
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselectivity of D-amino acid oxidase in the presence of ionic liquids†
Qingju Liua,
Chuanfang Zhaoa,
Jincheng Huang a,
Li Chena,
Kunhao Yanga,
Lingling Gonga,
Yuguo Dua,
Chuyi Yuc,
Li Wu*a,
Xiangjun Li*a and
Yujian He*ab
a,
Li Chena,
Kunhao Yanga,
Lingling Gonga,
Yuguo Dua,
Chuyi Yuc,
Li Wu*a,
Xiangjun Li*a and
Yujian He*ab
aSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing, 101408, PR China. E-mail: wuli@ucas.ac.cn; lixiangj@ucas.ac.cn; heyujian@ucas.ac.cn; Fax: +86 10 69672529; Tel: +86 10 69672529
bState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, PR China
cLaboratory of Molecular Recognition and Selective Synthesis, Institute of Chemistry Chinese Academy of Sciences, Beijing, 100191, PR China
First published on 7th July 2017
Abstract
In this paper, enantioselectivities of D-amino acid oxidase (DAAO) in ten ionic liquids were investigated in detail. Among the ionic liquids studied, 3-methylimidazolium formate ([MIM][COO]) was able to trigger enzyme activation with L-Ala as substrate. The enzyme activity of DAAO reached a maximum of 4.26 U mg−1 min−1 in the presence of 40% [MIM][COO], while no activity toward L-Ala was observed when DAAO was incubated with other ILs. Moreover, DAAO activities towards other L-amino acids such as L-Pro and L-Arg were also improved in [MIM][COO], which indicated that the enzymatic enantioselectivity of amino acids was lowered. This was further confirmed by capillary electrophoresis, circular dichrosim and fluorescence analysis. Molecular optimization supported that the protein–ionic liquid interactions modulated the structure of the enzyme, especially in [MIM][COO], leading to relaxation in the enantioselectivity of DAAO. This observation and exploration might open a new window for modulation enantioselectivity resolution in ionic liquids.
Introduction
As potential environmentally benign “green” solvents, ionic liquids (ILs) have garnered much attention. It is widely acknowledged that ILs are compounds that are composed only of ions but still exist in the liquid state at ambient conditions.1 Due to their favourable features, such as excellent ability to dissolve a variety of solutes, ILs are being considered as leading substitutes of conventional organic solvents.2,3 To perform enzymatic reactions in ILs can be promising because of their unique properties, particularly their physicochemical properties can be finely designed by appropriate combination of the cationic and anionic parts. Furthermore, these ions display a wide range of varied physicochemical properties which can significantly increase substrate specificity, improve enzyme selectivity or enhance the enzyme activity or stability.4,5In view of ILs' excellent properties for use as reaction solvents, extensive studies of biocatalytic reactions using ILs have been carried out. Up to date, substantial studies have focused on the role of ILs in enzymatic reaction either as the reaction media or as the extraction phase.2,6 The correlations between ionic constituents as well as the alkyl chain length of ILs and enzymatic activity and stability have been analysed in detail.3,7–9 The enzymatic behaviours have been improved in ILs (higher enantioselectivity and better recoverability and recyclability) due to high conversion rates.10–13 Interestingly, enzymatic reactions in the presence of ILs have provided useful information on the enzymes flexibility and stability, enzyme inhibition, behaviour of enzyme.14,15
In general, by optimization of the type and concentration of ILs, utilization of ILs as co-solvents greatly enhances the enzymatic activity and stability. However, little attention has been devoted to the control of substrate enantioselectivity. Only recently had the stereoselectivity and enantioselectivity of several enzymes in the presence of ILs began to be studied and investigated by several research groups.10–12
Enantiocomplementary enzymes favor opposite enantiomers because either the substrate or the active-site machinery is oriented differently.16 In an oxidation example, the active site of D-amino acid oxidase (DAAO) is a mirror image of that of L-amino acid oxidase (LAAO). Both enzymes catalyze amino acid oxidation, but with opposite enantiomer preference.17 However, there are some exceptions to this rule. For example, the LAAO from Bacillus carotarum 2Pfa accepts 10 L-amino acids and 7 D-amino acids as substrates (ie, D-Leu, D-Lys and D-Ser).18 Also, catalytic oxidation of L-Pro by DAAO at high concentrations has been confirmed.19 Moreover, it is acknowledged that the alteration of enzyme enantioselectivity can be achieved by reaction temperature,20 biomimetic encapsulation,21 directed evolution,22 site-directed mutagenesis.23,24 In addition, our recent work has shown that the DAAO can catalytically oxidize many L-amino acids by modulating pH of aqueous solution.25 Especially, there is only 15.99-fold (pH 8.0) and 14.18-fold (pH 9.8) drop for the enantioselectivity (kcat,D-AA/kcat,L-AA) of Pro and Ala, respectively. In contrast, it's reported that the initial rate of enzymatic deamination for D-amino acids is found to exceed that for L-amino acids by 3000 to 4000-fold under physiological condition.19
In the case of DAAO, Lutz-Wahl et al.26 have compared the activity and stability of free and immobilized DAAO from Trigonopsis variabilis in different water-soluble and water-insoluble ionic liquids as well as inorganic solvents. They found that the most promising ILs were [BMIM][BF4] and [MMIM][MMPO4]. However, the comparison of the enantioselectivity of DAAO was not reported in ILs yet. Altering enantioselectivity of DAAO has been regarded as one of the most remarkable topics in enzymatic reactions, since the stereochemical outcome of a given enzyme–substrate reaction is predetermined in specific target environment. Taking these findings into consideration, a comprehensive study of enzymic enantioselectivity in aqueous solution containing different ILs is necessary, which will provide valuable information to understand the behaviour of enzyme in varied ionic liquids.
In the present work, the enantioselectivities of DAAO in presence of ten ILs were investigated in detail. In particular, we observed the enzymatic activity towards L-Ala in presence of [MIM][COO] was sharply enhanced. Under optimization conditions of pH value and temperature in [MIM][COO], the kinetic parameters were determined. Additionally, capillary electrophoresis, circular dichroism and fluorescence analysis were undertaken to support the results. The present review intends to show the modulation of enantioselectivity of DAAO by ILs, and provide mechanistic insights into how different factors contribute to the relaxation of enzymatic enantioselectivity by effect of specific ion in various ILs.
Experimental
Regents and instruments
All L-amino acids, sodium 3,9-fluorenylmethyloxycarbonyl (FMOC) and 5-dichloro-2-hydroxybenzenesulfonate (DHBS), imidazole, 3-methylimidazole, 4-aminoantipyrine (4-AAP), were purchased from J&K Scientific. D-Amino acid oxidase from porcine kidney (pkDAAO, ≥8.2 units per mg solid) and peroxidase (POD) were obtained from Sigma-Aldrich. ILs were kindly supplied by Process Research Institute, Chinese Academy of Sciences.DAAO activity in presence of ILs with L-Ala as substrate
Enzymatic activity of DAAO in presence of ILs was determined by measuring the formation of H2O2 from L-Ala.27 The reaction was performed for 5 min at 25 °C. Reaction mixtures were carried out in tubes containing borate saline buffer (50 mM, pH = 8.5), 300 U L−1 POD, 0.275 nM 4-AAP, 5.5 mM DHBS, 0.1 mM FAD and 25 mM L-Ala, various amounts of ILs (0–50%, v/v), and enzyme at a concentration of 228 U L−1. Negative control experiments without ILs were performed. After addition of 500 μL trichloroacetic acid (10%, w/v), the mixture was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min. After that absorbance change was monitored at 512 nm using Ultraviolet spectrophotometer (Shimadzu UV-2550). One unit of activity was defined as the amount of H2O2 produced by 1 mg enzyme per minute under the experimental conditions. All measurements were carried out in 3 repetitions.
