
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of novel 1,2,4-triazole derivatives containing the quinazolinylpiperidinyl moiety and N-(substituted phenyl)acetamide group as efficient bactericides against the phytopathogenic bacterium Xanthomonas oryzae pv. oryzae†
Lan Yang and
Xiao-Ping Bao *
*
State Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Centre for Research and Development of Fine Chemicals, Guizhou University, Guiyang 550025, China. E-mail: baoxp_1980@aliyun.com; Tel: +86-0851-88292090
First published on 5th July 2017
Abstract
A series of novel 1,2,4-triazole derivatives (7a–7p) containing the quinazolinylpiperidinyl moiety and N-(substituted phenyl)acetamide group were designed, synthesized and evaluated for their antimicrobial activities in vitro. These compounds were fully characterized by 1H NMR, 13C NMR, HRMS and IR spectra. Notably, the structure of compound 7p was further confirmed through the single-crystal X-ray diffraction method. The obtained bioassay results indicated that most of these compounds exhibited good to excellent antibacterial activities against the rice bacterial pathogen Xanthomonas oryzae pv. oryzae (Xoo). For example, compounds 7e, 7g, 7n, 7l, 7i, 7k, 7a and 7h had EC50 (half-maximal effective concentration) values of 34.5, 38.3, 39.0, 46.0, 47.5, 54.6, 55.0 and 58.2 μg mL−1 against the bacterium, respectively, which were significantly lower than the control agent Bismerthiazol (85.6 μg mL−1). Additionally, antifungal experiments demonstrated that all the compounds did possess weak inhibition capabilities against three phytopathogenic fungi at 50 μg mL−1, except in the cases of compounds 7e and 7p against the fungus Gibberella zeae. The above experimental results proved that 1,2,4-triazole derivatives bearing both a quinazolinylpiperidinyl fragment and N-(substituted phenyl)acetamide unit are promising candidates for the development of new agricultural bactericides against the pathogenic bacterium Xoo, deserving further investigation in the future.
Introduction
Xanthomonas oryzae pv. oryzae (Xoo) and Ralstonia solanacearum (Rs) are pathogenic bacteria for rice bacterial leaf blight and tobacco bacterial wilt, respectively, which cause serious harm to global agricultural products and give rise to huge economic losses to farmers all over the world.1 Taking the phytobacterium Xoo as an example, it is one of the most destructive bacterial diseases for the rice crop, which occurs throughout all the growth stages of rice and triggers bacterial blight by invading vascular tissues,2 generally bringing about production losses by up to 50%.3 Additionally, the phytobacterium Rs is also one kind of highly devastating and widespread soil-borne plant pathogen.4 The most typical symptoms of tobacco plants infected by this pathogen are rapid yellowing and wilting of tobacco leaves.5 Although some commercial antibactericidal agents (i.e., Bismerthiazol and Thiodiazole copper) are currently available on the market for fighting against these two bacterical diseases, however, poor efficiency, high phytotoxicities, high residue levels, adverse effects on the natural environment and growing problems of antibacterial resistance related with the utilization of these bactericides are attracting increasing attention from agricultural chemists. Therefore, the search of new and more efficient antibacterial agents still remains an urgent task in the agrochemical field.1,2,4-Triazole derivatives are a class of important nitrogen-containing heterocyclic compounds, and many of them displayed remarkable antibacterial activities.6–8 Some marketed agrofungicides also contain the ring of 1,2,4-triazole, such as Triadimefon, Triadimenol, Diniconazole, Flusilazole and Difenoconazole (Fig. 1). Moreover, quinazoline and quinazolinone derivatives have attracted a great deal of attention from medicinal chemists because of their diverse biological activities, such as anticancer,9 anticonvulsant,10 anti-leishmanial & anti-proliferative,11 analgesic and anti-inflammatory activities.12 Until now, the method of “active substructure combination” is still quite useful for the development of new bioactive molecules,13,14 because it can provide multiple binding sites within a single molecule to interact with target proteins of pathogenic microorganisms. Fluquinconazole, a well-known agrofungicide, is just one good example of ingenious combination of quinazolinone and 1,2,4-triazole heterocycles. Moreover, the moiety of acetamide often occurs in bioactive molecules, such as fungicide Oxathiapiprolin and carbonic anhydrase inhibitor Acetazolamide.
