition of PVP disrupted a small fraction of the intermolecular interactions between PAA–PAA chains leading to the formation of a range of intermolecular interactions between PAA and PVP chains.
The thermal stability and phase transitions of the polymer coating measured using TGA and DSC under a N2 atmosphere is summarized in Table 2 (also see Fig. S3†). The higher decomposition and glass transition temperatures (385 °C and 53 °C) of pure PVP as compared with that of PAA (197 °C and 43 °C) suggested a higher thermal stability and stronger interactions between PVP chains. The decomposition of both PAA–PVP (1:1) (375 °C) and PAA–PVP (1:10) (380 °C) were close to that of pure PVP, suggesting the increased thermal stability and increased interactions as PAA complexed with PVP. The stronger interactions between PVP–PVP chains as well as between the PAA–PVP chains is consistent with earlier observations where the Ag–PVP nanoparticles was estimated to have the thinnest polymer coating and the reduction in Ag–PAA coating thickness upon the formation of PAA–PVP (1:1) complexes (see Table 1, based on TGA polymer weight loss).
| Samples | Td (°C) | Tg (°C) | Tm (°C) |
|---|---|---|---|
| Pure PAA | 197 | 43 | 149 |
| Pure PVP | 385 | 53 | 127 |
| PAA–PVP (1:1) |
375 | 151 | — |
| PAA–PVP (1:10) |
380 | 115 | — |
| PAA + PVP (1:1) |
335 | 84 | — |
| PAA + PVP (1:10) |
383 | 99 | — |
The lower decomposition temperature of the physically blended PAA + PVP (335 °C for 1:1) than the complexed PAA–PVP (375 °C for 1:1) was due to the weaker intermolecular interactions within the physical blends compared to that of the complexes. Both the chemical complexes and physical blends of PAA and PVP exhibited a single DSC glass transition temperature (see Fig. S3†), which indicates the presence of a miscible amorphous phase. The higher glass transition temperature in the complexes was attributed to the stronger interactions formed due to the complexation process. In addition, both pure PAA and PVP exhibited a melting transition at 149 °C and 127 °C, respectively. The absence of the melt transition in both the complexes and blends suggests the crystallinity in either PAA or PVP was reduced, which was attributed to the additional intermolecular interaction between the PAA and PVP. These results verified that our proposed polymer complexation was successful, leading to an overall improvement in the thermal stability of these polymers blends or complexes.
2.4. Dispersion of surface-modified nanoparticles in PDMS
Since the PDMS pre-polymer is known to be compatible with non-polar organic solvents, Ag nanoparticles were dispersed in toluene and blended with the viscous PDMS pre-polymer.22 To confirm the hypothesis of our proposed approach shown earlier in Fig. 1 that our Ag–PAA–PVP particles have improved dispersion characteristics, suspensions of as-synthesized Ag nanoparticles in toluene (50 mg particle in 2.5 mL toluene) after sonication for 30 min was studied. Obvious sedimentation appeared in the Ag–PAA mixture almost immediately after resuspension (see Fig. S4†). In contrast, the PAA–PVP- and PVP-modified Ag nanoparticles remained in suspension in toluene for much longer, which verifies the effective stabilization of the Ag particles by PAA–PVP modification. After the evaporation of toluene and addition of curing agent, conductive composites were prepared by spin coating the solution of surface-modified Ag particles dispersed in PDMS pre-polymers on glass substrates, followed by a curing at 80 °C for 3 h. Both Ag–PAA and Ag–PVP were not able to form the conductive PDMS composites once Ag loading exceeded 7 vol%. The inability to cure the PDMS pre-polymer at higher loadings of Ag–PAA or Ag–PVP was due to the formation of chemical coordinate bonds between the carboxyl and carbonyl groups of either PAA or PVP with the platinum-based hydrosilylation catalysts used for accelerated curing in PDMS. The inability to cure the PDMS at higher loadings of the conductive Ag nanoparticles would limit the performance range of these composites.The dispersion of our surface modified Ag nanoparticles in PDMS matrix was analyzed by observing the SEM micrographs of both the cross-section and top surface of our composites with a fixed loading of ∼7 vol% (see Fig. 5). Fig. 5a shows that the Ag–PAA/PDMS composite exhibited the poorest dispersion of Ag particles amongst the four samples. Ag particles aggregates (marked with yellow circles) and empty regions (marked with red circle) in PDMS were observed in the Ag–PAA/PDMS composite. Based on the differences in particle concentrations across the cross section and the top surface, distinct Ag–PAA particle sedimentation towards the bottom of the composite was observed (see Fig. 5a). In contrast, Ag–PVP had a much more uniform dispersion and existed as clusters in the order of hundreds of nanometers (Fig. 5b). For the PAA–PVP-modified Ag nanoparticles, Ag–PAA–PVP (1:1) particles in Fig. 5c showed further improvement of its dispersion with a much lower extent of aggregation than that of Ag–PVP particles (see Fig. 5b). The presence of these aggregates will give rise to a discontinuous percolation path and consequently lead to poor electrical conductivity. By increasing the PVP amount in our PAA–PVP modified Ag nanoparticles, we were able to significantly improve the dispersion of our fillers within PDMS. Ag–PAA–PVP (1:10) exhibited the best dispersion in PDMS, where a mostly single-particle dispersion was observed from the cross-section and top surface SEM images (see Fig. 5d).