000 × g for 10 min. After that absorbance change was monitored at 512 nm using Ultraviolet spectrophotometer (Shimadzu UV-2550). One unit of activity was defined as the amount of H2O2 produced by 1 mg enzyme per minute under the experimental conditions. All measurements were carried out in 3 repetitions.
Influence of temperature and pH
The influence of pH on DAAO activity with L-Ala as substrate was determined by incubating the reaction mixture at pH values ranging from 5.0 to 12.0, in the following mix buffer system: 50 mM sodium acetate (pH 4.0–5.5); 50 mM sodium phosphate (pH 6.0–8.0); 50 mM borate saline buffer (pH 8.0–9.0); 50 mM borate–NaOH buffer (pH 9.5–12.0). To check influence of the pH, DAAO was pre-incubated with predetermined pH mix buffer solution containing 40% (v/v) [MIM][COO] for 5 min at 25 °C, and activity was measured under standard assay condition as described above. The optimum temperature for DAAO activity with L-Ala as substrate was determined by conducting the assay at various temperatures from 20 to 80 °C. The thermostability of the enzyme was measured after pre-incubation of the enzyme in borate saline buffer (50 mM, pH 9.0) containing 40% (v/v) of [MIM][COO] at various temperature for 5 min.Circular dichroism (CD) spectroscopy
CD spectroscopic studies were performed using a Jasco-815 spectrophotometer (Jasco, Japan) equipped with a Peltier system for controlling the temperature. The sample was pre-equilibrated at 25 °C for 15 min and the scan speed was fixed for adaptive sampling (error F 0.01) with a response time of 1 s and 1 nm bandwidth. The secondary and tertiary structures of DAAO were monitored by using 1.0 cm path length cuvette. The concentrations of DAAO were 0.4 mg mL−1 in 50 mM borate saline buffer (pH 9.0) containing 40% (v/v) IL. Each sample spectrum was obtained by appropriate blank media without DAAO from the experimental enzyme spectrum.Fluorescence spectroscopy
Fluorescence measurements for DAAO were monitored using Jasco FP-750 instrument (Cremello, Italy) at 0.1 mg L−1 protein concentration in 50 mM borate saline buffer containing of various ILs (40%, v/v) (final pH 9.0). The experiments were performed at 25 °C by using a 1 cm sealed cell. Emission fluorescence spectra of Trp were recorded between 300 and 400 nm, with excitation at 280 nm; flavin emission were recorded from 475 to 600 nm, with excitation at 450 nm. A blank medium without enzyme was subtracted to eliminate influence of the fluorescence of the ILs and buffer components on the enzyme fluorescence spectrum. Both the change in fluorescence intensity and the shift in fluorescence maximum wavelength were recorded to monitor the unfolding transition; each spectrum being an average of six spectra.Computational details
The X-ray structure of D-amino acid oxidase download from Protein Data Base (http://www.rcsb.org, PDB code: 1C0P). The ligand FAD was extracted from 1C0P and used as reference, while the [MIM] derived from FAD. Atomic charges of ligands and proteins were calculated with the Gasteiger–Hückel protocol. Surflex-Dock program was used to search possible binding conformations of ligands. Default parameters were used in the Docking computation. FAD was docked into the protein, with a TotalScore 25.36, and the RMSD value was 0.646, which indicated that parameters used here was reasonable. With all the parameters, [MIM] was docked into the binding site of D-amino acid oxidase, with TotalScore of 3.42, which was much lower than that of FAD.Results and discussion
Activity of DAAO with L-Ala in presence of ILs
Imidazolium-based ILs such as [BMIM][BF4], [BMIM][PF6] are commonly and popularly used in study.28 Previous experiences and recent literature data have shown that imidazolium and pyridinium-based ILs are efficient and promising for enzymatic reaction,26,29 thus, ten kinds of imidazolium and pyridinium-based ILs were chosen for the comparison of the enantioselectivity of DAAO. The ten kinds of ILs used were 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]), 1-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]), 1-ethyl-3-methylimidazolium bromide ([EMIM]Br), 1-hexyl-3-methylimidazolium bromide ([HMIM]Br), 1-octyl-3-methylimidazolium bromide ([OMIM]Br), N-butylpyridinium bromide ([BPy]Br), N-methylpyridinium formate ([MPy][COO]), 1-butyl-3-methylimidazolium chloride ([BMIM]Cl), 3-methylimidazolium formate ([MIM][COO]) and 3-methylimidazolium propionate ([MIM][CH3CH2COO]), respectively, and their structures were shown in Fig. 1.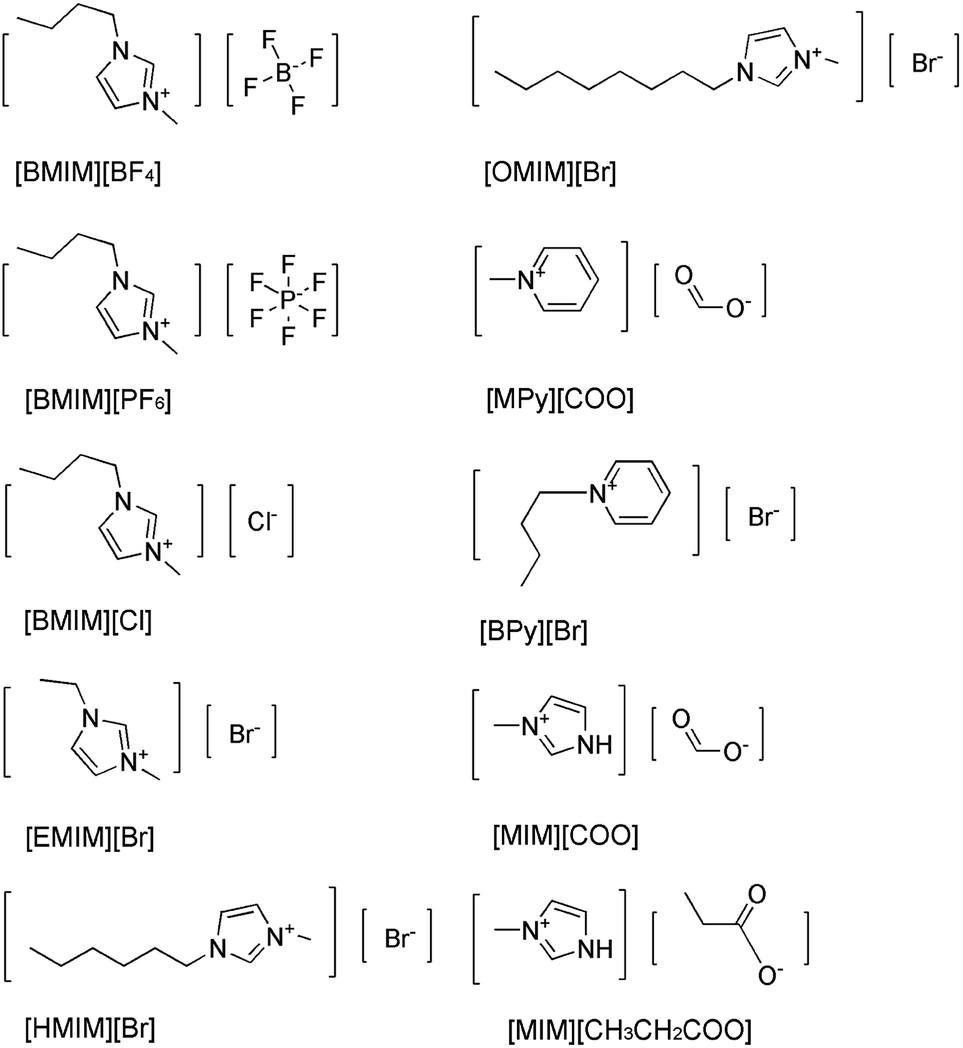 | ||
| Fig. 1 Structure of ILs used in this study. | ||
The activities of DAAO with L-Ala as substrate in various ILs were investigated. As summarized in Fig. 2, of all the ILs tested, no detectable activity of DAAO in [BMIM][BF4], [BMIM][PF6], [BMIM]Cl, [EMIM]Br, [HMIM]Br, [OMIM]Br, [BPy]Br, [MPy][COO] and [MIM][CH3CH2COO] was observed. Enantioselectivity of DAAO remained unchanged even at ratios of more than 40% (v/v) ILs. However, enzymatic activity towards L-Ala showed a sharp increase in [MIM][COO]. With the increase in ratio of [MIM][COO], DAAO activity towards L-Ala was enhanced accordingly. The enzyme activity reached to a maximum (4.26 U mg−1 min−1) in presence of 40% [MIM][COO]. When the ratio of [MIM][COO] was above 40%, the enzyme activity reached a plateau. The results suggested that [MIM][COO] disrupted the enantioselectivity of DAAO and improved DAAO activity on L-Ala.