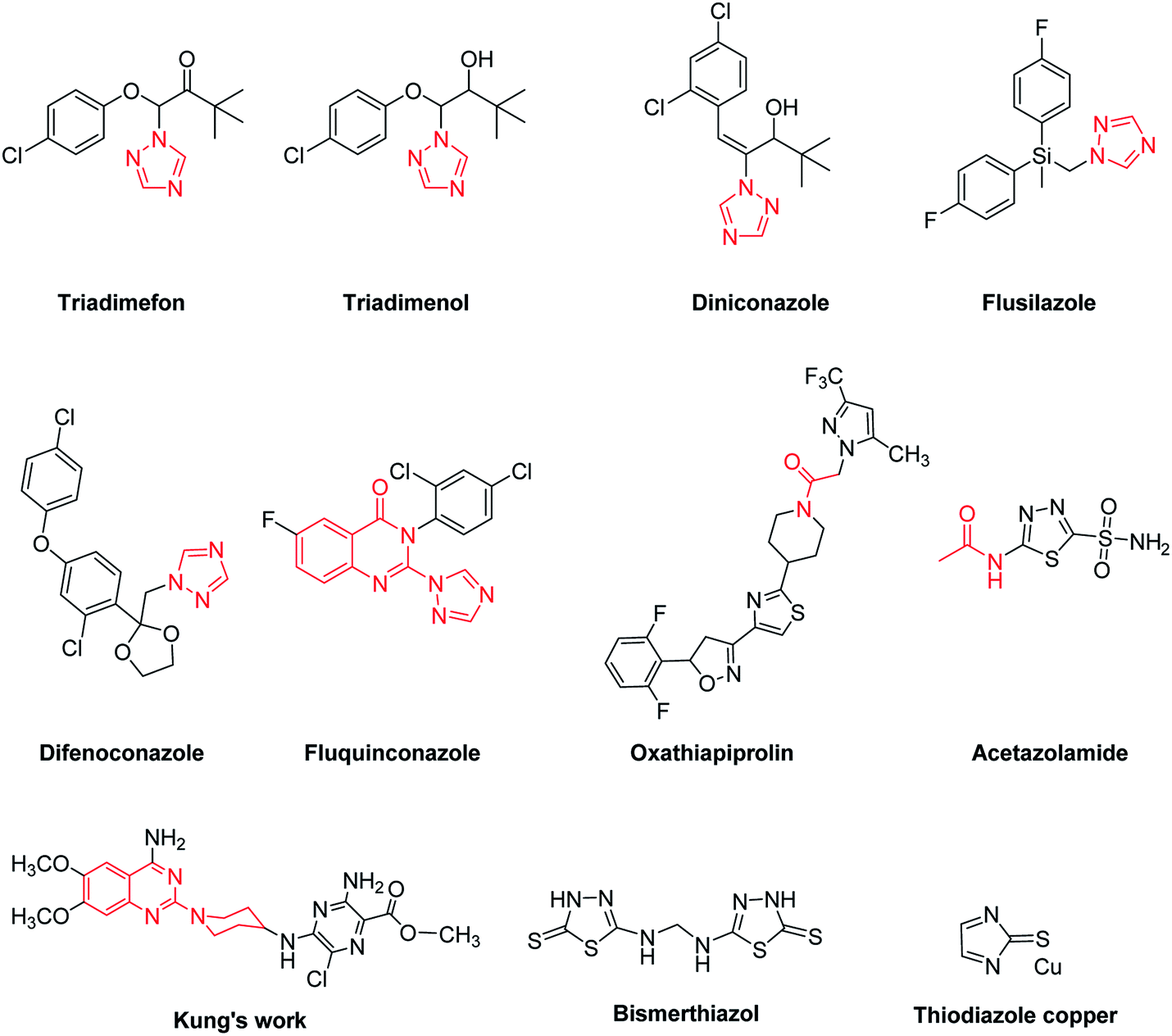 | ||
| Fig. 1 Chemical structures of some bioactive molecules. | ||
In 1999, the findings made by Kung et al. indicated that both quinazoline ring and piperidinyl linker in target compounds were indespensible for achieving effective antibacterial activities.15 Based on the above-mentioned considerations and our continuing interest in the development of 1,2,4-triazole-quinazoline/quinazolinone hybrid derivatives as effective antimicrobial agents,16–20 herein we introduced various N-(substituted phenyl)acetamide groups into quinazolinylpiperidinyl-modified 1,2,4-triazole ring (Fig. 2) and evaluated their inhibition activities against some important phytopathogenic bacteria and fungi.
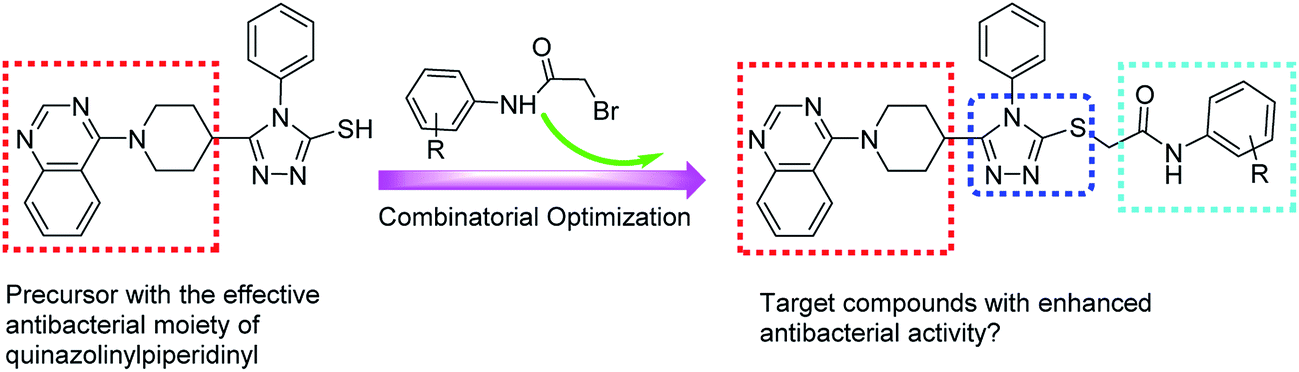 | ||
| Fig. 2 Design strategy for target compounds 7a–7p. | ||
Results and discussion
Chemistry and spectral analysis
The synthetic route of target compounds 7a–7p was depicted in Scheme 1. Briefly, quinazolin-4-one21 as starting material was reacted with SOCl2 to give 4-chloroquinazoline 1,22 which was then subjected to substitution reaction with methyl 4-piperidinecarboxylate to generate ester 2. Subsequently, the ester 2 was converted into the corresponding hydrazide 3 through reaction with N2H4·H2O. Once 3 in hand, it was reacted with phenyl isothiocyanate in ethanol to afford amidothiourea 4. After a ring closure reaction under basic conditions, triazole 5 was obtained in good yield. Finally, the triazole 5 and various N-(substituted phenyl)bromoacetamide derivatives (6a–6p) were reacted under CH3COCH3/K2CO3 system to give target compounds 7a–7p. Taking 7p as an example, the strong peak at 1688 cm−1 showed the presence of amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionality. In the DMSO-d6 solution of compound 7p, the singlet at 4.12 ppm was attributed to SCH2 proton resonance. The appearance of a broad peak around 10.01 ppm was assigned to amide NH, which rapidly vanished after D2O exchange. Additionally, a distinct singlet at 8.53 ppm was seen, resulting from the resonance of the position-2 (sp2 methine) of the quinazoline ring. In the 13C NMR spectrum, the CO carbon signal was found at 164.2 ppm. The high-resolution mass spectrum (HRMS) of 7p demonstrated an intense peak at m/z = 590.1293, attributed to the species of [M + H]+.