 | ||
| Fig. 5 Dispersion characteristics of 7 vol% of Ag nanoparticles with different surface characteristics embedded within the PDMS composites. Ag–PAA–PVP (1:10) particles shows the best dispersion in PDMS as shown from the cross-section SEM images of (a) Ag–PAA/PDMS, (b) Ag–PVP/PDMS, (c) Ag–PAA–PVP (1:1)/PDMS and (d) Ag–PAA–PVP (1:10)/PDMS composites. Insets are the SEM images taken from the top surface of the corresponding composites. | ||
In order to verify the particle distribution for the 4 samples, an elemental analysis of mainly Ag was obtained by EDS (see Table S2†). Whilst the measured Ag content on Ag–PAA/PDMS surface was only 0.9 at%, much higher Ag content of 14.3, 14.2 and 16.1 at% was measured from the surfaces of Ag–PVP/PDMS, Ag–PAA–PVP (1:1)/PDMS and Ag–PAA–PVP (1:10)/PDMS composites, respectively. These results were consistent with earlier SEM observations where the poor dispersion and phase separation of Ag–PAA led to lower measured Ag content at the surface. For the Ag–PAA–PVP (1:10)/PDMS composite, the elemental analysis indicates homogenous dispersion of particles in the PDMS matrix without any phase separation. Therefore, by modifying our Ag–PAA particles using our proposed method to form PAA–PVP complexes, we have successfully improved the particle dispersion and prevented severe agglomeration in the PDMS composites.
2.5. Electrical and sensing characteristics of the Ag–PDMS composites
The sensing characteristics of the Ag/PDMS composites is largely dependent on the electrical resistivity of the composites composed of surface-modified Ag nanoparticles and PDMS. Since PDMS composites of Ag–PAA and Ag–PVP cannot be formed at Ag loading >7 vol%, we proceeded to compare the volumetric resistivities of the 4 samples at 7 vol%. We observed that Ag–PAA–PVP (1:10)/PDMS composites yielded the lowest volumetric resistivity of 110 Ω cm (see Table 3). The relatively low resistivity of Ag–PAA–PVP (1:10)/PDMS composite was associated with the uniform dispersion of the particles in the matrix as observed in SEM images. The more uniform dispersion of Ag–PAA–PVP (1:10)/PDMS was attributed to the formation of a more stable polymer coating with a negative surface charge characteristic leading to particle stabilization via electrostatic repulsion. The more stable polymer coating is consistent with our earlier observations where the PAA–PVP interactions were the strongest (e.g., higher thermal stability, Table 2) leading to a more strongly bonded polymer coating with increased stability. The intrinsic conductivities and resistivities of the as-synthesized surface modified Ag nanoparticles were also measured (see Table S3†). The conductivity of Ag–PAA was 8.74 × 10−1 S cm−1, which was at the same order of magnitude as those of Ag–PAA–PVP (1:1) (8.18 × 10−1 S cm−1) and Ag–PAA–PVP (1:10) (7.72 × 10−1 S cm−1). In contrast, the measured conductivity of Ag–PVP was the highest at 4.92 × 101 S cm−1. The much higher conductivity of Ag–PVP nanoparticles is likely due to the improved packing of the nanoparticles that led to improved contact and reduced interfacial resistances. It was observed that although Ag–PVP exhibited the lowest linear resistivity (i.e. highest conductivity), the Ag–PVP/PDMS composites showed a much higher volumetric resistivity (180 Ω cm) than that of the PAA–PVP-modified Ag/PDMS composites (110–160 Ω cm). Considering that the key difference is the poor dispersion of Ag–PVP particles within PDMS, we have therefore attributed the relatively low resistivity of Ag–PAA–PVP (1:10)/PDMS composites (i.e., improved conductivity) to the improved dispersion of the particles in the PDMS matrix as observed in the SEM images.