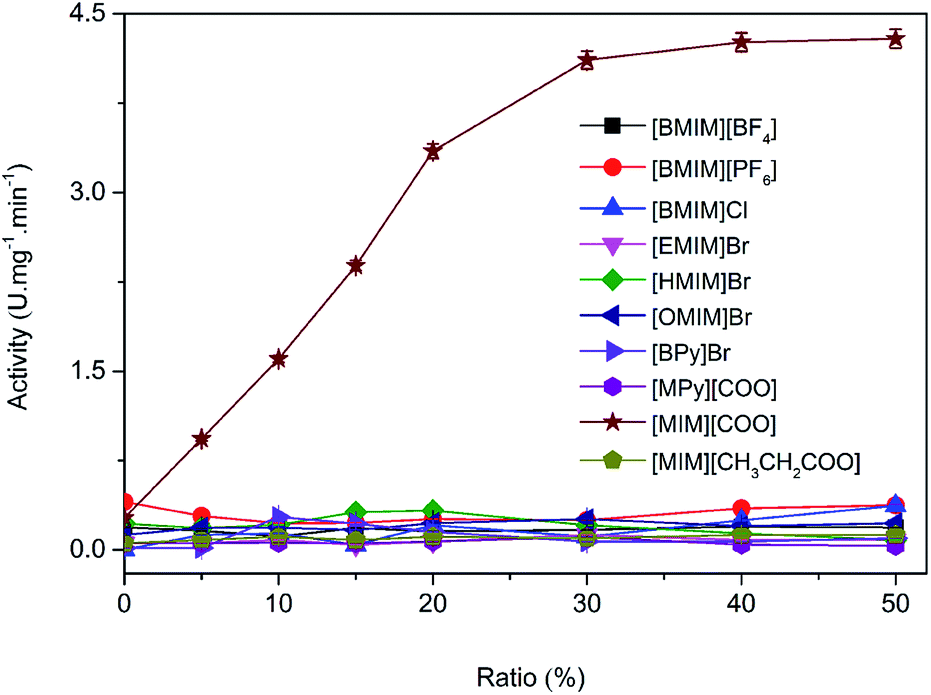 | ||
| Fig. 2 Enzymatic activity of DAAO in ILs with L-Ala as substrate. Reaction mixtures containing borate saline buffer (50 mM, pH = 8.5), ILs in a range of concentrations, 228 U L−1 DAAO, 300 U L−1 POD, 0.275 nM 4-AAP, 5.5 mM DHBS, 0.1 mM FAD and 25 mM L-Ala were incubated for 30 min at 25 °C. | ||
As we know, one of the most important factors influencing enzyme activity is pH. We employed four common buffer systems in a pH range of 5.0 to 12.0 for determination. Based on the preliminary study on pH-dependent DAAO activity for the optimization of pH was performed with 40% (v/v) [MIM][COO]. As Fig. 3a shown, enzymatic activity of DAAO with L-Ala as substrate was significantly improved with the increase of pH value of [MIM][COO]. It reached a maximum (4.78 U mg−1 min−1) at pH 9.0. When pH value of [MIM][COO] was above 9.0, the enzymatic activity of DAAO was sharply decreased. The enzymatic activity became almost negligible at pH 12.0. In view that temperature could influence the solubility of the reactants and reaction rates, the enzymatic activities altered with the change of temperature. Similarly, the optimal temperature for DAAO activity towards L-Ala was determined in the temperature range from 20 °C to 80 °C, as shown in Fig. 3b. It's observed that the activity of DAAO was enhanced with the increase of temperature, and the optimum temperature was about 37 °C. The enzymatic activity was gradually decreased with increase of temperature when the temperature was above 37 °C. Thus, DAAO activities towards various amino acids were assessed under the optimal conditions (pH 9.0 and 37 °C).
