
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoMoO4 as a novel heterogeneous catalyst of peroxymonosulfate activation for the degradation of organic dyes†
Yanan Fan,
Wenjie Ma,
Jianglong He and
Yunchen Du *
*
MIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: yunchendu@hit.edu.cn
First published on 21st July 2017
Abstract
Sulfate radical-based advanced oxidation processes (SR-AOPs) are receiving more and more attention for the removal of recalcitrant organic pollutants. In this study, we employ CoMoO4 as a novel heterogeneous catalyst for peroxymonosulfate (PMS) activation to release powerful sulfate radicals for the first time. The CoMoO4, prepared through a hydrothermal route and high-temperature calcination, displays a hierarchical microstructure assembled from ultrathin nanosheets and a large surface area (61.9 m2 g−1). Methylene blue (MB) is selected as a model organic pollutant, and it is found that the CoMoO4/PMS system can realize 100% degradation of MB in 40 min and maintain its removal efficiency during three recycling experiments. Such a catalytic performance of CoMoO4 is indeed superior to those of conventional Co3O4 and CoFe2O4. The effects of some potential influential factors, including reaction temperature, dosages of PMS and CoMoO4 and the initial pH value are systematically evaluated. More importantly, the CoMoO4/PMS system not only shows its universality in the degradation of other organic dyes (e.g. orange II and rhodamine B), but also exhibits considerable degradation efficiency under some actual water background conditions. The quenching experiments confirm that sulfate radicals are the main active species for the degradation of dyes, and XPS spectra reveal that Co sites on the surface of CoMoO4 are the primary active sites for the generation of sulfate radicals.
Introduction
Wastewater from textile, printing, and other industries usually contains various dyes in high-concentration, which can produce considerable adverse effects on eco-environments due to their toxicity and recalcitrance.1 In the past few decades, some conventional treatments, such as physical, chemical and biological techniques, have been utilized for environmental remediation, while their effectiveness in the removal of organic dyes is still far away from the expected performance.2,3 As an attractive alternative, advanced oxidation processes (AOPs) offer immense potential for overcoming the challenges related to the deep purification of wastewater owing to their excellent oxidation ability and mineralization efficiency.4,5 Hydroxyl radicals (˙OH) are the classic reactive species in AOPs, and their notable reactivity is almost applicable to all organic compounds.6 However, their practical applications always suffer from many limitations, e.g. sophisticated and cost-ineffective radical generation processes, pH adjustments and potential sludge generation.7–9 Recently, sulfate radicals (˙SO4−) have received intensive research interests as another powerful reactive radicals, because they can not only compensate some critical inadequacies of ˙OH, but also provide higher standard reduction potential (2.5–3.1 V) under neutral conditions than ˙OH (1.8–2.7 V).10 It is well known that ˙SO4− can be released from peroxymonosulfate (PMS) or persulfate (PS) by heating, UV irradiation, ultrasonication, and catalytic activation, where catalytic activation has its own advantages without the assistance of necessary equipment and high-energy input.6,11–13 Compared with PS, PMS is a preferable candidate for the generation of ˙SO4− in catalytic system, because its asymmetrical molecular structure and relatively large O–O bond length in free molecules will facilitate the activation process.14 Literature review reveals that homogeneous catalytic activation of PMS by some transition metal ions, especially Co2+, is very effective for the generation of ˙SO4−,15 while the secondary pollution caused by the high solubility and great toxicity of transition metal ions will severely hinder the application of homogeneous catalysis in water treatment.16 As a result, it is of great importance to develop high-performance heterogeneous catalysts to alleviate the potential secondary contamination from the homogeneous systems.To date, many kinds of heterogeneous catalysts, including zero-valence metal, oxides, supported catalysts, and carbon materials, have been continuously reported as promising PMS activators.8,17–20 Compared with free metal ions, immobilization of active sites in heterogeneous catalysts makes them lose the characteristic of single atom catalysis, resulting in inferior activities than those homogeneous catalysts. However, it is still found that some Co-containing heterogeneous catalysts can produce better catalytic performance in PMS activation than their analogous counterparts.21–23 For example, Lin and co-workers compared the catalytic activities of Prussian blue analogues (PBAs) with various metal ions (e.g. Co, Cu, Fe, Mn, Ni), and the results indicated that Co-containing PBAs exhibited excellent capability for activating PMS, while Cu, Fe and Ni based PBAs did not provide conspicuous catalytic activity to activate PMS;24 Ren et al. evaluated the performance of magnetic ferrospinel, including CoFe2O4, CuFe2O4, MnFe2O4 and ZnFe2O4, in PMS activation, and they found that CoFe2O4 had the highest catalytic performance among these ferrites towards PMS for the degradation of di-n-butyl phthalate.21 In most previous studies on heterogeneous PMS activation, there was a consensus on the possible reaction mechanism, that was, electron transfer between M2+/M3+ and HSO5−/˙SO4− would be responsible for the generation of ˙SO4−.25,26 Therefore, it may be an effective way to further enhance the effectiveness of PMS activation by employing a Co-containing heterogeneous catalyst with improved electron transfer features.
More recently, CoMoO4 has appeared as a novel and promising pseudo-capacitor, and its unique advantages, such as great redox activity, stable crystalline structure, high electrical conductivity, as well as fast transport between ions and electrons, are highly desirable for the excellent electrochemical performance.27–29 It is very exciting that these characteristics also cater to PMS activation, and thus the catalytic performance of CoMoO4 in the degradation of organic pollutants can be expected. Unfortunately, there are very few correlative literatures accessible. In this article, we employ CoMoO4 as a heterogeneous catalyst in PMS activation for the first time. The organic dye, methylene blue (MB), is selected as a model pollutant. It is found that CoMoO4 displays much better MB degradation than common CoFe2O4 and Co3O4, and its good catalytic activity is also versatile for the degradation of other dyes [orange II (OII) and rhodamine B (RhB)], even under some actual water background conditions. We believe that CoMoO4 can be an efficient and green heterogeneous catalyst for the degradation of organic pollutants, and this study provides new insights into the development of alternative catalysts for wastewater treatment.