:10)/PDMS composite shows the lowest volumetric resistivity (i.e. highest conductivity). Sheet resistance, thickness and volume resistivity measured at a current of 100 mA for various Ag–PDMS composites with 7 vol% of Ag particles
| Samples | Sheet resistance (Ω sq−1) | Thickness (μm) | Resistivity (Ω cm) | Conductivity (S cm−1) |
|---|---|---|---|---|
| Ag–PAA | 34414 |
55 | 189 | 5.29 × 10−3 |
| Ag–PVP | 23937 |
75 | 180 | 5.56 × 10−3 |
| Ag–PAA–PVP (1:1) |
35587 |
45 | 160 | 6.25 × 10−3 |
| Ag–PAA–PVP (1:10) |
31413 |
35 | 110 | 9.09 × 10−3 |
Next, to investigate the effects of filler content and evaluate the percolation threshold of our system, we varied the Ag loading in our Ag–PAA–PVP (1:10)/PDMS composite system. It was found that the resistivity decreased from 4303 Ω cm to 110 Ω cm as Ag loading increased from 3 to 7 vol% (see Fig. 6a). The resistivity of our composite was further reduced to 6.0 Ω cm when the filler loading was increased to 20 vol%, which is still much higher to that of bulk silver (∼1.65 × 10−6 Ω cm).23 However, it should be noted that the measured 6.0 Ω cm is much lower than that of the alternative carbon-based electrodes which has reported resistivities of 12000 Ω cm and 100 Ω cm for 0.4 wt% of carbon nanotubes and 1 wt% mixed carbon nanotube to graphene, respectively.9 At 25 vol% (i.e. 78.4 wt%), we observed a slight increase of the resistivity to 13.2 Ω cm, which is most likely due to particle phase separation at high loading leading to non-uniform composites.
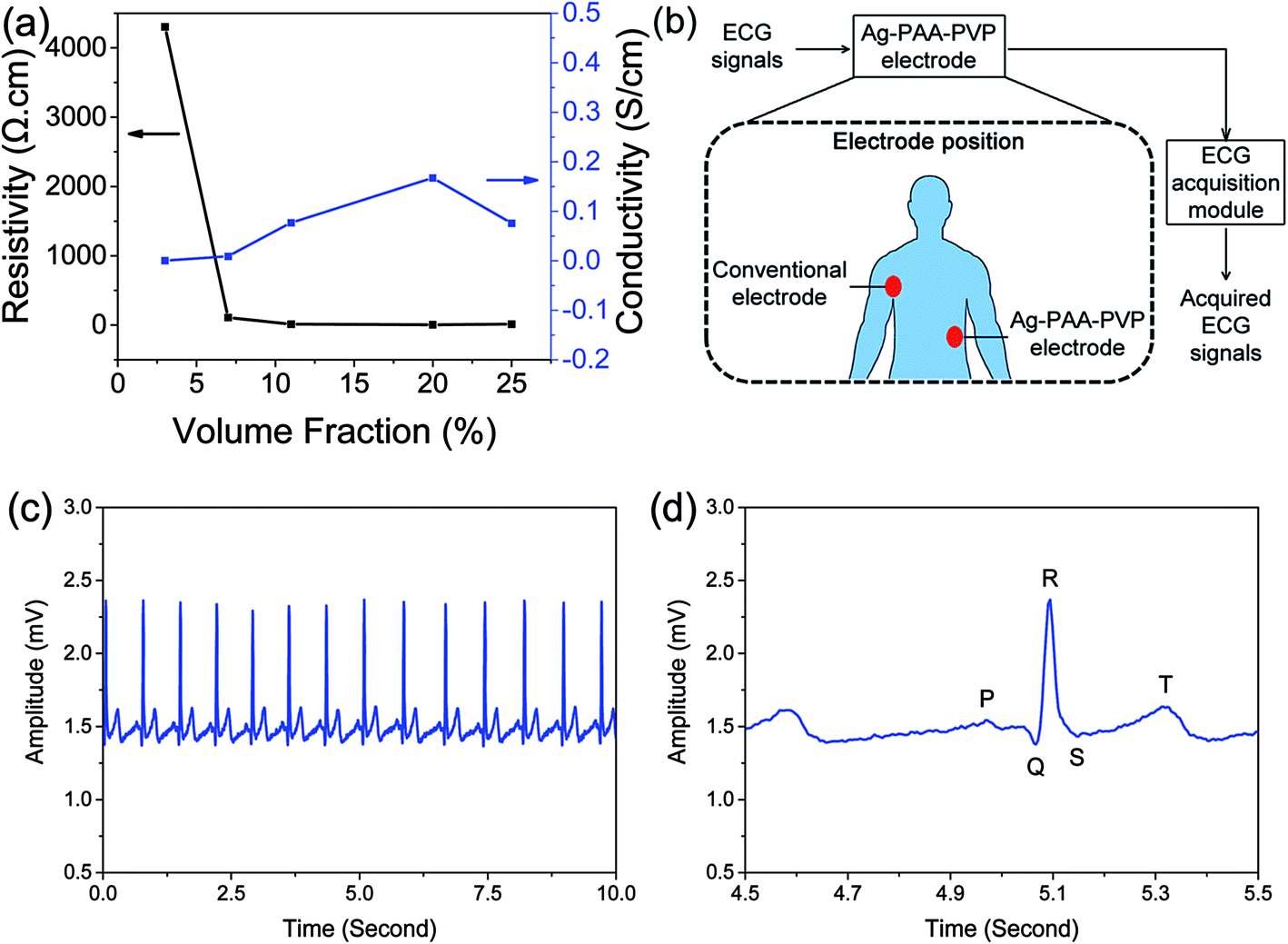 | ||
| Fig. 6 Electrical and ECG sensing measurements using our Ag–PDMS composites show that a conductive network of Ag particles was achieved and clear ECG signals were obtained. (a) Volume fraction of Ag particles required to reach the percolation threshold was ∼7 vol%. Volumetric resistivity and conductivity for various Ag volume fractions in Ag–PAA–PVP (1:10)/PDMS composite was measure to determine the percolation threshold. (b) The schematic of the ECG measurement method26 recording that was adopted to measure the (c) ECG signals using our Ag–PAA–PVP (1:10)/PDMS as an electrode, where (d) clear P, Q, R, S, T waves are observed in the magnified signal of 1 second. | ||
For our system, the percolation threshold is estimated to be ∼7 to 8 vol%, where below the threshold the network connectivity does not exist. The three-dimensional percolation threshold limit depends on the filler morphology, strength of interaction of individual particles within the agglomerate and dispersion characteristics of the filler agglomerates (or individual fillers). Generally a higher aspect ratio for the fillers leads to lower percolation thresholds, where spherical nanoparticles have a percolation threshold that can be 10 to 100 times higher than its high-aspect ratio counterpart.24 However, a potential benefit of spherical nanoparticles is that the higher mobility of spherical nanoparticles would allow conductive pathways that were lost upon deformation to be recovered in a different particle configuration. Our estimated percolation threshold of ∼7 to 8 vol% is consistent with other reports for metal nanoparticle-based composites where ranges from 10 to 16 vol% have been reported.24,25 Our lower measured threshold was attributed to two possible reasons: (1) the departure of our irregular agglomerated particles towards a more anisotropic form compared to that of a spherical morphology, and (2) improved uniformity in the distribution of our Ag nanoparticles. It should be noted here that the conductivity of the 7 vol% composite having reached the percolation threshold would meet the minimum conductivity requirements for ECG electrodes.