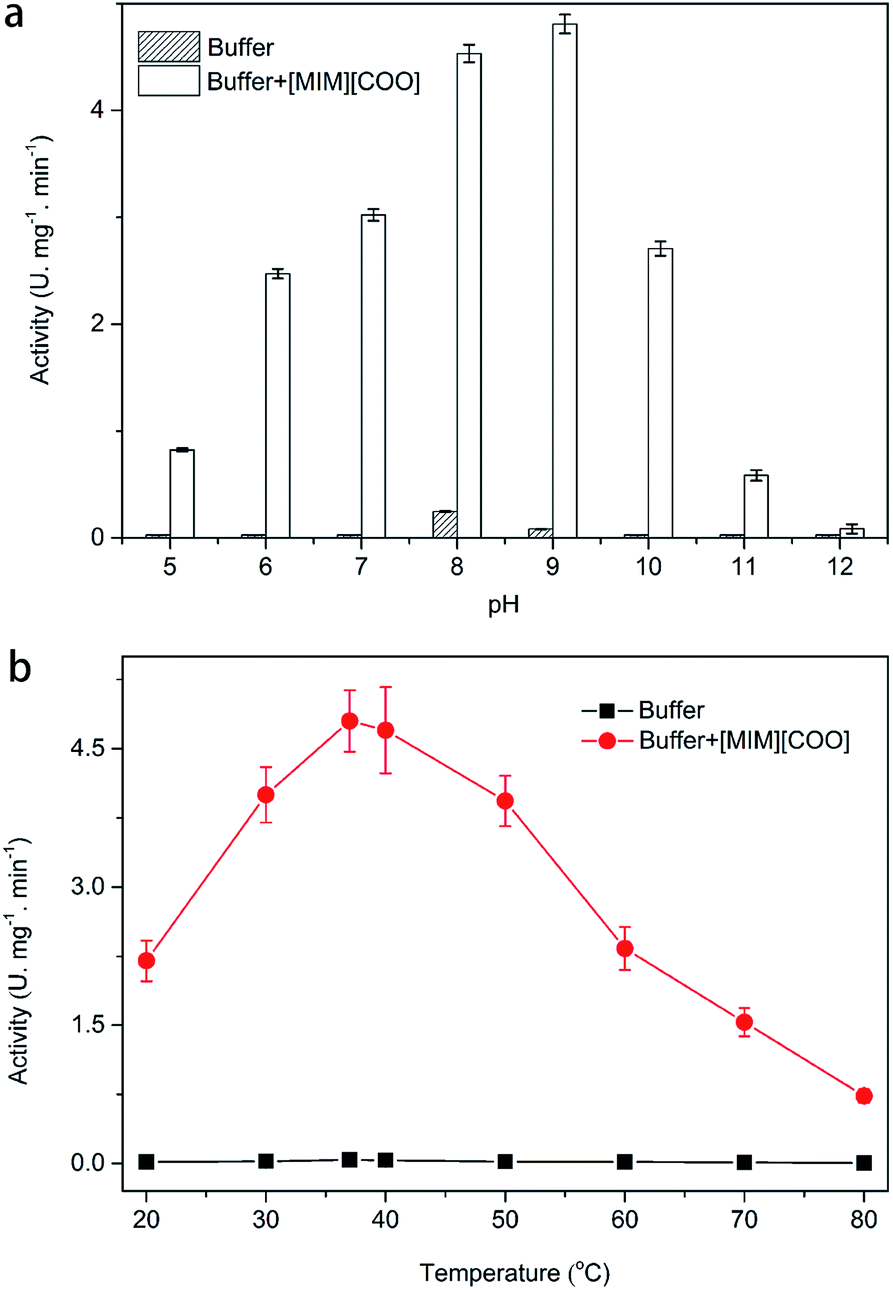 | ||
| Fig. 3 Optimization of pH (a) and temperature (b) for DAAO activities towards L-Ala. Reaction mixtures containing 40% [MIM][COO], 228 U L−1 DAAO, 300 U L−1 POD, 0.275 nM 4-AAP, 5.5 mM DHBS, 0.1 mM FAD and 25 mM L-Ala were incubated for 30 min. | ||
Then, the kinetic parameters were determined under the optimum conditions by analysis of Michaelis–Menten plots. DAAO showed a Vmax value of 6.1 ± 0.50 mM min−1 and Km value of 19.70 ± 0.30 mM with L-Ala as substrate in the presence of 40% (v/v) [MIM][COO] (Fig. 4). The results indicated that the performance of both binding and catalytic ability of DAAO towards L-Ala was remarkably improved in the presence of [MIM][COO].
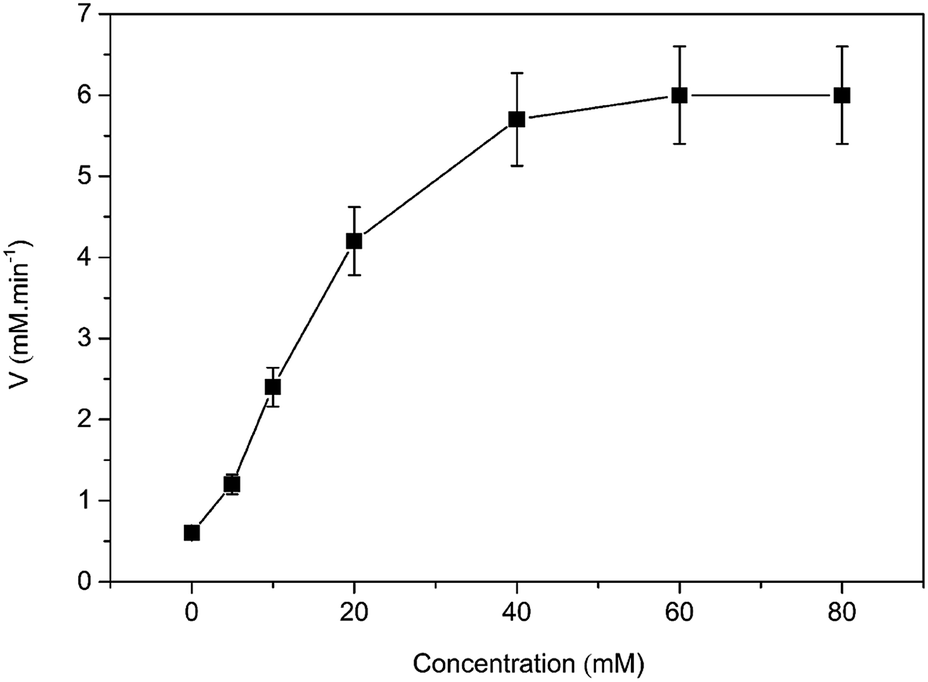 | ||
| Fig. 4 Michaelis–Menten plots of the DAAO with L-Ala as substrate in presence of 40% (v/v) [MIM][COO]. The experiment was carried out in borate saline buffer (50 mM, pH = 9.0) at 37 °C. | ||
Activity of DAAO with other L-amino acids as substrate in presence of ILs
To check whether the observation was occurred for other L-amino acids, the enzymatic activity of DAAO with other L-amino acids as substrate in the presence of [MIM][COO] was determined. By incubating the enzyme with 5, 20 and 40% (v/v) concentrations of [MIM][COO], enzymatic activities were determined in the standard assay conditions. Similar results were obtained and illustrated in Table 1. Noticeably, for L-Trp, L-Asn, L-Cys, the enzyme activity was slightly increased when the [MIM][COO] concentration increased (0.92 ± 0.08 U mg−1 min−1 for L-Trp, 0.89 ± 0.05 U mg−1 min−1 for L-Asn, 0.39 ± 0.03 U mg−1 min−1for L-Cys in 40% [MIM][COO]). In contrast, in the case of L-His, L-Thr, L-Gln and L-Arg, activities of DAAO were modestly improved. For example, activity of DAAO towards L-His (U mg−1 min−1) was increased from 0.37 ± 0.02 (5% [MIM][COO]) to 1.38 ± 0.11 (40% [MIM][COO]). More extraordinary, with the increase of [MIM][COO] ratio from 5% to 40%, enzymatic activities were significantly enhanced in presence of L-Pro, L-Phe, L-Leu, L-Val and L-Ser except for L-Ala and enzyme activity towards L-Pro was up to 4.06 U mg−1 min−1 in presence of 40% (v/v) [MIM][COO]. These results indicated that exposure to [MIM][COO] changed the general enantioselectivity of DAAO toward many amino acids.| Amino acids | Enzyme activity (U mg−1 mim−1) | ||
|---|---|---|---|
| 5% | 20% | 40% | |
| a NA: no activity. | |||
| L-Ala | 0.82 ± 0.09 | 3.46 ± 0.37 | 4.26 ± 0.09 |
| L-Pro | 0.16 ± 0.13 | 0.31 ± 0.02 | 4.06 ± 0.11 |
| L-Met | NAa | 1.38 ± 0.04 | 3.65 ± 0.44 |
| L-Phe | NA | 1.61 ± 0.06 | 3.28 ± 0.05 |
| L-Leu | NA | 0.45 ± 0.04 | 2.27 ± 0.57 |
| L-Val | NA | 0.44 ± 0.03 | 2.17 ± 0.07 |
| L-Ser | NA | 0.41 ± 0.03 | 2.05 ± 0.04 |
| L-His | NA | 0.37 ± 0.02 | 1.83 ± 0.11 |
| L-Thr | NA | 0.36 ± 0.33 | 1.79 ± 0.09 |
| L-Tyr | NA | 0.29 ± 0.05 | 1.46 ± 0.04 |
| L-Gln | NA | 0.75 ± 0.07 | 1.26 ± 0.09 |
| L-Arg | NA | 0.24 ± 0.04 | 1.22 ± 0.05 |
| L-Trp | NA | 0.48 ± 0.09 | 0.92 ± 0.08 |
| L-Asn | NA | 0.18 ± 0.01 | 0.89 ± 0.05 |
| L-Cys | NA | 0.08 ± 0.01 | 0.39 ± 0.03 |
Confirmation of catalytic oxidation and analysis of conformational change in presence of ILs
In order to solidify the above idea, CE analysis was performed to detect whether L-amino acids were converted into their D-isomers induced by [MIM][COO]. Compared with the signal of control, peak of D-Ala wasn't observed in the L-Ala solution containing 40% (v/v) [MIM][COO] within the reaction time, which indicated that the L-Ala was not transformed to its mirror-image configuration after [MIM][COO] treatment and [MIM][COO] indeed mediated the oxidation of L-Ala catalyzed by DAAO during the reaction time. The similar results were obtained through the CE analysis of other L-amino acids (ESI