Experimental
Materials synthesis and characterization
CoMoO4 was synthesized through a hydrothermal route according to previous literature.30 Briefly, 0.119 g (0.5 mmol) of CoCl2·6H2O, 0.877 g (0.071 mmol) of (NH4)6Mo7O24·4H2O and 0.3003 g (5 mmol) of urea were dissolved in 36 mL of deionized (DI) water. This mixture was vigorously stirred at room temperature for 30 min to form a homogeneous solution. The transparent solution was then transferred into a Teflon-lined stainless steel autoclave and sealed to heat at 120 °C for 12 h. The obtained purple particles were collected by centrifugation, and washed several times with DI water and ethanol till the filtrate became colourless. After drying at 60 °C for 12 h, the as-prepared precursor was further calcined at 500 °C for 3 h with a heating rate of 1 °C min−1 under air atmosphere to produce highly crystalline CoMoO4. For comparison, CoFe2O4 and Co3O4 were also prepared by co-precipitation method and high-temperature calcination under the same conditions.Powder X-ray diffraction (XRD) pattern of the as-prepared samples was obtained at room temperature with a D/MAXRC X-ray diffractometer using Cu Kα radiation source which operated at 45 kV and 40 mA. The structure and morphology of the sample was performed by scanning electron microscopy (SEM, Quanta 200S) and transmission electron microscopy (TEM, JEM-3000F). Brunauer–Emmett–Teller (BET) surface area data were measured by a QUADRASORB SI-KR/MP (Quantachrome, USA). Samples were normally treated for porosity measurement by degassing at 200 °C for 4 h with a N2 flow. X-ray photoelectron spectroscopy (XPS) was collected using PHI 5700 ESCA System with an Al Kα radiation as the X-ray source. The absorbance of different dye solution was determined by using a UV-Vis diffuse reflectance spectrophotometer (TU-1901). Total organic carbon (TOC) was determined by Analytik Jena AG MultiN/C 2100 TOC analyzer.
Catalytic test procedure
All the catalytic experiments were evaluated in 100 mL beaker containing 50 mL of pollutant solution at 25 ± 1 °C. In a typical run, 5.0 mg of catalysts was dispersed into 50 mL MB solution (100 mg L−1) under constant magnetic stirring (the agitation speed was fixed at 400 rpm) for about 30 min to achieve the adsorption–desorption balance. The oxidative process was initiated after the introduction of a certain amount of PMS (2.0 mM) [Note: PMS concentration was calculated from its actual content in commercially available oxone (KHSO5·0.5KHSO4·0.5K2SO4)]. At given time intervals, 0.4 mL of the reaction mixture was withdrawn and mixed with saturated Na2S2O3 solution to quench the reaction. The concentration of MB was analyzed using UV spectrophotometer at λ = 664 nm, and the corresponding degradation efficiencies were obtained according to the following equation:| Degradation efficiency (%) = [(C0 − Ct)/C0] × 100% | (1) |
Results and discussion
The crystalline structure of the as-prepared CoMoO4 is primarily studied by wide angle XRD (Fig. 1), and the diffraction peaks in the 2θ range of 10–60° can be well matched with β-CoMoO4 phase (JCPDS 21-0868). Although CoMoO4 can occur in the forms of α-phase and β-phase, high temperature (330–410 °C) will induce the transformation from α-CoMoO4 to β-CoMoO4.31 In our case, the calcination temperature is 500 °C, and thus the characteristic peaks of α-CoMoO4 (JCPDS 25-1434) are not detected in final product. In addition, the absence of characteristic peaks of possible impurities, such as Co3O4 (JCPDS 42-1467) and MoO3 (JCPDS 47-1320 and 47-1081) further validates the formation of high-purity β-CoMoO4 through hydrothermal treatment and high-temperature calcination.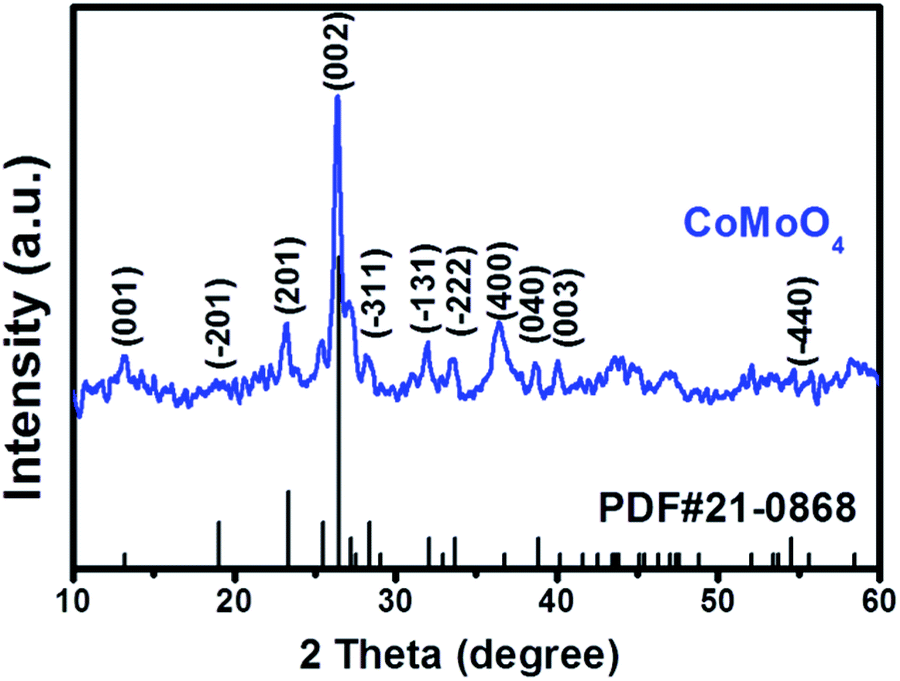 | ||
| Fig. 1 XRD pattern of the as-prepared CoMoO4. | ||
SEM and TEM measurements were conducted in order to confirm the morphology and microstructures of the as-prepared product. Before calcination in the muffle furnace, the precursor holds the relatively uniform spheres with wrinkle surface and an average diameter of about 2.5 μm, as shown in Fig. S1.† It can be clearly seen that each flower-like microsphere is assembled by numerous ultrathin nanosheets, and these curved and corrugated nanosheets are connected to each other, forming a stable hierarchical microstructure. Very interestingly, SEM image reveals that this hierarchical microstructure can be well preserved during the high-temperature calcination, and the as-prepared CoMoO4 still exhibits flower-like morphology except for a slight shrinkage in the average diameter (ca. 2.0 μm). It has to mention that this hierarchical microstructure in CoMoO4 may provide abundant exposure of active sites, which will be quite beneficial to the catalytic process. TEM results are consistent with SEM data, further confirming that these microspheres are constituted by sequential nanosheets with a relatively thin thickness (Fig. 2b and c). The high-resolution TEM (HRTEM) image of the edge region identifies the lattice fringe of these nanosheets of about 0.336 nm, corresponding to the (002) plane of β-CoMoO4. Fig. S2† shows N2 adsorption–desorption isotherm of CoMoO4. As observed, this isotherm performs its profile between standard II-type and IV-type isotherms, characteristic of disordered mesoporous structure in the material according to the IUPAC classification.32 The corresponding BET surface and pore volume are 61.9 m2 g−1 and 0.44 cm3 g−1, respectively, and these superior structural parameters after high-temperature calcination may be associated with its unique hierarchical microstructure (Fig. 2).