To demonstrate the practical applicability of the Ag/PDMS composite as a conductive electrode, the performance of the composite was evaluated by measuring the human electrocardiography (ECG) signals. The schematic for the setup of the ECG signal measurement experiment is shown in Fig. 6b. In brief, the conventional ECG adhesive electrode and the Ag–PAA–PVP (1:10)/PDMS electrode were respectively placed on right side of higher chest near armpit and left side of waist and the ECG signals were recorded by using the designed ECG acquisition module.26 The continuously recorded ECG signals for 10 seconds and the magnification of 1 second of ECG signal were acquired and respectively shown in Fig. 6c and d, and the P, Q, R, S, T waves of the ECG signal were also clearly obtained by using the Ag–PAA–PVP (1:10)/PDMS electrode. The P, Q, R, S and T waves of the ECG signal indicate the specifically terms, such as atrial depolarization, septal depolarization, early ventricular depolarization, late ventricular depolarization and repolarization of the ventricles, respectively. These experimental results indicated that the signal quality recorded by the proposed electrode potentially achieves the performance for recording the human ECG signals, without skin preparation or conductive gel usage. Furthermore, these results from our ECG experiments also suggests potential applications for the brain-machine interface technologies or neuroprosthetics development.26,27
3. Summary
In conclusion, a single-step process for modifying the existing PAA-functionalized Ag nanoparticles through an inter-polymer complexation between PAA and PVP was presented. After PAA–PVP modification, the morphology of Ag nanoparticles was similar as original Ag–PAA and displayed irregular shape and broad size distribution, which will be beneficial to a high conductivity of the conductive polymer composites when used as fillers. Interfacial characteristics of the PAA–PVP-modified nanoparticles system were investigated. The formation of intermolecular hydrogen bond between PAA and PVP, and the increased thermal stability and intermolecular interactions as PAA complexed with PVP were verified. The PAA–PVP-modified Ag nanoparticles showed an improved and best dispersion in PDMS with minimal particle aggregation or phase separation. The complex modification allowed the composite reached the Ag loading as high as 25 vol%, while the PAA or PVP-modified Ag nanoparticles-based PDMS composites cannot be cured once the loading exceeded 7 vol%. The fabricated Ag–PAA–PVP (1:10)/PDMS composites at 7 vol% with a volumetric resistivity of 110 Ω cm was preliminarily shown to be applicable as an ECG electrode. The reported modification method through the inter-polymer complexation can be extended to other systems to obtain a desirable filler loading without interfering with the polymer curing process.