Fig. S1†). As a consequence, it was concluded that the racemization of L-Ala did not occur during the reaction time in these experiments.Subsequently, the structure of DAAO in the presence of ILs was further studied to make clear the reason responsible for alteration in enantioselectivity. The protein conformation of DAAO before and after [MIM][COO] treatment was tested by CD spectrum analysis. Due to that a negative signal around 215 nm in CD spectrum was observed for DAAO in buffer, the conformation change of DAAO were monitored by the CD signal around 215 nm (taken as a reporter of the change in secondary structure). Changes in signal at 215 nm were shown in ESI Fig. S2.† Concerning the signal at 215 nm, there were no remarkable changes in the CD spectrum of the DAAO in presence of [BMIM][BF4], [BMIM][PF6] and [MIM][COO] (ESI Fig. S2a and b† and Fig. 5), suggesting that DAAO remained its native conformation. However, the CD spectrum of DAAO was moderately altered after [MPy][COO] treatment compared to that in buffer. A decrease was evident for the enzyme of CD signal in 215 nm (ESI Fig. S2c†), pointing to a decrease in secondary structure content. The analysis of CD spectrum of DAAO showed that the signal was almost disappeared after incubated with [BMIM]Cl, [EMIM]Br, [HMIM]Br, [OMIM]Br, [BPy][COO], [MIM][CH3CH2COO] (ESI Fig. S2d–i†). The structural change was accompanied by a sharp decrease of activity, which was consistent with previous reports that IL may lead to the secondary structure collapse in enzyme.30 But surprisingly, [MIM][COO] didn't result in conformational alteration of DAAO (Fig. 5), although [MIM][COO] induced enhancement of DAAO activities towards L-amino acids.
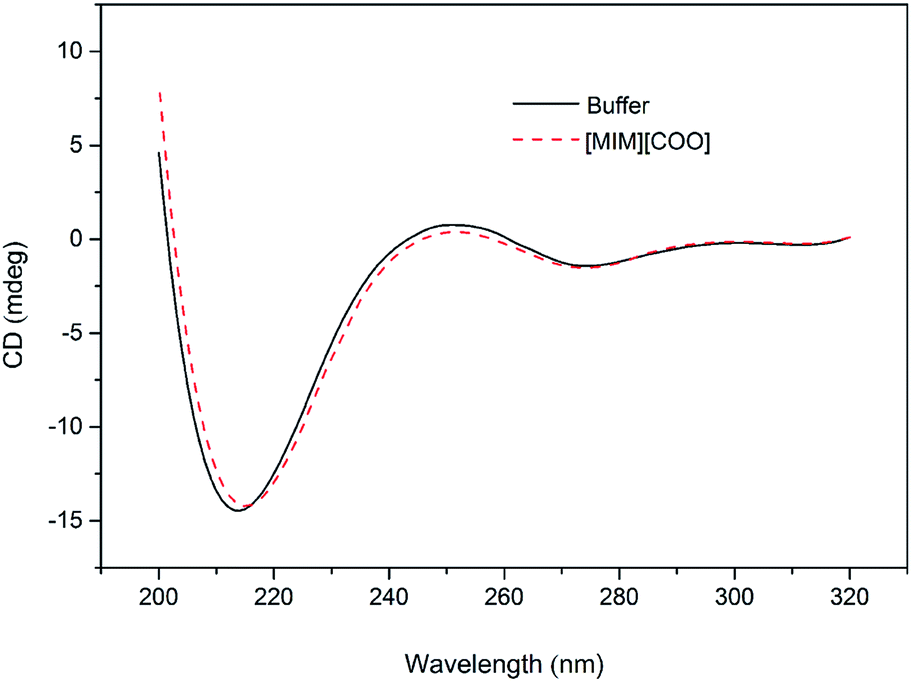 | ||
| Fig. 5 CD spectrum of DAAO in presence of ILs. The DAAO was pre-incubated with 40% (v/v) ILs at 25 °C for 15 min and the CD spectrum of DAAO were monitored by using 1.0 cm path length cuvette. The concentration of DAAO was 0.4 mg mL−1. | ||
Due to that there is a change in the maximum emission of Trp and flavin with the change of solvent environment in proteins containing fluorophore residues, thus different microenvironment surroundings of the fluorophore residues of DAAO can be monitored by observing changes in the maximal intensity of fluorescence (Imax) and the red shift of the maximal emission wavelength (λmax) of the Trp residues and flavin of the protein.31
To further explain the conformational change of DAAO in various ILs, fluorescence analysis for DAAO in the presence of ILs was carried out. The fluorescence spectra of DAAO after treatment with 40% (v/v) ILs were illustrated in Fig. 6 and ESI Fig. S3.† After incubation with 40% (v/v) [BMIM][BF4], [BMIM][PF6], [BMIM]Cl, [MPy][COO] and [MIM][COO] for 30 min, the enzyme exhibited the same λmax of Trp and flavin and the slightly decreases in Imax were observed, compared with that of in buffer. In contrast, with DAAO after 40% (v/v) [OMIM]Br treatment, a noticeable red-shift (about 9 nm) was observed in λmax of Trp, accompanied by a significant increase in Imax of Trp (ESI Fig. S3a†) and a sharp decrease in Imax flavin (ESI Fig. S3b†). As for DAAO in presence of 40% (v/v) [EMIM]Br, [HMIM]Br, [BPy]Br and [MIM][CH3CH2COO], there were remarkable decreases in Imax of Trp and flavin as compared to that of control. Moreover, there were red-shifts (about 13 nm) in λmax of Trp for [BPy]Br and [MIM][CH3CH2COO] treatment group. This suggested that [EMIM]Br, [HMIM]Br, [OMIM]Br, [BPy]Br and [MIM][CH3CH2COO] induced variation in protein structure, accompanied by an alteration of activity. It is evident that the addition of [MIM][COO] induces a tiny decrease in fluorescence intensity of DAAO, without any changes in the maximum emission wavelength and the shape of fluorescence spectra.