 | ||
| Fig. 2 SEM image (a), TEM images (b and c), and HR-TEM image (d) of CoMoO4. | ||
Fig. 3 shows the degradation of MB via catalytic oxidation process with different catalysts. As observed, sole PMS can work for the decoloration of MB solution, and 32.2% of MB is removed after 60 min. CoMoO4 fails to promise any MB removal in the absence of PMS, suggesting that the contribution from simple physical adsorption is negligible. It is very interesting that the removal of MB can be substantially reinforced with the presence of both PMS and CoMoO4, and complete degradation of MB can be achieved in 40 min. In previous reports, CoMoO4 is rarely studied as a catalyst for heterogeneous PMS activation, and thus the degradation of MB is also performed over some common Co-containing catalysts, e.g. CoFe2O4 and Co3O4, to evaluate the catalytic activity of CoMoO4. It is found that although these Co-containing catalysts can also accelerate the degradation of MB in the same system, the removal efficiencies in 60 min catalysed by CoFe2O4 and Co3O4 are only 54.1% and 42.2%, respectively, which are far behind that over CoMoO4. It is also observed that the degradation rate of MB in CoMoO4/PMS system is extremely fast in the first 5 min, implying that the active radicals may be released as soon as the introduction of PMS. As mentioned above, PMS activation is always accompanied by the electron transfer between active sites and HSO5−/˙SO4−, and thus the good performance of CoMoO4 should be linked with its better electrochemical properties as compared with CoFe2O4 and Co3O4.28,33 What merits a special note is that CoMoO4 not only provides high MB degradation efficiency, but also performs good mineralization capacity (Fig. S3†). After 60 min, TOC removal can reach 79.6%. By considering that Co sites are generally taken as the primary active sites for PMS activation, we also evaluate the catalytic activities of Co sites in Co3O4, CoFe2O4, and CoMoO4 with turn over number (TON, the number of chemical conversions of substrate molecules on a single catalytic site in unit time). In terms of the degradation efficiencies of MB in 40 min, the TON values for Co3O4, CoFe2O4, and CoMoO4 can be deduced as 9.25 × 10−3, 3.14 × 10−2, and 5.48 × 10−2, respectively (Table 1). It is undoubted that Co sites in CoMoO4 display the best catalytic ability. In general, the degradation of organic dye obeys the pseudo-first order kinetics, and thus the kinetics constant over different catalysts can be calculated by the following equation,
| ln(Ct/C0) = −kt | (2) |
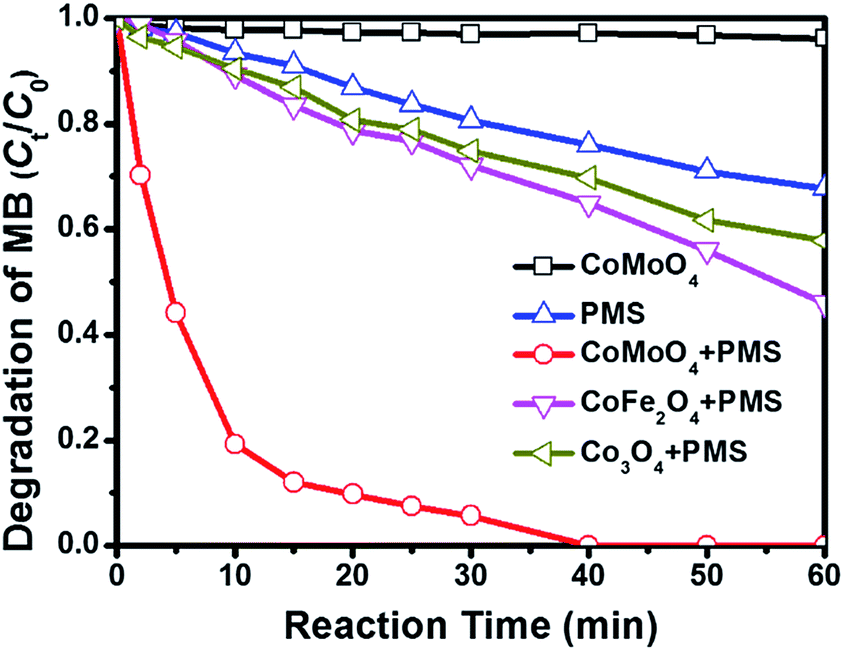 | ||
| Fig. 3 MB degradation by various catalytic PMS systems. Conditions: [MB]0 = 100 mg L−1, volume (MB) = 50 mL, [Oxone] = 2 mM, catalyst amount = 0.05 g (0.10 g L−1), temperature = 25 °C, without pH adjustment. | ||