O functionality. In the DMSO-d6 solution of compound 7p, the singlet at 4.12 ppm was attributed to SCH2 proton resonance. The appearance of a broad peak around 10.01 ppm was assigned to amide NH, which rapidly vanished after D2O exchange. Additionally, a distinct singlet at 8.53 ppm was seen, resulting from the resonance of the position-2 (sp2 methine) of the quinazoline ring. In the 13C NMR spectrum, the CO carbon signal was found at 164.2 ppm. The high-resolution mass spectrum (HRMS) of 7p demonstrated an intense peak at m/z = 590.1293, attributed to the species of [M + H]+.
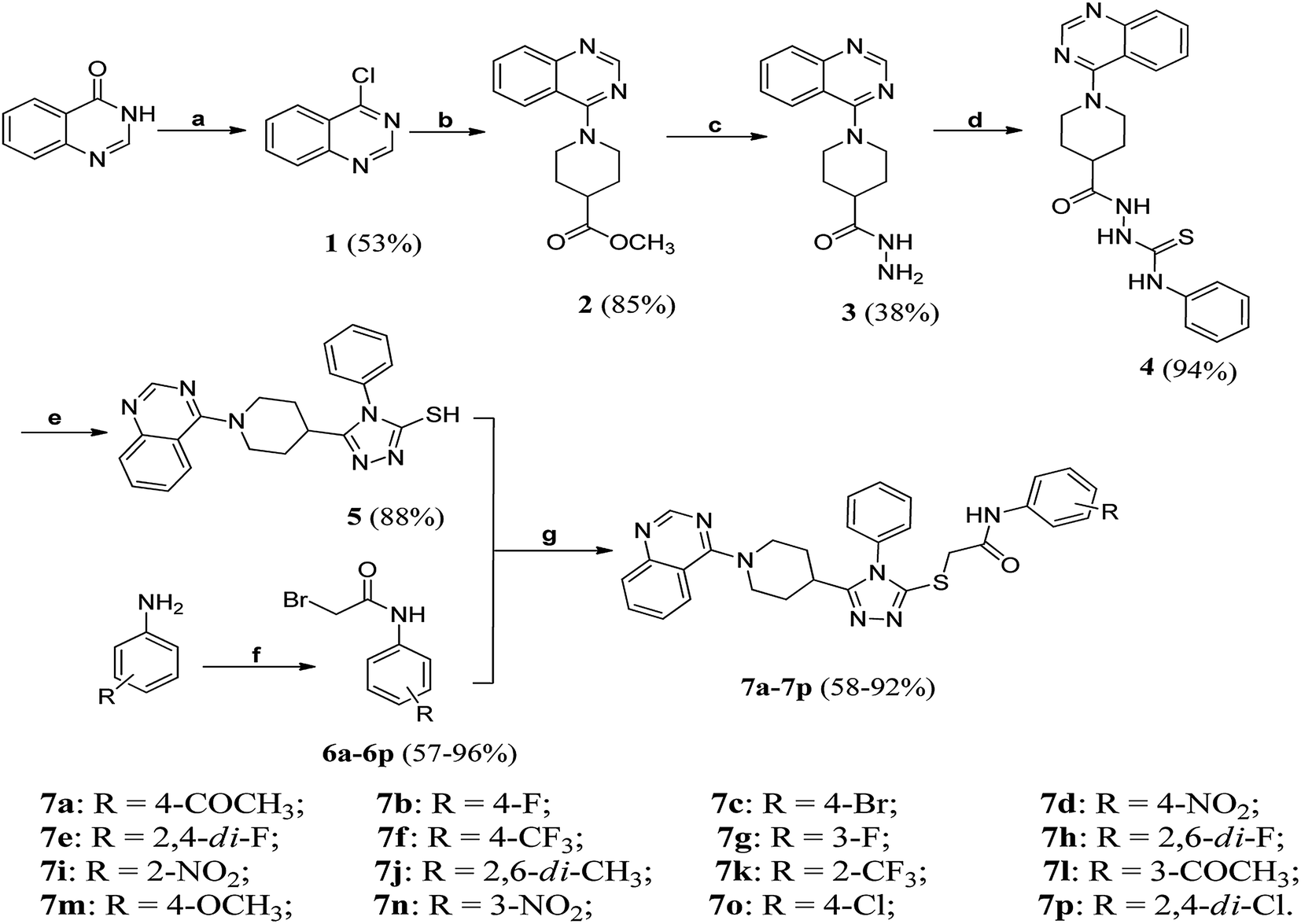 | ||
| Scheme 1 The synthesis of target compounds 7a–7p. Reagents and conditions: (a) SOCl2/DCE/DMF, 140 °C, 5 h; (b) methyl 4-piperidinecarboxylate/1,4-dioxane, r.t. 5 h to 130 °C, 4 h; (c) hydrazine hydrate/MeOH, r.t. 13 h; (d) PhNCS/EtOH, 100 °C, 5 h; (e) (1) 10% K2CO3, 110 °C, 5 h, (2) dilute HCl, pH = 7.0; (f) BrCH2COBr/Et3N/1,4-dioxane, r.t. 5 h; (g) CH3COCH3/K2CO3, 40 °C, 2–4 h. | ||
Crystal structure analysis
Fortunately, single-crystal of compound 7p suitable for X-ray diffraction analysis (Fig. 3) was obtained by slow evaporation of CH2Cl2–EtOH (2/1, v/v) solution of 7p at room temperature. Crystallographic parameters of 7p: colorless crystal, C29H25Cl2N7OS, Mr = 590.52, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.7640(5), b = 10.6660(5), c = 15.4487(9) (Å); α = 74.316(5), β = 82.742(5), γ = 82.883(4); V = 1373.09(13) Å3, T = 293 K, Z = 1, Dc = 0.714 g cm−3, F(000) = 306, reflections collected/independent reflections = 4826/3391, goodness-of-fit on F2 = 1.035, fine, R1 = 0.0447, wR2 = 0.1112. Crystallographic data for compound 7p (CCDC 1479247†).