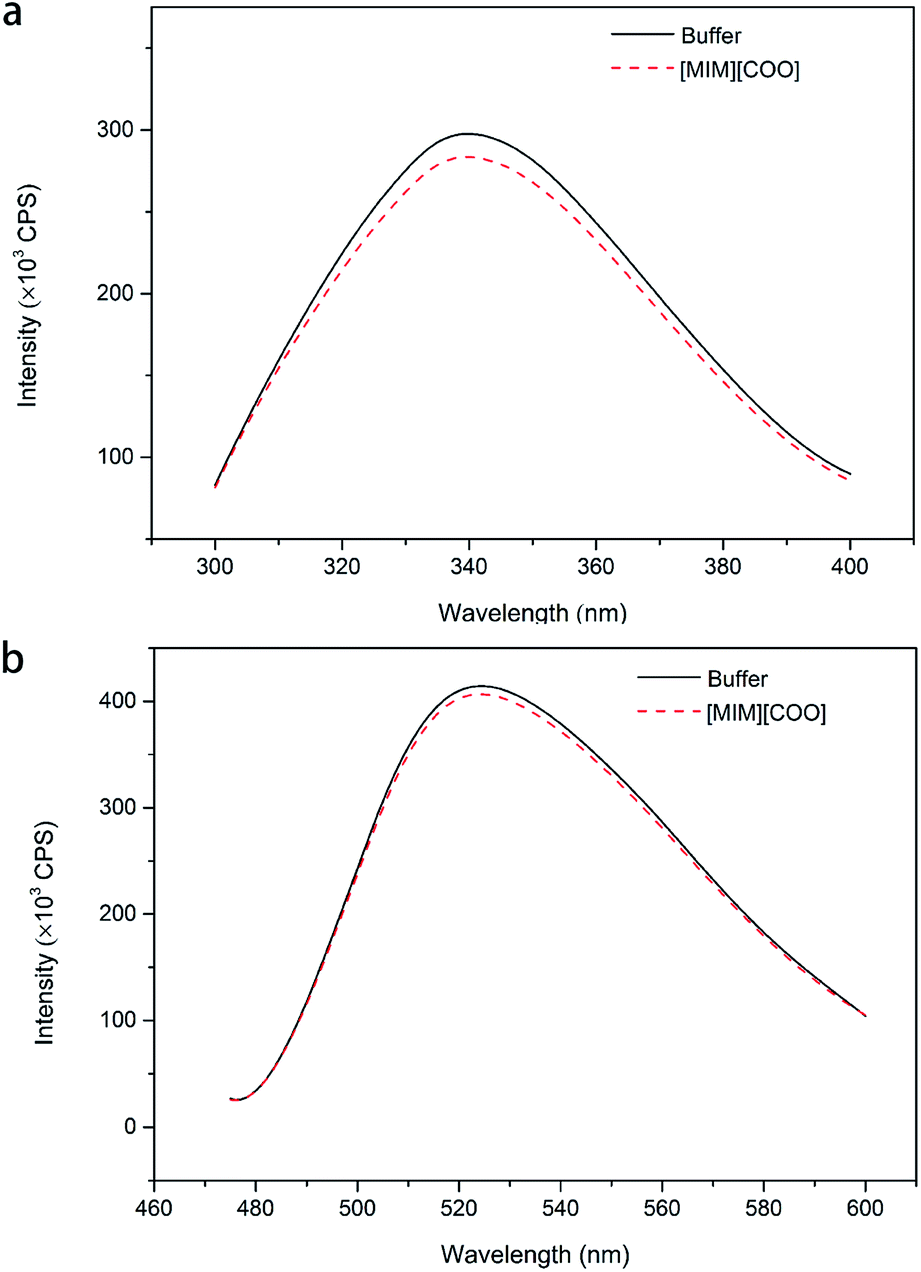 | ||
| Fig. 6 Fluorescence spectra of DAAO in the presence of ILs. Fluorescence spectra of Trp (a) and flavin (b) of DAAO in borate saline buffer (50 mM, pH 9.0) in the absence and presence of 40% IL (v/v) after incubation for 30 min at 25 °C. The excitation wavelength was 280 and 450 nm, respectively. | ||
Structural study of DAAO in ILs
With the same anion, DAAO activity towards L-Ala in the existence of [MIM][COO] was sharply improved compared to that of [MPy][COO]. Previously, it's found that ILs comprising the N-methylimidazolium group exhibits beneficial effects on activity than the one embracing the N-methylpyridinium group with the same anion.32,33 The examination of DAAO activity in presence of imidazole and N-methylimidazole showed that there was no noticeable alteration in substrate specificity of DAAO (ESI Fig. S4†). Therefore, it's inferred that [MIM]+ plays a critical role in the alteration of enantioselectivity.Data from CD and fluorescence showed no noticeable collapse of secondary structures and changes in the enzyme conformation induced by [MIM][COO] (Fig. 5 and 6).
A close look at the active site structure of DAAO is necessary to understand the improved catalytic performance of DAAO modulated by [MIM][COO]. The shape of the enzyme is a typical α/β fold (Fig. 7a). The key residues in binding site that are responsible for the catalytic activity of DAAO are Tyr 224, Tyr 228, Arg 283, Gly 313 and Gln 5334,35 (Fig. 7b). The first step in oxidative deamination involves the nucleophilic attack of the hydroxyl group of Tyr 224 and Tyr 228 to the carbonyl carbon on the substrate to form an enzyme–substrate intermediate. The guanidine group of Arg 283 forms a salt bridge with α-COO− of the substrate. The amino group of substrate is hydrogen bind with hydroxyl group of Tyr 224 and the backbone carbonyl of Gly 313 and Gln 53 which also form hydrogen bonds with bound water molecules.36 Moreover, D-Ala is tightly bound to the active site of the enzyme by noncovalent interaction, forming a quite stable transient intermediate. In contrast, the interactions of L-Ala with the enzyme were shown in Fig. 7c. The coordination positions of α-COO− are normally occupied by guanidine group from Arg 283, two hydroxyl group from the protein amino acid side chains of Tyr 224 and Tyr 228, and the amino group faces away from the carbonyl moiety of Gly 313 and Gln 53, thereby making the nucleophilic attack difficult (Fig. 7c).