4. Experimental methods
4.1. Materials
All the chemicals were used as received. Silver nitrate (99%), diethanolamine (DEA) (98%), polyvinylpyrrolidone (PVP) (Mw 10000 g mol−1), ethylene glycol (99.8%) were purchased from Sigma-Aldrich. Poly-acrylic acid (PAA) (Mw 5000 g mol−1 and Mw 50000 g mol−1) solutions were obtained from Polyscience Inc. Poly(dimethylsiloxane) (PDM) Sylgard 184 was provided by Dow Corning Corp.
4.2. Preparation of PAA-modified Ag particles
Silver particles stabilized with polymers (PAA or PVP) were synthesized. Ag–PAA was prepared using silver nitrate, PAA and DEA as the reducing agent.13 The typical procedure is as follows. Preparation of Ag–PAA was performed by mixing 30 mL deionized (DI) water, 24 g DEA, 1.2 g PAA (Mw 5000 g mol−1, 50 wt% in water), 0.6 g PAA (Mw 50000 g mol−1, 25 wt% in water) in a conical flask and stirring for 2 h at room temperature. Then 12 mL AgNO3 (12 g) aqueous solution was added quickly into the solution and the mixture was kept stirring for 22 h. The resultant Ag nanoparticles were then heated at ∼65 °C in a sonicating bath. Ag–PAA particles were collected by adding ethanol into the system and washing with the mixture of DI water and ethanol to remove the excess PAA.
4.3. Preparation of PVP-modified Ag particles
Ag–PVP particles were obtained by the reduction of silver nitrate by ethylene glycol in the presence of PVP.28 Ag–PVP particles were prepared by dissolving 6.0 g PVP in 50 mL ethylene glycol at 60 °C and 1.2 g AgNO3 was added into the solution. Then the mixture was heated to 120 °C for 1 h before cooling to room temperature. The Ag–PVP particles were precipitated with the addition of acetone. The precipitate was resuspended in ethanol and separated with centrifugation.4.4. Preparation of PAA–PVP-modified Ag particles
750 mg Ag–PAA was dispersed in 60 mL DI water under vigorously stirring and a 2 M HCl solution was dropped until the pH reached around 5.8. The low pH value allows the inter-polymer complexation to occur between PAA and PVP.20 The amount of PAA coated around the Ag nanoparticles was first determined using TGA (see Table 1) before we calculated the amount of PVP to be added. Then, 0.207 mmol (23 mg) (for 1:1) or 2.07 mmol (230 mg) (for 1:10) PVP was added and the mixture was kept stirring for 24 h, for 0.19 mmol (13.5 mg) of PAA. The obtained Ag–PAA–PVP (1:1) and Ag–PAA–PVP (1:10) particles were subjected to the same washing steps as that for Ag–PVP. The actual amount of PAA and PVP coated on the Ag-nanoparticles were measured using the TGA (see Table 1).
4.5. Chemical complexation of PAA and PVP
PAA and PVP chemical complexation mechanism was studied using only the polymers (i.e. without Ag particles), by dissolving pure PAA (mixture of Mw 5000 and Mw 50000 in the weight ratio of 4:1) in DI water, followed by the addition of 1 M NaOH to obtain a pH of 5.8. The hydrogen bonds between PAA is hypothesized to occur between the hydroxyl group on the carboxyl of PAA and the lone pair on the oxygen of the carbonyl group of PVP (see Fig. 4d). This is also why we have tuned the pH of the solution to 5.8, which was deduced based on the pKa of PAA of ∼4.5. We avoided lowering the pH by too much to prevent the local sintering of Ag nanoparticles, which would lead to more severe agglomeration and larger secondary particle sizes. Only when the pH value increases beyond ∼8 that all the carboxyl groups are ionized, hydrogen bond drive complexation between PAA and PVP would consequently be absent. Next, PVP was added with a molar ratio of PAA/PVP of 1:1 and 1:10. After stirring for 24 h, the solutions were freeze dried to obtain the polymer powders, which are designated as PAA–PVP (1:1) and PAA–PVP (1:10) according to the molar ratio of PAA to PVP. The amount of polymers used were 0.2 mmol (22.2 mg) of PVP (for 1:1) or 2.0 mmol of PVP (222 mg) (for 1:10) for 0.2 mmol (14.4 mg) of PAA.