It is well known that reaction temperature is a critical operating parameter in AOPs, which can affect the degradation rate of organic pollutants greatly.34–36 Date fitting (Fig. 4a) shows that the reaction temperature also plays an important role in determining the degradation rate of MB. When the reaction temperature is increased from 15 °C to 35 °C, the kinetic rate constants will be increased from 0.056 min−1 to 0.430 min−1 (Table S2†). Furthermore, the activation energy (Ea) of the reaction on the surface of CoMoO4 is evaluated by plotting ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k versus 1/T based on the Arrhenius equation and is determined to be 69.89 kJ mol−1 (Fig. 4b). The Ea value is much higher than that of diffusion-controlled reaction (10–13 kJ mol−1), indicating that the apparent reaction rate of this oxidation process is more dependent on the rate of intrinsic chemical reactions on the surface of CoMoO4 rather than the rate of mass transfer.37
k versus 1/T based on the Arrhenius equation and is determined to be 69.89 kJ mol−1 (Fig. 4b). The Ea value is much higher than that of diffusion-controlled reaction (10–13 kJ mol−1), indicating that the apparent reaction rate of this oxidation process is more dependent on the rate of intrinsic chemical reactions on the surface of CoMoO4 rather than the rate of mass transfer.37
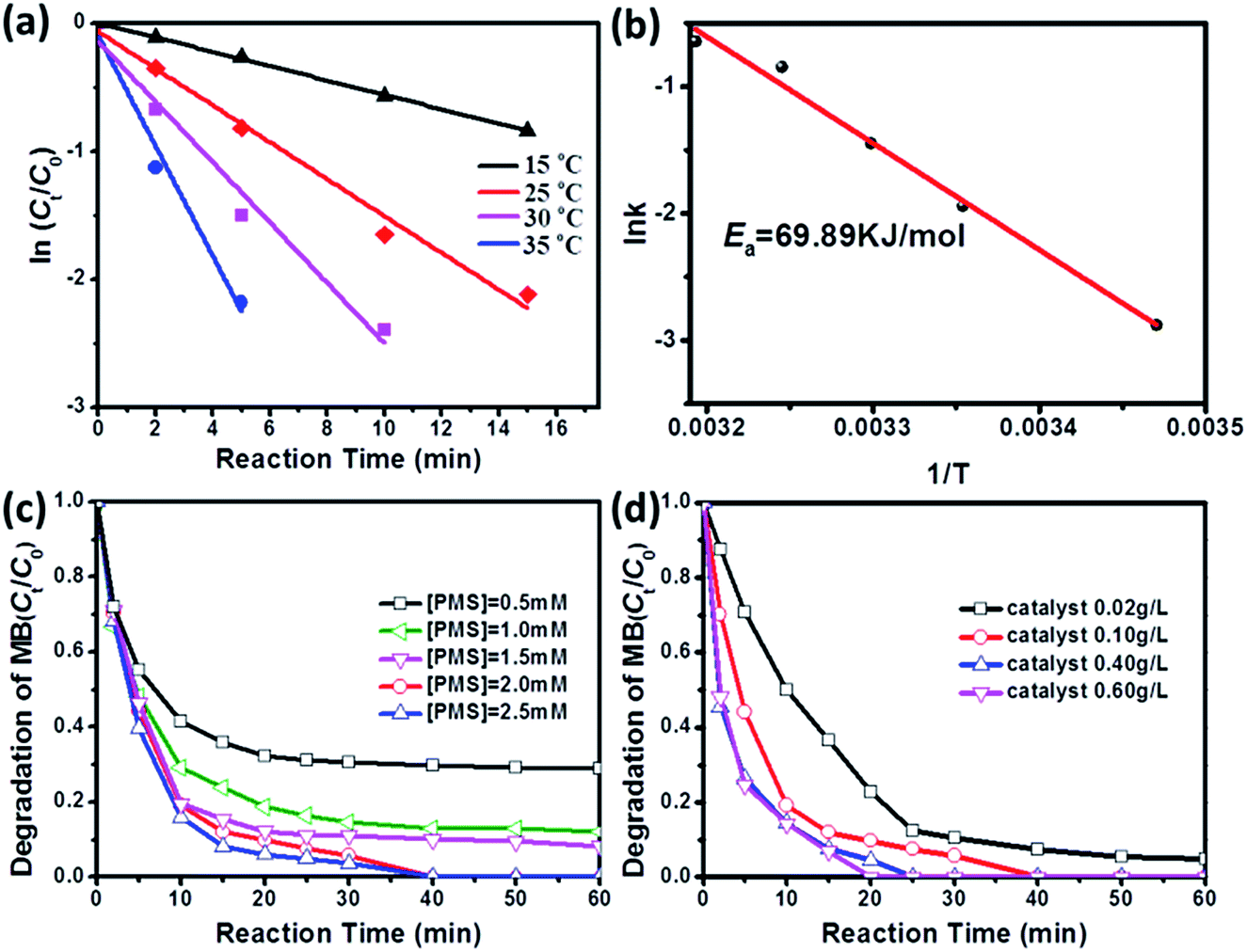 | ||
| Fig. 4 Kinetic curves (a) and the Arrhenius curve (b) of MB removal at different temperature in CoMoO4/PMS system. Conditions: [MB]0 = 100 mg L−1, volume (MB) = 50 mL, [Oxone] = 2 mM, catalyst amount = 0.05 g (0.10 g L−1), temperature = 25 °C, without pH adjustment. Effects of oxone concentration (c) and CoMoO4 dosage (d) on MB removal catalyzed by CoMoO4/PMS system. | ||
In addition, the effects of catalyst dosage and PMS dosage are further studied in CoMoO4/PMS system. As shown in Fig. 4c, with increased dosage of PMS, the degradation efficiency of MB will rise from 69.5% at 0.5 mM PMS to 96.3% at 2.5 mM in 30 min. It is reliable that increasing PMS concentration can make more HSO5− attach to the active sites of CoMoO4, which facilitates the generation of radical species. Interestingly, MB degradation shows a similar dependence on the dosage of CoMoO4 (Fig. 4d). When the dosage of CoMoO4 is set at 0.02 g L−1, the degradation efficiency of MB will be 95.3% in 60 min. For the dosage of 0.40 g L−1, MB can be completely eliminated in 25 min, and its kinetic rate constant (0.1597 min−1) is about 2.36 times higher than that at 0.02 g L−1 (0.0677 min−1). However, it is also found that the stimulation effect will be strongly restrained once the dosage of CoMoO4 is beyond 0.40 g L−1. According to previous literature,38–40 this phenomenon can be explained by the diffusion limitation in heterogeneous reactions. If the dosage of the catalysts exceeds the optimum value, the ineffective oxidant consumption on its surface will be accelerated and become dominant before radical species can react with organic pollutant molecules, leading to the constant or decreased degradation efficiency. In the studied interval of dosage, the diffusion limitation of CoMoO4 is determined as 0.40 g L−1, which is higher than those reported in some oxides and ferrites,41,42 suggesting that CoMoO4 may be a better candidate as heterogeneous catalysts for PMS activation.