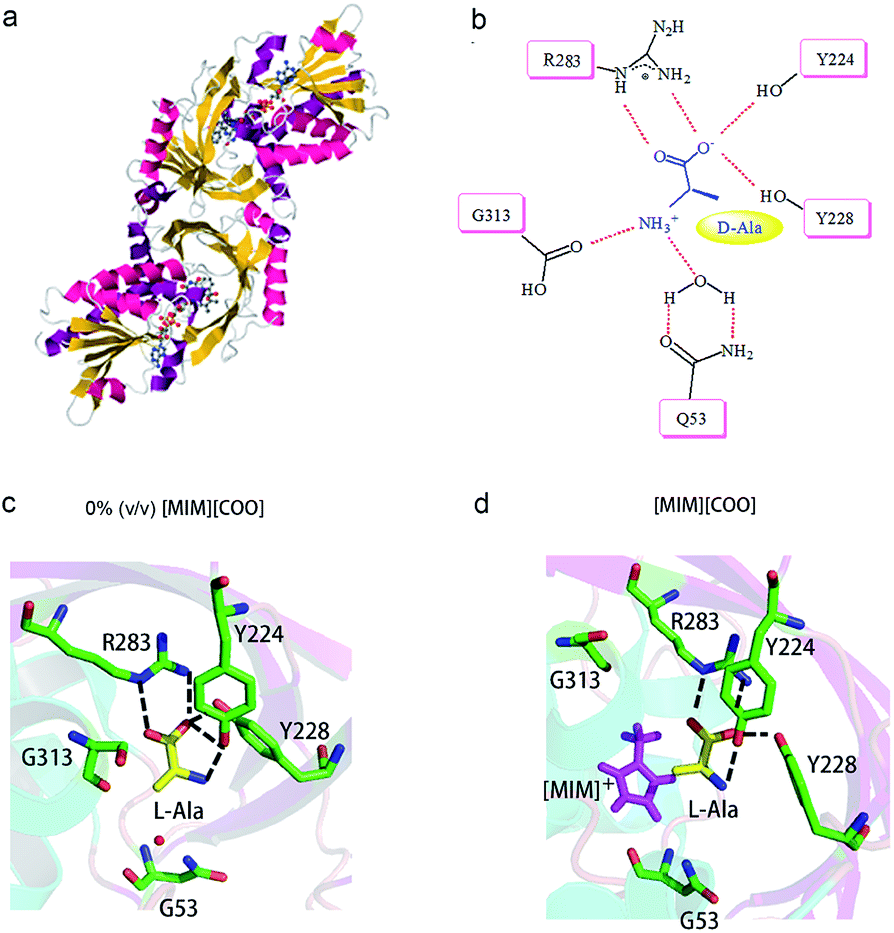 | ||
| Fig. 7 Schematic drawing of interaction of DAAO–L-Ala complex with [MIM][COO]. (a) Detail of DAAO structure (PDB entry 1KIF). (b) Interactions between D-Ala and DAAO. Schematic drawing of the active site cavity of DAAO in complex with L-Ala in absence (c) and presence (d) of [MIM][COO]. The active site residues were shown in stick representation coloured in green (carbon atoms), blue (nitrogen atoms) and red (oxygen atoms). The L-Ala molecule was shown in stick presentation, coloured in yellow (carbon atoms), blue (nitrogen atoms) and red (oxygen atoms). [MIM][COO] was shown in pink. H-bonds were shown as dashed lines. Figure was prepared with PyMol. | ||
Previous reports have shown that small structural changes in catalytic centre are important in biological and chemical processes.37,38 Water-miscible solvents can affect enzymatic performance by stripping off the essential water associated with the enzyme or penetrating into the micro-aqueous phase to interact with the enzyme by changing the protein dynamics, conformation and/or the enzyme's active site.39 Since the reaction system has sufficient aqueous component, water stripping seems to be unlikely. Thus, a more direct interaction of the ILs with the enzyme may be responsible for the alteration in enantioselectivity.
We mimicked the binding mode of [MIM]+ and D-amino acid oxidase with Surflex-Dock program in SYBYL8.0 software (Tripos Inc., St. Louis, MO, USA). The X-ray structure of D-amino acid oxidase download from Protein Data Base (http://www.rcsb.org PDB code: 1C0P). The FAD was extracted from 1C0P and used as reference, while the [MIM]+ derived from FAD. Direct interaction of ILs with the catalytic site of the enzyme occurred due to structural adaptation between the active site of DAAO and the IL, as shown in Fig. 7c and d and ESI Fig. S5.† The comparation of structure of the DAAO–L-Ala complex in buffer (Fig. 7c) and the DAAO–[MIM][COO]–L-Ala complex formed in presence of [MIM][COO] (Fig. 7d) showed that with existence of [MIM][COO], L-Ala occupied the same region of the binding pocket as that of the L-Ala in buffer and the overall structure of DAAO–[MIM][COO]–L-Ala is very similar to that previously reported in buffer. A large unoccupied patch of electron density in the entrance of the active site could be fitted with [MIM][COO]. It is reported that the reaction of Ala catalyzed by DAAO first dehydrogenates the amino acid to the corresponding imino acid, coupled with the reduction of FAD then hydrolyzes to the α-keto acid and ammonia.40 Thus, the transfer of α-proton to FAD and the attack of H2O to α-amino group play key roles in catalyzed oxidization of amino acid. In buffer, L-Ala is tightly bound to the active site of the enzyme by noncovalent interaction, forming a quite stable transient intermediate, in which α-NH3 group faces away from the carbonyl moiety. However, [MIM] cation can interact with surface residue (Tyr) through hydrophobic π–π interactions due to a permanent positive charge and a N-methyl group of the imidazolium ring, which agrees with the previous report that most of the cations interact with hydrophobic regions on the surface of the enzyme.41 Thus, in presence of [MIM][COO], the interaction between [MIM] cation and active site places L-Ala in a better attacking position to transfer of α-proton to FAD or the nucleophilic attack to the amino group by water.
In comparison, [MIM][CH3CH2COO] has the same cation as [MIM][COO], whereas it doesn't enhance the activity of DAAO towards L-Ala. A possible reason is that larger steric structure and longer linear chain of [MIM][CH3CH2COO] blocks the entrance of [MIM]+ to the active site area in the enzymatic reaction process. In contrast, with existence of [MIM][COO], the electrostatic interaction of [COO]− and [MIM]+ strengthens the interaction of [MIM]+ with region of the binding pocket of DAAO. The data from kinetic studies with L-Ala as a substrate show a dramatic improvement in the affinity of the substrate (Fig. 4), indicates that a slight relaxation of active centre for the substrate induced by [MIM][COO]. In addition, pH had a synergistic effect on the alteration substrate specificity of DAAO based the optimization of pH (Fig. 3a). This was in consistent with our previous data that pH could mediate change in enantioselectivity of DAAO.25
Conclusions
In this study, the activities of DAAO towards L-Ala in presence of ten ILs were checked to evaluate the enantioselectivity of DAAO. It's observed that the activity of DAAO with L-Ala as substrate in presence of [MIM][COO] was greatly enhanced and the enzyme activity of DAAO reached to a maximum of 4.26 U mg−1 min−1 in presence of 40% [MIM][COO]. Furthermore, DAAO activities towards other L-amino acids such as L-Pro, L-Phe, L-Leu, L-Val and L-Ser were also improved in [MIM][COO]. CE, CD and fluorescence analysis confirmed the observations. [MIM][COO] could induce alteration of the activities of DAAO towards L-amino acids than the other 10 ILs such as [MIM][CH3CH2COO]. The alteration might also be related to a slight conformational relaxation of active centre for the DAAO due to [MIM][COO] fitting into the binding pocket. Although the reason for the change of the activities of DAAO towards L-Ala need to be further explored, our investigation could offer a simple and effective method to predict the relaxation of the enantioselectivity of DAAO and thus guide the choice of a reaction system with the proper enantioselectivity.Conflict of interest
The authors declare no competing financial interest.Author contribution statement
Y. H. and X. L. conceived the idea and directed the work. Q. L. performed the experiments. C. Z. performed computational analysis. J. H. provided the ionic liquids. L. C. performed the CE analysis. K. Y., L. G., Y. D., & C. Y. helped to analyse the results. Q. L. and L. W. analysed the experiments and wrote the main manuscript text. All authors reviewed the manuscript.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (Grant No. 21272263), the State Key Laboratory of Natural and Biomimetic Drugs (No. K20140204), Universities of Chinese Academy of Sciences Grant (No. O8JT011J01), Fusion project of molecular science and education for Institute of Chemistry (Y52902HED2).Notes and references
- Z. Yang and W. B. Pan, Enzyme Microb. Technol., 2005, 37, 19–28 CrossRef CAS.