4.6. Physical blending of PAA and PVP
The physical blends of PAA and PVP were prepared by dissolving PAA in ethanol and followed by the addition of the fixed amounts of PVP (for molar ratio of PAA/PVP of 1:1 and 1:10) with continuous stirring for 24 h. The amount of polymers used were 0.2 mmol (22.2 mg) of PVP (for 1:1) or 2.0 mmol of PVP (222 mg) (for 1:10) for 0.2 mmol (14.4 mg) of PAA. In this case, we did not expect significant hydrogen bond interactions between PAA and PVP prepared in this non-acidic environment, since most of the carboxyl groups of PAA would not be protonated. The mixtures were concentrated using a rotary evaporator to remove the solvent before being re-dissolved in DI water. After freeze drying, the blends of PAA + PVP (1:1) and PAA + PVP (1:10) were obtained.
4.7. Direct writing of PAA-, PVP- and PAA–PVP-modified Ag particles
The as-prepared PAA-, PVP- and PAA–PVP-modified Ag nanoparticle inks with 40 wt% of silver were prepared by dispersing the particles in DI water. Subsequently, direct-writing of the inks was completed using an air-powered fluid dispenser (Ultimus V, Nordson EFD, USA). The silver lines were printed using glass slides as substrates through nozzles (Nordson EFD, USA) with an inner diameter of 150 μm.4.8. Fabrication of Ag/PDMS composites
Typically, the surface-modified Ag nanoparticles were dispersed in toluene (1 mg μl−1) and sonicated for 1 h to yield uniform dispersions. Then the dispersions were mixed with PDMS pre-polymer by manual stirring and sonicating. The solvent toluene was employed to lower the viscosity of PDMS mixture and improve the dispersion of Ag particles in the matrix. Subsequently, toluene was evaporated at ∼80 °C in the sonicating bath. PDMS curing agent was added at the weight ratio of 1:10 with the pre-polymer with manual stirring and the mixtures were degassed for 1 h in a vacuum chamber. Finally, the surfactant-modified Ag/PDMS composites were formed by spin-coating (1000 rpm, 60 s) of the liquid mixtures on substrates and the curing process at 80 °C for 3 h.
4.9. Ag particle characterization
The morphology and size distribution of the Ag nanoparticles was characterized by scanning electron microscopy (SEM, JSM-7600F, JEOL Ltd., Japan). The averaged hydrodynamic diameter of the particles was measured using the dynamic light scattering (DLS) technique (NanoBrook Omni, Brookhaven Corp., USA). Zeta potential measurements of the nanoparticles were conducted using the same instrument (NanoBrook Omni). For both DLS and zeta potential measurements, samples of nanoparticles were suspended in DI water (20 mg L−1).4.10. Characterization of surface-modified particles
Fourier transform infrared (FTIR) spectra of pure polymers and surface-modified particles were measured using a VERTEX 70 spectrometer (Bruker, Germany) from 4000 to 400 cm−1 at a resolution of 4 cm−1 and averaged over 256 scans. Pelletized samples for the FTIR measurements in transmission were prepared by mixing ∼1 mg of samples with 130 mg of KBr using a mortar and pestle, before being pressed into a pellet using a hydraulic press (Atlas 15 ton Manual Hydraulic Press, Specac, UK). Thermogravimetric analysis (TGA) of the samples (∼5 mg) was conducted using a TGA Q50 analyzer (TA Instruments, USA) under a N2 atmosphere from 30 to 600 °C for a ramp rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were made using a TA Instrument Q100 (TA Instruments, USA) in an N2 atmosphere. The samples (3–5 mg) were loaded and sealed into aluminum pans with lids. Empty aluminum pan and lid were used as a reference sample. The heating process for the samples was set as (1) equilibrate at −50 °C, ramp at 20 °C min−1 to 200 °C and isothermal for 2 min, (2) ramp at 20 °C min−1 to −50 °C and isothermal for 2 min, (3) ramp at 10 °C min−1 to 200 °C.4.11. Dispersion of Ag particles in PDMS
Micrographs of Ag–PDMS composites were taken using the SEM to evaluate the dispersion characteristics of Ag particles. Elemental composition analysis of the composite surface was performed with an energy dispersive X-ray spectrometer (EDS) (Oxford Instruments Ltd., Bucks, UK) connected to the SEM. The composite samples were prepared by sputtering with gold at a discharge current of 20 mA for 60 s prior to SEM observation.4.12. Electrical properties of Ag–PDMS and printed Ag nanoparticles