As reported in many previous studies, sulfate radical-based AOPs can work in a wider pH range as compared to conventional Fenton reaction.15,43,44 This deduction is also applicable to CoMoO4/PMS system (Fig. 5a). It is observed that the degradation efficiency of MB increases sharply as the initial pH value rises from 3.0 to 11.0. However, when the pH value further increases to 12.50, the degradation rate is decreased reversely. The corresponding kinetic rate constants (k) at different initial pH values are deduced in Fig. 5b, which clearly demonstrates the effect of initial pH value on MB degradation catalyzed by CoMoO4/PMS system. This phenomenon can be explained from two aspects: (1) pH value has a great impact on the speciation of PMS, and acidic condition can induce the formation of strong hydrogen bond between H+ and O–O bond in PMS,6,8 which will obviously inhibit the interaction between PMS and CoMoO4; (2) the hydrogen bond will be continuously weakened with increased pH value, and meanwhile, hydroxyl radicals will be inevitably introduced into the catalytic system under alkaline condition, which can not only attack organic pollutants, but also react with other species to stimulate PMS activation.45,46 As a result, the degradation of MB will be reinforced in the pH range of 3.0–11.0. However, excessive high basicity (e.g. pH = 12.5) will produce less oxidative SO52− through the deprotonation of HSO5−,47 leading to an inferior MB degradation.
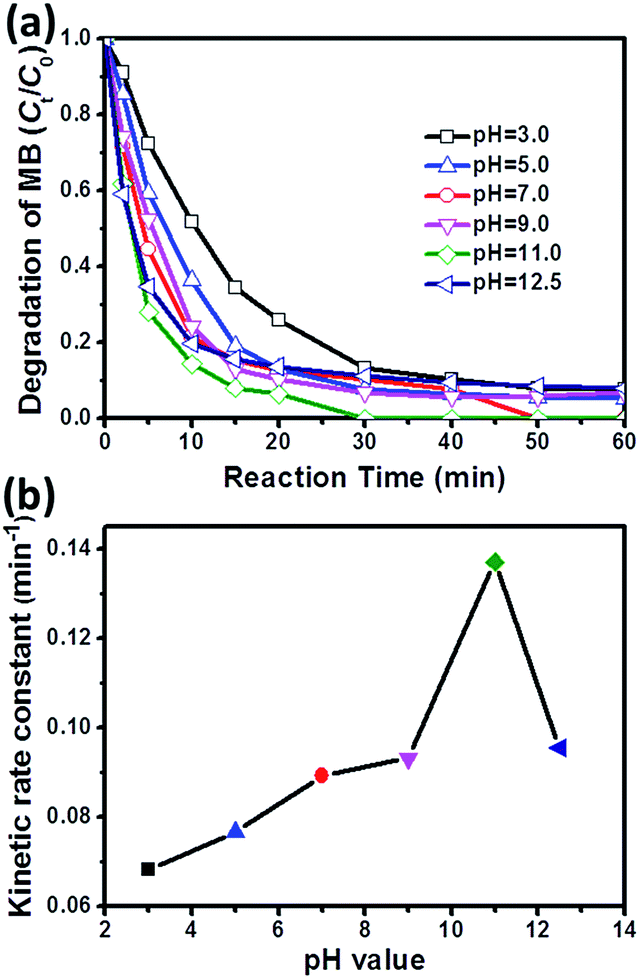 | ||
| Fig. 5 Effect of initial pH value on MB degradation (a) and kinetic rate constant (b) in CoMoO4/PMS system. Conditions: [MB]0 = 100 mg L−1, volume (MB) = 50 mL, [Oxone] = 2 mM, catalyst amount = 0.05 g (0.10 g L−1), temperature = 25 °C. | ||
It has been reported that both ˙SO4− and ˙OH can be identified in conventional PMS activation system, because ˙OH is a typical product from the reaction between ˙SO4− and H2O.43 Therefore, the classical quenching tests are carried out to figure out the specific reaction mechanism in the system of MB degradation, where tert-butyl alcohol (TBA) is selected as a particular scavenger for ˙OH and MeOH is utilized as a universal scavenger for both ˙OH and ˙SO4−.40,48 As shown in Fig. 6a and b, the degradation efficiency of MB displays an anticipated decrease with increasing the concentration of TBA or MeOH, and more importantly, MeOH always plays a more