, a = 8.7640(5), b = 10.6660(5), c = 15.4487(9) (Å); α = 74.316(5), β = 82.742(5), γ = 82.883(4); V = 1373.09(13) Å3, T = 293 K, Z = 1, Dc = 0.714 g cm−3, F(000) = 306, reflections collected/independent reflections = 4826/3391, goodness-of-fit on F2 = 1.035, fine, R1 = 0.0447, wR2 = 0.1112. Crystallographic data for compound 7p (CCDC 1479247†).
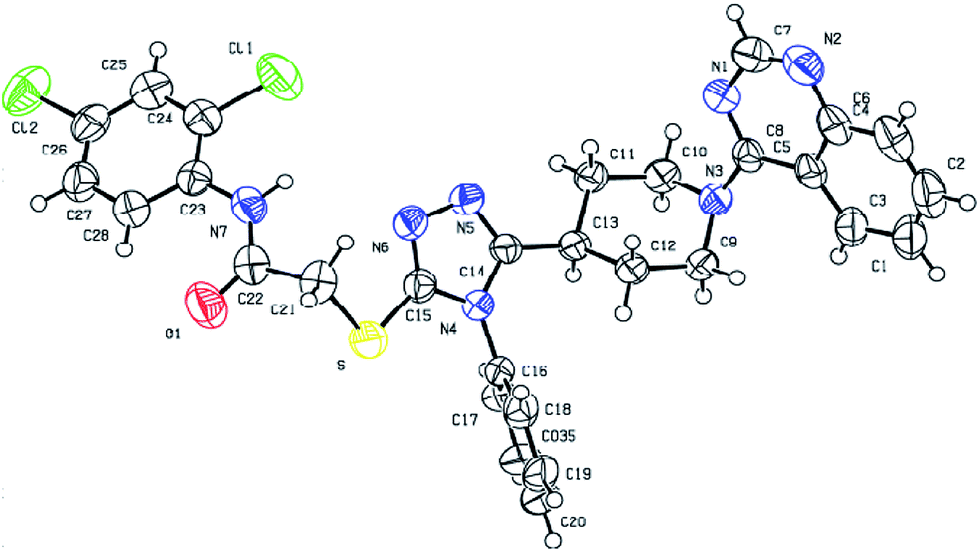 | ||
| Fig. 3 Crystal structure of compound 7p. | ||
Antibacterial activity
The turbidimetric assay23–25 was conducted to evaluate antibacterial activities of target compounds 7a–7p and intermediate 5 against two phytopathogenic bacteria Xoo and Rs. Meanwhile, two commercial agricultural bactericides (namely Bismerthiazol (BMT) and Thiodiazole copper (TDC)) were utilized as the control agents. As shown in Table 1, most of the target compounds exhibited comparable or even better antibacterial activity against the pathogen Xoo. For example, compounds 7e, 7g, 7l, 7n and 7h had the inhibition rate of 77.8%, 66.7%, 66.9%, 65.3% and 64.4% against this bacterium at 100 μg mL−1, respectively, which was more active than the control agent BMT (54.1%). Moreover, compounds 7a, 7b, 7c, 7d, 7f, 7i, 7k, 7m and 7p were found to possess similar inhibition capability against the pathogen Xoo (relative to BMT), at 200 and 100 μg mL−1. Importantly, intermediate 5 only exhibited very weak antibacterial activity towards this bacterium (with the inhibition rate of 32.0% and 17.9% at the above two concentrations, respectively), which adequately proved the necessarity for introduction of N-(substituted phenyl)acetamide group into target compounds and the rationality of our initial design strategy depicted in Fig. 2. Different with the bacterium Xoo, a large majority of the target compounds did not have noticeable antibacterial activity against the pathogen Rs, excluding compounds 7a, 7i, 7n and 7p. Notably, 2-nitro substituted compound 7i was found to possess a potent inhibition activity against the bacteriumn Rs (84.4%) even at 100 μg mL−1, which was far better than two control agents BMT and TDC (34.1% and 26.0%, respectively).| Compd | Inhibition ratea (%) | |||
|---|---|---|---|---|
| Xoo | Rs | |||
| 200 μg mL−1 | 100 μg mL−1 | 200 μg mL−1 | 100 μg mL−1 | |
| a The average of three trials.b Commercial agricultural bactericides Bismerthiazol (BMT) and Thiodiazole copper (TDC) were used as control.c NT = not tested. | ||||
| 5 | 32.0 ± 0.2 | 17.9 ± 1.3 | 17.3 ± 3.1 | 16.6 ± 2.2 |
| 7a | 77.4 ± 1.0 | 55.8 ± 2.6 | 80.0 ± 2.5 | 48.9 ± 2.1 |
| 7b | 77.5 ± 3.7 | 54.0 ± 3.3 | 44.6 ± 1.9 | 25.4 ± 2.6 |
| 7c | 74.7 ± 2.4 | 63.8 ± 0.9 | 55.5 ± 4.2 | 24.1 ± 4.7 |
| 7d | 72.2 ± 2.7 | 61.6 ± 4.6 | 39.0 ± 4.2 | 24.3 ± 1.4 |
| 7e | 88.9 ± 5.3 | 77.8 ± 1.3 | 23.0 ± 0.5 | 17.9 ± 0.9 |
| 7f | 72.7 ± 3.3 | 58.6 ± 0.9 | 58.9 ± 0.7 | 41.4 ± 1.2 |
| 7g | 83.7 ± 2.3 | 66.7 ± 2.5 | 42.0 ± 1.4 | 20.6 ± 0.7 |
| 7h | 69.9 ± 1.1 | 64.4 ± 1.8 | 48.5 ± 3.1 | 35.2 ± 1.0 |
| 7i | 74.0 ± 2.2 | 61.6 ± 6.4 | 100 ± 1.5 | 84.4 ± 4.5 |
| 7j | 40.1 ± 1.7 | 27.2 ± 1.3 | 41.0 ± 2.3 | 23.2 ± 2.2 |
| 7k | 76.1 ± 1.2 | 63.5 ± 1.9 | 52.8 ± 9.1 | 46.1 ± 0.1 |
| 7l | 75.8 ± 3.2 | 66.9 ± 4.7 | 26.6 ± 0.7 | 18.8 ± 0.2 |
| 7m | 67.6 ± 2.2 | 54.6 ± 0.9 | 39.3 ± 2.1 | 25.3 ± 3.6 |
| 7n | 73.2 ± 1.7 | 65.3 ± 3.8 | 67.0 ± 2.7 | 40.7 ± 5.6 |
| 7o | 62.4 ± 1.2 | 48.4 ± 0.6 | 44.4 ± 0.2 | 34.8 ± 5.3 |