- M. Moniruzzaman, N. Kamiya and M. Goto, Org. Biomol. Chem., 2010, 8, 2887–2899 CAS.
- M. C. Bubalo, K. Hanousek, K. Radosevic, V. G. Srcek, T. Jakovljevic and I. R. Redovnikovic, Ecotoxicol. Environ. Saf., 2014, 101, 116–123 CrossRef PubMed.
- M. Naushad, Z. A. ALOthman, A. B. Khan and M. Ali, Int. J. Biol. Macromol., 2012, 51, 555–560 CrossRef CAS PubMed.
- J. L. L. Carter, M. Bekhouche, A. Noiriel, L. J. Blum and B. Doumeche, ChemBioChem, 2014, 15, 2710–2718 CrossRef CAS PubMed.
- A. Chefson and K. Auclair, ChemBioChem, 2007, 8, 1189–1197 CrossRef CAS PubMed.
- D. Ajloo, M. Sangian, M. Ghadamgahi, M. Evini and A. A. Saboury, Int. J. Biol. Macromol., 2013, 55, 47–61 CrossRef CAS PubMed.
- M. F. Machado, R. P. Queiros, M. D. Santos, L. G. Fidalgo, I. Delgadillo and J. A. Saraiva, World J. Microbiol. Biotechnol., 2014, 30, 487–494 CrossRef CAS PubMed.
- H. S. Kim, D. Eom, Y. M. Koo and Y. G. Yingling, Phys. Chem. Chem. Phys., 2016, 18, 22062–22069 RSC.
- H. Zhao and S. V. Malhotra, Biotechnol. Lett., 2002, 24, 1257–1260 CrossRef CAS.
- M. Goldfeder, M. Egozy, V. S. Ben-Yosef, N. Adir and A. Fishman, Appl. Microbiol. Biotechnol., 2013, 97, 1953–1961 CrossRef CAS PubMed.
- H. W. Yu, J. C. Wu and C. C. Bun, Chirality, 2005, 17, 16–21 CrossRef CAS PubMed.
- C. C. Barron, B. J. D. Sponagle, P. Arivalagan and G. B. D'Cunha, Enzyme Microb. Technol., 2017, 96, 151–156 CrossRef CAS PubMed.
- A. P. S. Brogans and J. P. Halleet, J. Am. Chem. Soc., 2016, 138, 4494–4501 CrossRef PubMed.
- H. Zhao, J. Chem. Technol. Biotechnol., 2016, 91, 25–50 CrossRef CAS PubMed.
- P. F. Mugford, S. M. Lait, B. A. Keay and R. J. Kazlauskas, ChemBioChem, 2004, 5, 980–987 CrossRef CAS PubMed.
- A. Mattevi, G. Tedeschi, L. Bacchella, A. Coda, A. Negri and S. Ronchi, Struct. Anal., Folding Des., 1999, 7, 745–756 CrossRef CAS.
- G. M. Brearley, C. P. Price, T. Atkinson and P. M. Hammond, Appl. Microbiol. Biotechnol., 1994, 41, 670–676 CrossRef CAS.
- K. Yagi and M. Nishikimi, J. Biochem., 1968, 64, 371–376 CrossRef CAS PubMed.
- X. Jin, B. K. Liu, Z. Ni, Q. Wu and X. F. Lin, Enzyme Microb. Technol., 2011, 48, 454–457 CrossRef CAS PubMed.
- S. Emond, D. Guieysse, S. Lechevallier, J. Dexpert-Ghys, P. Monsan and M. Remaud-Simeon, Chem. Commun., 2012, 48, 1314–1316 RSC.
- M. Ivancic, G. Valinger, K. Gruber and H. Schwab, J. Biotechnol., 2007, 129, 109–122 CrossRef CAS PubMed.
- Y. Ijima, K. Matoishi, Y. Terao, N. Doi, H. Yanagawa and H. Ohta, Chem. Commun., 2005, 877–879 RSC.
- S. Bartsch, R. Kourist and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2008, 47, 1508–1511 CrossRef PubMed.
- Q. Liu, L. Chen, Z. Zhang, B. Du, Y. Xiao, K. Yang, L. Gong, L. Wu, X. Li and Y. He, Sci. Rep., 2017, 7, 2994 CrossRef PubMed.
- S. Lutz-Wahl, E. M. Trost, B. Wagner, A. Manns and L. Fischer, J. Biotechnol., 2006, 124, 163–171 CrossRef CAS PubMed.
- Y. Nishiya and T. Imanaka, FEBS Lett., 1998, 438, 263–266 CrossRef CAS PubMed.
- A. Kurata, H. Senoo, Y. Ikeda, H. Kaida, C. Matsuhara and N. Kishimoto, Extremophiles, 2016, 20, 415–424 CrossRef CAS PubMed.
- A. I. Akhmetshina, O. R. Gumerova, A. A. Atlaskin, A. N. Petukhov, T. S. Sazanova, N. R. Yanbikov, A. V. Nyuchev, E. N. Razov and I. V. Vorotyntsev, Sep. Purif. Technol., 2017, 176, 92–106 CrossRef CAS.
- S. Ghaedizadeh, R. Emamzadeh, M. Nazari, S. M. M. Rasa, S. H. Zarkesh-Esfahani and M. Yousefi, Biochem. Eng. J., 2016, 105, 505–513 CrossRef CAS.
- L. Caldinelli, S. Iametti, A. Barbiroli, F. Bonomi, L. Piubelli, P. Ferranti, G. Picariello, M. S. Pilone and L. Pollegioni, J. Biol. Chem., 2004, 279, 28426–28434 CrossRef CAS PubMed.
- J. L. Kaar, A. M. Jesionowski, J. A. Berberich, R. Moulton and A. J. Russell, J. Am. Chem. Soc., 2003, 125, 4125–4131 CrossRef CAS PubMed.
- L. Suresh, P. Onkara, P. S. V. Kumar, Y. Pydisetty and G. V. P. Chandramouli, Bioorg. Med. Chem. Lett., 2016, 26, 4007–4014 CrossRef CAS PubMed.
- M. Bakke and N. Kajiyama, Appl. Biochem. Biotechnol., 2004, 112, 123–131 CrossRef CAS PubMed.
- A. Nueangaudom, K. Lugsanangarm, S. Pianwanit, S. Kokpol, N. Nunthaboot and F. Tanaka, Phys. Chem. Chem. Phys., 2012, 14, 2567–2578 RSC.
- M. G. Sarower, S. Okada and H. Abe, ScienceAsia, 2009, 35, 150–155 CAS.
- A. D. Mesecar, B. L. Stoddard and D. E. Koshland, Science, 1997, 277, 202–206 CrossRef CAS PubMed.
- D. E. Koshland, Nat. Med., 1998, 4, 1112–1114 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, J. Biol. Chem., 1988, 263, 8017–8021 CAS.
- M. S. Pilone, Cell. Mol. Life Sci., 2000, 57, 1732–1747 CrossRef CAS PubMed.
- E. M. Nordwald, J. G. Plaks, J. R. Snell, M. C. Sousa and J. L. Kaar, ChemBioChem, 2015, 16, 2456–2459 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04687a |
| This journal is © The Royal Society of Chemistry 2017 |