The sheet resistance and volumetric resistivity of the Ag–PDMS composite films were measured by a four-point probe system (CMT-SR2000N, AIT, Korea). For the sheet resistance measurement, a current flow (100 mA) was passed between the outer two probes and the induced voltage between the two inner probes was measured. The volumetric resistivity was calculated by multiplying the sheet resistance by the thickness of the sample film. Tungsten carbide probes with the probe spacing of 1 mm and tip diameter of 100 μm were used. Linear resistivity measurements of the printed Ag nanoparticle lines were measured using a Keithley 4200-SCS semiconductor characterization system (Keithley, USA) equipped with a two-point probe station (Summit 11000, Cascade Microtech, USA). For the measurement, a voltage (0.5 V) was applied and the change in current (I) was recorded to calculate the resistance of each printed line. The linear resistivity and conductivity were then calculated.4.13. ECG measurement
The wireless EEG acquisition module has been developed in our previous studies.29,30 In brief, it composed of a biosignal amplifier (INA2126, Texas Instruments, USA), an acquisition component (AD8609, Analog Devices, USA), a microprocessor component (MSP430, Texas Instruments), and a wireless transmission component (BM0403, Unigrand Ltd., Taiwan), was used to detect the EEG signals. The preamplifier, band-pass filter (0.5–50 Hz), and an analog-to-digital converter (ADC) were embedded in this system to amplify and filter the EEG signals. The gains of amplifier and acquisition component were set to 5500, with a frequency band (0.5 to 50 Hz). The 12 bit ADC was used to digitize the EEG signals and the sampling rate amplified and filtered is set to 256 Hz. For the microprocessor component, the probed EEG signals were digitally stored. A moving average filter (60 Hz) was used to prevent the power-line interference while the wireless transmission. In addition, the module was fully compliant with the specifications for a Bluetooth v2.0+ enhanced data rate (EDR) and a printed circuit board (PCB) antenna. For human ECG signal recording, the conventional ECG adhesive electrode (Kendall Medi-Trace 200, Chicopee, MA, USA) and the Ag–PAA–PVP (1:10)/PDMS electrode were respectively attached on right side of higher chest near armpit and left side of waist, and the ECG signals were recorded at least for 10 seconds. All experimental protocols were approved by the Research Ethics Committee for Human Subject Protection Office, National Chiao Tung University (NCTU) in Taiwan. Protocol #NCTU-REC-104-033-e: “Aiming for the top university program: The Developing System of Brain Computer Interface”. The methods were carried out in accordance with the approved NCTU guidelines. Informed consent was obtained from all subjects. The ECG data were further output and analyzed by using MATLAB (Mathworks Inc., Natick, MA).
Acknowledgements
We acknowledge the support of the SUTD Digital Manufacturing and Design (DManD) Centre which is supported by the National Research Foundation Singapore (Project number RGDM1530203).References
- B. Zhu, Z. Niu, H. Wang, W. R. Leow, H. Wang, Y. Li, L. Zheng, J. Wei, F. Huo and X. Chen, Small, 2014, 10, 3625–3631 CrossRef CAS PubMed.
- D. Son, J. Lee, S. Qiao, R. Ghaffari, J. Kim, J. E. Lee, C. Song, S. J. Kim, D. J. Lee, S. W. Jun, S. Yang, M. Park, J. Shin, K. Do, M. Lee, K. Kang, C. S. Hwang, N. Lu, T. Hyeon and D.-H. Kim, Nat. Nanotechnol., 2014, 9, 397–404 CrossRef CAS PubMed.
- M. S. White, M. Kaltenbrunner, E. D. Glowacki, K. Gutnichenko, G. Kettlgruber, I. Graz, S. Aazou, C. Ulbricht, D. A. M. Egbe, M. C. Miron, Z. Major, M. C. Scharber, T. Sekitani, T. Someya, S. Bauer and N. S. Sariciftci, Nat. Photonics, 2013, 7, 811–816 CrossRef CAS.
- Z. Yang, J. Deng, X. Chen, J. Ren and H. Peng, Angew. Chem., Int. Ed., 2013, 52, 13453–13457 CrossRef CAS PubMed.
- H. C. Jung, J. H. Moon, D. H. Baek, J. H. Lee, Y. Y. Choi, J. S. Hong and S. H. Lee, IEEE Trans. Biomed. Eng., 2012, 59, 1472–1479 CrossRef PubMed.
- K. I. Jang, S. Y. Han, S. Xu, K. E. Mathewson, Y. Zhang, J. W. Jeong, G. T. Kim, R. C. Webb, J. W. Lee and T. J. Dawidczyk, Nat. Commun., 2014, 5, 4779 CrossRef CAS PubMed.
- S. M. Lee, J. H. Kim, H. J. Byeon, Y. Y. Choi, K. S. Park and S.-H. Lee, J. Neural Eng., 2013, 10, 036006 CrossRef PubMed.