| 7p | 66.7 ± 1.3 | 55.3 ± 1.1 | 55.6 ± 1.2 | 52.4 ± 3.1 |
| BMTb | 69.4 ± 1.6 | 54.1 ± 0.8 | 81.9 ± 3.3 | 34.1 ± 5.2 |
| TDCb | NTc | NTc | 48.8 ± 0.6 | 26.0 ± 0.9 |
Inspired by the above experimental results, the EC50 (half-maximal effective concentration) values of compounds 7a–7p and intermediate 5 against the pathogen Xoo were further determined. As shown in Table 2, nearly all of the target compounds did exhibit comparable or much lower EC50 values relative to control BMT, except compounds 7j, 7o and intermediate 5. Among them, compounds 7e, 7g, 7n, 7l, 7i, 7k and 7a had an EC50 value of 34.5, 38.3, 39.0, 46.0, 47.5, 54.6 and 55.0 μg mL−1, respectively, which was obviously better than control BMT (85.6 μg mL−1). A preliminary structure–activity relationship analysis was conducted, which showed that the presence of electron-donating substituents on the benzene ring (e.g., 4-OCH3 and 2,6-di-CH3) was unfavorable for their antibacterial activities against the pathogen Xoo. For example, compound 7j incorporating electron-donating and sterically hindered 2,6-dimethylphenyl substitution gave the weakest activity (EC50 = 233.0 μg mL−1) among this class of compounds. On the whole, the presence of strongly electron-withdrawing substituents (such as 2,4-di-F, 3-F, 3-NO2, 3-COCH3, 2-NO2, 2-CF3 and 4-COCH3) helped to enhance antibacterial activity against the pathogen Xoo. Interestingly, the position of the substitutions (concerning these electron-withdrawing substitutions) in the benzene ring also produced a remarkable influence on their antibacterial activities against the Xoo, such as compounds 7e vs. 7h, 7g vs. 7b, 7n vs. 7d and 7k vs. 7f.
| Compd | EC50a (μg mL−1) | Toxic regression equation | Correlation coefficient (r) |
|---|---|---|---|
| a The average of three trials.b The commercial bactericide Bismerthiazol (BMT) was used as control agent. | |||
| 5 | 332.9 ± 12.2 | y = 1.6086x + 0.9426 | 0.9552 |
| 7a | 55.0 ± 3.7 | y = 1.1005x + 3.0844 | 0.9791 |
| 7b | 75.8 ± 1.1 | y = 1.7358x + 1.7373 | 0.9823 |
| 7c | 78.6 ± 2.3 | y = 1.9210x + 1.3585 | 0.9922 |
| 7d | 77.4 ± 1.0 | y = 1.6224x + 1.9359 | 0.9945 |
| 7e | 34.5 ± 1.6 | y = 1.5664x + 2.5902 | 0.9939 |
| 7f | 85.7 ± 4.3 | y = 1.7409x + 1.6347 | 0.9931 |
| 7g | 38.3 ± 0.4 | y = 1.2438x + 3.0307 | 0.9853 |
| 7h | 58.2 ± 3.9 | y = 1.0924x + 3.0723 | 0.9836 |
| 7i | 47.5 ± 2.5 | y = 1.0712x + 3.2042 | 0.9808 |
| 7j | 233.0 ± 0.6 | y = 1.6841x + 1.0132 | 0.9750 |
| 7k | 54.6 ± 2.3 | y = 1.2129x + 2.8932 | 0.9955 |
| 7l | 46.0 ± 3.4 | y = 1.2846x + 2.8637 | 0.9476 |
| 7m | 93.7 ± 2.3 | y = 1.6639x + 1.7189 | 0.9948 |
| 7n | 39.0 ± 1.4 | y = 0.9623x + 3.4693 | 0.9854 |
| 7o | 116.0 ± 2.5 | y = 1.8645x + 1.1506 | 0.9926 |
| 7p | 75.1 ± 1.8 | y = 1.0959x + 2.9444 | 0.9945 |
| BMTb | 85.6 ± 1.6 | y = 1.5786x + 1.9496 | 0.9852 |
Compared with some of the arylimine derivatives containing the 3-aminoethyl-2-[(p-trifluoromethoxy)anilino]-4(3H)-quinazolinone moiety reported by Song et al.,24 compounds 7e, 7g and 7n had weaker but comparable activities against the Xoo on the basis of their EC50 values. Notably, relative to some quinazoline derivatives containing the 1,2,4-triazolylthioether moiety reported by our group,16 the above three compounds exhibited significantly better antibacterial activities against the Xoo.