aggressive role in inhibiting MB degradation as compared with TBA. For example, when the concentration of TBA increases from 0 to 300 mM, the degradation efficiency of MB will decrease from 100% to 90.3% in 40 min. In contrast, the degradation efficiency of MB decreases from 100% to 77.2% in the presence of MeOH (300 mM). It is worth noting that the extremely high MeOH concentration (300 mM) does not suppress MB degradation completely, implying that CoMoO4/PMS can release sufficient radicals to react with MB. To rule out the contribution of radicals completely, we further perform the degradation of MB in absolute MeOH solution, and only 11.4% of MB can be removed in 60 min (Fig. S6†), which suggests that the degradation of MB through non-radical pathway is rather limited.49,50
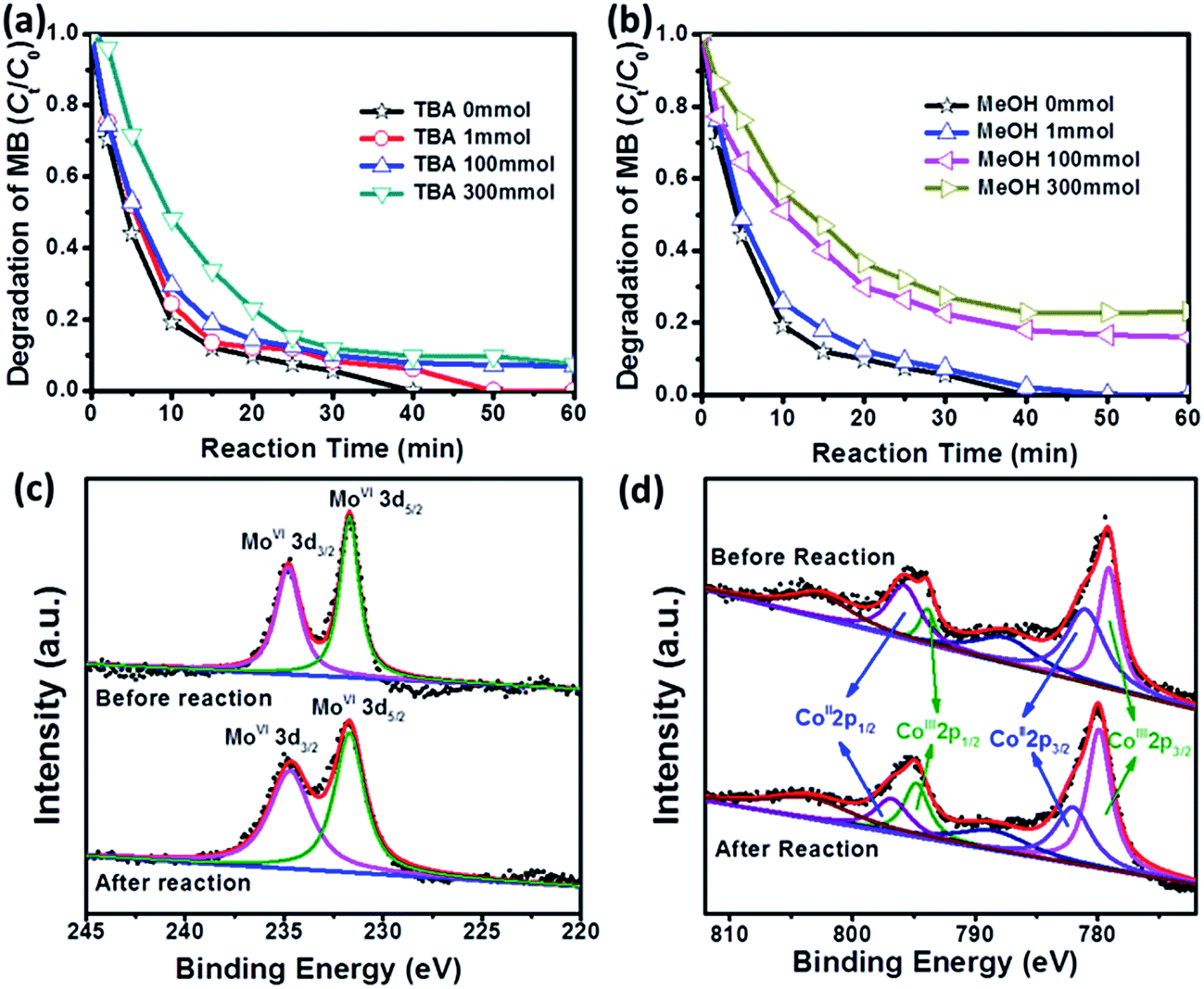 | ||
| Fig. 6 Effect of (a) TBA and (b) MeOH on MB degradation by CoMoO4/PMS system. Conditions: [MB]0 = 100 mg L−1, volume (MB) = 50 mL, [Oxone] = 2 mM, catalyst amount = 0.05 g (0.10 g L−1), temperature = 25 °C, without pH adjustment. XPS patterns of Mo 3d (c), Co 2p (d) in fresh and used CoMoO4 sample. | ||
The existing states of elements Co and Mo in fresh and used CoMoO4 are characterized by XPS to discern the primary functional sites for PMS activation. As shown in Fig. 6c, Mo 3d spectrum of fresh CoMoO4 exhibits two peaks with binding energy at 234.7 eV and 231.0 eV, which can be assigned to Mo 3d3/2 and Mo 3d5/2, respectively.51,52 After degradation, the profile of Mo 3d in used CoMoO4 is quite similar to that of fresh CoMoO4, and no peak shift or additional signal can be detected, indicating that Mo sites keep their initial state during the catalytic process. In contrast, the profiles of Co 2p in fresh and used CoMoO4 are a little different. In order to better illustrate the variation, the fitting curves of Co 2p1/2 and Co 2p3/2, as well as their satellite peaks in fresh and used CoMoO4 are presented in Fig. 6d, where the peaks at binding energies of 779.9 eV and 794.8 eV with the satellite signal at 788.5 eV are due to Co(III), and peaks at binding energies of 782.0 eV and 796.8 eV with the satellite signal at 803.5 eV are characteristic of Co(II).53,54 It is clear that the proportion of Co(III) increases from 39.9% in fresh catalyst to 59.8% in used catalyst. This phenomenon means that Co sites in CoMoO4 still play the primary functional sites for PMS activation.