- C. T. Lin, L. D. Liao, Y. H. Liu, I. J. Wang, B. S. Lin and J. Y. Chang, IEEE Trans. Biomed. Eng., 2011, 58, 1200–1207 CrossRef PubMed.
- T. Kim, J. Park, J. Sohn, D. Cho and S. Jeon, ACS Nano, 2016, 10, 4770–4778 CrossRef CAS PubMed.
- E. Bovero, K. E. A. Magee, E. C. Young and C. Menon, Macromol. React. Eng., 2013, 7, 624–631 CrossRef CAS.
- M. Park, J. Im, M. Shin, Y. Min, J. Park, H. Cho, S. Park, M.-B. Shim, S. Jeon and D.-Y. Chung, Nat. Nanotechnol., 2012, 7, 803–809 CrossRef CAS PubMed.
- Y. Hu, T. Zhao, P. Zhu, Y. Zhu, X. Shuai, X. Liang, R. Sun, D. D. Lu and C. P. Wong, J. Mater. Chem. C, 2016, 4, 5839–5848 RSC.
- B. Y. Ahn, E. B. Duoss, M. J. Motala, X. Guo, S.-I. Park, Y. Xiong, J. Yoon, R. G. Nuzzo, J. A. Rogers and J. A. Lewis, Science, 2009, 323, 1590–1593 CrossRef CAS PubMed.
- A. Panáček, R. Prucek, J. Hrbáč, T. j. Nevečná, J. Šteffková, R. Zbořil and L. Kvítek, Chem. Mater., 2014, 26, 1332–1339 CrossRef.
- W. J. Hyun, S. Lim, B. Y. Ahn, J. A. Lewis, C. D. Frisbie and L. F. Francis, ACS Appl. Mater. Interfaces, 2015, 7, 12619–12624 CAS.
- K. Park, G. Tuttle, F. Sinche and S. L. Harper, Arch. Pharmacal Res., 2013, 36, 125–133 CrossRef CAS PubMed.
- L. Wang, Z. Wang, X. Zhang, J. Shen, L. Chi and H. Fuchs, Macromol. Rapid Commun., 1997, 18, 509–514 CrossRef CAS.
- S. Chunhui, P. Mu and Y. Runzhang, Int. J. Hydrogen Energy, 2008, 33, 1035–1039 CrossRef.
- L. D. S. Yadav, Infrared (IR) Spectroscopy, in Organic Spectroscopy, Springer, Netherlands, 2005, pp. 52–106 Search PubMed.
- C. Maltesh, P. Somasundaran, R. Kulkarni and S. Gundiah, Langmuir, 1991, 7, 2108–2111 CrossRef.
- C. Lau and Y. Mi, Polymer, 2002, 43, 823–829 CrossRef CAS.
- R. Shvartzman Cohen, Y. Levi Kalisman, E. Nativ Roth and R. Yerushalmi Rozen, Langmuir, 2004, 20, 6085–6088 CrossRef CAS PubMed.
- M. Grouchko, A. Kamyshny, C. F. Mihailescu, D. F. Anghel and S. Magdassi, ACS Nano, 2011, 5, 3354–3359 CrossRef CAS PubMed.
- Y. Kim, J. Zhu, B. Yeom, M. Di Prima, X. Su, J. G. Kim, S. J. Yoo, C. Uher and N. A. Kotov, Nature, 2013, 500, 59–63 CrossRef CAS PubMed.
- L. Rivière, N. Causse, A. Lonjon, E. Dantras and C. Lacabanne, Polym. Degrad. Stab., 2016, 127, 98–104 CrossRef.
- K. Gramann, J. T. Gwin, D. P. Ferris, K. Oie, T. P. Jung, C. T. Lin, L. D. Liao and S. Makeig, Rev. Neurosci., 2011, 22, 593–608 Search PubMed.
- L. D. Liao, C. T. Lin, K. McDowell, A. E. Wickenden, K. Gramann, T. P. Jung, L. W. Ko and J. Y. Chang, Proc. IEEE, 2012, 100, 1553–1566 CrossRef CAS.
- J. Jiang, B. Bao, M. Li, J. Sun, C. Zhang, Y. Li, F. Li, X. Yao and Y. Song, Adv. Mater., 2015, 28, 1420–1426 CrossRef PubMed.
- L. D. Liao, I. J. Wang, S. F. Chen, J. Y. Chang and C. T. Lin, Sensors, 2011, 11, 5819–5834 CrossRef PubMed.
- L. D. Liao, C. Y. Chen, I. J. Wang, S. F. Chen, S. Y. Li, B. W. Chen, J. Y. Chang and C. T. Lin, J. Neuroengineering Rehabil., 2012, 9, 5 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04657j |
| This journal is © The Royal Society of Chemistry 2017 |