Antifungal activity
The mycelial growth rate method24,26 was utilized to assess antifungal activities of target compounds 7a–7p against three phytopathogenic fungi (namely Phytophthora infestans, Gibberella zeae and Verticillium dahliae). As displayed in Table 3, all the compounds were proven to have no noticeable inhibition activity against the above fungi at 50 μg mL−1, except that moderate inhibition activities were found for compounds 7e and 7p against the fungus G. zeae.| Compd | Inhibition ratea (%) | ||
|---|---|---|---|
| P. infestans | G. zeae | V. dahliae | |
| a The average of three trials.b The commercial fungicide Hymexazol was used as control agent. | |||
| 7a | 0 | 14.4 ± 6.7 | 6.1 ± 2.4 |
| 7b | 3.7 ± 1.8 | 19.3 ± 2.5 | 22.0 ± 2.4 |
| 7c | 0 | 0 | 14.1 ± 2.0 |
| 7d | 0 | 12.3 ± 2.5 | 0 |
| 7e | 7.4 ± 1.8 | 30.9 ± 4.4 | 26.7 ± 2.0 |
| 7f | 3.1 ± 2.1 | 19.3 ± 5.1 | 16.0 ± 2.0 |
| 7g | 3.1 ± 1.1 | 7.0 ± 1.3 | 7.6 ± 2.4 |
| 7h | 4.3 ± 1.0 | 14.2 ± 0.8 | 15.2 ± 2.0 |
| 7i | 8.0 ± 7.0 | 19.1 ± 0.8 | 32.3 ± 2.0 |
| 7j | 15.1 ± 2.2 | 11.9 ± 6.2 | 4.2 ± 2.0 |
| 7k | 7.4 ± 3.2 | 9.2 ± 2.6 | 18.2 ± 2.4 |
| 7l | 0 | 0 | 21.9 ± 2.4 |
| 7m | 5.2 ± 0.2 | 14.8 ± 6.9 | 22.5 ± 2.4 |
| 7n | 0 | 14.8 ± 4.9 | 6.0 ± 2.0 |
| 7o | 11.1 ± 0.4 | 18.9 ± 0.6 | 30.3 ± 2.6 |
| 7p | 11.6 ± 4.1 | 36.7 ± 0.7 | 37.9 ± 2.0 |
| Hymexazolb | 69.2 ± 3.2 | 48.4 ± 2.4 | 84.7 ± 0.8 |
Experimental
Instruments
All the chemicals were obtained from commercial suppliers and used without further purification (unless otherwise stated). Melting points were determined on a XT-4 binocular microscope (Beijing Tech Instrument Co., China). IR spectra were recorded on a Shimadzu IR Prestige-21 spectrometer in KBr disk. 1H and 13C NMR spectra were measured on a JEOL-ECX 500 NMR spectrometer at room temperature using TMS as an internal standard and chemical shift (δ) was expressed in parts per million (ppm). HRMS-ESI spectra were recorded on Thermo Scientific Q Exactive series. The X-ray crystallographic data were collected based on a Bruker Smart Apex CCD area detector diffractometer (Bruker, Germany) with Mo-Kα radiation.Synthesis of intermediate 2
The 1,4-dioxane solution (20 mL) of newly-prepared 4-chloroquinazoline 1 (1.0 g, 6.08 mmol) and methyl 4-piperidinecarboxylate (0.83 mL, 6.08 mmol) was stirred for 5 h at room temperature and then heated at reflux for 4 h. After cooling to room temperature, the formed precipitate was filtrated off, washed with 1,4-dioxane and dried to give 2 (1.4 g) as a white solid. Yield: 85%; mp 181–182 °C; 1H NMR (500 MHz, DMSO-d6): 8.79 (s, 1H), 8.12 (d, J = 10.0 Hz, 1H), 7.98 (t, J = 5.0 Hz, 1H), 7.93 (d, J = 10.0 Hz, 1H), 7.67 (t, J = 5.0 Hz, 1H), 4.59 (d, J = 15.0 Hz, 2H), 3.67 (d, J = 10.0 Hz, 2H), 3.61 (s, 3H), 2.92–2.86 (m, 1H), 2.08–2.04 (m, 2H), 1.84–1.76 (m, 2H); 13C NMR (125 MHz, DMSO-d6): 174.5, 162.6, 148.9, 140.8, 136.1, 127.9, 127.8, 127.7, 119.7, 112.6, 52.3, 49.0, 28.3; ESI-HRMS m/z: [M + H]+ calcd for C15H18N3O2: 272.1394; found: 272.1394.Synthesis of intermediate 3
A mixture of ester 2 (300 mg, 1.11 mmol) and hydrazine hydrate (1 mL) in methanol (3 mL) was stirred at room temperature for 13 h, and the formed white precipitate was filtered off, washed with methanol and dried to afford 3 (114 mg). Yield: 38%; mp 173–175 °C; 1H NMR (500 MHz, DMSO-d6): 9.03 (s, 1H), 8.58 (s, 1H), 7.93 (d, J = 10.0 Hz, 1H), 7.78–7.75 (m, 2H), 7.53–7.49 (m, 1H), 4.26 (d, J = 15.0 Hz, 2H), 4.16 (s, 2H), 3.16–3.10 (m, 2H), 2.44–2.37 (m, 1H), 1.79–1.74 (m, 4H); 13C NMR (125 MHz, DMSO-d6): 174.0, 164.4, 154.2, 151.9, 133.2, 128.5, 126.1, 125.8, 116.4, 49.5, 28.8; ESI-HRMS m/z: [M + H]+ calcd for C14H18N5O: 272.1506; found: 272.1505.Synthesis of intermediate 4
Intermediate 3 (304 mg, 1.12 mmol) and phenyl isothiocyanate (0.16 mL, 1.34 mmol) were added to EtOH (12 mL), and the above mixture was heated at reflux for 5 h. After cooling to room temperature, the formed solid was filtered off, washed with ethanol and dried to generate 4 (428 mg) as a white solid. Yield: 94%, mp 195–196 °C; 1H NMR (500 MHz, DMSO-d6): 9.94 (s, 1H), 9.54 (s, 1H), 8.59 (s, 1H), 7.97 (d, J = 10.0 Hz, 1H), 7.80–7.76 (m, 2H), 7.53–7.12 (m, 6H), 4.28 (d, J = 10.0 Hz, 2H), 3.16 (t, J = 10.0 Hz, 2H), 2.61–2.56 (m, 1H), 1.94–1.75 (m, 4H); 13C NMR (125 MHz, DMSO-d6): 174.3, 144.7, 144.1, 139.7, 137.7, 135.0, 134.5, 131.3, 130.8, 129.8, 129.2, 128.5, 126.2, 122.2, 49.5, 28.5; ESI-HRMS m/z: [M + H]+ calcd for C21H23N6OS: 407.1649; found: 407.1651.Synthesis of intermediate 5
An aqueous solution of potassium carbonate (10%, 4 mL) containing 4 (100 mg, 0.26 mmol) was heated to reflux for 5 h. After cooling to room temperature, the above solution was neutralized with dilute HCl to pH = 7.0, and the resulted precipitate was filtered off, washed with water and dried to give triazole 5 (88 mg) as a white solid. Yield: 88%, mp 142–144 °C; 1H NMR (500 MHz, DMSO-d6): 13.70 (s, 1H), 8.55 (s, 1H), 7.89 (d, J = 5.0 Hz, 1H), 7.76–7.75 (m, 2H), 7.57–7.53 (m, 3H), 7.49–7.44 (m, 3H), 4.16 (d, J = 10.0 Hz, 2H), 3.10–3.05 (m, 2H), 2.82–2.76 (m, 1H), 1.83–1.79 (m, 4H); 13C NMR (125 MHz, DMSO-d6): 168.4, 164.2, 155.0, 154.1, 151.8, 134.3, 133.2, 130.2, 130.1, 129.1, 128.5, 126.1, 125.8, 116.4, 49.0, 33.0, 29.5; ESI-HRMS m/z: [M + H]+ calcd for C21H21N6S: 389.1543; found: 389.1545.General procedure for the synthesis of intermediates 6a–6p