According to the above experimental results, the possible catalytic mechanism of radicals generation in the decomposition of MB can be depicted as follows.55,56 Firstly, the complex Co(II)–(OH)OSO3− is generated by the complexation between PMS and Co sites on the surface of CoMoO4 as shown in eqn (3). By means of one-electron transfer inside the complex, ˙SO4− is generated and new hydroxyl groups are formed (eqn (4)). Co(III) then oxidized PMS to ˙SO5− that further attaches to the hydroxyl groups, producing the surface peroxo species, and reaching a lower transient valence (eqn (5)). Then, the surface peroxo species combine with each other, and recycled hydroxyl groups bonded to Co(II) are obtained (eqn (6) and (7)).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Co(II)–−OH + HSO5− → Co(II)–(OH)OSO3− + OH− Co(II)–−OH + HSO5− → Co(II)–(OH)OSO3− + OH−
| (3) |
|
Co(II)–(OH)OSO3− → Co(III)–−OH + SO4˙−
| (4) |
|
Co(III)–−OH + HSO5− → Co(II)–˙OOSO3− + H2O
| (5) |
|
2Co(II)–˙OOSO3− + 2H2O → 2Co(II)–−OH + O2 + 2SO4˙− + 2H+
| (6) |
|
2Co(II)–˙OOSO3− + 2H2O → 2Co(II)–−OH + O2 + S2O82− + 2H+
| (7) |
To test the generality of CoMoO4/PMS system, two additional dyes, RhB and OII, are further selected as model pollutants. As shown in Fig. S7,† sole PMS does not account for the obvious degradation for both RhB and OII, while CoMoO4/PMS removes over 80% of dyes in 30 min, confirming that this heterogeneous activation system can work for the degradation of different types of organic dyes. In addition, we also perform the degradation of MB in two actual water bodies (tap water and surface water), whose compositions are listed in Table S3.† It is very interesting that CoMoO4/PMS system can maintain its high MB removal efficiency in these actual water bodies, and its performance is always superior to that of CoFe2O4/PMS (Fig. S8†). In view of the results in this study, we believe that CoMoO4 can be a promising heterogeneous catalyst for PMS activation with a bright prospect in practical application.
Conclusions
In this study, CoMoO4 is employed as a novel heterogeneous catalyst for PMS activation for the first time. The as-prepared CoMoO4 through hydrothermal route and high-temperature calcination displays hierarchical microstructure that is assembled by ultrathin nanosheets and high surface area (61.9 m2 g−1). CoMoO4 exhibits excellent catalytic performance in activating PMS for MB degradation, which is much better than conventional Co3O4 and CoFe2O4. Radical quenching tests reveal that radical pathway is dominant in the degradation of MB, and both sulfate and hydroxyl radicals will be released from PMS activation. The results of XPS spectra confirm that Co sites on the surface of CoMoO4 are primarily active sites. More importantly, it is found that the excellent catalytic performance of CoMoO4 is not only versatile for the degradation of different dyes, but also available in some actual water bodies. We believe CoMoO4 may be an advanced alternative to conventional heterogeneous catalysts for PMS activation in advanced oxidation processes.Acknowledgements
This work is supported by the financial support from Natural Science Foundation of China (21676065) and the Natural Science Foundation of Heilongjiang Province (B201405).References
- S. K. Ling, S. Wang and Y. Peng, J. Hazard. Mater., 2010, 178, 385–389 CrossRef CAS PubMed.
- T. Robinson, G. McMullan, R. Marchant and P. Nigam, Bioresour. Technol., 2001, 77, 247–255 CrossRef CAS PubMed.
- K. Pazdzior, A. Klepacz-Smolka, S. Ledakowicz, J. Sojka-Ledakowicz, Z. Mrozinska and R. Zylla, Chemosphere, 2009, 75, 250–255 CrossRef CAS PubMed.
- X. G. Duan, H. Q. Sun, Y. X. Wang, J. Kang and S. B. Wang, ACS Catal., 2015, 5, 553–559 CrossRef CAS.
- Y. J. Yao, H. Chen, C. Lian, F. Y. Wei, D. W. Zhang, G. D. Wu, B. J. Chen and S. B. Wang, J. Hazard. Mater., 2016, 314, 129–139 CrossRef CAS PubMed.
- Z. Wang, Y. Du, Y. Liu, B. Zou, J. Xiao and J. Ma, RSC Adv., 2016, 6, 11040–11048 RSC.
- J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz and J. J. Rodriguez, Environ. Sci. Technol., 2005, 39, 9295–9302 CrossRef CAS PubMed.
- Y. Du, W. Ma, P. Liu, B. Zou and J. Ma, J. Hazard. Mater., 2016, 308, 58–66 CrossRef CAS PubMed.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, RSC Adv., 2015, 5, 7628–7636 RSC.
- Z. Huang, H. Bao, Y. Yao, J. Lu, W. Lu and W. Chen, J. Chem. Technol. Biotechnol., 2016, 91, 1257–1265 CrossRef CAS.
- Y.-H. Guan, J. Ma, X.-C. Li, J.-Y. Fang and L.-W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- M. Feng, R. Qu, X. Zhang, P. Sun, Y. Sui, L. Wang and Z. Wang, Water Res., 2015, 85, 1–10 CrossRef CAS PubMed.
- Z. Fang, X. Qiu, J. Chen and X. Qiu, Appl. Catal., B, 2010, 100, 221–228 CrossRef CAS.
- X. Duan, Z. Ao, H. Sun, L. Zhou, G. Wang and S. Wang, Chem. Commun., 2015, 51, 15249–15252 RSC.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- Z. Xu, J. Lu, Q. Liu, L. Duan, A. Xu, Q. Wang and Y. Li, RSC Adv., 2015, 5, 76862–76874 RSC.
- Y. B. Ding, L. H. Zhu, A. Z. Huang, X. R. Zhao, X. Y. Zhang and H. Q. Tang, Catal. Sci. Technol., 2012, 2, 1977–1984 CAS.
- W.-D. Oh, S.-K. Lua, Z. Dong and T.-T. Lim, J. Hazard. Mater., 2015, 284, 1–9 CrossRef CAS PubMed.
- Q. Yang, H. Choi, Y. Chen and D. D. Dionysiou, Appl. Catal., B, 2008, 77, 300–307 CrossRef CAS.
- P. R. Shukla, S. Wang, H. Sun, H. M. Ang and M. Tade, Appl. Catal., B, 2010, 100, 529–534 CrossRef CAS.