Intermediates 6a–6p were synthesized using a modified method reported by Barker et al.27 A mixture of substituted aniline (3.11 mmol) and dry triethylamine (1.5 mL) in dry 1,4-dioxane (6 mL) was stirred at 0 °C for 5 min, and then bromoacetyl bromide (10.54 mmol) dissolved in the dried dioxane (6 mL) was added dropwise to the above solution and continued to stir for 5 h at room temperature. Finally, the reaction mixture was poured into cold water and the resulted precipitate was filtered off, washed with water and dried to afford amides 6a–6p in 57–96% yield.General procedure for the synthesis of target compounds 7a–7p
A mixture of triazole 5 (50 mg, 0.13 mmol), potassium carbonate (18 mg, 0.13 mmol) and the corresponding amide intermediates 6a–6p (0.13 mmol) was added to acetone (4 mL) and then stirred at 40 °C until the reaction was complete as indicated by TLC (2–4 h). Subsequently, the reaction mixture was poured into cold water and the resulted precipitate was filtered off, washed with water and dried to give compounds 7a–7p in 58–92% yield.Antibacterial bioassay
Antibacterial activities of target compounds 7a–7p were determined against two phytopathogenic bacteria (Xoo and Rs), based on the turbidimetric method.23 Firstly, the tested compounds were prepared with two concentrations of 200 and 100 μg mL−1. Pure DMSO in sterile distilled water was used as blank control, and commercially available bactericide Bismerthiazol (BMT) and Thiodiazole copper (TDC) were used as positive control. About 40 μL of solvent NB (3 g of beef extract, 5 g of peptone, 1 g of yeast powder, 10 g of glucose, 1 L of distilled water, pH = 7.0–7.2) containing the bacterium Xoo or Rs was added to the mixed solvent system including 4 mL of solvent NB and 1 mL of 0.1% Tween-20 containing tested compound or BMT/TDC. The above test tube was incubated at 30 ± 1 °C and continuously shaken at 180 rpm for three days. The bacterial growth was monitored by measuring the optical density at 600 nm (OD600), given by turbiditycorrected values = ODbacterium − ODno bacterium, I = (Ctur − Ttur)/Ctur × 100%. The Ctur represented the corrected turbidity value of bacterial growth of untreated NB (blank control), and Ttur represented the corrected turbidity value of bacterial growth of tested compound-treated NB. The I represented the inhibition rate of tested compound against the bacterium.Secondly, antibacterial activities of target compounds 7a–7p were assessed against the pathogen Xoo under five different concentrations (namely 200, 100, 50, 25 and 12.5 μg mL−1) to obtain their EC50 values, which were statistically determined by probit analysis with the probit package of the SPSS 17.0 software.
Antifungal bioassay
Mycelial growth rate method26 was utilized to assess antifungal activities of target compounds against three phytopathogenic fungi (namely P. infestans, G. zeae and V. dahliae). The tested compounds were prepared at 50 μg mL−1 in DMSO solution. The above solution was added into sterilized Petri dishes, which contained about 10 mL molten potato dextrose agar (PDA). Subsequently, a 4 mm-diameter of mycelial plug was cut from the fungal colony and placed at the center of PDA plate at 28 ± 1 °C for 4 days. Antifungal assays were conducted in triplicate for each compound. Additionally, pure DMSO and commercially available fungicide (Hymexazol) were utilized as negative and positive control, respectively.The inhibition rate (I) of the tested compound was determined based on the following formula:
| I = (C − T)/(C − 0.4) × 100% |
Conclusion
In conclusion, a class of novel 1,2,4-triazole derivatives containing both quinazolinylpiperidinyl moiety and N-(substituted phenyl)acetamide group were designed and synthesized as agricultural bactericides. The bioassay results showed that some of the target compounds possessed good to excellent antibacterial activities against phytopathogenic bacterium Xoo. For example, compounds 7e, 7g, 7i, 7l and 7n had EC50 values of 34.5–47.5 μg mL−1, which was significantly better than commercial bactericide Bismerthiazol (85.6 μg mL−1). The above studies proved that 1,2,4-triazole derivatives containing quinazolinylpiperidinyl heterocycle and acetamide unit were promising candidates for the development of new and more efficient agricultural bactericides against the pathogen Xoo, deserving further investigation in the future.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21362003) and the Agricultural Research Projects of Guizhou Province (No. 20093010).References
- F. P. Carvalho, Environ. Sci. Policy, 2006, 9, 685–692 CrossRef.
- D. O. Niňo-Liu, P. C. Ronald and A. J. Bogdanove, Mol. Plant Pathol., 2006, 7, 303–324 CrossRef PubMed.
- N. Huang, E. R. Angeles, J. Domingo, G. Magpantay, S. Singh, G. Zhang, N. Kumaravadivel, J. Bennett and G. S. Khush, Theor. Appl. Genet., 1997, 95, 313–320 CrossRef CAS.
- M. Salanoubat, S. Genin, F. Artiguenave, J. Gouzy, S. Mangenot, M. Arlat, A. Billault, P. Brottier, J. C. Camus, L. Cattolico, M. Chandler, N. Choisne, C. Claudel-Renard, S. Cunnac, N. Demange, C. Gaspin, M. Lavie, A. Moisan, C. Robert, W. Saurin, T. Schiex, P. Siguier, P. Thébault, M. Whalen, P. Wincker, M. Levy, J. Weissenbach and C. A. Boucher, Nature, 2002, 415, 497–502 CrossRef CAS PubMed.
- P. Wang, M. Gao, L. Zhou, Z. Wu, D. Hu, J. Hu and S. Yang, Bioorg. Med. Chem. Lett., 2016, 26, 1136–1139 CrossRef CAS PubMed.
- C. Li, J. C. Liu, Y. R. Li, C. Gou, M. L. Zhang, H. Y. Liu, X. Z. Li, C. J. Zheng and H. R. Piao, Bioorg. Med. Chem. Lett., 2015, 25, 3052–3056 CrossRef CAS PubMed.
- A. M. Vijesh, A. M. Isloor, P. Shetty, S. Sundershan and H. K. Fun, Eur. J. Med. Chem., 2013, 62, 410–415 CrossRef CAS PubMed.
- Y. Wang, G. L. V. Damu, J. S. Lv, R. X. Geng, D. C. Yang and C. H. Zhou, Bioorg. Med. Chem. Lett., 2012, 22, 5363–5366 CrossRef CAS PubMed.
- K. Juvale, J. Gallus and M. Wiese, Bioorg. Med. Chem., 2013, 21, 7858–7873 CrossRef CAS PubMed.
- A. S. El-Azab and K. E. H. ElTahir, Bioorg. Med. Chem. Lett., 2012, 22, 327–333 CrossRef CAS PubMed.
- S. Kumar, N. Shakya, S. Gupta, J. Sarkar and D. P. Sahu, Bioorg. Med. Chem. Lett., 2009, 19, 2542–2545 CrossRef CAS PubMed.
- A. M. Alafeefy, A. A. Kadi, O. A. Al-Deeb, K. E. H. El-Tahir and N. A. Al-jaber, Eur. J. Med. Chem., 2010, 45, 4947–4952 CrossRef CAS PubMed.
- Y. J. Zhu, X. F. Guo, Z. J. Fan, L. Chen, L. Y. Ma, H. X. Wang, Y. Wei, X. M. Xu, J. P. Lin and V. A. Bakulev, RSC Adv., 2016, 6, 112704–112711 RSC.
- A. Guan, C. Liu, X. Yang and M. Dekeyser, Chem. Rev., 2014, 114, 7079–7107 CrossRef CAS PubMed.
- P. P. Kung, M. D. Casper, K. L. Cook, L. Wilson-Lingardo, L. M. Risen, T. A. Vickers, R. Ranken, L. B. Blyn, J. R. Wyatt, P. D. Cook and D. J. Ecker, J. Med. Chem., 1999, 42, 4705–4713 CrossRef CAS PubMed.
- B. R. Yan, X. Y. Lv, H. Du, M. N. Gao, J. Huang and X. P. Bao, Chem. Pap., 2016, 70, 983–993 CAS.
- D. Pan, H. Du, X. Lv and X. Bao, Chin. J. Org. Chem., 2016, 36, 818–825 CrossRef CAS.
- B. Yan, X. Lv, H. Du and X. Bao, Chin. J. Org. Chem., 2016, 36, 207–212 CrossRef CAS.
- J. Liu, Y. Liu, J. Jian and X. Bao, Chin. J. Org. Chem., 2013, 33, 370–374 CrossRef CAS.
- X. Bao, J. Liu, F. Zhang, L. Zou and J. Jian, Chin. J. Synth. Chem., 2013, 21, 200–203 Search PubMed.
- M. M. Endicott, E. Wick, M. L. Mercury and M. L. Sherrill, J. Am. Chem. Soc., 1946, 68, 1299–1301 CrossRef CAS PubMed.
- A. J. Bridges, H. Zhou, D. R. Cody, G. W. Rewcastle, A. McMichael, H. D. H. Showalter, D. W. Fry, A. J. Kraker and W. A. Denny, J. Med. Chem., 1996, 39, 267–276 CrossRef CAS PubMed.
- W. M. Xu, F. F. Han, M. He, D. Y. Hu, J. He, S. Yang and B. A. Song, J. Agric. Food Chem., 2012, 60, 1036–1041 CrossRef CAS PubMed.
- X. Wang, P. Li, Z. Li, J. Yin, M. He, W. Xue, Z. Chen and B. Song, J. Agric. Food Chem., 2013, 61, 9575–9582 CAS.
- X. Wang, J. Yin, L. Shi, G. Zhang and B. Song, Eur. J. Med. Chem., 2014, 77, 65–74 CrossRef CAS PubMed.
- C. J. Chen, B. A. Song, S. Yang, G. F. Xu, P. S. Bhadury, L. H. Jin, D. Y. Hu, Q. Z. Li, F. Liu, W. Xue, P. Lu and Z. Chen, Bioorg. Med. Chem., 2007, 15, 3981–3989 CrossRef CAS PubMed.
- J. M. Hung, H. J. Arabshahi, E. Leung, J. Reynisson and D. Barker, Eur. J. Med. Chem., 2014, 86, 420–437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data (including melting point, 1H & 13C NMR, HRMS and IR) of target compounds 7a–7p and spectra (1H & 13C NMR, HRMS) of intermediates 2–5 as well as target compounds. CCDC 1479247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04819j |
| This journal is © The Royal Society of Chemistry 2017 |