- Y. M. Ren, L. Q. Lin, J. Ma, J. Yang, J. Feng and Z. J. Fan, Appl. Catal., B, 2015, 165, 572–578 CrossRef CAS.
- P. Nfodzo and H. Choi, Chem. Eng. J., 2011, 174, 629–634 CrossRef CAS.
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 88, 462–469 CrossRef CAS.
- K.-Y. A. Lin, B.-J. Chen and C.-K. Chen, RSC Adv., 2016, 6, 92923–92933 RSC.
- S. Indrawirawan, H. Sun, X. Duan and S. Wang, J. Mater. Chem. A, 2015, 3, 3432–3440 CAS.
- Y. Ji, C. Dong, D. Kong and J. Lu, J. Hazard. Mater., 2015, 285, 491–500 CrossRef CAS PubMed.
- C. T. Cherian, M. V. Reddy, S. C. Haur and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 918–923 CAS.
- D. Guo, H. Zhang, X. Yu, M. Zhang, P. Zhang, Q. Li and T. Wang, J. Mater. Chem. A, 2013, 1, 7247–7254 CAS.
- M.-C. Liu, L.-B. Kong, C. Lu, X.-M. Li, Y.-C. Luo and L. Kang, Mater. Lett., 2013, 94, 197–200 CrossRef CAS.
- M. Q. Yu, L. X. Jiang and H. G. Yang, Chem. Commun., 2015, 51, 14361–14364 RSC.
- X. Xu, J. Shen, N. Li and M. Ye, J. Alloys Compd., 2014, 616, 58–65 CrossRef CAS.
- Y. Du, T. Liu, B. Yu, H. Gao, P. Xu, J. Wang, X. Wang and X. Han, Mater. Chem. Phys., 2012, 135, 884–891 CrossRef CAS.
- M.-C. Liu, L.-B. Kong, X.-J. Ma, C. Lu, X.-M. Li, Y.-C. Luo and L. Kang, New J. Chem., 2012, 36, 1713–1716 RSC.
- H. Sun, S. Liu, G. Zhou, H. M. Ang, M. O. Tade and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466–5471 CAS.
- B. Yang, Z. Tian, B. Wang, Z. Sun, L. Zhang, Y. Guo, H. Li and S. Yan, RSC Adv., 2015, 5, 20674–20683 RSC.
- S. Zhang, Q. Fan, H. Gao, Y. Huang, X. Liu, J. Li, X. Xu and X. Wang, J. Mater. Chem. A, 2016, 4, 1414–1422 CAS.
- L. Xu and J. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CrossRef CAS PubMed.
- X. Duan, K. O'Donnell, H. Sun, Y. Wang and S. Wang, Small, 2015, 11, 3036–3044 CrossRef CAS PubMed.
- S. L. Luo, L. Duan, B. Z. Sun, M. Y. Wei, X. X. Li and A. H. Xu, Appl. Catal., B, 2015, 164, 92–99 CrossRef CAS.
- L. J. Xu, W. Chu and L. Gan, Chem. Eng. J., 2015, 263, 435–443 CrossRef CAS.
- K.-Y. A. Lin and B.-J. Chen, Chemosphere, 2017, 166, 146–156 CrossRef CAS PubMed.
- P. H. Shi, R. J. Su, S. B. Zhu, M. C. Zhu, D. X. Li and S. H. Xu, J. Hazard. Mater., 2012, 229, 331–339 CrossRef PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- M. G. Antoniou, A. A. de la Cruz and D. D. Dionysiou, Appl. Catal., B, 2010, 96, 290–298 CrossRef CAS.
- S. Yuan, P. Liao and A. N. Alshawabkeh, Environ. Sci. Technol., 2014, 48, 656–663 CrossRef CAS PubMed.
- J. Zou, J. Ma and J. Zhang, Environ. Sci. Technol., 2014, 48, 4630–4631 CrossRef CAS PubMed.
- C. Tan, N. Gao, Y. Deng, J. Deng, S. Zhou, J. Li and X. Xin, J. Hazard. Mater., 2014, 276, 452–460 CrossRef CAS PubMed.
- Y.-H. Guan, J. Ma, Y.-M. Ren, Y.-L. Liu, J.-Y. Xiao, L.-q. Lin and C. Zhang, Water Res., 2013, 47, 5431–5438 CrossRef CAS PubMed.
- T. Zhang, W. Li and J.-P. Croue, Environ. Sci. Technol., 2011, 45, 9339–9346 CrossRef CAS PubMed.
- X. Duan, H. Sun, Y. Wang, J. Kang and S. Wang, ACS Catal., 2015, 5, 553–559 CrossRef CAS.
- N. S. McIntyre, D. D. Johnston, L. L. Coatsworth, R. D. Davidson and J. R. Brown, Surf. Interface Anal., 1990, 15, 265–272 CrossRef CAS.
- X. Xia, W. Lei, Q. Hao, W. Wang and X. Wang, Electrochim. Acta, 2013, 99, 253–261 CrossRef CAS.
- G. Chen, X. Si, J. Yu, H. Bai and X. Zhang, Appl. Surf. Sci., 2015, 330, 191–199 CrossRef CAS.
- Y. Ding, L. Zhu, A. Huang, X. Zhao, X. Zhang and H. Tang, Catal. Sci. Technol., 2012, 2, 1977–1984 CAS.
- Y. Yao, Y. Cai, F. Lu, F. Wei, X. Wang and S. Wang, J. Hazard. Mater., 2014, 270, 61–70 CrossRef CAS PubMed.
- T. Zhang, H. Zhu and J.-P. Croue, Environ. Sci. Technol., 2013, 47, 2784–2791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM image of precursor; N2 adsorption/desorption isotherm of CoMoO4; MB degradation in recycling experiments, MeOH solution, and actual water bodies; RhB and OII degradations catalyzed by CoMoO4/PMS; the compositions of tap water and surface water. See DOI: 10.1039/c7ra04761d |
| This journal is © The Royal Society of Chemistry 2017 |